DOI:
10.1039/C1OB06497E
(Communication)
Org. Biomol. Chem., 2012,
10, 36-39
Expeditious diastereoselective construction of a thiochroman skeleton via a cinchona alkaloid-derived catalyst†
Received
1st September 2011
, Accepted 22nd September 2011
First published on 22nd September 2011
Abstract
An example of diastereoselective and enantioselective synthesis of thiochroman derivatives through a sulfa-Michael–Michael cascade sequence is disclosed. This is a significant complement of the quinine-thiourea catalyzed sulfa-Michael–Michael cascade reaction. The densely functionalized target thiochromans were obtained in high diastereoselectivities, and with high to excellent enantioselectivities.
Introduction
Asymmetric organocatalysis is nowadays recognized as a fundamental synthetic strategy for efficient assembly of chiral molecules.1 The rapid growth of organocatalysis over the last decade has been warranted by many factors such as efficiency, cost-effectiveness, low environmental impact, and operational simplicity. All of these features have generated a remarkable scientific competition, which has guided asymmetric organocatalysis towards excellent levels of development and opened new synthetic opportunities that were considered inaccessible a few years ago. The extraordinary pace of innovation and progress has been mainly dictated by the discovery of generic catalytic modes that have enabled previously unknown transformations. Consequently, venerable chemical transformations that lead to the preparation of useful chiral building blocks have been generally chosen as benchmarks for developing novel, more effective catalysts and asymmetric transformations. Within this context, the organocatalytic cascade reaction has recently emerged as a powerful tool to facilitate a rapid increase in molecular complexity and diversity from simple and readily available starting materials by reducing the steps of manual operation as well as the generation of waste.2 Of the catalytic strategies developed for enantioselective cascade transformation, bifunctional organocatalysts bearing both hydrogen-bond donor and Brønsted base have generated a focus of attention in recent years.3
The chemistry of sulfur based six-membered rings, thiopyrans, has, to date, been less extensively studied than that of the analogous pyrans.4 Although not particularly common throughout nature, this class of sulfur-containing heterocycles is of synthetic and biological interest. 3,4-Dihydro-2H-1-benzothiopyrans, more commonly known as thiochromans, exhibit anti-inflammatory, anti-pyretic, anti-depressant, and analgesic activities.4 For example, compound A, CH4986399 (Fig. 1), exhibited high clinical efficacy in tamoxifen and fulvestrant resistant ER-positive breast cancer patients.5 Compound B was discovered as a 5-HT1A receptor agonist for use in depression, schizophrenia and Parkinson's disease.6 Therefore, these complex polycyclic frameworks have become targets of interest in the synthetic organic community. Despite these advances, organocatalytic asymmetric methods for the construction of thiochromans remain largely unexplored.
 |
| | Fig. 1 Examples of biologically active thiochroman derivatives. | |
Wang et al. reported an example of the synthesis of densely functionalized thiochromans via cascade thio-Michael–Michael reactions of trans-3-(2-mercaptophenyl)-2-propenoic acid ethyl esters with nitroalkenes (Fig. 2, A).8 Although it is a highly enantioselective approach, the diastereomeric synthesis of thiochroman derivatives continues to be a substantial challenge. In this context, we document here a different asymmetric catalytic strategy for the diastereoselective synthesis of thiochromans through a new catalyst and a sulfa-Michael–Michael cascade sequence (Fig. 2, B). In particular, the one-pot cascade process affords enantio-enriched thiochromans with the creation of three new stereogenic centers in remarkably high efficiency and stereoselectivity. Undoubtedly, this method is a significant complement to previous synthetic contributions.
 |
| | Fig. 2 Overview of the H-bonding mediated sulfa-Michael–Michael cascade reactions. | |
Results and discussion
To probe the feasibility of the proposed cascade reaction, trans-3-(2-mercaptophenyl)-2-propenoic acid ethyl ester 2a was treated with trans-β-nitrostyrene 1a in the presence of catalyst I (Fig. 3) in CH2Cl2 at room temperature. In contrast to quinine thiourea catalyst (Fig. 2, A), there is a chiral center inversion on C9 of I. Catalyst I is also a bifunctional catalyst, which bears a hydroxyl group and a tertiary amine. It is surprising that 3a with a novel diastereomeric structure was finally produced (Table 1, entry 1, 85%). Meanwhile, 3a was obtained in 68% ee and 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 d.r.. We envisioned that chiral inversion of C6 is the main element for new diastereomer formation. We conclude that the dihedral angle between two functional groups would be a decisive factor. It is associated with the size of quinoline at the C9 position which can directly affect the rotation between C9 and C8. To validate our hypothesis, a series of cinchona alkaloid derivatives were synthesized and applied to this reaction. Accordingly, some bulky groups, such as OMe, OEt, OiPr, OcHex, and OTr, were introduced to the C6′ position of catalyst I to finally generate catalysts II–VI (Fig. 3). The investigation based on catalysts II–VI revealed that OiPr is the most appropriate size. In this case, catalyst IV promoted the reaction smoothly to furnish the desired diastereomer 3a in 78% yield and with good selectivity (16:1 d.r.; 82% ee) (Table 1, entry 4). More hindered catalysts (V and VI) and less hindered catalysts (II and III) gave relatively low enantioselectivities (73–76% ee) (Table 1, entries 2–3 and 5–6). Further investigation on quinoline structure was also examined. Catalyst VII (Fig. 3, cupreine) has a one more –OH group at C6′. Unfortunately, this acidic –OH group, as a potential H-bonding donor, did not improve the stereo-selective performance and afforded a non-desired diastereomer (entry 7). If the –OH group (C9 position) was protected, reactions were sluggish and only simple Michael adducts were generated (entries 8 and 9). In addition, catalyst X (quinidine) was also examined, but no obvious improvement was observed (entry 10, 82%, 20:1 d.r., −76% ee). To understand the role of the –OH group in catalyzing the reaction, we performed density functional theory (DFT) calculations under the generalized gradient approximation (GGA). It was found that the reaction between 1a and 2a is modestly exothermic with a thermochemical energy of −7.4 kcal mol−1. In the absence of the catalyst IV, the reactants approach each other, forming a transition state that allows the H2a atom of 2a to migrate to C2a of 2a assisted by the –NO2group of 1a, leading to the completion of the cycloaddition 3a. The calculated activation barrier of 29.1 kcal mol−1 is considerably high, suggesting the reaction is kinetically difficult. In the presence of the catalyst IV, however, we identified a transition state participated by 1a, 2a, and IV. Here, H2a transfer from the H–S bond to C2a of 2a is facilitated via H exchange mediated by the –OH group of the catalyst IV as indicated in the optimized structure shown in Fig. 4. Consequently, the activation barrier is substantially reduced to 15.9 kcal mol−1. Upon completion of the reaction, the conformation of the catalyst IV is recovered with the H atom of the –OH group pointing to the NIV atom to take advantage of the H-bonding. To verify our hypothesis, we did the following reactions. 1) catalyst IX (–OH group at C9 was protected by –OBn, Fig. 3) was synthesized and applied to this reaction. However, the experimental result showed that the reaction only generated the Michael adduct and no desired product was finally achieved (Table 1, entry 9). 2) Catalyst VIII (–OH group at C9 was protected by –OBn, but another –OH group was introduced to C6 position, Fig. 3) was also synthesized and applied to this reaction. Surprisingly, the experimental result showed that the reaction is sluggish and no desired product was detected. Based on the above experimental results, we predict that the position of this –OH is very critical for catalytic activation. 3) In addition, we synthesized an epi-quinine thiourea catalyst XI (a pseudo-enantiomer of quinine-thiourea catalyst XII,9Fig. 3). The catalyst XI has a bulky and more acidic thiourea group at the C9 position. Interestingly, this thiourea catalyst XI caused a sluggish reaction and gave no desired product (Table 1, entry 12). In summary, we found that the –OH group at C9 was a unique factor and played an irreplaceable catalytic function.
:1 d.r.. We envisioned that chiral inversion of C6 is the main element for new diastereomer formation. We conclude that the dihedral angle between two functional groups would be a decisive factor. It is associated with the size of quinoline at the C9 position which can directly affect the rotation between C9 and C8. To validate our hypothesis, a series of cinchona alkaloid derivatives were synthesized and applied to this reaction. Accordingly, some bulky groups, such as OMe, OEt, OiPr, OcHex, and OTr, were introduced to the C6′ position of catalyst I to finally generate catalysts II–VI (Fig. 3). The investigation based on catalysts II–VI revealed that OiPr is the most appropriate size. In this case, catalyst IV promoted the reaction smoothly to furnish the desired diastereomer 3a in 78% yield and with good selectivity (16:1 d.r.; 82% ee) (Table 1, entry 4). More hindered catalysts (V and VI) and less hindered catalysts (II and III) gave relatively low enantioselectivities (73–76% ee) (Table 1, entries 2–3 and 5–6). Further investigation on quinoline structure was also examined. Catalyst VII (Fig. 3, cupreine) has a one more –OH group at C6′. Unfortunately, this acidic –OH group, as a potential H-bonding donor, did not improve the stereo-selective performance and afforded a non-desired diastereomer (entry 7). If the –OH group (C9 position) was protected, reactions were sluggish and only simple Michael adducts were generated (entries 8 and 9). In addition, catalyst X (quinidine) was also examined, but no obvious improvement was observed (entry 10, 82%, 20:1 d.r., −76% ee). To understand the role of the –OH group in catalyzing the reaction, we performed density functional theory (DFT) calculations under the generalized gradient approximation (GGA). It was found that the reaction between 1a and 2a is modestly exothermic with a thermochemical energy of −7.4 kcal mol−1. In the absence of the catalyst IV, the reactants approach each other, forming a transition state that allows the H2a atom of 2a to migrate to C2a of 2a assisted by the –NO2group of 1a, leading to the completion of the cycloaddition 3a. The calculated activation barrier of 29.1 kcal mol−1 is considerably high, suggesting the reaction is kinetically difficult. In the presence of the catalyst IV, however, we identified a transition state participated by 1a, 2a, and IV. Here, H2a transfer from the H–S bond to C2a of 2a is facilitated via H exchange mediated by the –OH group of the catalyst IV as indicated in the optimized structure shown in Fig. 4. Consequently, the activation barrier is substantially reduced to 15.9 kcal mol−1. Upon completion of the reaction, the conformation of the catalyst IV is recovered with the H atom of the –OH group pointing to the NIV atom to take advantage of the H-bonding. To verify our hypothesis, we did the following reactions. 1) catalyst IX (–OH group at C9 was protected by –OBn, Fig. 3) was synthesized and applied to this reaction. However, the experimental result showed that the reaction only generated the Michael adduct and no desired product was finally achieved (Table 1, entry 9). 2) Catalyst VIII (–OH group at C9 was protected by –OBn, but another –OH group was introduced to C6 position, Fig. 3) was also synthesized and applied to this reaction. Surprisingly, the experimental result showed that the reaction is sluggish and no desired product was detected. Based on the above experimental results, we predict that the position of this –OH is very critical for catalytic activation. 3) In addition, we synthesized an epi-quinine thiourea catalyst XI (a pseudo-enantiomer of quinine-thiourea catalyst XII,9Fig. 3). The catalyst XI has a bulky and more acidic thiourea group at the C9 position. Interestingly, this thiourea catalyst XI caused a sluggish reaction and gave no desired product (Table 1, entry 12). In summary, we found that the –OH group at C9 was a unique factor and played an irreplaceable catalytic function.
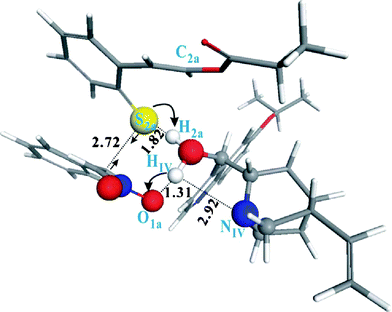 |
| | Fig. 4 The fully optimized transition state structure for the cascade reaction of 1a and 2a catalyzed by IV. | |
With regard to high stereo-control, further optimization was performed by examining other parameters, such as solvent, temperature and reaction concentration (Table 2). In contrast, CH2Cl2 was revealed as the best media. For the purpose of high enantioselectivity, we evaluated the reaction temperature (Table 2, entries 9–13). A higher stereo-control was achieved when the temperature was locked at 0 °C (Table 2, entry 12, 25:1 d.r., 90% ee). A lower temperature (−30 °C) slowed down the reaction but without any obvious improvement in stereo-control (Table 2, entry 13, 79%, 25:1 d.r., 89% ee). If the concentration decreased from 0.2 M to 0.1 M or 0.05 M, improved results were not observed (Table 2, entries 10 and 11, 83% ee and 82% ee, respectively).
Table 2 Substrate scope
Having established the optimal conditions, we then explored the generality of this cascade process. Remarkably, this enantio- and diastereo-selective cascade process served as a reliable synthetic protocol for the preparation of densely functionalized thiochromans (Table 2). Significantly, three new stereocenters were efficiently created in good to high enantioselectivities, and excellent diastereoselectivities in a one-pot process. It was found that an extensive range of nitroalkenes can efficiently participate in the process (Table 2, 3a–o), irrespective of the electronic nature and the substitution pattern of the aromatic system. For example, the reaction takes place with aromatic systems that possess electron-neutral (Table 2, 3a), -withdrawing (Table 2, 3b–f), or -donating (Table 2, 3g–l) groups at the o, m, or p positions of the nitroalkenes without substantial loss in yields (86–94%) or stereoselectivities (80–90% ee, 15:1 to 99:1 d.r.). However, if there is an –OH group at the p position of phenyl ring (Table 2, 3p), a moderate result (66% ee and 2:1 d.r.) was obtained. The loss of stereoselectivity may be caused by the additional hydrogen bonding between the –OH group and catalyst IV, which might affect the relative orientation of nucleophile and electrophile in this reaction. Moreover, a heteroaromatic nitroolefin (Table 2, 3n) can also be tolerated. A good ee value and diastereoselectivity were obtained in the presence of a less reactive aliphatic nitroalkene (Table 2, 3o, 84% ee and 15:1 d.r.). The absolute configuration of the products was determined by single-crystal X-ray diffraction analysis (Fig. 5, a sulfonate ester 3q) (for structure, see Supporting Information†).7
 |
| | Fig. 5 X-ray crystal structure of 3q. | |
Conclusions
In conclusion, we have developed a novel quinine derivative as an effective catalyst to promote a one-pot cascade reaction for the enantioselective and diastereoselective synthesis of thiochroman derivatives. This is an important complement of the previous method of quinine-thiourea catalyzed thio-Michael–Michael cascade reaction. The densely functionalized target thiochromans were obtained in good to high yields, moderate to high diastereoselectivities, and good to high enantioselectivities. Further applications of this mode, with regard to other transformations, are under investigation in our laboratory.
Acknowledgements
We gratefully acknowledge the National University of Singapore and Singapore Ministry of Education for financial support of this work (Academic Research Grants: R143000408133, R143000408733, R143000443112 and R143000480112).
Notes and references
- For selected books of organocatalysis, see:
(a)
A. Berkessel and H. Groger, Asymmetric Organocatalysis, Wiley, Weinheim 2005 Search PubMed;
(b)
P. I. Dalko, Enantioselective Organocatalysis, Wiley, Weinheim, 2007 Search PubMed;
(c)
M. T. Reetz, B. List, S. Jaroch, H. Weinmann, Organocatalysis, Springer, 2007 Search PubMed;
(d)
B. List, Asymmetric Organocatalysis, Springer, 2009 Search PubMed.
- For recent selected examples of organocascade reactions, see:
(a) Y. Yamamoto, N. Momiyama and H. Yamamoto, J. Am. Chem. Soc., 2004, 126, 5962 CrossRef CAS;
(b) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS;
(c) J. W. Yang, M. T. H. Fonseca and B. List, J. Am. Chem. Soc., 2005, 127, 15036 CrossRef CAS;
(d) M. Marigo, T. Schulte, J. Franzen and K. A. Jørgensen, J. Am. Chem. Soc., 2005, 127, 15710 CrossRef CAS;
(e) D. Enders, M. R. M. Huttl, C. Grondal and G. Raabe, Nature, 2006, 441, 861 CrossRef CAS;
(f) W. Wang, H. Li, J. Wang and L. Zu, J. Am. Chem. Soc., 2006, 128, 10354 CrossRef CAS;
(g) R. Rios, H. Sunden, I. Ibrahem, G.-L. Zhao, L. Eriksson and A. Córdova, Tetrahedron Lett., 2006, 47, 8547 CrossRef CAS;
(h) H. Sunden, I. Ibrahem, G.-L. Zhao, L. Eriksson and A. Córdova, Chem.–Eur. J., 2007, 12, 574 CrossRef;
(i) H. Li, L. Zu, H. Xie, J. Wang, W. Jiang and W. Wang, Angew. Chem., Int. Ed., 2007, 46, 3732 CrossRef;
(j) Y. Hayashi, T. Okano, S. Aratake and D. Hazelard, Angew. Chem., Int. Ed., 2007, 46, 4922 CrossRef CAS;
(k) J. L. Vicario, S. Reboredo, D. BadNa and L. Carrillo, Angew. Chem., 2007, 119, 5260 CrossRef;
(l) M. Lu, D. Zhu, Y. Lu, Y. Hou, B. Tan and G. Zhong, Angew. Chem., Int. Ed., 2008, 47, 10187 CrossRef CAS;
(m) C. Cassini, X. Tian, E. C. Escudero-Adan and P. Melchiorre, Chem. Commun., 2011, 47, 233 RSC;
(n) G. Bencivenni, L.-Y. Wu, A. Mazzanti, B. Giannichi, F. Pesciaioli, M.-P. Song, G. Bartol and P. Melchiorre, Angew. Chem., Int. Ed., 2009, 48, 7200 CrossRef CAS;
(o) X. Companyó, A. Zea, A.-N. R. Alba, A. Mazzanti, A. Moyano and R. Rios, Chem. Commun., 2010, 46, 6953 RSC;
(p) Q. Wei and L.-Z. Gong, Org. Lett., 2010, 12, 1008 CrossRef CAS;
(q) K. Juhl and K. A. Jørgensen, Angew. Chem., Int. Ed., 2003, 42, 1498 CrossRef CAS;
(r) K. Jiang, Z.-J. Jia, S. Chen, L. Wu and Y.-C. Chen, Chem.–Eur. J., 2010, 16, 2852 CrossRef CAS;
(s) Y. J. Gao, Q. Ren, L. Wang and J. Wang, Chem.–Eur. J., 2010, 16, 13068 CrossRef CAS;
(t) Y. J. Gao, Q. Ren, H. Wu, M. G. Li and J. Wang, Chem. Commun., 2010, 46, 9232 RSC;
(u) Q. Ren, Y. J. Gao and J. Wang, Chem.–Eur. J., 2010, 16, 13594 CrossRef CAS;
(v) Y. J. Gao, Q. Ren, S. W. Ang and J. Wang, Org. Biomol. Chem., 2011, 9, 3691 RSC;
(w) Y. J. Gao, Q. Ren and W. Y. Siau, Chem. Commun., 2011, 47, 5819 RSC;
(x) W. Y. Siau and J. Wang, Catal. Sci. Technol., 2011 10.1039/C1CY00271F;
(y) Y. Hayashi, T. Itoh and Ha. Ishikawa, Angew. Chem., Int. Ed., 2011, 50, 3920 CrossRef CAS;
(z) S.-K. Xiang, B. Zhang, L. Zhang, Y. Cui and N. Jiao, Chem. Commun., 2011, 47, 5007 RSC;
(a
a) S.-K. Xiang, B. Zhang, L. Zhang, Y. Cui and N. Jiao, Chem. Commun., 2011, 47, 8097 RSC.
- For selected reviews on hydrogen bonding and bifunctional organocatalysis, see:
(a) P. R. Schreiner, Chem. Soc. Rev., 2003, 32, 289 RSC;
(b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713 CrossRef CAS;
(c) X. H. Yu and W. Wang, Chem.–Asian J., 2008, 3, 516 CrossRef CAS;
(d) H. Miyabe and Y. Takemoto, Bull. Chem. Soc. Jpn., 2008, 81, 785 CrossRef CAS;
(e) S. J. Connon, Chem. Commun., 2008, 2499 RSC.
- For reviews regarding the synthesis and biological application of thiochromans, see:
(a) S. W. Schneller, Adv. Heterocycl. Chem., 1975, 18, 59 CrossRef CAS;
(b)
A. H. Ingall, Comprehensive Heterocyclic Chemistry, Vol 3, A. S. Boulton and A. Mckillop, ed.; Oxford: Pergamon Press, 1984; p855 Search PubMed;
(c)
A. H. Ingall, in Comprehensve Heterocyclic Chemistry II, A. S. Boulton and A. McKkillop, ed.; Pergamon Press: Oxford, 1996; Vol. 5, p501 Search PubMed;
(d) S. W. Schneller, Adv. Heterocycl. Chem., 1975, 18, 59 CrossRef CAS;
(e) A. R. Katrizky and A. J. Bonlton, Adv. Heterocycl. Chem., 1975, 18, 76 Search PubMed;
(f) K. Fukamizu, Y. Miyake and Y. Nishibayashi, J. Am. Chem. Soc., 2008, 130, 10498 CrossRef CAS;
(g) R. Dodda, J. J. Goldman, T. Mandal, C.-G. Zhao and G. A. Broker, E. R. T., Adv. Synth. Catal., 2008, 350, 537 CrossRef CAS;
(h) R. Dodda, T. Mandal and C.-G. Zhao, Tetrahedron Lett., 2008, 49, 1899 CrossRef CAS.
-
(a) Y. Kanbe, M.-H. Kim, M. Nishimoto, Y. Ohtake, T. Tsunenari, K. Taniguchi, I. Ohizumi, S.-I. Kaiho, Y. Nabuchi, S. Kawata, K. Morikawa, J.-C. Jo, H.-A. Kwon, H.-S. Lim and H.-Y. Kim, Bioorg. Med. Chem. Lett., 2006, 16, 4090 CrossRef CAS;
(b) T. Yoneya, K. Taniguchi, R. Nakamura, T. Tsunenari, I. Ohizumi, Y. Kanbe, K. Morikawa, S.-I. Kaiho and H. Yamada-Okabe, Anticancer Res., 2010, 30, 873 CAS.
- L. Zimmer, G. Fournet, B. Joseph, G. Guillaumet and D. L. Bars, Nucl. Med. Biol., 2003, 30, 541 CrossRef CAS.
- CCDC 790476 (compound 3g) contains the supplementary crystallographic data for this paper..
- J. Wang, H. Xie, H. Li, L. Zu and W. Wang, Angew. Chem., Int. Ed., 2008, 47, 4177 CrossRef CAS.
-
Quinine-thiourea catalyst XII (Fig. 3) was reported in Ref. 8 for the enantioselective synthesis of another major diastereomer. Currently, the reaction mechanism and transitional state for making different diastereomers of this reaction are still unknown. Based on experimental results and computational study, we predicted that the size, strength, and orientation of hydrogen bonding acceptor (–OH and thiourea) could be the major factors for stereoselective control.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here. 
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :
:
