 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled immobilisation of active enzymes on the cowpea mosaic virus capsid†
Alaa A. A.
Aljabali‡
a,
J. Elaine
Barclay
a,
Nicole F.
Steinmetz
b,
George P.
Lomonossoff
a and
David J.
Evans§
*a
aDepartment of Biological Chemistry, John Innes Centre, Norwich Research Park, Norwich, NR4 7UH, UK. E-mail: dave.evans@jic.ac.uk; Tel: +44-(0)1603-450018
bDepartments of Biomedical Engineering, Radiology and Materials Science and Engineering, Case Western Reserve University, 10900 Euclid Ave., Cleveland, OH 44106, USA
First published on 26th July 2012
Abstract
Immobilisation of horseradish peroxidase (HRP) and glucose oxidase (GOX) via covalent attachment of modified enzyme carbohydrate to the exterior of the cowpea mosaic virus (CPMV) capsid gave high retention of enzymatic activity. The number of enzymes bound per virus was determined to be about eleven for HRP and 2–3 for GOX. This illustrates that relatively large biomacromolecules can be readily coupled to the virus surface using simple conjugation strategies. Virus-biomacromolecule hybrids have great potential for uses in catalysis, diagnostic assays or biosensors.
1. Introduction
Enzyme–nanoparticle conjugates that exploit the catalytic activity of the bound enzyme have been reported for various applications,1,2 especially for biosensing.3–5 For example, the enzymes horseradish peroxidase (HRP) and glucose oxidase (GOX) have been sequentially co-immobilised in polyelectrolyte multilayers on silica nanoparticles to facilitate the overall enzymatic reaction: the GOX layer generates hydrogen peroxide in the presence of β-D-glucose that diffuses toward the HRP layer to be reduced to water in the presence of o-dianisidine (HRP substrate).6 Enzymes have been attached also to carbon nanotubes, using both covalent and noncovalent approaches,7,8 gold nanoparticles9 and magnetic nanoparticles.10 Enzymes possess unique catalytic properties compared to chemical catalysts, including biocompatibility, higher efficiency and specific selectivity toward substrates. The ability to immobilise enzymes on a support is of importance as it enables easy separation of reaction products from the enzyme, facilitates the construction of porous multilayers and the development of micro-fluidic and lab-on-a-chip type devices.11,12 The advantages of immobilisation of enzymes onto virus nanoparticles include multiple attachment sites, defined geometry and the potential for the construction of permeable, multilayer catalytic arrays.Here we describe how cowpea mosaic virus (CPMV) can be used to construct enzyme-modified functional virus particles. The well-studied enzymes HRP and GOX were employed to prepare EnzymeCPMV conjugates by their covalent coupling to the external surface of wild-type CPMV. CPMV is an icosahedral virus with dimensions in the nanoscale (approximate diameter 28 nm), the structure of which is known to near-atomic resolution.13 The virions comprise 60 copies each of two different types of coat protein, the small (S) subunit with one domain and the large (L) subunit with two domains; the three domains together form the asymmetric unit. The multiple copies of the asymmetric unit provide regularly spaced attachment units, facilitating the coupling and presentation of different moieties on the exterior surface.14–17 HRP is a haemoprotein enzyme of molecular weight 40 kDa that catalyses the reaction of hydrogen peroxide with certain organic, electron-donating substrates to yield highly coloured products. GOX is a flavoenzyme that catalyses the oxidation of β-D-glucose to D-glucono-1,5-lactone and hydrogen peroxide, using molecular oxygen as the electron acceptor. The enzyme consists of two identical subunits, of 80 kDa, bound together by disulfide linkages.18 It is widely used in diagnostic assays for the determination of glucose concentration in physiological fluids. Although a range of coupling strategies of enzyme/protein-to-virus could be employed, we chose to follow, as proof-of-principle, the modified-carbohydrate approach. Both HRP and GOX contain a significant amount of carbohydrate. Thus, mild oxidation of the enzyme sugar residues with sodium metaperiodate generates reactive aldehyde groups that are coupled to linking groups that can subsequently be conjugated to the external surface of CPMV.
2. Experimental
2.1 Materials
All general reagents, HRP (type VI-A, Product code P6782), GOX (from Aspergillus niger), D-glucose, adipic acid dihydrazide (ADH) and 5 M sodium cyanoborohydride in 1 M NaOH were purchased from Sigma-Aldrich, UK, and used without further purification. 1-Ethyl-3-(3-dimethlyaminopropyl)carbodiimide hydrochloride (EDC) was purchased from Novabiochem and N-hydroxysuccinimide (NHS) from Fluka. Amplex Red Glucose/Glucose Oxidase assay kit was obtained from Invitrogen.2.2 Instruments and techniques
UV-visible spectra were recorded on a Perkin Elmer Lambda 25 spectrophotometer using UVWINLab software. Transmission electron microscopy (TEM) images were obtained on a FEI Tecnai 20 TEM, FEI UK Ltd, Cambridge, using carbon-coated copper EM grids (400 mesh, Agar Scientific). Dynamic light scattering (DLS) was measured on a DynaPro Titan, Wyatt Technology Corporation (laser wavelength 830 nm, scattering angle 20°). Zeta potential was measured on a Malvern Instruments Zetasizer-Nano ZS: 1 mL of 0.5 mg mL−1 EnzymeCPMV particles dispersed in 10 mM sodium phosphate buffer pH 7.2. Zeta cells were equilibrated at 21 °C for two minutes before recording three measurements each of ten runs. Data was collected with automatic attenuation selected and analysed using the Smoluchowski module. Samples for mass spectrometry were mixed in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio with the matrix (2,4,6-trihydroxyacetophenone, ∼2 to 3 mg mL−1 in 70% aqueous acetonitrile) and 1 μL was spotted onto a ground steel MALDI target plate (Bruker Daltonics, Bremen, Germany). After drying, the sample was analysed on a Bruker Ultraflex TOF/TOF using a laser power of approx. 25% and spectra were summed from ca. 30 × 15 laser shots. The instrument was calibrated using Bruker standards (ca. 200 laser shots) and data were processed in FlexAnalysis (Bruker).
:1 ratio with the matrix (2,4,6-trihydroxyacetophenone, ∼2 to 3 mg mL−1 in 70% aqueous acetonitrile) and 1 μL was spotted onto a ground steel MALDI target plate (Bruker Daltonics, Bremen, Germany). After drying, the sample was analysed on a Bruker Ultraflex TOF/TOF using a laser power of approx. 25% and spectra were summed from ca. 30 × 15 laser shots. The instrument was calibrated using Bruker standards (ca. 200 laser shots) and data were processed in FlexAnalysis (Bruker).
Agarose gel electrophoresis of 5–10 μg of CPMV particles suspended in 10 mM sodium phosphate buffer pH 7.0 with 2 μL of loading dye (Coomassie staining solution or MBI Fermentas dye) were analysed on 1.2% (w/v) agarose gel in an electric field of 60 V for 1–2 hours. For ethidium bromide staining (of nucleic acid), 0.1 μg mL−1 (4–5 μL) in 1× TBE buffer was added to the gel. Particles were visualised on a UV transilluminator at 302 nm using Gene Genius Bio Imaging System with software Gene Snap (Syngene). For coat protein staining, gels were treated with Coomassie staining solution (50% (v/v) methanol; 10% (v/v) acetic acid; 0.25% (w/v) Coomassie Brilliant Blue G-250) for 1 hour followed by destaining solution (50% (v/v) methanol; 20% (v/v) acetic acid in Milli-Q water) overnight. Sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) was performed on 5–10 μg of virus in 10 mM sodium phosphate buffer pH 7.0 mixed with 3 μL of 4× RunBlue LDS Sample Buffer (Expedeon), samples were heated for 5–10 minutes at 100 °C in a thermoblock in order to denature the protein. The samples were analysed on 12% TEO-CI SDS RunBlue precast gels (Expedeon) under non-reducing conditions at 180 V for 30–70 minutes using 600–700 mL of 20:1 dilution of 20× RunBlue SDS Running Buffer (Expedeon). The bands when stained with 20 mL of InstantBlue (Expedeon) were visible after a few minutes. Gels were washed with Milli-Q water prior to imaging. Gel images were recorded using camera or scanner.
2.3 Synthetic methods
:10. Dye-labelled enzyme was dialysed against 10 mM sodium phosphate buffer for 2 days using 5 kDa cut-off membrane, with a buffer change approximately every 12 hours, to remove all unbound dye.
GOX–ADH CPMV: The Amplex Red Glucose/Glucose Oxidase assay kit was used according to the manufacturer's instruction. The reaction was protected from light at all times.
3. Results and discussion
3.1 Enzyme oxidation (activation) with periodate
Sodium metaperiodate is used to cleave bonds between adjacent carbons that carry hydroxyl groups in the polysaccharide residues of glycoproteins, forming reactive aldehyde residues suitable for coupling with amines or hydrazide-containing molecules. HRP contains 21% by mass carbohydrate according to the PDB data bank (1H58). The reaction cleaves the carbon–carbon bond between two adjacent (vicinal) secondary hydroxyls to create two terminal aldehyde groups.24 Herein, the approach was used to oxidize hydroxyl-containing carbohydrate within the HRP and GOX to create reactive aldehyde groups, which were then conjugated to ADH and subsequently coupled to activated carboxylate ester groups on the CPMV external surface (Fig. 1). Periodate modification of the enzyme carbohydrate has no effect on reactivity.24 On the external surface of CPMV there are 420 carboxylates of which 180 have been shown to be readily addressable by small molecules.22,23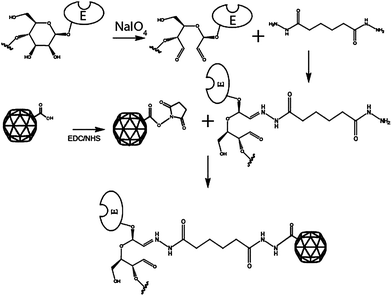 | ||
| Fig. 1 Enzymes such as HRP and GOX, that are glycoproteins, are oxidised with sodium metaperiodate to produce reactive aldehydes. The aldehyde reacts with ADH to form a hydrozone-linked enzyme-ADH conjugate. The conjugate reacts with ester-activated carboxylate on the surface of CPMV to form an amide bond. | ||
3.2 EnzymeCPMV
The best method for coupling was found to be that of ADH-modified enzyme carbohydrate to the CPMV surface. EnzymeCPMV was obtained in a yield of 60–70% based on the initial virus concentration as determined by UV-vis spectrophotometry. Particle integrity was confirmed by agarose gel electrophoresis and TEM. Various alternative coupling strategies for enzyme CPMV conjugation were investigated, not all requiring carbohydrate modification, (including the Schiff base method,24 streptavidin–biotin,25 benzaldehyde26 and N-succinimidyl-S-acetylthiopropionate27 (see ESI†)) but these were not as efficient. The number of enzymes bound per virus was calculated by fluorescent labelling of the enzyme as summarised in Table S1(ESI†).Intact virions of EnzymeCPMV were characterised by native gel electrophoresis according to their migration towards the anode. HRPCPMV derivatives from the various coupling strategies exhibited different mobility patterns (Fig. S1, ESI†). This behaviour arises because of a combination of size and charge effects perturbing the mobility through the gel. However, all migrate to a different extent than wild-type (negative control) and positive controls, confirming that the CPMV has been modified and that it remains intact after bioconjugation. To confirm that the enzyme was covalently attached to the virus capsid, the activated enzyme was incubated with CPMV under the same conditions but in the absence of any coupling reagents. After the same purification protocol, agarose gel electrophoresis showed no mobility shift in comparision to unmodified CPMV particles and, in addition, no enzymatic activity was detected. This was consistent with no physically adsorbed enzymes being present on the virus capsid.
Successful bioconjugation of the enzyme to the virus capsid was also demonstrated by comparison of the SDS-PAGE of denatured modified and unmodified CPMV particles (Fig. 2). In addition to unmodified S and L proteins, SDS-PAGE of HRP–ADHCPMV revealed two extra bands for the modified particles at approximately 62 and 80 kDa (Fig. 2, lane 3), corresponding to conjugation of one enzyme (∼40 kDa) to each of the S (24 kDa) and L (41 kDa) proteins, respectively. Attempts to confirm by mass spectrometry coupling of HRP to each of the virus subunits were unsuccessful, probably due to the trypsin digestion being unable to cleave proteins in the presence of the enzyme haem group.28 SDS-PAGE analysis of HRP–ADHCPMV also revealed the presence of unmodified S and L coat protein indicating that not all of the virus protein subunits are coupled to the enzyme. This is probably because the dimensions of the bound enzyme molecules are sufficient to occlude some of the potential binding sites. Control experiments which involved incubating the enzyme, both carbohydrate unmodified and activated, with the virus in the absence of the linking reagent revealed no extra bands on SDS-PAGE gels, eliminating the possibility of non-covalent association between the virus and the enzyme. To estimate the number of HRP molecules per virus, HRP–ADH enzyme was fluorescently labelled with DyLight488–NHS–ester by coupling the dye to the two accessible lysine-amine residues on the enzyme surface.21 After purification, the labelled enzyme was conjugated to the virus capsid. Dye-labelled particles (DyLight488)–HRP–ADHCPMV were purified on sucrose gradients, dialysed for 2–3 days and the recovered particles were found to be fluorescent as shown in Fig. 2B, further confirmation that the enzyme is coupled to the virus capsid. The number of dyes per HRP was determined spectrophotometrically to be 2 ± 0.45 and the number of HRP on each virus was estimated to be 14 ± 3, (Fig. S2 and S3, ESI†). Zeta potential measurements for suspensions of the HRP–ADHCPMV particles in buffer, −36.7 ± 2.1 mV, are considerably more negative than that for CPMV of ca. −12 ± 1.5 mV, consistent with HRP being bound to the external virus surface. NHS–esterCPMV, as a positive control, has a zeta potential value of −19.5 ± 1.2 mV.
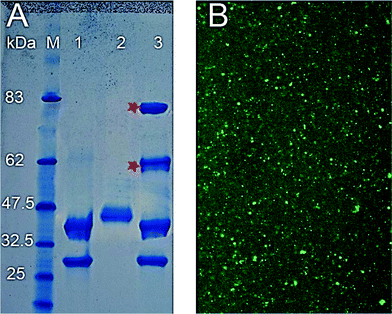 | ||
| Fig. 2 (A) Coomassie stained 4–12% SDS-PAGE of 1. CPMV; 2. HRP–ADH; 3. HRP–ADHCPMV. Asterisk denotes bands examined by mass spectrometry, M contains prestained protein markers with their size in kDa indicated. (B) (DyLight488)–HRP–ADHCPMV dried on coverslip and viewed by Leica SP2 inverted confocal microscope. | ||
A uranyl acetate stained transmission electron micrograph (TEM) image for HRP–ADHCPMV (Fig. 3) revealed intact, monodisperse, CPMV particles of ca. 38.5 ± 0.5 nm in diameter, which is in agreement with the hydrodynamic diameter of ∼40 ± 0.3 nm measured by DLS, (Fig. 3). This confirms that the particles are modified; the increase in particle size of ∼10 nm is again consistent with enzyme being bound to the exterior surface. The TEM images show that any difference in shape of the HRP–ADHCPMV particles compared to wild-type virus as a consequence of enzyme binding is very minor. Furthermore, TEM was used to detect the remaining, unreacted aldehyde groups on the HRP–ADHCPMV by staining with AgNO3 (Fig. 4). It has been reported that the silver staining technique (also known as Tollens' reagent) can be used for selective staining of exposed polysaccharides in the cell wall. The specificity of this staining is dependent on the production of aldehyde groups after periodate oxidation of the polysaccharide.29 It is proposed that the remaining unreacted aldehyde groups, present due to incomplete reaction of the aldehyde groups with ADH, on the HRP–ADHCPMV react with silver nitrate, causing a deposition of silver at specific sites. The pattern observed in the TEM images as shown in Fig. 4A does not resemble any patterns obtained from the negative and positive controls as shown in Fig. 4B–D. The black dots (metallic silver) in Fig. 4A are localised on the spherical particles of the same size of modified virus particles. This confirms that the oxidised enzyme is coupled to the virus capsid. Metallic silver was not seen on any other part of the grids.
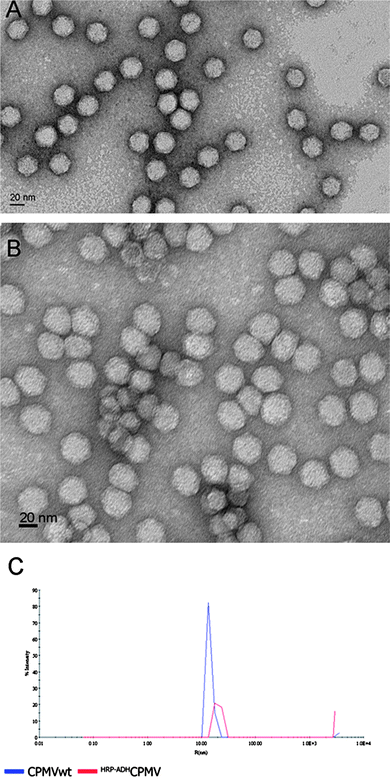 | ||
| Fig. 3 TEM image of CPMV (A) and of HRP–ADHCPMV particles (B), both stained with 2% uranyl acetate and the corresponding DLS measurement (C). Both methods show a particle diameter of ∼40 nm for HRP–ADHCPMV particles consistent with successful coupling of HRP to the virus capsid. | ||
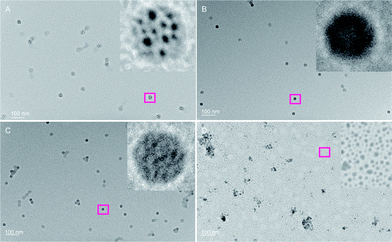 | ||
| Fig. 4 TEM images of HRP–ADHCPMV stained with 1% AgNO3 solution. Particles were washed thoroughly with Milli-Q water after deposition on TEM grids and after the addition of 5–10 μL of 1% AgNO3 solution. (A) HRP–ADHCPMV; inset confirms localization of metallic silver on the virus capsid; (B) CPMV; (C) NHS–esterCPMV; (D) HRP–ADH. | ||
The enzymatic activity of the immobilised HRP was investigated in the presence of H2O2 and TMB as substrates. Incubation with HRP–ADHCPMV particles resulted in the production of various degrees of blue colour depending on the enzyme concentration; in contrast to CPMV particles that did not produce a colour change. The CPMV-immobilised HRP retained its catalytic activity. When enzymatic activity was measured on particles after storage at 4 °C for 7 days, approximately the same activity was observed indicating that no structural changes to the HRP–ADHCPMV had occurred during this period.
To quantify the number of active enzyme molecules coupled to the virus, HRP–ADHCPMV calibration curves were obtained using different standard solutions of free HRP, prepared by serial dilution, suspended in the same buffer as the virus. The HRP concentration was determined independently as described above. Each enzyme concentration was prepared and used on the same day, 36 activity measurements for each enzyme concentration were recorded and the absorbance was averaged. The HRP concentration was plotted versus the absorbance of the substrate (TMB) at 450 nm. The data was fitted by linear regression with a slope of y = 4.635 × 105 (R2 = 0.9709). The absorbance recorded at 450 nm for HRP–ADHCPMV (treated as unknown) was 0.9069 and from the calibration curve (Fig. S3, ESI†) the corresponding HRP concentration was 1.83 μg. After measuring the CPMV concentration by UV-vis, the number of immobilised enzymes was calculated to be approximately 11 ± 1 per virus. This assumes that HRP activity on the virion is the same as the HRP activity in solution (i.e., no enzymatic activity loss). The estimated number of enzymes per virus is very similar, within experimental error, to the number determined by fluorescent labelling. This indicates that, very importantly, all the HRP molecules attached to the surface of CPMV have retained their full catalytic activity.
From the number of carboxylates addressable by small molecules,22,23 on purely chemical grounds it should be possible to attach 180 enzymes to a virus particle. However, given the larger size of the enzyme molecules this is likely to be an over-estimate. In an attempt to calculate the maximum number of HRP molecules that could be fitted on the virus surface, we assumed that CPMV is a solid sphere of radius 14 nm and that HRP is a solid sphere with a radius of gyration of 2.65 nm, as calculated by molecular dynamics simulation based on its crystal structure.30 Using Tables of Spherical Codes31 we calculate that a maximum of 130 HRP molecules could be fitted around the virus on geometrical grounds. Therefore, the finding that we have attached 11 HRP molecules per particle indicates that we have obtained coverage of about 8.5% of the maximum predicted on strictly geometric grounds. This suggests that there may be room for improvement in the number of enzymes that can be displayed on the virus. However, the estimate of the maximum number possible is not precise because neither CPMV nor HRP are perfect spheres, the reactive carboxyls are not evenly distributed over the virus surface and we have no information about the orientation of the enzyme molecules on the virus surface. Therefore, the overall CPMV surface area might not represent that actually available for enzyme coupling. Further, other effects, such as electrostatic repulsion, are likely to reduce the number of bound enzymes and probably help to explain the lower numbers of bound enzyme actually observed; however, we have no way of readily calculating the electrostatic effects.
3.3 GOX–ADHCPMV
Glucose oxidase (GOX) from the fungus Aspergillus niger is a highly specific enzyme for D–glucose; it is a dimeric protein with a molecular weight of 160 kDa. GOX was selected for the possible development of a selective glucose sensor, which might be useful for blood glucose monitoring. The enzyme was activated via the periodate method as previously described and the ADH linker was attached to the enzyme before coupling to CPMV. The integrity of the modified particles was confirmed by agarose gel electrophoresis and TEM (Fig. S4, ESI†). According to their migration in an electric field towards the anode, the NHS–esterCPMV particles migrate more slowly than wild-type and GOX–ADHCPMV. Furthermore, SDS-PAGE (Fig. 5) revealed an extra band for the modified particles of approximately 65–70 kDa mass, which possibly corresponds to the small coat protein and one monomer of the enzyme. The particle size in buffer as measured by DLS shows an increase in average hydrodynamic diameter to ca. 38 nm following enzyme coupling (Fig. S5, ESI†).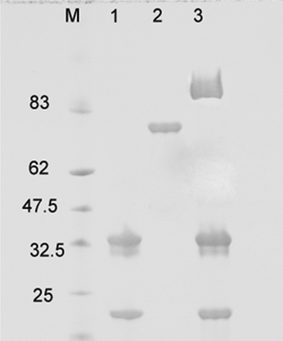 | ||
| Fig. 5 SDS-PAGE of GOX–ADHCPMV. Lane 1, CPMV; 2, ADH–GOX; 3, GOX–ADHCPMV. M is prestained protein marker. | ||
To quantify the number of enzymes coupled to the virus in GOX–ADHCPMV, calibration curves were obtained using different standard solutions of free GOX prepared from stock solution. The reactivity was determined photometrically using an Amplex Red Glucose/Glucose Oxidase Assay Kit according to the manufacturer's instructions. In the case of the Amplex Red assay, the added hydrogen peroxide reacts with 10-acetyl-3,7-dihydroxyphenoxazine (the Amplex Red reagent) in the presence of HRP to form the oxidation product resorufin. Resorufin has an absorption peak at approximately 571 nm at a pH of 7.4.
The free GOX concentration was determined from serial dilution values. Each enzyme concentration was prepared and used on the same day, 36 activity measurements for each enzyme concentration were recorded and the absorbance was deducted from the empty well values and averaged. The GOX concentration was plotted versus the absorbance of the substrate (Amplex Red) at 571 nm. The data was fitted by linear regression with a slope of y = 0.0852 (R2 = 0.9161). From the absorbance recorded at 571 nm for GOX–ADHCPMV (treated as unknown) and from the calibration (Fig. S6, ESI†) the corresponding free GOX concentration was 3.598 μg. By comparison to the concentration of CPMV particles from UV-vis (0.0546 mg), the number of immobilised GOX enzymes was calculated as approximately 2–3 ± 1 per virus, assuming that GOX activity on the virion is the same as GOX activity in solution (i.e. that there is no loss of enzymatic activity on binding). Presumably the lower number of molecules bound in the case of GOX compared with HRP at least partly reflects the fact that the GOX is considerably larger (160 kDa for the dimer) than HRP (40 kDa).
4. Conclusions
The enzymes HRP and GOX have been covalently coupled to the CPMV capsid external surface via the carbohydrate residues on the enzyme surface. This shows that relatively large biomacromolecules can be conjugated to the virus surface by simple conjugation strategies without destroying their biological activity. The number of enzymes bound to the surface of the virus was determined to be ∼11 HRP and 2–3 GOX, respectively. The HRP–ADHCPMV and GOX–ADHCPMV particles have potential uses as building blocks for catalytic devices, diagnostic assays or biosensors. We are currently investigating the construction of EnzymeCPMV arrays on solid surfaces.Although the current manuscript deals exclusively with the coupling of enzymes to the surface of CPMV, the methods developed are applicable to other proteins. For example, the immunogenicity of antigens is known to be increased by displaying multiple copies on the surface of a larger carrier structure. The methods reported here, which preserve the activity of the attached proteins, could be adapted for this purpose and therefore make a contribution towards the development of novel vaccines.
Acknowledgements
This work was supported by the Biotechnology and Biological Sciences Research Council, UK (Core Strategic Grant to the John Innes Centre, G.P.L. and D.J.E. and JIC DTG, A. A. A. A.). Grant Calder (JIC) is acknowledged for assistance with fluorescence microscopy. We thank Dr Richard Morris (JIC) for performing the calculations to determine the maximum number of HRP molecules that can be packed around CPMV.References
- E. Katz and I. Willner, Angew. Chem., Int. Ed., 2004, 43, 6042–6108 CrossRef CAS.
- C. M. Niemeyer, Angew. Chem., Int. Ed., 2001, 40, 4128–4158 CrossRef CAS.
- P. W. Barone, S. Baik, D. A. Heller and M. S. Strano, Nat. Mater., 2005, 4, 86–92 CrossRef CAS.
- V. G. Gavalas, S. A. Law, J. C. Ball, R. Andrews and L. G. Bachas, Anal. Biochem., 2004, 329, 247–252 CrossRef CAS.
- Y. Xiao, F. Patolsky, E. Katz, J. F. Hainfeld and I. Willner, Science, 2003, 299, 1877–1881 CrossRef CAS.
- P. Pescador, I. Katakis, J. L. Toca-Herrera and E. Donath, Langmuir, 2008, 24, 14108–14114 CrossRef CAS.
- J. J. Gooding, R. Wibowo, J. Liu, W. Yang, D. Losic, S. Orbons, F. J. Mearns, J. G. Shapter and D. B. Hibbert, J. Am. Chem. Soc., 2003, 125, 9006–9007 CrossRef CAS.
- X. Yu, D. Chattopadhyay, I. Galeska, F. Papadimitrakopoulos and J. F. Rusling, Electrochem. Commun., 2003, 5, 408–411 CrossRef CAS.
- P. Yanez-Sedeno and J. M. Pingarron, Anal. Bioanal. Chem., 2005, 382, 884–886 CrossRef CAS.
- F. Caruso and C. Schüler, Langmuir, 2000, 16, 9595–9603 CrossRef CAS.
- P. J. Halling, R. V. Ulijn and S. L. Flitsch, Curr. Opin. Biotechnol., 2005, 16, 385–392 CrossRef CAS.
- A. Basso, P. Braiuca, C. Ebert, L. Gardossi and P. Linda, J. Chem. Technol. Biotechnol., 2006, 81, 1626–1640 CrossRef CAS.
- T. Lin and J. E. Johnson, Adv. Virus Res., 2003, 62, 167–239 CrossRef CAS.
- D. J. Evans and N. F. Steinmetz, Org. Biomol. Chem., 2007, 5, 2891–2902 Search PubMed.
- E. Strable and M. G. Finn, in Current Topics in Microbiology and Immunology, ed. M. Manchester and N. F. Steinmetz, Springer-Verlag, Berlin, 2009, pp. 1–21 Search PubMed.
- D. J. Evans, Inorg. Chim. Acta, 2010, 363, 1070–1076 CrossRef CAS.
- D. J. Evans and G. P. Lomonossoff, in Current Topics in Microbiology and Immunology, ed. K. Palmer and Y. Gleba, Springer-Verlag, Berlin, 2012 DOI:10.1007/82_2011_184.
- J. J. O'Malley and J. L. Weaver, Biochemistry, 1972, 11, 3527–3532 CrossRef CAS.
- H. B. Dunford and J. S. Stillman, Coord. Chem. Rev., 1976, 19, 187–251 CrossRef CAS.
- C. Rota, C. F. Chignell and R. P. Mason, Free Radical Biol. Med., 1999, 27, 873–881 CrossRef CAS.
- A. Basu, T. G. Shrivastav and K. P. Kariya, Clin. Chem., 2003, 49, 1410–1412 CAS.
- N. F. Steinmetz, G. P. Lomonossoff and D. J. Evans, Langmuir, 2006, 22, 3488–3490 CrossRef CAS.
- A. A. A. Aljabali, J. E. Barclay, J. N. Butt, G. P. Lomonossoff and D. J. Evans, Dalton Trans., 2010, 39, 7569–7574 RSC.
- G. T. Hermanson, Bioconjugate Techniques, Academic Press, London, 2008 Search PubMed.
- N. F. Steinmetz, G. Calder, G. P. Lomonossoff and D. J. Evans, Langmuir, 2006, 22, 10032–10037 CrossRef CAS.
- F. M. Brunel, J. D. Lewis, G. Destito, N. F. Steinmetz, M. Manchester, H. Stuhlmann and P. E. Dawson, Nano Lett., 2010, 10, 1093–1097 CrossRef CAS.
- N. F. Steinmetz, D. J. Evans and G. P. Lomonossoff, ChemBioChem, 2007, 8, 1131–1136 CrossRef CAS.
- M. Kvaratskhelia, C. Winkel, M. T. Naldrett and R. N. F. Thorneley, J. Plant Physiol., 1999, 154, 273–282 CrossRef CAS.
- J. S. Hanker and B. I. Giammara, Stain Technol., 1983, 58, 184 Search PubMed.
- M. Laberge, Q. Huang, R. Schweitzer-Stenner and J. Fidy, Biophys. J., 2003, 84, 2542–2552 CrossRef CAS.
- N. J. A. Sloane, with the collaboration of R. H. Hardin and W. D. Smithand others, Tables of Spherical Codes, www.research.att. com/%E2%88%BCnjas/packings/.
Footnotes |
| † Electronic supplementary information (ESI) available: Alternative conjugation strategies, agarose gel electrophoresis of CPMV and CPMV–HRP conjugates, UV-vis spectrum of HRP–ADHCPMV, agarose gel electrophoresis of GOX–ADHCPMV particles and corresponding TEM image, calibration curves for HRP–ADHCPMV and GOX–ADHCPMV, DLS data for GOX–ADHCPMV are made available. See DOI: 10.1039/c2nr31485a |
| ‡ Present address: Department of Cardiovascular Medicine, University of Oxford, Level 6 West Wing, John Radcliffe Hospital, Headley Way, Headington, Oxford, OX3 9DU, UK. |
| § Present address: From August 2012: Department of Chemistry, University of Hull, Cottingham Road, Hull, HU6 7RX, UK. E-mail: E-mail: david.evans@hull.ac.uk |
| This journal is © The Royal Society of Chemistry 2012 |