
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The generation of compartmentalized nanoparticles containing siRNA and cisplatin using a multi-needle electrohydrodynamic strategy†
Maria F.
Pina
a,
Wai
Lau
b,
Kathrin
Scherer
c,
Maryam
Parhizkar
b,
Mohan
Edirisinghe
b and
Duncan
Craig
 *a
*a
aUniversity College London School of Pharmacy, 29-39 Brunswick Square, London WC1N 1AX, UK. E-mail: duncan.craig@ucl.ac.uk
bDepartment of Mechanical Engineering, University College London, Torrington Place, London WC1E 7JE, UK
cMRC Laboratory for Molecular Cell Biology, University College London, Gower Street, London WC1E 6BT, UK
First published on 13th April 2017
Abstract
This study outlines a novel manufacturing technique for the generation of compartmentalized trilayered nanoparticles loaded with an anti-cancer agent and siRNA as a platform for the combination treatment of cancers. More specifically, we describe the use of a multi-needle electrohydrodynamic approach to produce nanoparticles with high size specificity and scalable output, while allowing suitable environments for each therapeutic agent. The inner polylactic-glycolic-acid (PLGA) layer was loaded with cisplatin while the middle chitosan layer was loaded with siRNA. The corresponding polymeric solutions were characterized for their viscosity, surface tension and conductivity, while particle size was determined using dynamic light scattering. The internal structure was studied using transmission electron microscopy (TEM) and Structured Illumination Microscopy (SIM). The inclusion of cisplatin was studied using electron dispersive spectroscopy (EDS). We were able to generate nanoparticles of approximate size 130 nm with three distinct layers containing an outer protective PLGA layer, a middle layer of siRNA and an inner layer of cisplatin. These particles have the potential not only for uptake into tumors via the enhanced permeability and retention (EPR) effect but also the sequential release of the siRNA and chemotherapeutic agent, thereby providing a means of overcoming challenges of targeting and tumor drug resistance.
Introduction
The potential of using combination regimes of different chemotherapeutic agents for the treatment of cancers1 is now a widely used approach for increasing the efficacy of treatment for multi-resistant tumors. More recently, strategies involving the combination of anticancer drugs and small interference RNA (siRNA) have emerged; the use of siRNA allows the silencing of specific drug-resistance genes, thereby reducing tumor resistance to the chemotherapeutic agent.2–6 Although the separate administration of the two actives has proved to be advantageous, studies have recently indicated that if the agents can be delivered simultaneously using a single carrier system (co-delivery) this may enhance the therapeutic efficiency of these formulations.7–9 For example, Sun and co-authors were able to demonstrate that the co-delivery of Plk1 siRNA and paclitaxel had a greater inhibitory effect on cell proliferation than the two actives administered separately.10 In this case, the authors used amphiphilic polymers that self-assembled into micellar nanoparticles creating a hydrophobic core and a cationic shell that allowed the simultaneous loading of paclitaxel and siRNA, respectively. Similarly, Saad et al. developed a multifunctional cationic liposomal delivery system containing doxorubicin in the aqueous core and two types of siRNA (for silencing of pump and non-pump drug resistance) electrostatically bound to the surface.11 A further general approach to deliver drug-siRNA combinations involves the use of polymeric carriers such as poly(lactic-co-glycolic) acid (PLGA), polycaprolactone (PCL), chitosan, polyethylene glycol (PEG), polyethylenimine (PEI) and poly(2-aminoethyl ethylene phosphate) (PPEEA). In general, polymeric nanoparticles are prepared via double emulsion solvent evaporation techniques and have either a hydrophobic (PLGA or PCL) or a hydrophilic (PEG) matrix that allows the encapsulation of small molecules and cationic polymers (PEI, PPEEA or chitosan) that enhance the loading efficiency of the anionic siRNA.2,12 Inorganic delivery vectors such as mesoporous silica nanoparticles4,5 and gold nanorods13 have also been use to delivery such combination therapies.Despite the interest in developing co-administered systems, combination systems present a number of unresolved challenges. True compartmentalization in distinct, stable environments is difficult to achieve, particularly when there are significant differences in the physicochemical properties of the two actives such as molecular weight, hydrophobicity and metabolic stability. This is especially important for siRNA systems due to the environmental requirements associated with maintaining stability. More specifically, siRNA is sensitive to nuclease degradation and its hydrophilic and anionic nature makes it unable to cross cellular membranes. Therefore, siRNA-mediated silencing strongly depends on the design of powerful delivery systems to (i) protect siRNA molecules from enzymatic degradation, (ii) be internalized by the cells and (iii) release of siRNA in the cell cytoplasm where the RNAi machinery is located. A widely used approach for siRNA delivery is the complexation with cationic polymers namely chitosan which has the advantage of being biodegradable and biocompatible, characteristics that are highly desired in a drug delivery system.14,15 In addition, incompatibility between siRNA and anticancer drugs can result in both chemical and physical instability.16 Finally, one needs to consider whether sequential release is required. In the case of siRNA-chemotherapeutic systems, after the cellular uptake of the nanocarrier, the siRNA cargo should ideally be released prior to the drug, allowing enough time for the siRNA sensitizing effect to take place before the drug is released.17,18 For example, Yadav et al. observed maximum therapeutic effect when paclitaxel was administered 24 h after the cells being treated with P-glycoprotein (Pgp) siRNA as this permitted an efficient Pgp knock-down.19 Similar results were shown by Beh and co-workers where maximum cytotoxicity of paclitaxel was observed 48 h after Bcl-2 siRNA treatment.20 To date, there is no carrier system available that is able to fulfil the requirements of complete individualization of the actives, environments conducive to siRNA stability and rational sequential release.
The size range of the nanoparticles used in cancer therapy should also be considered. Most of the tumors are known to have an underdeveloped and thus show leaky vasculature, leading to the process known as enhanced permeability and retention (EPR), whereby macromolecules or nanoparticles can escape the circulation into the tumor and accumulate in the tumor site due to inefficient drainage by the lymphatic system.21 The EPR effect can be observed in most human cancers with the exception of hypovascular tumors such as prostate or pancreatic cancers. Particles having a size between 100–200 nm have the highest potential for prolonged circulation since they are small enough to avoid uptake by the liver but sufficiently large to avoid renal filtration.22 For example, Unezaki et al. showed the extravasation of PEG-liposomes with a size of ∼130 nm in tumour tissues but not in healthy tissue.23
Here we report the use of an innovative multi-needle electrohydrodynamic (EHD) device that is able to prepare multi-layered nanoparticles that potentially fulfil the requirements of true compartmentalization with appropriate environments, sequential release and suitable size. In brief, the device allows controlled co-flow of up to four different solutions through a capillary nozzle under the influence of an electric field.24 The electric charge generated competes with the surface tension of the droplet, causing it to break up into smaller droplets (micro or nano) that undergo solvent evaporation during collection.25,26 The device operates at ambient temperature, does not require the use of surfactants (as often required for other nanoparticle preparation techniques) and can generate layered particles with a controlled size distribution at a rate of 109–1017 per minute per nozzle.27,28 The technique has shown potential in preparing nanoparticles with three and four layers24 hence it is entirely appropriate to explore the use of the method in preparing the sophisticated multi-layered nanoparticles required for siRNA-based combination therapy. A point of concern when using EHD to prepare nanoparticles containing biologically active compounds is their degradation under the applied electric field. However, the available literature indicates that a significant amount of work has been performed on electrospun nanofibers containing nucleic acids, including siRNA.29–31 These authors have shown that, despite the harsh processing conditions during electrospinning, the bioactivity of siRNA was retained and able to provide a silencing efficiency of 61–81%.32 Considering that the technical aspects of electrospinning are very similar to electrospraying it is reasonable to suggest that siRNA will remain active in the prepared nanoparticles, although further work will be required to establish this prior to in vivo studies.
In this work, we aim to prepare three-layered nanoparticles with an ideal size for drug delivery containing PLGA in the inner (drug-loaded) and outer layers and siRNA-loaded chitosan in the middle layer, as indicated schematically in Fig. 1. All the polymeric carriers are non-toxic, biodegradable and biocompatible. The inner layer of PLGA was loaded with the anti-cancer drug cisplatin as to achieve a controlled release formulation from the hydrophobic matrix of the polymer. Chitosan is a cationic polymer which forms polyelectrolyte complexes with negatively charged biomolecules such as siRNA, hence allowing protection of the nucleic acid.33,34 The outer layer of PLGA protects the two inner compartments and allows the particle to be taken up intact. To prevent opsonisation and particle agglomeration, PEGylation or equivalent would be desirable but this may be achieved reasonably easily once the technology is established and hence is not considered further here. Our intention is to develop a novel platform for the manufacture and characterization of complex combination nanocarrier systems using the multi-needle electrohydrodynamic system; a successful outcome to this study will result in a generalizable delivery approach that may be applied to a wide range of combination therapeutic approaches.
 | ||
| Fig. 1 Schematic representation of a compartmentalized three-layered particle for combination cancer therapy containing an anti-cancer drug (cisplatin, in this study) in the inner layer, a silencing agent (siRNA) in the middle layer and a coating protective layer. | ||
Experimental
Materials
Chitosan medium molecular weight (Molecular weight, Mw = 190![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–310000 g mol−1) and chitosan low molecular weight (Mw = 50000–190000 g mol−1), both with a degree of deacetylation (DD) of 75–85%, were purchased from Sigma Aldrich (Dorset, UK). Poly-(D,L-lactic-co-glycolic acid), PLGA, 50:50 Resomer® RG 503 H (Mw = 24000–38000 g mol−1) was purchased from Evonik (Darmstadt, Germany). Small interfering RNA (siRNA) universal negative control fluorescently labelled with Cyanine 5 (Cy5) was purchased from Sigma Aldrich (UK). Cisplatin was obtained from Enzo (Exeter, UK). 2′7′ difluorofluorescein (Oregon Green®) and ProLong Diamond® antifade mountant were obtained from Molecular Probes, Life Technologies (Oregon, USA). Amicon Ultra-4 centrifugal filter units with ultracel-3 membrane Mw cut-off of 50 kDa were obtained from Merck Millipore (Cork, Ireland). HyPure molecular biology grade was purchased from GE Healthcare Life Sciences (Utah, USA). Tween® 80, dimethyl carbonate (DMC), acetic acid, N,N-dimethylacetamide (DMAC) and ethanol absolute analytical grade were obtained from Sigma Aldrich (Dorset, UK).
000–310000 g mol−1) and chitosan low molecular weight (Mw = 50000–190000 g mol−1), both with a degree of deacetylation (DD) of 75–85%, were purchased from Sigma Aldrich (Dorset, UK). Poly-(D,L-lactic-co-glycolic acid), PLGA, 50:50 Resomer® RG 503 H (Mw = 24000–38000 g mol−1) was purchased from Evonik (Darmstadt, Germany). Small interfering RNA (siRNA) universal negative control fluorescently labelled with Cyanine 5 (Cy5) was purchased from Sigma Aldrich (UK). Cisplatin was obtained from Enzo (Exeter, UK). 2′7′ difluorofluorescein (Oregon Green®) and ProLong Diamond® antifade mountant were obtained from Molecular Probes, Life Technologies (Oregon, USA). Amicon Ultra-4 centrifugal filter units with ultracel-3 membrane Mw cut-off of 50 kDa were obtained from Merck Millipore (Cork, Ireland). HyPure molecular biology grade was purchased from GE Healthcare Life Sciences (Utah, USA). Tween® 80, dimethyl carbonate (DMC), acetic acid, N,N-dimethylacetamide (DMAC) and ethanol absolute analytical grade were obtained from Sigma Aldrich (Dorset, UK).
Solution preparation and characterization
PLGA and chitosan solutions of different compositions were prepared and electrosprayed so as to select those giving a better jet stability that allowed the preparation of three-layered nanoparticles. PLGA solutions were prepared in DMC at 3 and 6% (w/v); chitosan solutions were prepared at a concentration of 0.05% (w/v) in 0.05% (v/v) aqueous acetic acid solution. Two different additives, Tween® 80 (at a concentration of 2% v/v) and ethanol (at two different concentrations of 20 and 30% v/v) were added to the chitosan solution to reduce surface tension and electrical conductivity, respectively. The solutions showing the most promising properties were electrosprayed and the existence of a three-layered structure was evaluated. The same solutions were then used to prepare loaded siRNA particles. SiRNA fluorescently labelled with Cy5 was used to prepare siRNA-chitosan solutions at a ratio of 1:10. Solutions were prepared by adding 10 μL of a 100 μM siRNA to 490 μL of HyPure molecular biology grade water. This solution was then added to 500 μL of chitosan 0.05% w/v and gently mixed just before spraying. For fluorescence microscopy imaging, 1–2 mg of Oregon Green® was added to the PGLA solution and left stirring overnight. PLGA-cisplatin solutions were prepared at 0.1% w/v drug loading in 40:60 DMAC/DMC. All solutions were prepared at the ambient temperature. The viscosity, conductivity and surface tension properties of each solution were measured prior spraying after calibrating the equipment (see below) used.
Four-needle coaxial electrohydrodynamic (EHD)
A four-needle coaxial EHD device was used to prepare three-layered nanoparticles (schematic diagram of the device shown in Fig. 2). PLGA solution was used in the outer and inner needles while chitosan, and subsequently chitosan-siRNA solution, was introduced through the middle needle. Four needles with inner diameters of 0.3, 0.69, 1.37 and 2.4 μm were used. Flow rates and voltage were optimised for each solution and were as follow (from the inner to the outer needles): 20, 20, 50 μL min−1 for the polymeric particles; 15, 15, 35 μL min−1 for the particles loaded with siRNA; 15, 10, 35 μL min−1 for the cisplatin loaded particles and 15, 10, 50 μL min−1 for the siRNA/cisplatin loaded systems. DMC was used in the outermost needle at a rate varying from 100 and 50 μL min−1. The voltage applied was 20.4 ± 1.1 kV for the polymeric and siRNA-loaded particles, but significantly higher for the cisplatin and siRNA/cisplatin loaded systems which was around 28.5 ± 0.7 kV. Particles were collected in a petri dish filled with absolute ethanol at a distance of 150 mm for the polymeric and siRNA-loaded particles and 260 mm for cisplatin and siRNA/cisplatin loaded systems. A copper ring was placed in the extremity of the coaxial four-needle arrangement to help focus the jet.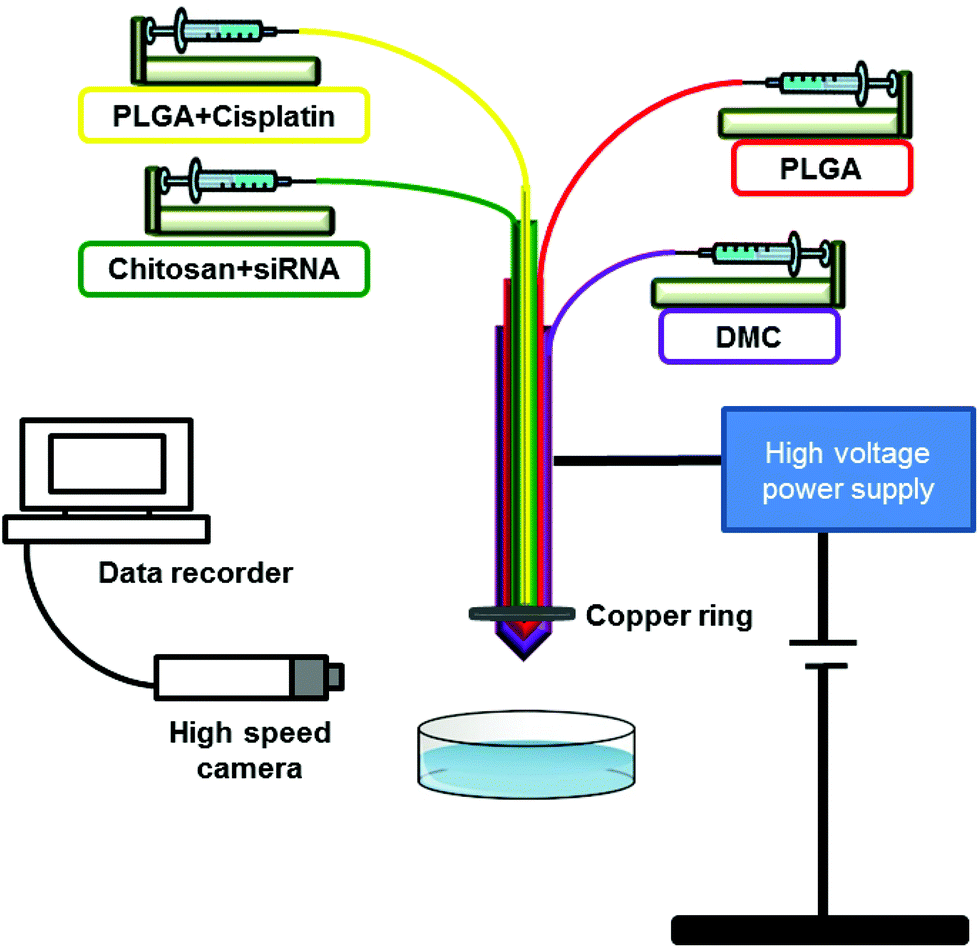 | ||
| Fig. 2 Schematic representation of the experimental four-needle coaxial electrohydrodynamic set-up used in this work. | ||
Dynamic light scattering (DLS)
Intensity mean hydrodynamic size and zeta potential of the nanoparticles were measured on a Malvern Zetasizer-NanoZS (Malvern Instruments, Malvern, UK) with a He–Ne laser (wavelength of 632.8 nm). The measurements were carried out at a scattering angle of 173 at 25 °C and ethanol was used as the dispersion medium.Encapsulation efficiency
The loading efficiency of siRNA and cisplatin was determined by measuring the free siRNA and cisplatin concentration in the recovered medium after particle centrifugation using a Sigma 3-16KL centrifuge (Sigma, Osterode am Harz, Germany) at 3000 rpm/10 min. Amicon Ultra-4 centrifugal filter units with ultracel-3 membrane Mw cut-off of 50 kDa were used. The siRNA concentration on the solution collected after centrifugation (non-encapsulated siRNA) was measured using NanoDrop® Lite Spectrophotometer (Thermo Scientific, Wilmington, USA). The solution recovered after centrifugation of polymeric nanoparticles (without siRNA and cisplatin) was used as blank. The cisplatin concentration was measured using UV spectroscopy at 300 nm. The siRNA encapsulation efficiency (%) was given by the difference between the total amount of siRNA added for the nanoparticles preparation and the siRNA collected in solution after centrifugation, to the total of siRNA added.15 The same formula was applied to determine the encapsulation efficiency of cisplatin.Scanning electron microscopy (SEM)
SEM images of the nanoparticles surface were acquired using a FEI Quanta 200F (FEI, Eindhoven, Netherlands). To improve electrical conductivity prior to examination, samples were coated with gold using a Quorum Q150 T sputter gold coater (Quorum Technologies, Lewes, UK).Transmission electron microscopy (TEM)
TEM images were collected using a Philips CM 120 Bio-Twin (FEI, Oregon, USA). A drop of the nanoparticles suspension (as collected during the EHD preparation process) was placed on a copper grid and imaged without any staining.Super-resolution imaging using structured illumination microscopy (SIM)
SiRNA loaded samples for SIM imaging and analysis were centrifuged using a Sigma 3-16KL centrifuge (Sigma, Osterode am Harz, Germany) at 3000 rpm for 10 min, washed with 1 mL of absolute ethanol and centrifuged again at 3000 rpm for 5 min. All steps were carried out at 5 °C. Ultra-4 centrifugal filter units with ultracel-3 membrane Mw cut-off of 50 kDa were used. A drop of the siRNA loaded samples collected in ethanol (100%) was put on a clean glass coverslip (#1.5, 0.17 mm thickness, Marienfield, Lauda-Königshofen, Germany), evenly spread out using the pipette tip and the excess ethanol was allowed to evaporate so that the nanoparticles stick to the glass. The coverslips were then mounted on a microscopy glass slide using ProLong Diamond®, as mounting medium. The mounted samples were allowed to cure at ambient temperature for a minimum of 24 h and sealed using liquid plaster (Germolene®, Bayer, UK) before SIM imaging.SIM images were acquired using a commercial Zeiss ELYRA PS.1 microscope equipped with a pco.edge sCMOS camera (2048 × 2048 pixels). A Zeiss 63× Plan-Apochromat DIC M27 oil objective lens (1.4 NA, 0.19 mm working distance) was used for all image acquisitions. Fluorescence was excited using a 488 nm (BP 495-550 + LP750 emission filter) and 642 nm (LP 655 emission filter) laser at 1.8 mW power, sequentially, and an exposure time of 200 ms was used. SIM gratings with a period of 28 μm and 34 μm were used with the 488 nm and 642 nm laser respectively. Sixteen-bit images (62.58 μm × 62.58 μm, 1942 × 1942 pixels) were acquired using 3 rotations and 5 phase shifts of the gratings in each slice of the Z-stack (2.63 μm total range, 110 nm steps).
All SIM images were processed using the Structured Illumination algorithm within the Zeiss acquisition software (ZEN Black). The 488 nm channel was processed selecting 3D and using the Automatic setting. The 642 nm channel was analysed in the 3D mode as well, but using the Manual settings and selecting Maximum Isotropy to remove some high frequency noise. Resolutions between ∼99–130 nm were obtained.
Following the Structured Illumination processing all images were corrected for daily chromatic shifts between the 488 and 642 nm channel by applying a channel alignment table obtained from processing a 4 μm z-stack of 200 nm TetraSpeck beads (Molecular Probes) using the Channel Alignment tool (affine alignment) in ZEN black.
Energy dispersive spectroscopy – scanning electron microscopy (EDS–SEM)
Samples were mounted directly on SEM holders, carbon coated and examined with a S-3400N microprobe (Hitachi, Illinois, USA). An Oxford Instruments X-sight Energy Dispersive Spectrometer (EDS) and INCA analytical software package were used to collect and process compositional data. The excitation voltage was set as 10 kV and the beam current at 50 nA for a working distance of 10 mm.Results and discussion
The preparation of multi-layered nanoparticles with a controlled size distribution and morphology using EHD primarily relies on having a stable cone-jet mode during the process. To achieve a stable cone-jet, the complex inter-dependence between the solution properties (concentration, viscosity, surface tension and electrical conductivity) and processing parameters (flow rate, voltage and collection distance) should be carefully considered and interconnected. Below we present both the solution optimization steps and processing parameters we adjusted to improve the stability of the jet during the spraying process of the different solutions.Solutions optimization and characterization for the preparation of the polymeric nanoparticles
As previously mentioned earlier, we aim to prepare three-layered nanoparticles containing PLGA-chitosan-PLGA. PLGA is a hydrophobic polymer and solutions were prepared in dimethyl carbonate (DMC) at concentrations of 3 and 6% w/v. On the other hand, chitosan is only soluble in slightly acidic aqueous conditions and therefore solutions were prepared in 0.05% v/v aqueous acetic acid. When using EHD it is important that the solutions properties are similar in terms of their surface tension and conductivity but should ideally be immiscible since this can facilitate the formation of individualized layers. In this case, PLGA and chitosan solutions have completely distinct properties which can lead to difficulties during spraying. Table 1 presents the characterization of the different chitosan solutions prepared with medium and low molecular weight and with different additives (ethanol and Tween® 80), aimed to achieve a desirable stable cone-jet mode. The properties of the PLGA solutions are also shown.000–190000 g mol−1) and medium (190000–310000 g mol−1) molecular weight and PLGA (24000–38000 g mol−1) at different concentrations were tested
| Solution | Chitosan Mw | Concentration [%] | Additives (v/v) | Properties | |||
|---|---|---|---|---|---|---|---|
| Viscosity [mPa. s] | Surface tension [mN m−1] | Conductivity [μS m−1] | |||||
| N/A – not applicable. | |||||||
| Chitosan | A | Medium | 0.05 | Ethanol (20%) | 12.5 ± 0.5 | 37.1± 0.3 | 73 |
| B | Medium | 0.05 | Ethanol (20%) + Tween 80 (2%) | 8.5 ± 0.4 | 31.7 ± 0.5 | 191 | |
| C | Low | 0.05 | Ethanol (30%) | 9.5 ± 0.3 | 36.5± 0.6 | 48.2 | |
| PLGA | D | N/A | 6 | N/A | 3.0 ± 0.1 | 30.4 ± 0.5 | 0 |
| E | N/A | 3 | N/A | 1.4 ± 0.3 | 29.4 ± 0.3 | 0 | |
We started by adding 20% ethanol to the medium Mw chitosan solution (0.05% w/v chitosan in 0.05% v/v acetic acid) to reduce both surface tension and conductivity (solution A). Attempts to electrospray this solution were not successful since a stable jet could not be achieved and fiber formation was observed due to the high viscosity of the solution. In the next step, Tween® 80 at 2% v/v was added (solution B). The addition of Tween® 80 reduced surface tension (compared to solution A) but it substantially increased the electrical conductivity of the solution. Higher conductivity can lead to disentanglement of the polymer network during electrospraying resulting in a very unstable jet.35 In order to find a good balance between these three properties, we reduced the viscosity of solution A by using low molecular weight chitosan at the same concentration (0.05% w/v) and used 30% v/v ethanol instead of Tween® 80 2% v/v which allowed the reduction of both surface tension (compared to solution A) and electrical conductivity (compared to solution B). Solution C was therefore selected for further experiments. PLGA solutions at a concentration of 6 and 3% w/v for the inner and outer layers were also tested, although the higher concentration (solution D) led to the formation of fibres, thus solution E (3% PLGA) was then used for the two layers in the particle preparation.
Processing parameters
Once the solution properties were optimised, other parameters such as flow rate, voltage applied, collection medium and distance of collection, also needed to be considered. The values for each parameter have been described in the methodology section. We noted that although the properties of the solutions have been carefully optimized, the stability of the jet still needed further improvement due to the large electrical conductivity differences between the two solutions. Therefore, as an attempt to isolate the solution with higher conductivity (chitosan solution), often referred to as the driving liquid/solution, we used pure DMC in the fourth needle of the coaxial EHD, which significantly enhanced the jet stability during processing. DMC is a green solvent with low hazard properties for human health and environment in comparison to other solvent alternatives that mainly evaporates during electrospraying without causing any disruption to the particle formation.36 As a result, the amount of DMC in the final particles is expected to be residual and have minimal impact to the human health. We also found that the use of a copper ring placed in the tip of the coaxial needle aided the stabilization of the electrospraying process (video 1 in ESI†). Another parameter to account for is the collection medium. When hydrophobic polymers are used, agglomeration can be an issue, as we observed when collecting the samples in distilled water where a thin film of particles formed on the surface of the medium. To overcome this issue, we used ethanol as the collection medium which allowed a well dispersed suspension to be collected.In comparison to other available chemical or physical methods used to prepare nanoparticulate systems, numerous advantages make EHD an attractive technique for drug delivery purposes. These include: (i) processing at room temperature; (ii) particles are dried during the process (no requirement for an additional drying step); (iii) control of the particle size and morphology by adjusting process parameters and solution characteristics. Furthermore, this manufacturing technique is low cost and durable, with a particle production rate of more than 109–1017 per minute per nozzle achievable which is a well-known limitation of other conventional methods, i.e., emulsion-solvent evaporation and lipid based techniques.
Characterization of the unloaded, mono- and dual-loaded nanoparticles
After optimizing the properties of the solutions and adjusting the processing parameters, nanoparticles containing chitosan solution C in the middle layer and PLGA solution E in the inner and outer layers were prepared and characterized in terms of their size, morphology and internal structure. The subsequent step involved loading of the middle chitosan layer with a negative control fluorescently labelled siRNA-Cy5, and the inner PLGA layer with cisplatin to show proof of concept that each active ingredient can be incorporated in the desired layer for further combination therapy applications.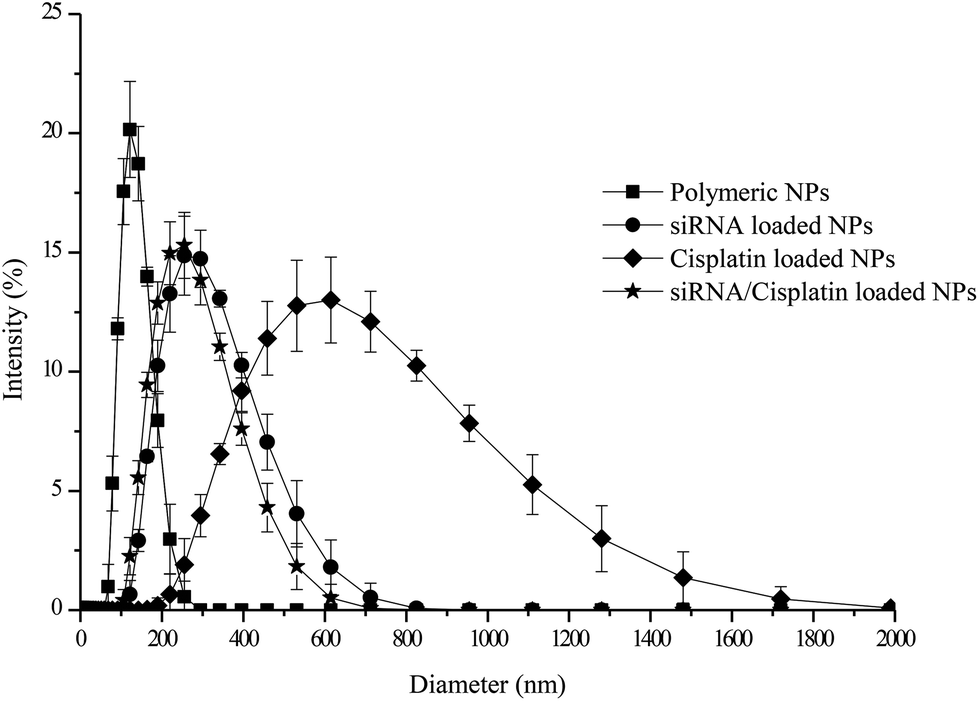 | ||
| Fig. 3 Size distribution profiles obtained by dynamic light scattering measurements for the polymeric, individually loaded (siRNA and cisplatin) and siRNA/cisplatin loaded nanoparticles. | ||
The encapsulation efficiency (EE) of siRNA and cisplatin into the particles was found to be 71.9 ± 5.5% and 83.8 ± 3.5%, respectively. Studies have reported a loading efficiency of siRNA in chitosan-PLGA nanoparticles to vary between 4.3% and 77.7% depending on the amount of chitosan added to prepare the particles, with the highest EE observed with a higher percentage of chitosan present.37 One of the main advantages when using EHD to prepare nano and microparticles is its high encapsulation efficiency when compared to other preparation techniques;38 this appears to be the case in the current study as well.
 | ||
| Fig. 4 Scanning electron microscopy images of polymeric (a, b), siRNA (c, d), cisplatin (e, f) and siRNA/cisplatin loaded particles (g, h). Micrographs were recorded at two different magnifications and scale bars are displayed in each image. | ||
Fig. 5 below shows the TEM images for the polymeric (a, b), mono-loaded siRNA (c, d) and cisplatin (e, f) systems and siRNA/cisplatin loaded particles (g, h). TEM is an essential characterization tool for directly imaging nanomaterials to obtain quantitative measures of particle size, size distribution, and morphology. Amplitude and phase variations in the transmitted beam provide imaging contrast that is a function of the sample thickness (the amount of material that the electron beam must pass through) and the sample material (heavier atoms scatter more electrons and therefore have a smaller electron mean free path than lighter atoms). Therefore, successful imaging of nanoparticles using TEM depends on the contrast of the sample relative to the background. In this study, all the materials were carbon-based with little contrast between them. However, it is still possible to identify a three-layered internal structure in all samples. PLGA is a more electron dense polymer and therefore has a dark appearance in TEM while chitosan appears as a lighter layer in the middle of the two PLGA layers. However, to confirm the existence of individualized layers a florescent-based microscopic technique was necessary as shown below.
 | ||
| Fig. 5 Transmission electron microscopy images of polymeric (a, b), mono-loaded siRNA (c, d) and cisplatin (e, f) systems and siRNA/cisplatin loaded particles (g, h). Micrographs were collected without any staining at two different magnifications. Scale bars are shown in each image. | ||
Amongst all the prepared systems, cisplatin-loaded NPs (e, f) again showed a much less uniform spherical shape compared to the rest of the samples. SiRNA-loaded NPs (single and dual systems) showed a homogeneous spherical shape. Due to its polycationic nature, chitosan may form polyelectrolyte complexes with negatively charged molecules, such as may be the case with siRNA.15
 | ||
| Fig. 6 Super resolution-structured illumination microscopy images (Z-stack) of the particles labelled with PLGA-Oregon Green® (green) and siRNA-Cy5 (orange). The Z-step size was 110 nm. Scale bar for all images is 0.2 μm long. | ||
With SIM it is possible to record images at different focal planes enabling the entire volume of the sample to be visualized. Z-stacks are generated by incrementally stepping through the sample using a piezoelectric motor. Furthermore, Z-stacks represent a very straightforward means to analyze the entirety of thin fluorescent samples. This allows 3D reconstruction of images which provides helpful visual information at a higher resolution level.41 A 3D video of a single particle is supplied in ESI (video 2†).
SIM imaging allowed us to resolve the outer and the middle layer of the nanoparticles. The resolution of this technique is not sufficient though to resolve the core of the nanoparticles as it is limited to approximately twice the diffraction resolution limit of a fluorescence microscope. Other super-resolution techniques such as Stimulated Emission Depletion (STED)42 microscopy or Stochastic Optical Reconstruction Microscopy (STORM)43 are known to provide a higher resolution than SIM and might therefore be useful tools to study the composition of these multilayer nanoparticles, and particularly their core, in future studies.
 | ||
| Fig. 7 Detection of cisplatin in the three layered nanoparticles by EDS-SEM. (a) SEM image showing the characterized particle (location highlighted by red outline); (b) EDS elemental spectrum composition showing the presence of platinum when the selected particle was analyzed. Three-layered polymeric particles (non-loaded) were used as a negative control (c) and (d). | ||
Conclusions
The study has outlined the manufacture and characterization of trilayered nanoparticles containing distinct environments for the combined administration of low molecular weight chemotherapeutic agents and resistance-reducing nucleic acids, using cisplatin and fluorescently labelled siRNA as models for the two therapeutic agents. The parameters associated with successful particle manufacture (viscosity, electrical conductivity and surface tension) have been explored, while size and surface charge measurements have allowed the effect on agent incorporation on particle characteristics to be ascertained. A range of imaging techniques has been employed that have allowed the confirmation of the presence of the agents in their respective layers as well as allowing the architecture of the trilayered systems to be studied. It was found that the particles were of a size commensurate with EPR uptake and the loading efficiency was high. Overall, the study has shown that it is possible to produce truly compartmentalized nanoparticles which are capable of encapsulating agents with very different structures and environmental requirements in a manner that lends itself to larger scale production. This in turn suggests that multi-needle EHD represents a potentially highly useful and practical platform for combination therapy approaches.Acknowledgements
Maria F. Pina thanks Maplethorpe Postdoctoral Fellowships of the University of London. Maryam Parhizkar is supported by an EPSRC grant (EP/L026287/1).References
- V. T. Devita, R. C. Young and G. P. Canellos, Cancer, 1975, 35, 98–110 CrossRef PubMed.
- Y. B. Patil, S. K. Swaminathan, T. Sadhukha, L. Ma and J. Panyam, Biomaterials, 2010, 31, 358–365 CrossRef CAS PubMed.
- L. Zhang, Z. Lu, Q. Zhao, J. Huang, H. Shen and Z. Zhang, Small, 2011, 7, 460–464 CrossRef CAS PubMed.
- H. Meng, W. X. Mai, H. Zhang, M. Xue, T. Xia, S. Lin, X. Wang, Y. Zhao, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2013, 7, 994–1005 CrossRef CAS PubMed.
- H. Meng, M. Liong, T. Xia, Z. Li, Z. Ji, J. I. Zink and A. E. Nel, ACS Nano, 2010, 4, 4539–4550 CrossRef CAS PubMed.
- A. M. Chen, M. Zhang, D. Wei, D. Stueber, O. Taratula, T. Minko and H. He, Small, 2009, 5, 2673–2677 CrossRef CAS PubMed.
- B. Spankuch, E. Kurunci-Csacsko, M. Kaufmann and K. Strebhardt, Oncogene, 2007, 26, 5793–5807 CrossRef CAS PubMed.
- B. Spänkuch, S. Heim, E. Kurunci-Csacsko, C. Lindenau, J. Yuan, M. Kaufmann and K. Strebhardt, Cancer Res., 2006, 66, 5836–5846 CrossRef PubMed.
- J. A. MacDiarmid, N. B. Amaro-Mugridge, J. Madrid-Weiss, I. Sedliarou, S. Wetzel, K. Kochar, V. N. Brahmbhatt, L. Phillips, S. T. Pattison, C. Petti, B. Stillman, R. M. Graham and H. Brahmbhatt, Nat. Biotech., 2009, 27, 643–651 CrossRef CAS PubMed.
- T.-M. Sun, J.-Z. Du, Y.-D. Yao, C.-Q. Mao, S. Dou, S.-Y. Huang, P.-Z. Zhang, K. W. Leong, E.-W. Song and J. Wang, ACS Nano, 2011, 5, 1483–1494 CrossRef CAS PubMed.
- M. Saad, O. B. Garbuzenko and T. Minko, Nanomedicine, 2008, 3, 761–776 CrossRef CAS PubMed.
- Y. Patil and J. Panyam, Int. J. Pharm., 2009, 367, 195–203 CrossRef CAS PubMed.
- Y. Xiao, R. Jaskula-Sztul, A. Javadi, W. Xu, J. Eide, A. Dammalapati, M. Kunnimalaiyaan, H. Chen and S. Gong, Nanoscale, 2012, 4, 7185–7193 RSC.
- X. Liu, K. A. Howard, M. Dong, M. Ø. Andersen, U. L. Rahbek, M. G. Johnsen, O. C. Hansen, F. Besenbacher and J. Kjems, Biomaterials, 2007, 28, 1280–1288 CrossRef CAS PubMed.
- H. Katas and H. O. Alpar, J. Controlled Release, 2006, 115, 216–225 CrossRef CAS PubMed.
- J. Jiang, S.-j. Yang, J.-c. Wang, L.-j. Yang, Z.-z. Xu, T. Yang, X.-y. Liu and Q. Zhang, Eur. J. Pharm. Biopharm., 2010, 76, 170–178 CrossRef CAS PubMed.
- V. Tsouris, M. K. Joo, S. H. Kim, I. C. Kwon and Y.-Y. Won, Biotechnol. Adv., 2014, 32, 1037–1050 CrossRef CAS PubMed.
- J. Li, Y. Wang, Y. Zhu and D. Oupický, J. Controlled Release, 2013, 172, 589–600 CrossRef CAS PubMed.
- S. Yadav, L. van Vlerken, S. Little and M. Amiji, Cancer Chemother. Pharmacol., 2009, 63, 711–722 CrossRef CAS PubMed.
- C. W. Beh, W. Y. Seow, Y. Wang, Y. Zhang, Z. Y. Ong, P. L. R. Ee and Y.-Y. Yang, Biomacromolecules, 2009, 10, 41–48 CrossRef CAS PubMed.
- F. Danhier, O. Feron and V. Préat, J. Controlled Release, 2010, 148, 135–146 CrossRef CAS PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615–627 CrossRef CAS PubMed.
- S. Unezaki, K. Maruyama, J. I. Hosoda, I. Nagae, Y. Koyanagi, M. Nakata, O. Ishida, M. Iwatsuru and S. Tsuchiya, Int. J. Pharm., 1996, 144, 11–17 CrossRef CAS.
- S. Labbaf, H. Ghanbar, E. Stride and M. Edirisinghe, Macromol. Rapid Commun., 2014, 35, 618–623 CrossRef CAS PubMed.
- N. Bock, T. R. Dargaville and M. A. Woodruff, Prog. Polym. Sci., 2012, 37, 1510–1551 CrossRef CAS.
- I. G. Loscertales, A. Barrero, I. Guerrero, R. Cortijo, M. Marquez and A. M. Gañán-Calvo, Science, 2002, 295, 1695–1698 CrossRef CAS PubMed.
- M. W. Chang, E. Stride and M. Edirisinghe, J. R. Soc., Interface, 2010, 7, S451–S460 CrossRef CAS PubMed.
- S. Labbaf, S. Deb, G. Cama, E. Stride and M. Edirisinghe, J. Colloid Interface Sci., 2013, 409, 245–254 CrossRef CAS PubMed.
- P.-o. Rujitanaroj, Y.-C. Wang, J. Wang and S. Y. Chew, Biomaterials, 2011, 32, 5915–5923 CrossRef CAS PubMed.
- M. Chen, S. Gao, M. Dong, J. Song, C. Yang, K. A. Howard, J. Kjems and F. Besenbacher, ACS Nano, 2012, 6, 4835–4844 CrossRef CAS PubMed.
- S. Lee, G. Jin and J.-H. Jang, J. Biol. Eng., 2014, 8, 30 CrossRef PubMed.
- H. Cao, X. Jiang, C. Chai and S. Y. Chew, J. Controlled Release, 2010, 144, 203–212 CrossRef CAS PubMed.
- H. Ragelle, G. Vandermeulen and V. Préat, J. Controlled Release, 2013, 172, 207–218 CrossRef CAS PubMed.
- S. Mao, W. Sun and T. Kissel, Adv. Drug Delivery Rev., 2010, 62, 12–27 CrossRef CAS PubMed.
- K. Sung and C. S. Lee, J. Appl. Phys., 2004, 96, 3956–3961 CrossRef CAS.
- P. Tundo and M. Selva, Acc. Chem. Res., 2002, 35, 706–716 CrossRef CAS PubMed.
- X. Yuan, B. Shah, N. Kotadia, J. Li, H. Gu and Z. Wu, Pharm. Res., 2010, 27, 1285–1295 CrossRef CAS PubMed.
- L. Zhang, J. Huang, T. Si and R. X. Xu, Expert Rev. Med. Devices, 2012, 9, 595–612 CrossRef CAS PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- M. G. L. Gustafsson, L. Shao, P. M. Carlton, C. J. R. Wang, I. N. Golubovskaya, W. Z. Cande, D. A. Agard and J. W. Sedat, Biophys. J., 2008, 94, 4957–4970 CrossRef CAS PubMed.
- A. Ostrowski, D. Nordmeyer, A. Boreham, C. Holzhausen, L. Mundhenk, C. Graf, M. C. Meinke, A. Vogt, S. Hadam, J. Lademann, E. Rühl, U. Alexiev and A. D. Gruber, Beilstein J. Nanotechnol., 2015, 6, 263–280 CrossRef PubMed.
- S. W. Hell and J. Wichmann, Opt. Lett., 1994, 19, 780–782 CrossRef CAS PubMed.
- E. Betzig, G. H. Patterson, R. Sougrat, O. W. Lindwasser, S. Olenych, J. S. Bonifacino, M. W. Davidson, J. Lippincott-Schwartz and H. F. Hess, Science, 2006, 313, 1642–1645 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr01002h |
| This journal is © The Royal Society of Chemistry 2017 |