Investigating the structural evolution of thiolate protected gold clusters from first-principles
Yong
Pei
*a and
Xiao Cheng
Zeng
*b
aDepartment of Chemistry, Key Laboratory of Environmentally Friendly Chemistry and Applications of Ministry of Education, Xiangtan University, Hunan Province, China 411105. E-mail: ypnku78@gmail.com
bDepartment of Chemistry and Nebraska Center for Materials and Nanoscience, University of Nebraska-Lincoln, Lincoln, Nebraska 68588, USA. E-mail: xzeng1@unl.edu
First published on 23rd April 2012
Abstract
Unlike bulk materials, the physicochemical properties of nano-sized metal clusters can be strongly dependent on their atomic structure and size. Over the past two decades, major progress has been made in both the synthesis and characterization of a special class of ligated metal nanoclusters, namely, the thiolate-protected gold clusters with size less than 2 nm. Nevertheless, the determination of the precise atomic structure of thiolate-protected gold clusters is still a grand challenge to both experimentalists and theorists. The lack of atomic structures for many thiolate-protected gold clusters has hampered our in-depth understanding of their physicochemical properties and size-dependent structural evolution. Recent breakthroughs in the determination of the atomic structure of two clusters, [Au25(SCH2CH2Ph)18]q (q = −1, 0) and Au102(p-MBA)44, from X-ray crystallography have uncovered many new characteristics regarding the gold–sulfur bonding as well as the atomic packing structure in gold thiolate nanoclusters. Knowledge obtained from the atomic structures of both thiolate-protected gold clusters allows researchers to examine a more general “inherent structure rule” underlying this special class of ligated gold nanoclusters. That is, a highly stable thiolate-protected gold cluster can be viewed as a combination of a highly symmetric Au core and several protecting gold–thiolate “staple motifs”, as illustrated by a general structural formula [Au]a+a′[Au(SR)2]b[Au2(SR)3]c[Au3(SR)4]d[Au4(SR)5]e where a, a′, b, c, d and e are integers that satisfy certain constraints. In this review article, we highlight recent progress in the theoretical exploration and prediction of the atomic structures of various thiolate-protected gold clusters based on the “divide-and-protect” concept in general and the “inherent structure rule” in particular. As two demonstration examples, we show that the theoretically predicted lowest-energy structures of Au25(SR)8− and Au38(SR)24 (–R is the alkylthiolate group) have been fully confirmed by later experiments, lending credence to the “inherent structure rule”.
 Yong Pei | Yong Pei received his BSc degree from the Deptartment of Chemistry, Xiangtan University, China (Hunan) in 2001. He obtained a PhD degree from Nanjing University, China (Nanjing) in 2006. After post-doctoral research at the University of Nebraska_Lincoln, he joined the Chemistry faculty of Xiangtan University in 2010. His current research interests include the methodological development of global search algorithms for complicated ligand protected metal clusters and theoretical studies on the structure, optical, catalytic, and magnetic properties of metal nanoparticles. |
 Xiao Cheng Zeng | Xiao Cheng Zeng is Ameritas Distinguished University Professor at the University of Nebraska-Lincoln, USA, a fellow of the American Association for the Advancement of Science, a fellow of the American Physical Society, a former John Simon Guggenheim fellow and a recipient of the American Chemical Society Midwest Award. He has published 296 papers in refereed journals (h-index: 41), and supervised 20 graduate students (14 PhD and 6 MS) and 20 postdoctoral fellows. Zeng received his BS from Peking University, PhD from Ohio State University and performed postdoctoral research at University of Chicago and UCLA. |
1 Introduction
The strong relativistic effect of Au results in more significant s–d hybridization than other coinage metals, which leads to many novel structures and properties for nanosized gold clusters. For gas-phase gold clusters (AuNq, q = 0, ±1, N ≤ 60), significant advances have been made through combined experimental and theoretical efforts over the past two decades, which provide deeper insights into their size-dependent atomic structures, and thereby the critical information needed for understanding the structure–size–property relationship.1–10 The combination of global optimization algorithms and density functional theory11,12 (DFT) calculation has been proven to be a powerful tool to generate structures of low-energy gold clusters, as well as for the determination of the lowest-energy structure by comparing computed photoelectron spectra with the experimental spectra. For example, unique structural transitions for gold cluster anions in the size range of N = 14–20 have been revealed, e.g., from the flat cage to hollow cage and to tetrahedral pyramid.1–3 However, the determination of the atomic structure for ligand-protected gold clusters is still a grand challenge to both experimentalists and theoreticians, largely due to the complexity of the ligand shell.The thiolate-protected gold nanoparticles (RS-AuNPs) or nanoclusters entail a distinctive quantum confinement effect, as well as many size-dependent physicochemical properties and functionalizations, such as magnetism, catalysis, enhanced photoluminescence, sizeable optical absorption or HOMO–LUMO gap, electrochemical properties, and high stability at magic numbers.13–17 The first experimental method for synthesizing RS-AuNPs was demonstrated by Brust et al. in 1994.18 Although many variants of the original method were subsequently developed,19 the key route is essentially the same, i.e., to involve the reduction of high valence Au salts. However, the RS-AuNPs synthesized from these methods are usually a mixture of different sizes, which require further separation for structural and compositional analysis. During the past fifteen years or so, most experimental efforts were devoted to resolving the chemical composition of RS-AuNPs.20–54 In particular, gold nanoparticles in the size range of subnanometre to ∼2 nm have attracted most interest. Due to the difficulty in crystallization of RS-AuNPs, few structures of RS-AuNPs have been fully resolved, which has greatly hindered an in-depth understanding of the structure–property relationship of RS-AuNPs.
The gold–phosphane–halide clusters are one of few examples of ligand-protected gold clusters whose structures have been well resolved in experiments.55–58 The Au cores in these gold–phosphane–halide clusters exhibit ordered and uniform packing pattern so that a typical structural pattern for these clusters can be described as a symmetric Au core plus protecting ligands. Mingos et al. pointed out that the coordination of ligands to gold clusters may promote a more favorable hybridization among metal–atom orbitals and result in stronger radial metal–metal bonding, leading to a major structural transition of the gold clusters.59 Theoretical efforts for the exploration of structures of RS-AuNPs date from 1999. Before 2008, due to the lack of a precise atomic structure of any RS-AuNPs, earlier theoretical models of RS-AuNPs, e.g. Au38(SR)24, mainly followed the “conventional” structural pattern attained based on gold–phosphane–halide clusters, which is typically a combination of a ligand and an intact symmetric Au core.60 More specifically, the Au atoms were assumed to arrange into a compact core and the –RS ligand sticks to the Au core on the atop, bridge, or triangle site. Such a conventional model of Au–SR linkage has prevailed for a long time in the study of the interfacial structure of self-assembled thiol monolayers on gold surfaces.61 Although some theoretical studies have suggested that the ligand/Au core interfacial structure in RS-AuNPs could be quite different from the conventional model of metal–ligand linkage, no general structural rule was proposed due largely to the lack of experimentally determined structures.62,63
The successful crystallization of Au102(p-MBA)44 has been a huge motivation for recent RS-AuNP research, especially due to the finding of unexpected atomic structure.64 For the first time, a clear picture of the gold–sulfur bonding in an RS-AuNP was revealed, i.e., the –SR group is not merely adsorbed on the surface of an Au core to form a single Au–S linkage; rather, it can strongly disturb the surface structure of the Au core and lead to the formation of novel gold-thiolate protecting units (coined as “staple motif”) on a highly symmetric Au core. It is worthy of quoting one sentence in a perspective article by Whetten et al. on the structure of Au102(p-MBA)44:65 “The known properties of nanoscale clusters can now be rationalized in terms of atomic ordering.” The term “atomic ordering” was also applicable in describing another successfully crystallized thiolate-protected gold cluster, Au25(SCH2CH2Ph)18−.66,67 On the basis of the known atomic structures of Au102(p-MBA)44 and Au25(SCH2CH2Ph)18−, a plausible “inherent structural rule” about the formation of RS-AuNPs has been introduced, namely, any RS-AuNP can be viewed as a combination of a highly symmetric AuN-core with several protection staple motifs, as represented by a general structural formula [Au]a+a′[Au(SR)2]b[Au2(SR)3]c[Au3(SR)4]d[Au4(SR)5]e where a, a′, b, c, d and e are integers that satisfy certain constraints.63 This view is also consistent with the “divide-and-protect” concept.63 Over the past few years, this structural rule has been successfully applied to the structural prediction of several thiolate-protected gold clusters. In this review article, our focus will be placed on recent theoretical progress in the structural prediction of thiolate (–RS) protected gold clusters on the basis of the “inherent structure rule”, combined with other structural search methods and density functional theory (DFT) calculations.
This article is organized as follows: Section 2 discusses the major difficulties encountered for the structural prediction of thiolate-protected gold clusters. Section 3 summarizes earlier studies of the Au38(SR)24 cluster, followed by a summary of the recent breakthrough in the structure determination of Au102(p-MBA)44 and Au25(SCH2CH2Ph)18−, and by an illustration of the “inherent structure rule” that will be utilized for the structural prediction of various thiolate-protected gold clusters. Section 4 elucidates some details about the intrinsic connection between the cluster's geometric structure and electronic properties, including the origin of electronic magic numbers. Finally, we briefly review the structure–activity relationship for the thiolate-protected gold clusters with an example of catalytic oxygen activation. Note that in this article we mainly focus on the structural aspects of thiolate-protected gold clusters: other properties derived from the electronic effects such as optical absorption are not fully discussed. The readers can refer to recently published review articles by Aikens, Jiang, and Häkkinen for more information about the electronic structures and optical absorption spectra of thiolate-protected gold clusters.68–70
2. Challenges in structural prediction for RS-AuNPs
2.1 Potential energy surface and global optimization algorithms
The potential energy surface (PES) can be described by a mathematical potential function whose values are the potential energies of a system. For the PES of a molecule or a cluster, the atomic positions are represented as the variables of the potential function, while the values of the function are the corresponding potential energies. Two forms of potential functions of gold have been widely used in the study of gold clusters: (1) a parameterized classical many-body potential (such as Sutton–Chen71 and Gupta72 potential functions) and (2) the total energy calculated using ab initio and density-functional theory methods. For the former, the potential function is represented by an analytical mathematical function including a few pre-defined parameters. A major advantage of such a parameterized potential function (or force field) is its high computation efficiency, and this form of potential function has been commonly used in the study of the structures of gold clusters since the 1990s.73,74 A disadvantage of the classical potential function is that the pre-defined parameters are inadequate to fully address electronic effects in gold clusters. The quantum mechanical methods such as density functional theory methods11,12 yield more genuine PES and have been generally used in studying structures and properties of small-sized gold clusters since the 1970s. The foremost information that can be obtained from solving the Schödinger equation is the electronic energy for given atomic positions of molecules and clusters. To reduce computational costs, the Born–Oppenheimer (BO) approximation is usually applied, which assumes the positions of nuclei are fixed while computing electronic properties.75 The resulting hyper-PES as a function of the atomic positions is the so-called BO-PES.The lowest point on a PES is referred to as the global minimum. For a cluster, the global minimum corresponds to the most stable structure of the cluster, which is generally believed to be the observed structure in experiments. The search for the global minimum on the PES is therefore equivalent to the determination of the most stable structure of a cluster. However, locating the global minimum on a complicated hyper-PES is a grand challenge due to the existence of a huge number of local minima for medium- to large-sized clusters. The genetic-algorithm (GA),76 simulating annealing (SA),77 and basin-hopping (BH)78 are three popular methods for seeking the global minimum of an atomic cluster. The GA method explores the cluster structure by modeling some aspects of biological evolution. For example, a population of clusters evolves toward low energy through mutation and mating of structures, along with the selection of those with low potential energy. As a result, new configurations are produced via “genetic manipulation” combined with a local optimization algorithm such as the conjugate gradient method. SA method takes advantage of the relatively less complex free-energy landscape at high temperatures and attempts to follow the free energy global minimum as the temperature is decreased. A difficulty with the SA approach is that, if the free-energy global minimum changes at low temperatures where dynamical relaxation is slow, the algorithms will become confused by the structure corresponding to the high temperature free energy global minimum. The BH method, originally proposed by Wales and Doye, has been widely used in predicting the global-minimum structure of bare gold clusters AuN− with N up to 58.2–10 In the BH method, the transformation maps the potential function onto a series of plateaus where the barriers between local minima can be removed, as shown in Scheme 1. A series of Monte Carlo random walks are normally used to explore the PES, combined with a local optimization method to locate local minima. Once a local minimum is located, the next step is to perform a hopping (or a random walk) to explore new configurations on the PES.
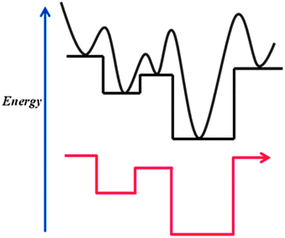 | ||
| Scheme 1 A schematic diagram illustrating the energy transformation for a one-dimensional potential energy surface. The black curve is the original potential energy surface and the red line is the transformed energy basin map. | ||
However, direct application of the three global optimization methods has encountered computational difficulties in the case of thiolate-protected gold clusters due largely to the presence of organic ligands, which dramatically increases the computational cost and time required to locate the global minimum. In addition, many global optimization methods typically require random moves of atomic positions rather than a fraction of ligand. In reality, the organic ligands tend to stay outside the metal core, which also requires modification of the algorithm to take such physical effects into account. Hence, the “inherent structure rule” for the thiolate-protected gold clusters summarized from recent experiments65–67 can be viewed as an insight-based, highly efficient, and low-cost search method to seek the true global minima of thiolate-protected gold clusters without the expensive enumeration of cluster structures.
2.2 Information required for predicting the structure of a complex RS-AuNP
To predict the lowest-energy structure of small-sized gas-phase gold clusters from theory, low-lying isomers (typically several hundred) are attained using a combined global optimization method and DFT calculations.2–10 Moreover, comparison with experimental data such as a photoelectron spectrum of anionic clusters is a key step to determine the structures of the most stable clusters. For medium-sized thiolate-protected gold clusters, the generation of low-lying structures from DFT calculations is computationally very expensive. Furthermore, relative energies predicted from DFT calculations are also affected by the exchange-correlation functional and basis set. Therefore, additional evidence is needed to support the predicted low-energy clusters. For example, the UV-vis absorption spectra have been often used to provide information on the optical absorption edge, feature absorption peaks, and the shape of optical curves, which are very sensitive to the structure and composition of the cluster. The optical absorption curves can therefore provide one set of experimental evidences for validating theoretical predictions. In principle, UV-vis absorption spectra originate from the excitation of electrons from the ground state, including the singlet-to-singlet transition and singlet-to-triplet transitions. Theoretically, the excitation energy of a thiolate gold cluster can be computed using time-dependent DFT (TDDFT) methods.79 For nanosized gold clusters, it has been found that the excitation of electrons involves mainly singlet-to-singlet transitions. Therefore, good agreement between the theoretical excitation energy curve and the experimental optical absorption spectrum is necessary to validate the theoretically predicted thiolated gold cluster.In addition, the experimental powder X-ray diffraction (XRD) curve is an auxiliary data to further validate the theoretically predicted cluster structure. The theoretical XRD curve can be obtained using the Debye formula with the atomic distance (for the cluster studied) as the input:
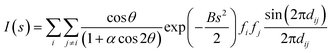
3. Structural prediction of thiolate-protected gold clusters from the “inherent structure rule”
3.1 Earlier structural models of Au38(SR)24 and the “divide-and-protect” concept
The Au38(SR)24 cluster deserves special mention because many theoretical predictions were published long before the experimental confirmation, which reflects the progress in the research of the structure of RS-AuNPs. In 1996, Luedtke and Landman reported the first theoretical study of the structural, dynamic, and thermodynamic properties of passivated gold nanocrystallites AuN(SR)M, with N = 140, 201, 459, and 586, using classical molecular mechanics (MM) and force field based molecular dynamics (MD) modeling.80 They showed that the coordination of thiol molecules on the surface of finite-sized gold crystallites may differ from that on bulk surfaces, depending on the size and geometry of the crystallites. It was also suggested that the gold nanocrystallites maintain an intact structure with the presence of thiol ligands. Wilson and Johnston introduced an alternative Au cluster/thiol–ligand interaction model based on the MM method to describe the surface passivation effects on gold clusters.81 It was suggested that surface passivation by thiol ligands results in changes in the order of stability of Au clusters compared to Au clusters observed in the gas-phase. In particular, the icosahedron-based core structure was predicted to be more stable than fcc-like cuboctahedral or truncated octahedral geometries for thiol-protected Au38 and Au55 clusters.The Au38(SR)24 cluster also appears to be the first thiolate-protected gold cluster studied via ab initio calculations. The composition of Au38(SR)24 was first reported by Whetten and co-workers in 1997 from their mass spectrometric experiments.14,20 However, the internal atomic structure has puzzled both experimentalists and theorists for more than ten years. Since 1999, more theoretical efforts have been made to explain the unique stability and structure of Au38(SR)24 from ab initio calculations. In the first structural model proposed by Häkkinen et al., a truncated octahedron Au38 containing 6 square Au4 units on the Au core surface was constructed with 24 –RS groups evenly distributed on the surface of the Au core. The structural relaxation based on a plane-wave density-functional theory and pseudopotential method (Born–Oppenheimer local-spin-density molecular dynamics) led to a local-minimum structure with the high-symmetry Au38 core unchanged.60 Additional electronic structure analysis of the optimized cluster revealed notable electron transfer from gold to sulfur. The Au core was positively charged. Majumder and Larsson and their co-workers employed a similar ligand adsorption pattern to study the interactions between thiol groups and nano-sized gold clusters via ab initio calculations. They found the structure of the Au core was only slightly affected by the thiolate ligands.82,83
Garzón et al. demonstrated that the thiol monolayer may have a much stronger structural effect on the Au core through observing the structural relaxation of an Au38(SR)24 model based on the DFT calculations.62 Interestingly, the S-head of the thiolate ligands was found to significantly penetrate into the Au core, strongly affecting not only the symmetry of the Au core but also the core/ligand interfacial structure. A mixture of ligand motifs on the cluster surface, including –RS–Au–SR– and –RS–Au–SR–Au–SR– motifs, were proposed for the first time. In fact, a novel gold-substrate sulfur-headgroup interfacial structure for the self-assembled monolayers on Au(111), named as “staple motif” was also recognized at the time,84 which co-indicated the strong bonding interactions between gold and sulfur at the interface. These results called for new physical pictures to understand the interfacial structure of RS-AuNPs.
Inspired by earlier theoretical predictions of Au38(SR)24 structure and concurrent experimental findings of novel –RS–Au–RS– motifs on the Au surface, Häkkinen et al. introduced in 2006 a new concept to understand the structure of thiol group-protected gold nanoparticles, namely, the “divide-and-protect” concept.63 This concept was proposed based on the optimization of a structural model reported previously by using the generalized gradient approximation (GGA) in the Perdew–Burke–Ernzerhof (PBE) form. A dramatic structural change was observed. The new structure contains an Au core (with Oh point-group symmetry) of 14 atoms and six planar, ring-like gold-thiolate capping units, as shown in Fig. 1. In view of this structure, the Au atoms can be divided into two groups: 14 Au atoms formed the Au core in the Au(0) state (metallic state), and the remaining 24 Au formed an Au(I) thiolate species (in the oxidation state) capping the Au core. This new structure is closely related to the Garzón Au38 model,62 which also incorporates the strong etching effects of thiol groups on the Au core in RS-AuNPs. Although the relative stability between the two models depends on the exchange correlation functional used, the finding of the formation of stable [AuSR]4 units capped on a symmetric Au core not only supports the “divide-and-protect” concept, but also offers a new picture for understanding the bonding and structure of thiolate-protected gold nanoparticles. In 2007, a similar ‘core-in-cage’ model was also proposed to understand the structure of another magic-number RS-AuNP, Au25(SR)18−,85 which will be discussed in detail in the next section.
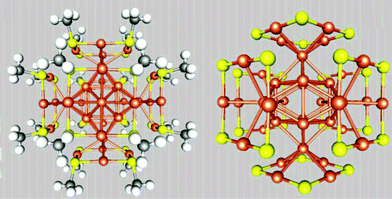 | ||
| Fig. 1 Structural model for Au38(SMT)24 proposed by Häkkinen et al.. Au, orange-brown; S, yellow; P, red; Cl, green; C, dark gray; H, white. The right hand model has the MT groups removed. Reprinted (adapted) with the permission of ref. 63. Copyright 2006 American Chemical Society. | ||
3.2 Two breakthroughs: the total structure determination of Au102(p-MBA)44 and Au25(SCH2CH2Ph)18−
Until 2007, Au39(PPh3)14Cl62+ held the record of the largest ligand-protected gold cluster that was successfully crystallized.57 In October 2007, a research group led by Kornberg determined the atomic structure of an all-thiol-protected gold nanoparticle with 102 Au atoms, namely, Au102(p-MBA)44, based on the single-crystal X-ray diffraction at 1.1 Å resolution.64 Subsequently, a series of theoretical structural analyses of Au102(p-MBA)44 were performed by Gao et al., Han et al., and Choi et al., independently.86–88 Choi et al. classified the Au atoms in Au102(p-MBA)44 into three different shells through radial distance analysis: (1) the inner core consists of 39 Au atoms within 5.3 Å from the center of cluster; (2) the second shell consists of 40 Au atoms within a spherical shell of 6.1–6.4 Å from the center of cluster. The Au atoms in the second shell are connected to the S terminal of the –RS–Au–SR– staple motifs through a single Au–S bond. (3) The remaining 23 Au atoms occupy the outermost shell within a radius of 7.9–8.3 Å from the center, which form staple motif protection units.87Gao et al. provided an alternative view of the multi-layer structure of Au102(p-MBA)44. From their structural analysis, the embedded Au102 cluster was decomposed into a multi-layered structure described as Au54(penta-star)@Au38(ten wings)@Au10(two pentagon caps) as shown in Fig. 2.86 The inner layer was an Au54 ‘penta-star’ consisting of five twinned Au20 tetrahedral subunits. Note that the tetrahedron Au20 is a magic-number structure of a free-standing gold cluster. Gao et al. pointed out that although a perfect Td Au20 tetrahedral cluster is highly stable due to closure of the electronic shell, five perfect Td Au20 clusters cannot completely form a perfect penta-star because of the deficiency in the solid-angle if the vertices of five Td Au20 clusters are connected through the midpoint of the opposing edge. A stand-alone 54 Au-atom penta-star must be energetically unfavorable due to the large strain. Indeed, in the definition of the penta-star 54 atom Au core, the Au atom in part of the –RS–Au–SR– staple motif is taken into account. That is, each of the five vertices of the Au54 penta-star is a part of the –RS–Au–SR– staple motif covering the Marks decahedron Au core. Ultimately, the formation of a highly stable Au102(p-MBA)44 nanocluster is a manifestation of a delicate (energetic) balance between local thiolate–gold interactions (in the form of staple motifs) with the growth mode compatible with the underlying Marks decahedral Au49 core.
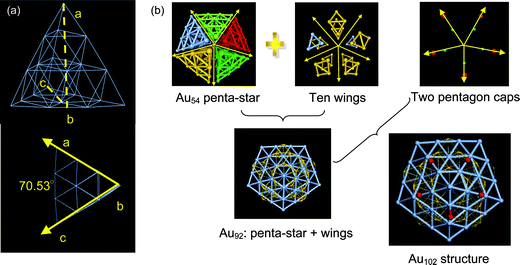 | ||
| Fig. 2 Structural decomposition of an Au102 cluster. (a) Perfect tetrahedral Td Au20; (b) Graphitic anatomy of embedded Au102 structure. An Au54 penta-star consists of five twinned Au20 tetrahedral subunits. Reprinted (adapted) with permission from ref. 86. Copyright 2008 American Chemical Society. | ||
The successful experimental determination and subsequent theoretical analysis of the atomic structure of Au102(p-MBA)44 provides new knowledge about the structure of RS-AuNPs. The most important insight from these studies is that the interfacial structure in an RS-AuNP may be more complicated than previously expected: Au atoms on the surface of the Au core are lifted by thiol groups to form a certain number of protecting gold-thiolate staple-like motifs. If one studies the historic progress of the interfacial chemistry of thiol self-assembled monolayer (SAM) on Au surfaces, similar behavior of Au-SR interactions at the planar interface can be found.84 As Au nanoparticles have larger curvatures than planar surfaces, much stronger interactions are expected at the interface between the thiol-ligands and Au core, consistent with previous theoretical models of Au38(SR)24. Another important finding is that the structure of Au102(p-MBA)44 also supports the “divide-and-protect” concept.63 However, the structure and number of staple motifs are still difficult to predict for an RS-AuNP like Au38(SR)24. The same problem exists for the determination of the geometry of the Au core in RS-AuNPs. Does an Au core always form a highly symmetric structure like the one in Au102(p-MBA)44? Clearly, more atomic structures of RS-AuNPs are needed in order to derive more generic structural rules.
The determination of atomic structure of Au25(SR)18− is the second experimental breakthrough. For a long time, Au25(SR)18− had been incorrectly identified as Au38(SR)24.21,37,89 In 2005, Tsukuda and co-workers corrected the mislabeled composition by electrospray ionization mass spectrometry (ESI-MS) measurement90 of a series of electrophoretically fractionated NPs. The structural composition of the cluster is ascertained as Au25(SR)18. Extensive tests of the synthesis conditions and measurements affirm that the Au25(SG)18 (where SG denotes glutathione) is a stable magic-number cluster. In the meantime, theoretical efforts had also been made toward understanding the structure and magic-number nature of Au25(SG)18. In collaboration with Tsukuda, Nobusada et al. reported the first DFT calculation of the structure and electronic properties of Au25(SR)18+ with R being simplified by a methyl group (MT).85 In their theoretical study, the model still followed the conventional viewpoint of ligand protection on the metal clusters. Nobusada et al. constructed two types of Au25-core, one with a face-centered-cubic (FCC) structure with six Au(111) facets consisting of eight gold atoms (FCC-Au25-core), and another with a vertex-shared bi-icosahedral structure (SES-Au25-core). The selection of SES-Au25-core is partially motivated by the finding of a vertex-shared bi-icosahedral Au25-core in [Au25(PPh3)10(SR)5Cl2]2+.91 With both types of Au25-core, the 18 thiol ligands were manually placed on the Au25-core followed by a DFT optimization based on the Lee–Yang–Parr correlation functional (B3LYP). Surprisingly, the optimized structures from DFT showed the similar feature that part of the Au on the surface of the Au25-core were etched by thiol groups to form gold-thiolate protecting units as that observed previously by Häkkinen et al. and Garzón et al. in their study of Au38(SR)24.62,63 The most stable structure (FCC-2) with the lowest energy exhibited a clearly ‘core-in-cage’ structure, i.e., an inner Au7-core surrounded by Au12(SMT)12 and two Au3(SMT)3 rings.
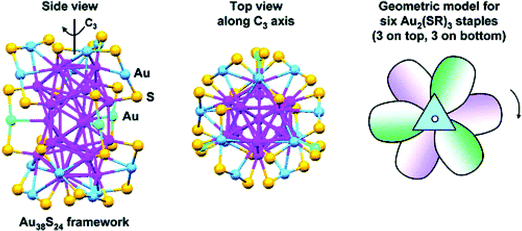
Shortly after (in 2008), Akola et al. proposed an entirely new structural model92 for Au25(SMT)18−. The optimal structure model suggested by Akola et al. has an icosahedral Au13-core that is protected by six –RS–Au–RS–Au–RS– staple motifs via the formation of 12 Au–S bonds at the core/ligand interface. The DFT calculation indicates the new model is much more stable than Nobusada's Au25 model, as shown in Fig. 3. The calculated binding energy of each –RS–Au–RS–Au–RS– complex to the Au13 core is 3.5 eV, much higher than that based on the FCC-2 model of Nobusada et al. (1.6 eV per (AuSCH3)6 oligomer). The simulated powder-XRD curve and UV-vis absorption spectra of various models also indicated that the combined icosahedrons Au13-core and six –RS–Au–RS–Au–RS– staple motifs are in the best agreement with the experimental curves. Moreover, the absorption spectrum calculated from the TDDFT method suggested a HOMO–LUMO gap of 1.3 eV, in good agreement with the experimental measurement.
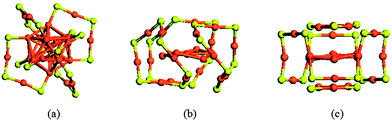 | ||
| Fig. 3 (a) Akola's new structure model of Au25(SMT)18−. (b) and (c) Nobusada's ‘core-in-cage’ models reported in 2007. The methyl groups are removed in all models. Reprinted (adapted) with permission from ref. 92. Copyright 2008 American Chemical Society. | ||
During the same time, the crystal structure of the Au25(SCH2CH2Ph)18−[Oct4N]+ salt was successfully resolved independently by two experimental research groups.66,67 Remarkably, the Au13-core and semi-ring staple motif structures of Au25(SCH2CH2Ph)18− were in excellent agreement with the theoretical prediction. The discovery of the highly symmetric Au core with six extended –RS–Au–RS–Au–RS– motifs, as well as the finding of the atomic structure of Au102(p-MBA)44, has stimulated considerable theoretical interest in exploring the structure and electronic properties of various RS-AuNPs.
3.3 Learning from nature: structural rules for thiolate-protected gold clusters
Based on the resolved atomic structures of Au102(p-MBA)44 and Au25(SCH2CH2Ph)18−, some generic structural features can be identified. First, both clusters exhibit a quasi-highly symmetric Au core, i.e. D5h Au79-core and Ih Au13-core, respectively. Second, the Au cores are protected by a certain number of staple-like motifs through Au–S linkages, but the type and number of staple motifs are different. Due to the higher curvature of the icosahedral Au13-core, the six protecting staple motifs are all in extended or dimeric form (–RS–Au–RS–Au–RS–) in Au25(SCH2CH2Ph)18−, whereas the shorter monomeric –RS–Au–RS– staple motifs are dominant in Au102(p-MBA)44 due to the relatively smaller curvature of the Au79-core.Jiang et al. performed a series of computational studies based on DFT/PBE calculations to understand how thiolate binds to gold atoms at a cluster surface.93 In their first model, two isolated methyl thiol groups were closely placed on a truncated octahedral Au38 surface. After structural relaxation, a gold atom was lifted by two pre-adsorbed thiol groups and formed a monomeric –SR–Au–SR– motif. This investigation indicated that the formation of gold thiolate species is an energetically favorable process on the curved gold cluster. The energy analysis of a series of staple-covered Au38(MT)x clusters for x = 6–24 indicated the adsorption energy per –SMT group decreases quickly with the increase of coverage of the –SMT group when x was less than 12. The maximum coverage for isolated staple motifs on an Au38 core is reached when x is about 20. With a further increase in the number of thiol ligands beyond 20, the dimerization of “staple” motifs (with a surface Au atom bonded to two terminals of –SR–Au–SR– motifs) was observed. Structural optimization of configurations of staple motifs and the Au core based on a simulated annealing approach yielded a more optimal structure whose electronic energy was 1.6 eV lower than the Häkkinen's model (2006).63 In contrast to Häkkinen's model, the cyclic ring gold–thiolate motifs were no longer observed and the core structure was rather disordered. However, the agreement between the computed and measured optical absorption spectra was not so good. Shortly after, Jiang et al. proposed an improved structural model for Au38(MT)24, which was composed of a more symmetric Au12-core and a series of monomeric and dimeric staple motifs. This new model yielded a further decrease in the total electronic energy, which was more stable than their earlier one by ∼1.3 eV.94
Given the experimentally determined and theoretically predicted atomic structures of Au25(SCH2CH2Ph)18−, Au38(SR)24, and Au102(p-MBA)18−, we have suggested a generic structural formula for RS-AuNPs similar to the “divide-and-protect” concept.95 In view of the fact that both Au25(SCH2CH2Ph)18− and Au102(p-MBA)18 can be decomposed into a highly symmetric AuN core and a series of monomeric and dimeric protecting staple motifs, and that the staple motifs protect the symmetric AuN-core through the formation of Au–S linkages on the outmost shell of the Au core, we introduce a general structure formula for RS-AuNP (Aum(SR)n) as [Au]a+a′[Au(SR)2]b[Au2(SR)3]c, where a, a′, b and c are integers. The [Au]a+a′ is the interior Au core and it satisfies a condition that the number of core ‘surface’ Au atoms (a′) equals the sum of the end-points of the exterior motifs (2b + 2c), that is, each core surface Au atom is protected by one terminal of the staple motif. The values of a, a′, b and c must satisfy a + a′ + b + 2c = m and 2b + 3c = n (constraint conditions). For example, the structure formula of Au102(SR)44 and Au25(SR)18− can be rewritten as [Au]39+40[Au(SR)2]19[Au2(SR)3]2 and [Au]1+12[Au2(SR)3]6, respectively. Note that for Au102(SR)44, one constraint condition, namely, a′ = 2b + 2c, is not fully satisfied (as a′ = 40, while 2b + 2c = 42). This exception case is due to the fact that two Au atoms on the surface of the Au core are bonded with two S-terminals each. Tsukuda and co-workers proposed similar structural rules in their joint experimental and theoretical study of Au38(SR)24.47 They proposed three simple structural principles: (1) a highly symmetric Au core; (2) the number of dimeric staple motifs increases with the decrease of cluster size; and (3) each surface Au atom is bound by one S-terminal of the staple motifs. Hereafter, we refer the structural formula and the three structural principles as the “inherent structure rule” for constructing a structural model of thiolate-protected gold clusters.
Based on the “inherent structure rule”, five sets of structural divisions are suggested for Au38(SR)24: (i) [Au]2+24[Au(SR)2]12, (ii) [Au]3+22[Au(SR)2]9[Au2(SR)3]2, (iii) [Au]4+20[Au(SR)2]6[Au2(SR)3]4, iv)[Au]5+18[Au(SR)2]3[Au2(SR)3]6 and (v) [Au]6+16[Au2(SR)3]8, all satisfying the constraint conditions.95 Since the structures of the staple motif units are pre-defined, the structural prediction of Au38(SR)24 is simplified into a search for a reasonable structure for [Au]2+24, [Au]3+22, [Au]4+20, and [Au]5+18 that can match the geometry of the protecting staple motifs flawlessly. A set of initial [Au]a+a′ (a + a′ = 22–26) core structures are then built and they are covered by certain numbers of staple motifs, e.g. [Au(SR)2] and [Au2(SR)3]. The DFT optimizations are then applied to relax the proposed structures. We find that an isomeric structure whose group division is [Au]5+18[Au(SR)2]3[Au2(SR)3]6 exhibits exceptional stability, as shown in Fig. 4a. The Au23-core can be viewed as a bi-icosahedral structure with two Au13 icosahedrons fused by an Au3 face (Fig. 4b). The six [Au2(SR)3] motifs are distributed evenly on two icosahedral Au13 subunits, with an additional three [Au(SR)2] motifs bridging the middle of Au core. The DFT calculation indicates that this new structure is 2.04 eV lower in energy than the lowest-energy structure previously reported. Electronic structure analysis further shows a HOMO–LUMO gap of about 0.9 eV. Further simulations of thw XRD curve and UV-vis absorption spectrum for the new structure indicates good agreements between experimental and theoretical results (Fig. 4c). The prediction of a face-fused bi-icosahedral Au core in Au38(SR)24 is interesting due to its close relation with the known icosahedral Au13-core in Au25(SCH2CH2Ph)18− and reinforces the notion of a highly symmetric Au core for stabilizing the cluster structure. On the other hand, the ratio of monomeric and dimeric staple motifs (1.58) in the newly predicted structure is also within the two benchmark values corresponding to Au25(SCH2CH2Ph)18− (1.39) and Au102(p-MBA)18− (2.32), a trend also noticed by Tsukuda et al.47 All these analyses suggest this structural model should be very close to the realistic structure of Au38(SR)24.
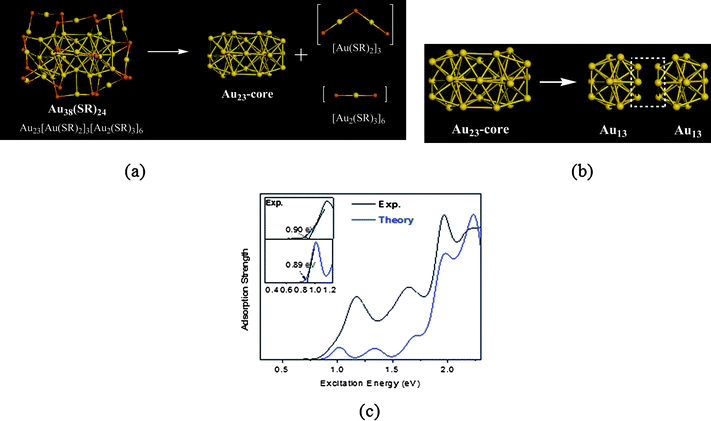 | ||
| Fig. 4 (a) Face-fused structural model predicted for Au38(SR)24; (b) Structural pattern of bi-icosahedral Au23-core. (c) Comparison of theoretical and experimental UV-vis absorption spectra. Reprinted (adapted) with the permission from ref. 95. Copyright 2008 American Chemical Society. | ||
In collaboration with Tsukuda and Häkkinen, Aikens et al. made a further improvement to the predicted structural model by making a number of different orientations of staple motifs.96 It was found from previous experimental studies that the Au38(SG)24 exhibits strong circular dichroism (CD) signals which are five times stronger than those from Au25(SG)18.14,19,97 The strong CD signals were assigned to metal-based transitions, which possibly included contributions from both the Au core and ligand-layer Au atoms. In our originally proposed Au38 model, the longer staple motif units are arranged in an idealized C3h symmetry and the whole cluster exhibits a C3h point-group symmetry. The TDDFT computation of the CD spectrum based on the C3h model yields a rather weak rotatory strength (less than 50 esu2 cm2), which is inconsistent with the experimental observation. By changing the orientations of the staple motifs, Aikens and co-workers found a new D3 symmetric structure with six –RS–Au–SR–Au–SR– motifs arranged in a zigzag form, which has an electronic energy ∼0.3 eV lower than the previously reported model,95 based on both LDA-Xα and PBE calculations with a triple-zeta polarized basis set. The computation of the CD spectrum of the improved model shows an increased rotatory strength below 2.2 eV.96 The highest rotatory strength is seen at nearly 1.95 eV (∼350 esu2 cm2), in agreement with the experimental observation.14,97
Jin and co-workers recently reported an improved synthesis method from which highly monodisperse, phenylethylthiolate-capped Au38(SC2H4Ph)24 clusters can be made (the yield increased to ∼25%). This opened up a new possibility of crystallization of the clusters. Indeed, shortly after Aikens's publication, the crystallization of the atomically monodisperse Au38(SC2H4Ph)24 nanoparticles in a mixed solution of toluene and ethanol solution was achieved.98 The X-ray crystallography revealed that the crystalline structure of Au38(SC2H4Ph)24 belongs to a triclinic space group P1 and the unit cell contains a pair of enantiomeric clusters. Atomic structure analysis indicated that the Au38(SC2H4Ph)24 is composed of a face-fused bi-icosahedron Au23-core and covered by six dimeric and three monomeric staple motifs, in good agreement with theoretical predictions.95,96 However, the crystalline structure shows a slightly different arrangement of the dimeric staple motifs on the icosahedral Au13 unit from the theoretical models, that is, the six dimeric staples are arranged in a staggered fashion (Fig. 5) with an inversion center in the fused Au3 plane.
 | ||
| Fig. 5 Atomic structure of Au38(SC2H4Ph)24 resolved from the single-crystal XRD. Reprinted (adapted) with permission from ref. 98. Copyright 2010 American Chemical Society. | ||
3.4 Structural models for Au12(SC2H4Ph)9+, Au18(SR)14, Au20(SC2H4Ph)16, Au44(SC2H4Ph)282− and Au144(SR)60
The experimental confirmation of the theoretically predicted structure of Au38(SR)24 lends credence to the “inherent structure rule”. Moreover, the confirmation of the bi-icosahedral Au23-core in Au38(SR)24 validates the speculation of “atomic ordering” in Au cores of all highly stable thiolate-protected gold clusters.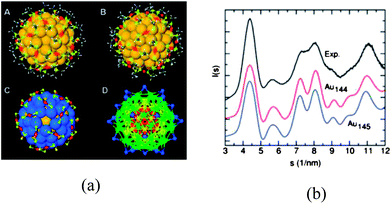 | ||
| Fig. 6 (a) Relaxed structure of Au144(SR)60 viewed through a 5-fold (A) and a 3-fold (B) symmetry axis. Yellow: Au in the Au114 core; orange: Au in the RS–Au–SR unit; bright yellow: S; gray: C; white, H. (C) Arrangement of the RS–Au–SR units covering the 60-atom surface of the Au114 core (blue). (D) The 144 gold atoms shown in different shells. (b) Comparison of experimental and theoretical powder-XRD curves. Reprinted (adapted) with permission from ref. 100. Copyright 2009 American Chemical Society. | ||
Jiang et al. first investigated a tetrahedron-core-based Au8(SR)6 cluster that is composed of a tetrahedron Au4 core and two dimeric staple motifs wrapping around two faces of the tetrahedron with the formation of four Au–S linkages.101 However, their DFT optimizations resulted in a somewhat open structure with two dimeric staple motifs capping opposing edges of the tetrahedron Au core with a 90° Au–S–Au angle. Although their electronic structure calculations suggested a quite large HOMO–LUMO gap of 3.23 eV for the cluster, it was thought to be chemically unfavorable due to the open structure of the cluster (which may be prone to chemical attacks). The model was later revised by capping two opposite edges of the tetrahedron with the monomic staple motifs, e.g. [Au(SR)2]. The optimized structure of neutral [Au]4[Au(SR)2]2 or Au6(SR)4 has the tetrahedral Au4-core with a slightly bent S–Au–S bond (∼160°) in the staple motif. The cluster has a HOMO–LUMO gap of 2.40 eV and is thought to be a more realistic model compared to Au8(SR)6 due to the half-unprotected core. In their subsequent study of effects of the length of staple motifs on the stabilization of the smaller thiolate-protected gold clusters, Jiang et al. presented a new tetrahedron Au4-core-based cluster, Au10(SR)8.102 In this new cluster, the tetrahedron Au4-core is wrapped by two extended trimeric staple motifs ([Au3(SR)4]). Geometric optimization and electronic calculation indicate that Au10(SR)8 is a good candidate that can accommodate a small tetrahedron Au4-core.
To investigate the possible existence of an octahedron Au core in certain thiolate-protected gold clusters, Jiang et al. constructed an octahedron Au6-core and used three dimeric staple motifs to cover the Au6-core. The cluster is referred to as Au12(SR)9.102 To remove the unpaired electron, a cation state of the cluster is investigated. After structural optimization, the optimized octahedral Au8-core in Au12(SMT)9+ is changed to D3d point-group symmetry (within a 0.06 Å tolerance), and its six facets are effectively wrapped by the thiolate ligands as shown in Fig. 7a and b. Topologically, the Au12S9 framework in the optimized Au12(SMT)9+ can be related to the familiar trefoil knot (Fig. 7c). Shortly after Jiang's theoretical prediction, an experimental isolation of Au12(SR)9 complexes (SR = N-acetylcysteine) was reported.103
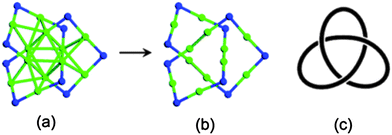 | ||
| Fig. 7 (a) and (b) are the framework of Au12(SMT)9+ proposed by Jiang et al. (c) A representation of cluster structure with a trefoil knot. Reprinted (adapted) with the permission from ref. 102. Copyright 2009 American Chemical Society. Au, green; S, blue. | ||
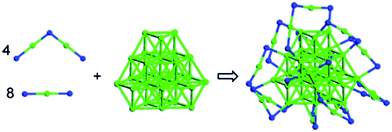 | ||
| Fig. 8 Structural model of Au44(SMT)182− constructed from the monomeric and dimeric staple motifs. Reprinted (adapted) with permission from ref. 105. Copyright 2010 American Chemical Society. | ||
Theoretical efforts have been made to explain the packing style of the Au atoms in Au20(SCH2CH2Ph)16. In view of the relatively low Au![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :SR ratio in Au20(SR)16, both Pei et al. and Jiang et al. proposed the possible existence of much-extended staple motifs, e.g. –RS–Au–RS–Au–RS–Au–RS– or [Au3(SR)4] in Au20(SCH2CH2Ph)16.101,108 As such, the structural formula is expanded as [Au]a+a′[Au(SR)2]b[Au2(SR)3]c [Au3(SR)4]d, where the [Au2(SR)3] type of staple motif was proposed for the first time. Considering the constraint conditions, only one division of [Au]0+8[Au3(SR)4]4 is allowed. Regarding the packing pattern of the Au core, Jiang et al. and Pei et al. suggested different models. Four structural forms of the Au8 core, including the cube, Td, cage, and fcc forms were proposed by Jiang et al. (see Fig. 9a).101 Pei et al. suggested a much looser structure for the Au8-core with a prolated shape, which can be viewed as the fusion of two tetrahedron Au4 units through edges.108 Three near-degenerate isomeric structures are attained (Iso1–Iso3 as shown in Fig. 9b) based on the edge-fused Au8-core. The electronic energy calculations (at the DFT/PBE level with the TZP basis set) with the –R group being simplified by a –CH3 group indicate that Pei et al.'s model is slightly lower in energy by ∼0.4 eV. Further calculations of the optical properties of the cluster indicate that the prolated Au8-core-based model (Iso2) nearly reproduces the optical absorption gap and major peaks in the measured UV-vis absorption curve, as well as the overall patterns of the measured powder-XRD curve (Fig. 9c). The prolated Au8-core-based model is considered as a leading candidate for the structure of Au20(SR)16.
:SR ratio in Au20(SR)16, both Pei et al. and Jiang et al. proposed the possible existence of much-extended staple motifs, e.g. –RS–Au–RS–Au–RS–Au–RS– or [Au3(SR)4] in Au20(SCH2CH2Ph)16.101,108 As such, the structural formula is expanded as [Au]a+a′[Au(SR)2]b[Au2(SR)3]c [Au3(SR)4]d, where the [Au2(SR)3] type of staple motif was proposed for the first time. Considering the constraint conditions, only one division of [Au]0+8[Au3(SR)4]4 is allowed. Regarding the packing pattern of the Au core, Jiang et al. and Pei et al. suggested different models. Four structural forms of the Au8 core, including the cube, Td, cage, and fcc forms were proposed by Jiang et al. (see Fig. 9a).101 Pei et al. suggested a much looser structure for the Au8-core with a prolated shape, which can be viewed as the fusion of two tetrahedron Au4 units through edges.108 Three near-degenerate isomeric structures are attained (Iso1–Iso3 as shown in Fig. 9b) based on the edge-fused Au8-core. The electronic energy calculations (at the DFT/PBE level with the TZP basis set) with the –R group being simplified by a –CH3 group indicate that Pei et al.'s model is slightly lower in energy by ∼0.4 eV. Further calculations of the optical properties of the cluster indicate that the prolated Au8-core-based model (Iso2) nearly reproduces the optical absorption gap and major peaks in the measured UV-vis absorption curve, as well as the overall patterns of the measured powder-XRD curve (Fig. 9c). The prolated Au8-core-based model is considered as a leading candidate for the structure of Au20(SR)16.
![(a) Au8-core suggested by Jiang et al. [ref. 101]. (b) Low-energy isomer structures predicted by Pei et al. [ref. 108]. (c) Comparison of the theoretical UV–vis adsorption spectra of Iso1–Iso3 with experiments. Reprinted (adapted) with permission from ref. 101 and 108. Copyright 2009 American Chemical Society.](/image/article/2012/NR/c2nr30685a/c2nr30685a-f9.gif) | ||
| Fig. 9 (a) Au8-core suggested by Jiang et al. [ref. 101]. (b) Low-energy isomer structures predicted by Pei et al. [ref. 108]. (c) Comparison of the theoretical UV–vis adsorption spectra of Iso1–Iso3 with experiments. Reprinted (adapted) with permission from ref. 101 and 108. Copyright 2009 American Chemical Society. | ||
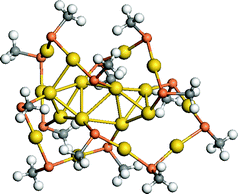 | ||
| Fig. 10 (a) Structural model for Au18(SR)14 with –R is simplified by a methyl group. S atoms are in red, C atoms are in gray, and H atoms are in white. Au atoms in the dimer and trimer motifs are in orange and the core Au atoms are in yellow. Reprinted (adapted) with permission from ref. 110. Copyright 2012 Royal Society of Chemistry. | ||
The calculated UV-vis absorption spectra and powder-XRD curve based on the theoretical model for the Au18(SG)14 cluster indicate reasonable agreement with the experimental measurement. In particular, the calculated CD spectra have two positive and negative peaks in the 1.5–3.5 eV range, which are also in agreement with the experimental curve.111 Nonetheless, by examining the energetics of the Au18(SR)14 + 2(AuSR)4 → Au20(SR)16 reaction, where (AuSR)4 is a cyclic tetramer and Au20(SR)16 is Pei et al.'s model,108 Garzón et al. found an even higher stability by 0.45 eV for the Au20(SR)16 cluster in the presence of cyclic (AuSR).
3.5 ‘Staple fitness’ and force-field based “divide-and-protect” scheme
The “inherent structure rule” and the “divide-and-protect” concept dramatically increase the likelihood of predicting the correct structural model for a thiolated gold nanoparticle. However, two more detailed questions arise when applying the rule and concept: (1) for a given symmetric Au core, several combinations of the arrangement of staple motifs are possible. How can one assign the most favorable arrangement? (2) There still exist many possibilities in constructing an AuN-core that satisfies the proposed structural formula. How to construct more sensible Au core structures is another important question.Jiang and Pei and their co-workers independently addressed the two questions in their recent theoretical studies of thiolate-protected gold clusters, concomitant with their structural predictions of two newly synthesized RS-AuNPs, Au19(SR)13 and Au24(SR)20, respectively. Jiang pointed out that a way to address the first question can be the concept of staple fitness.112 For a given highly symmetric AuN-core, the surface Au atoms on the core can be viewed as one-to-one dots that can bond to one S-atom terminal of the staple motifs. Since the staple motifs protect the Au core via S-atom terminals, a constraint must be enforced on the distance between the pair of surface Au atoms. For the monomeric and dimeric staple motifs, the head-to-tail distance usually falls into the range of 3.5 to 5.5 Å. Hence, two nearest-neighbor Au atoms with a distance of typically 2.8 Å cannot be connected by a staple motif. Therefore, one can set the nearest-neighbor distance in the Au core as a minimum distance constraint. By applying this constraint on distance, many combinations can be filtered out (a “pruning” process). The surviving combinations after the pruning process are ranked according to the total pair distance (TPD), which is defined as the sum of all inter-pair distances for a given combination of N pared-up dots. Taking the icosahedral Au13-core as an example, on its surface the smallest nearest-neighbour Au–Au distance is 2.81 Å and the greatest Au–Au distance is 5.34 Å. There are 368 combinations of staple motifs within these two distance limits. The 368 combinations are then ranked by their TPD from high to low. The combinations of staple motifs having the greatest TPD are found to be in good agreement with the experimental structure. A similar approach can be applied to derive the arrangements of staple motifs on a bi-icosahedral Au23-core for Au38(SR)24 from the nine theoretical models proposed previously by Pei et al. and by Aikens et al.. The TPD measurement clearly indicates slight differences between the nine models.95,96 The model with the greatest TPD value is indeed in good agreement with the experimental observation. These analyses suggest that the staple-fitness approach appears to be an efficient way to assign the most favorable mode of staple motif arrangement on a given Au core.
The staple-fitness method112 has also been applied to predict the structure of a recently synthesized thiolate-protected gold cluster, Au19(SR)13. This cluster was isolated by Jin and co-workers using a kinetically controlled size-focusing synthesis and its structural composition was confirmed by mass spectroscopy characterizations.113 By assigning the Au atoms into the core and staple motifs based on the proposed structural formula and constraints, two scenarios for the numbers of monomeric and dimeric motifs can be deduced: [Au0+12][Au(SR)2]5[Au2(SR)3] and [Au1+10] [Au(SR)2]2[Au2(SR)3]3. By applying the staple-fitness method to a series of constructed highly symmetric Au cores, the lowest-energy structure derived from the fitness combination of staple motifs includes two monomeric staple motifs (which cap the two opposite concave regions of a vertex-truncated Au11-core) and three dimer motifs (which protect the convex regions) (Fig. 11a and b). The simulated XRD curve (Fig. 11c) based on the optimized cluster is in good agreement with the experimental one, and the computed optical gap (1.3 eV) is also close to the experimental value (1.5 eV). In light of the predicted core structure of Au19(SR)13, the vertex-truncated icosahedral Au11-core is closely related to that in Au25(SR)18. The Au19(SR)13 cluster is considered as an important intermediate towards the formation of Au25(SR)18 from smaller clusters.
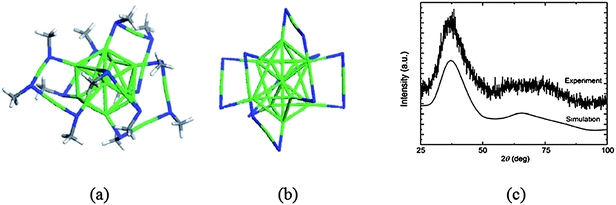 | ||
| Fig. 11 Structural model for Au19(SR)13 (a) with or (b) without the representation of methyl groups. The S, C, H and Au atoms are in dark blue, gray, white and green, respectively. (c) Comparison of XRD curves from experimental measurement and theoretical simulation. Reprinted (adapted) with permission from ref. 112. Copyright 2011 John Wiley & Sons, Inc. | ||
To address the question of how to efficiently search for a reasonable structure of an Au core, we have recently proposed a classical force-field based “divide-and-protect” method.114 The key steps involved in the force-field based “divide-and-protect” method are illustrated in Scheme 2. Any Aum(SR)n cluster can be viewed as an AuN-core fully protected by different staple motifs (i.e., the “divide-and-protect” concept). Since the length and number of staple motifs are defined in the extended structural formula, e.g. Aua+a′[Au(SR)2]b[Au2(SR)3]c[Au3(SR)4]d…, where Aua+a′ represents the Au core and b, c and d denote the number of different-sized staple motifs, the selection of different protecting staple motifs for a given cluster depends strongly on the ratio Au:SR. Typically, more extended staple motifs such as [Au3(SR)4] and [Au4(SR)5] etc. are only incorporated into the structural division when the ratio Au:SR is relatively small (e.g. <1.25). As structures of staple motifs are pre-defined, the structural prediction of RS-AuNPs turns into a search for the proper Au core structure (Aua+a′) that matches the protecting staple motifs in a flawless fashion. The degrees of freedom for a cluster are thus reduced from 3m + 5n (with R = CH3) to 3(a + a′), for which the computational cost is dramatically reduced.
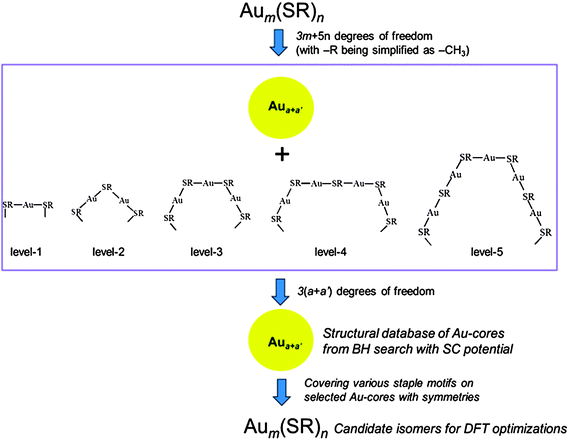 | ||
| Scheme 2 Illustration of the force-field based divide-and-protect method. Reprinted (adapted) with permission from ref. 114 @ Copyright 2012 American Chemical Society. | ||
To seek the best Au core structure, the combined basin-hopping algorithm and empirical Sutton–Chen potential for Au–Au interactions are suggested. The empirical potential is much more efficient in generating highly symmetric Au core structures with less computational costs than ab initio methods. As reported previously, the classical potentials favor geometric packing of the gold atoms. The combination of the BH algorithm and the Sutton–Chen potential can quickly enumerate numerous desirable and highly symmetric Au core structures. From the generated structural database, one can therefore pick up some typical structures that satisfy the constraint conditions described in Section 3.3 for further assembly with the pre-defined staple motifs. The force-field based “divide-and-protect” approach has been validated with three benchmark models, Au25(SR)18−, Au38(SR)24 and Au102(SR)44 whose Au core is Ih-Au13, D3h-Au23, and D5h-Au79, respectively. The stable local minima, generated from the combined BH search and SC potential, are quite stable. Note that the DFT-based global search is less efficient for generating an Au core database due to the lack of symmetric Au cluster structures from the DFT-based search.
The force-field-based divide-and-protect approach has also been applied to determine the structure of another recently synthesized cluster, Au24(SR)20.114 Because the ratio Au:S of Au24(SR)20 is slightly less than that of Au20(SR)16, which possesses a trimeric staple motif due to the relative small Au:S ratio (1.25), more extended staple motifs such as tetrameric and pentameric motifs ([Au4(SR)5] and [Au5(SR)6]) should be incorporated into the structural divisions. By applying the “inherent structure rule”, five structural divisions Au8[Au3(SR)4]2[Au5(SR)6]2, Au8[Au3(SR)4][Au4(SR)5]2[Au5(SR)6], Au8[Au4(SR)5]4, Au8[Au(SR)2][Au5(SR)6]3 and Au8[Au2(SR)3][Au4(SR)5][Au5(SR)6]2 are considered, all containing an Au8-core. Several Au8-core structures are thus generated via the BH search with the SC potential. It is found that the D2d symmetric Au8-core protected by four pentameric staple motifs exhibits exceptional stability according to DFT calculations using different exchange-correlation functionals (PBE, TPSS,115 and M06 (ref. 116)). This cluster also gives the best agreement between theoretical and experimental optical absorption spectra and XRD patterns among different isomer structures. Interestingly, the predicted lowest-energy structure of Au24(SR)20 has two tetrameric [Au3(SR)4] and two pentameric [Au5(SR)6] motifs interlocked like a linked chain from one end to another of the prolate Au8-core, as shown in Fig. 12. One Au atom in the pentameric [Au5(SR)6] motif is coordinated to four Au atoms in a tetrameric [Au3(SR)4] motif, which is termed a catenane-like staple motif. Topologically, the predicted structure can be also viewed as a combination of two sets of symmetric interlocked oligomers Au5(SR)4 and Au7(SR)6. In fact, a recently proposed growth mechanism for RS-AuNPs indicates that Aun(SR)n−1 oligomers are likely to be formed during the initial growth of RS-AuNPs from the reduction of homoleptic Au(I) thiolates.117
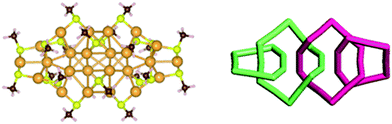 | ||
| Fig. 12 Predicted structure model for Au24(SR)16 (left panel). The topological structural model with methyl groups are removed for clarity (right panel). The Au, S, C, and H atoms are in khaki, yellow, grey and white, respectively (left panel). Reprinted (adapted) with permission from ref. 114. Copyright 2012 American Chemical Society. | ||
In contrast to all previously determined or predicted structures of RS-AuNPs, the catenane-like staple motifs are only seen in the homoleptic Au(I) thiolates, which can exhibit either catenane, helix, or crown configurations.118–120 The catenane-like staple motif in Au24(SR)20 is therefore considered as an intermediate structure that is part of the structural transition from a polymer chain-like form to a core-stacked form for thiolate-protected gold clusters. Here we define the core-stacked RS-AuNP by Aum(SR)n with m > n. The finding of catenane-like staple motifs in Au24(SR)20 suggests that in the low Au:SR ratio limit (i.e., approaching to 1:1), the interlocked staple motifs may become a prevalent conformation in RS-AuNPs. In fact, those isomers of Au24(SR)20 without the formation of catenane structures generally have higher energies, indicating the inter-locked structure is energetically favorable for thiolated gold clusters with a low Au:SR ratio.114 Moreover, Au24(SR)20 has a similar bi-tetrahedron Au8-core as that of Au20(SR)16. The tetrahedron Au4-core has been predicted theoretically and observed experimentally for several small ligand-protected clusters such as Au10(SR)8 (ref. 101) (with Au:SR = 1.25) and Au4(PPh3)42+.121 On the other hand, a bi-pyramidal Au6-core that has been previously predicted for Au12(SR)9+ (with Au:SR = 1.33) and revealed in Au6(PPh3)62+ can also be viewed as edge-fusion of two Au4 units.101,122 These analyses suggest that a major structural transition for the Au core may occur when the Au:SR ratio approaches the 1:1 limit. The close-packed tetrahedral Au4 is thought of as a common unit for the Au core in small-sized RS-AuNPs with a relatively small Au:SR ratio (e.g., <1.39).114
3.6 Structure of heteroatom-doped Au25(SR)18q (q = −2, −1, 0, 1, 2)
Heteroatom doping is an important way to modify the properties of metal clusters. For gas-phase gold clusters, a number of heterometal-doped Au clusters have been investigated via joint theoretical and experimental studies.123,124 In the case of thiolate-protected gold clusters, Jiang et al. studied theoretically a set of bi-metal M@Au24(SMT)18q clusters where M was the dopant metal atom that replaced the central Au atom in the 13-atom icosahedral core.125,126 Different charge states (q = −2, −1, 0, 1, 2) were considered to preserve the 8e closed electron-shell according to the superatom model (see Section 4.1 below). It was found that metal atoms such as Pd, Pt, Ni, Cu, and Ag etc. led to stable cluster structures with the icosahedral core structure being well preserved. In particular, doping with a Pt atom (i.e. Pt@Au24(SMT)182−) yields a large HOMO–LUMO gap that is slightly larger than that of Au25(SCH2CH2Ph)18−. Shortly after Jiang's theoretical prediction, several mass spectroscopy experiments showed that the Au25(SCH2CH2Ph)18− nanocluster can be doped by a single Pd (or Ag atom), becoming Pd@Au24(CH2CH2Ph)18−.127–131 Moreover, the one or two Pd-atom-doped Pdx@Au38–x(SCH2CH2Ph)24 nanoclusters were also synthesized.127,129 Kacprzak et al. and Walter et al. performed independent theoretical studies to determine the possible position of the doped metal atom in M@Au24(SMT)18q. They found that the energetically most favorable position for the Pd atom was in the center of the icosahedral core, as supported by the good agreement between measured and computed optical absorption curves.132,133 Other metal atoms such as Ag and Cr were suggested to replace Au atoms in the metal-thiolate ligand shell.3.7 Structures of fragment clusters: AumSn− and Au21(SR)14−
Recent MALDI-MS experiments showed that fragments of Au25(SR)18− led to the formation of a series of high-abundance AumSn− clusters.134 Among them, the mono-anionic clusters Au25S12−, Au23S11− and Au27S13− exhibit high stability. The formation of complex patterns of fragmented species includes many AumSn+ and AumSn− clusters. A high abundance (magic number) of smaller AumSn− clusters such as Au13S8−, Au12S8− and Au6S4− was also observed in the fast-atom-bombardment MS of Au25(SR)18− in both positive and negative charge modes and in the ion-mobility MS of Au25(SR)18−.135–137Using the combined DFT and BH algorithm, a unique core-in-cage structure was found for Au19S12−, Au23S11−, Au25S12− and Au27S13− (Fig. 13a).138 In such a core-in-cage structure, S atoms form the vertices of the cage and are connected by the Au atoms at the edges. The rest of the Au atoms form a symmetric core inside the cage. The core-in-cage structure of gold-sulfide clusters is distinct from the thiolate-protected gold clusters. A detailed study of the structural evolution of binary gold-sulfide cluster anions (AumSn−) in the smaller sizes was reported by us using a similar theoretical method.139 Highly stable AumSn− species such as Au6S4−, Au9S5−, Au9S6−, Au10S6−, Au11S6−, Au12S8− and Au13S8− as detected in the ion mobility MS experiment of Au25(SCH2CH2Ph)18 were found to possess unique symmetric hollow cage structures such as quasi-tetrahedron, pyramidal, quasi-triangular prism, or quasi-cuboctahedron, respectively (Fig. 13b). The formation of these polyhedron structures were attributed to the high stability of the linear S–Au–S unit. An “edge-to-face” growth mechanism was also proposed to understand the structural evolution of small AumSn− clusters from the quasi-tetrahedron to quasi-cuboctahedron structures.
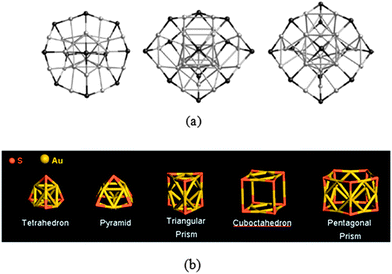 | ||
| Fig. 13 (a) Core-in-cage models for Au23S11−, Au25S12−, and Au27S13−. Adapted from ref. 138 @ Copyright 2011 John Wiley & Sons, Inc. (b) Hollow cage-like structures for Au6S4−, Au9S5−, Au9S6−, Au12S8−, and Au15S12−. Reprinted (adapted) with permission from ref. 139. Copyright 2009 American Chemical Society. | ||
The Au21(SCH2CH2Ph)14− was another magic-number cluster that was detected in MS experiments under several conditions.127,135,136 From its structural composition, it can be viewed as the loss of an [AuSR]4 fragment from the Au25(SR)18− cluster. The structure of Au21(SR)14− was examined recently based on DFT calculations from a step-by-step removal of an AuSR fragment from Au25(SR)18− according to the proposed structural principles.140 A mechanism for the replacement of the dimeric motif [Au2(SR)3] by the monomeric [Au(SR)2] motif was proposed as an energetically favorable process according to DFT calculations, which led to a structural model with an icosahedral Au13-core covered by four monomeric and two dimeric staple motifs.
4. Understanding the “magic” stability of thiolate-protected gold clusters
An interesting question related to the stability of thiolate-protected gold clusters is why the “staple motif” is common in these clusters. The formation of the staple motif units is largely caused by the strong Au–S interactions. With the decrease of the cluster's size, the increased curvature of the gold cluster renders the surface Au atom more reactive to the thiol groups. As demonstrated by Jiang's DFT calculations,93 the formation of a staple motif protection unit is a spontaneous process when two thiol groups are closely adsorbed on a curved gold cluster surface. The high curvature of the nano-sized gold cluster is thus a key for the favorable formation of staple motif units. However, numerous experimental studies demonstrated a few clusters such as Au25(SR)18, Au38(SR)24 and Au102(SR)44etc. exhibited extraordinary stabilities, or “magic” stability. To understand the intrinsic stability or “magic” stability of these clusters, both the electronic effects (the superatom model) and the thermodynamic factors have been taken into consideration.4.1 Superatom model
The superatom model for metal clusters with a closed electron-shell structure was first proposed to explain the high abundance of certain magic-number alkali metal clusters (N = 2, 8, 18, 20, 40, and 58) observed in the MS experiments.141 The model predicts that free valence electrons in a metal cluster may occupy a set of orbitals that belong to an entire group of atoms rather than individual atoms separately (also known as a homogeneous electron gas or “jellium” model). The superatom model has been used to explain the electronic structures and high stabilities of certain ligand-protected gold clusters (such as Au25(SR)18−, Au102(SR)44 and gold–phosphane–halide clusters) by Häkkinen et al. in 2008.142The total number of free valence electrons (n*) associated with a thiolate-protected gold cluster AuM(SR)Nq can be counted based on the formula n* = M − N − q, where M is the number of Au(6s1) electrons, N and q are the number of electron-withdrawing ligands (such as a thiol group) and the net charge of the cluster, respectively. From this electron counting formula, Au25(SR)18− and Au102(SR)44 have 8 and 58 free valence electrons, respectively, which are indeed the electronic magic numbers that correspond to a closed electron–shell within the framework of the spherical jellium model. Similarly, Au12(SR)9+ and Au44(SR)282− clusters also possess magic numbers of free valence electrons (2e and 18e, respectively) on the basis of the spherical jellium model. Earlier MS experiments by Whetten et al. revealed the existence of several magic-number clusters with 5, 8, 14, 22 and 28 kDa Au cores.13,99 Among them, the 5, 8 and 28 kDa cores were assigned to Au25, Au38, and Au144, respectively. Recently, a structural formula for the 14 kDa nanocluster was identified as Au68(SR)34 based on MALDI-TOF MS measurements.143 The Au68(SR)34 cluster possesses a 38e electron shell according to the superatom model. Through an analysis of the angular momentum of Kohn–Sham orbitals, Häkkinen et al. presented evidence for the existence of a set of superatom orbtials in thiolate-protected gold clusters.142 The reorganization of the electronic structures of the gold core upon passivation was shown in Au102(SR)44 from the 3S + 2D + 1H band of states.
Nonetheless, not all experimentally produced thiolate-protected gold clusters exhibit magic numbers of free valence electrons as described by the spherical jellium model. The Au38(SR)24 is a highly stable cluster that has been detected in many experiments under different reaction conditions, but the electron counting rule suggests it has fourteen free valence electrons that occupy the superatom orbitals. Similar situations were found for many other clusters such as Au18(SR)14 (4e), Au19(SR)13 (6e), Au20(SR)14 (4e), Au24(SR)16 (4e) and Au144(SR)60 (84e). The prediction of non-spherical Au cores in these clusters can be understood from a modified electron shell model. According to the ellipsoidal electron shell structure proposed by Clemenger,144 a spherical superatom electron shell for a metal cluster can be further divided into several ellipsoidal subshells. An ellipsoidal electronic shell may explain the formation of a metal cluster with a non-spherical shape. The prolate shape of the Au cores in Au20(SR)14, Au24(SR)20 and Au38(SR)24 is consistent with this explanation.
Besides providing an explanation of the magic-numbers, electronic structures, and geometries of thiolate-protected gold clusters, the superatom model can be also used to understand the optical absorption, circular dichroism, and EPR spectra of the clusters. Based on both the superatom and ligand band orbitals, Aikens and co-workers reported a detailed theoretical analysis of the electronic excitation modes of several ligand-protected gold clusters such as Au25(SR)18 and Au38(SR)24.68,96,145
4.2 Thermodynamic and chemical analyses of RS-AuNPs
Although the superatom model can provide an explanation of the magic numbers and sizable HOMO–LUMO gaps of thiolated-gold clusters, it does not include the thermodynamic effect that may be another important factor that controls the stability of clusters. Thermodynamic and chemical analyses of the stability of the Au102(SR)44 cluster were performed by Reimers et al. and Han et al. based on DFT calculations.146,147 By examining the thermodynamic stabilities of various structural models such as Au79(MT)40, [Au79(MT)40]−, Au102(p-MBA)44, Au100(MT)42, Au102(MT)44, and Au104(MT)46 through dissociation energy analysis, Reimers et al. found that the thermodynamic stability of these clusters varies considerably without an obvious correlation with the electronic shell closure.146 The strong local chemical effects were suggested to regulate the chemical properties of the thiolate-protected gold clusters, in lieu of global electronic structures as described by the superatom model. Han et al. arrived at similar conclusions in their thermodynamic analysis of a series of thiolate-protected gold clusters with Au core sizes ranging from 98 to 106.147 They suggested that the magic-number and high stability of the thiolate-protected gold cluster Au102(SMT)44 are not entirely due to the 58e closed electron shell or a relatively large HOMO–LUMO gap, because some other clusters with a similar Au core structure and a similar arrangement of staple motifs but without such a closed electron shell (e.g. n* ≠ 58) can possess an even larger HOMO–LUMO gap than Au102(SMT)44. Three factors were proposed to account for the high stability of Au102(SMT)44, namely, effective staple-motif formation, high stability against dissociation, and a large HOMO–LUMO gap.5. Structure- and size-dependent catalytic activity of thiolate-protected gold clusters
It is well known that the bonding between metal and ligand generally involves electron transfer between the ligand and the metal. Such electron transfer enables the metal to carry a small amount of charge, which may significantly enhance the catalytic activity of metal clusters.148 As an RS-AuNP consists of an inner Au core and outer staple motifs, the different charge state of the Au atoms in the ligand layer and in the gold core implies that they may undergo different degrees of chemical attack. To date, few theoretical and experimental studies of the catalytic mechanism of RS-AuNPs are reported. Several recent experimental studies show that Au25(SR)18− is an active catalyst in many organic reactions such as hydrogenation, styrene oxidation and O2 activations.149–152 The discovery of the conversion of anionic Au25(SCH2CH2Ph)18− to neutral Au25(SCH2CH2Ph)18via air oxidation is very interesting because bare gold clusters in this size range are inert to O2 activation.151 Jin and co-workers proposed that the charged Au13-core is a major active site for O2 activation.149,150 However, this mechanism has not been fully supported by theoretical calculations. Based on DFT calculations, Lopez-Acevedo et al. studied the catalytic activity of several ligand-protected gold clusters including Au25(SMT)18−, Au38(SMT)24, Au102(SMT)44 and Au144(SMT)60.152 Their DFT calculations indicate that the adsorption energy of an O2 molecule on the Au13-core is enhanced by the removal of one or two ligands from the original thiolate-protected gold clusters, and is strongly dependent on the cluster size. The intact clusters without the removal of ligands indeed resist the O2 adsorption. A nearly linear relationship between the HOMO–LUMO gap and the O2 adsorption energy is derived. The electronic quantum-size effects (particularly the magnitude of the HOMO–LUMO gap) along with the defects on the clusters were concluded to be two critical factors for binding and reducing oxygen to a more active form. As many experimental observations show that the ligand shell is preserved during the catalytic reactions,149−152 further theoretical studies on the catalytic mechanism of such ligand-protected gold clusters are needed to understand the role of cluster structure and ligands in various catalytic reactions.6. Concluding remarks
The determination of the atomic structure of thiolate-protected gold clusters is of critical importance to the understanding of the physical and chemical properties of these special cluster species. Two recent breakthroughs in the determination of the atomic structures of [Au25(SCH2CH2Ph)18]q (q = −1, 0) and Au102(p-MBA)44 from X-ray crystallography provide deeper insights into the structural properties of these complex clusters, as well as two benchmark systems for researchers to develop generic “inherent structure rules” for this class of clusters. The “inherent structure rule” has been found to be quite reliable in previous theoretical predictions of structural patterns in various synthesized thiolate-protected gold clusters. Theoretical studies have predicted a possible structural transition in thiolate-protected gold clusters with an Au:SR ratio close to 1:1, a phenomenon correlated with the initial nucleation stage in the growth of clusters. In particular, the prediction of much extended catenane-like staple motifs in Au24(SR)20 as well as common tetrahedral Au4-units as the Au core in Au12(SR)9+, Au20(SR)14 and Au24(SR)20 supports the existence of a major structural transition for thiolate-protected gold clusters at relatively small sizes. Moreover, the predicted truncated icosahedral Au12-core and bi-icosahedral Au23-core in Au19(SR)13 and Au38(SR)24, respectively, can be viewed as a manifestation of the structural evolution of Au cores from small-to-medium sizes. Despite notable progress in the predictive capability of atomic structures for thiolate-protected gold clusters, many research issues still need to be addressed. Among others, the most important need is continuous tests of the reliability of theoretical predictions. In Table 1, we provide a summary of the structural properties (determined from either experiments and/or theoretical predictions) of various alkylthiolate-protected gold clusters reported in the literature. To date, only two theoretical models, i.e. Au25(SR)18− and Au38(SR)24, have been successfully confirmed by experiments. Further experimental verification for the existence of extended “staple motifs” such as [Au3(SR)4], and [Au5(SR)6] are greatly needed to support theoretical predictions. Moreover, it remains unclear whether the theoretical basis of the “inherent structure rule” is universal for all types of thiol ligand-protected gold clusters such as Au44(SPh)242− and Au36(SPh)23 with relatively stronger acidic –SPh groups. Another issue that remains to be addressed is the dynamic process of nucleation during the growth of thiolate-protected gold clusters from the reduced form of gold(I)–thiolate polymers. Aikens and co-workers have theoretically studied this process by examining the electron and hydride addition to gold(I)–thiolate oligomers (AuN(SR)N, N = 3–7).153 The addition of a hydride ion to gold(I)–thiolate oligomers led to the production of free thiols and Au(0)-containing chain-like oligomers, which is considered as a possible intermediate for the growth of nanoclusters. Nevertheless, how these Au(0)-containing intermediates further grow into a nanoparticle is still largely unknown. In particular, the origin of the preferential formation of the magic-number Au25(SR)18− cluster in most synthesis conditions deserves special attention from theorists. Finally, the catalytic mechanism and active sites on the clusters deserve focused study in the future. Recent experiments have revealed that thiolate-protected gold clusters are active in many organic catalytic reactions such as hydrogenation, styrene oxidation and O2 activation.149–151 Few theoretical studies have been reported on the origin of catalytic activity and the structure–activity relationship of these clusters. With more atomic structures of thiolate-protected gold clusters being determined, a better understanding of the structure–activity selectivity relationship is expected in the years to come.
| RS-AuNPs |
N
Au:NS |
Au core | Type and number of staple motifs | n* | H–L Gap (eV) | ||||
|---|---|---|---|---|---|---|---|---|---|
| –SR–Au–SR– | –SR–Au–SR–Au–SR– | –SR–Au–SR–Au–SR–Au–SR– | –SR–Au–SR–Au–SR–Au–SR–Au–SR– | –SR–Au–SR–Au–SR–Au–SR–Au–SR–Au–SR– | |||||
| Au10(SR)10ref. 119 | 1.00 | n/a | [0 | 0 | 0 | 0 | 0] | 0 | 2.70 |
| Au24(SR)20ref. 114 | 1.20 | Au8 | 0 | 0 | 2 | 0 | 2 | 4 | 1.47 |
| Au20(SR)16ref. 108 | 1.25 | Au8 | 0 | 0 | 4 | 0 | 0 | 4 | 2.10 |
| Au18(SR)14ref. 110 | 1.29 | Au8 | 0 | 2 | 2 | 0 | 0 | 4 | 1.63 |
| Au12(SR)9+ref. 102 | 1.33 | Au6 | 0 | 3 | 0 | 0 | 0 | 2 | 1.70 |
| Au25(SR)18−ref. 66, 67 and 92 | 1.39 | Au13 | [0 | 6 | 0 | 0 | 0] | 8 | 1.33 |
| Au19(SR)13ref. 112 | 1.46 | Au11 | 2 | 3 | 0 | 0 | 0 | 6 | 1.50 |
| Au21(SR)14ref. 127, 135 and 136 | 1.50 | Au13 | 4 | 2 | 0 | 0 | 0 | 7 | 1.84 |
| Au44(SR)282−ref. 104 and 105 | 1.57 | Au28 | 8 | 4 | 0 | 0 | 0 | 18 | 1.60 |
| Au36(SR)23ref. 106 | 1.57 | Unknown | 13 | 0.90 | |||||
| Au38(SR)24ref. 21, 22 and 25 | 1.58 | Au23 | [3 | 6 | 0 | 0 | 0] | 14 | 0.90 |
| Au40(SR)24ref. 154 | 1.67 | Unknown | 16 | 1.00 | |||||
| Au68(SR)34ref. 143 | 2.00 | Unknown | 34 | 1.20 | |||||
| Au102(SR)44ref. 64 | 2.32 | Au79 | [19 | 2 | 0 | 0 | 0] | 58 | 0.50 |
| Au144(SR)60ref. 48 and 100 | 2.40 | Au114 | 30 | 0 | 0 | 0 | 0 | 84 | 0 |
| Au333(SR)79ref. 155 | 4.22 | Unknown | 254 | 0 | |||||
Acknowledgements
YP is supported by the Natural Science Foundation of China (Grant No. 21103144) and Academic Leader Program in Xiangtan University (10QDZ34). XCZ is supported by grants from US NSF (EPS-1010094), ARL (W911NF1020099), and a seed grant by the Nebraska Center for Energy Sciences Research.References
- P. Gruene, D. M. Rayner, D. M. Redlich, A. F. G. van der Meer, J. T. Lynon, G. Meijer and A. Fielicke, Science, 2008, 321, 674–676 CrossRef CAS.
- J. Li, X. Li, H.-J. Zhai and L.-S. Wang, Science, 2003, 299, 864–867 CrossRef CAS.
- S. Bulusu, X. Li, L.-S. Wang and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 8326–8330 CrossRef CAS.
- H. Häkkinen and U. Landman, Phys. Rev. B: Condens. Matter, 2000, 62, R2287–2290 CrossRef.
- F. Furche, R. Ahlrichs, P. Weis, C. Jacob, S. Glib, T. Bierweiler and M. M. Kappes, J. Chem. Phys., 2002, 117, 6982–6990 CrossRef CAS.
- W. Huang and L.-S. Wang, Phys. Rev. Lett., 2009, 102, 153401-1–153401-4 Search PubMed.
- M. Ji, X. Gu, X. Li, X. Gong, J. Li and L.-S. Wang, Angew. Chem., Int. Ed., 2005, 44, 7119–7123 CrossRef CAS.
- A. Lechtken, D. Schooss, J. R. Stairs, M. N. Blom, F. Furche, N. Morgner, O. Kostko, B. von Issendorff and M. M. Kappes, Angew. Chem., Int. Ed., 2007, 46, 2944–2948 CrossRef CAS.
- W. Huang, M. Ji, C.-D. Dong, X. Gu, L.-M. Wang, X. G. Gong and L.-S. Wang, ACS Nano, 2008, 2, 897–904 CrossRef CAS.
- N. Shao, W. Huang, Y. Gao, L.-M. Wang, X. Li, L.-S. Wang and X. C. Zeng, J. Am. Chem. Soc., 2010, 132, 6596–6605 CrossRef CAS.
- P. Hohenberg and W. Kohn, Phys. Rev., 1964, 136, B864–871 CrossRef.
- W. Kohn and L. J. Sham, Phys. Rev., 1965, 140, A1133–1138 CrossRef.
- R. L. Whetten, J. T. Khoury, M. M. Alvarez, S. Murthy, I. Vezmar, Z. L. Wang, P. W. Stephens, C. L. Cleveland, W. D. Luedtke and U. Landman, Adv. Mater., 1996, 8, 428–433 CrossRef CAS.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, K. I. Vezmar, R. Whetten, W. G. Gullen and P. N. First, J. Phys. Chem. B, 1997, 101, 7885–7891 CrossRef CAS.
- S. Chen, R. S. Ingram, M. J. Hostetler, J. J. Pietron, R. W. Murray, T. G. Schaaff, J. T. Khoury, M. M. Alvarez and R. L. Whetten, Science, 1998, 280, 2098–2101 CrossRef CAS.
- A. C. Templeton, W. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- R. Sardart, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef.
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801–802 RSC.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- M. M. Alvarez, J. T. Khoury, T. G. Schaaff, M. N. Shafigullin, I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 1997, 101, 3706–3712 CrossRef CAS.
- T. G. Schaaff, G. Knight, M. N. Shafigullin, R. F. Borkman and R. L. Whetten, J. Phys. Chem. B, 1998, 102, 10643–10646 CrossRef CAS.
- T. P. Bigioni, R. L. Whetten and O. Dag, J. Phys. Chem. B, 2000, 104, 6983–6986 CrossRef CAS.
- R. H. Terrill, T. A. Postlethwaite, C. H. Chen, C. D. Poon, A. Terzis, A. D. Chen, J. E. Hutchison, M. R. Clark, G. Wignall, J. D. Londono, R. Superfine, M. Falvo, C. S. Johnson, E. T. Samulski and R. W. Murray, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- S. J. Green, J. J. Stokes, M. J. Hostetler, J. Pietron and R. W. Murray, J. Phys. Chem. B, 1997, 101, 2663–2668 CrossRef CAS.
- R. S. Ingram, M. J. Hostetler, R. W. Murray, T. G. Schaaff, J. T. Khoury, R. L. Whetten, T. P. Bigioni, D. K. Guthrie and P. N. First, J. Am. Chem. Soc., 1997, 119, 9279–9280 CrossRef CAS.
- S. J. Green, J. J. Pietron, J. J. Stokes, M. J. Hostetler, H. Vu, W. P. Wuelfing and R. W. Murray, Langmuir, 1998, 14, 5612–5619 CrossRef CAS.
- M. J. Hostetler, J. E. Wingate, C. J. Zhong, J. E. Harris, R. W. Vachet, M. R. Clark, J. D. Londono, S. J. Green, J. J. Stokes, G. D. Wignall, G. L. Glish, M. D. Porter, N. D. Evans and R. W. Murray, Langmuir, 1998, 14, 17–30 CrossRef CAS.
- A. C. Templeton, M. J. Hostetler, C. T. Kraft and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 1906–1911 CrossRef CAS.
- W. P. Wuelfing, S. M. Gross, D. T. Miles and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 12696–12697 CrossRef CAS.
- S. W. Chen and R. W. Murray, J. Phys. Chem. B, 1999, 103, 9996–10000 CrossRef CAS.
- J. F. Hicks, D. T. Miles and R. W. Murray, J. Am. Chem. Soc., 2002, 124, 13322–13328 CrossRef CAS.
- D. Lee, R. L. Donkers, G. L. Wang, A. S. Harper and R. W. Murray, J. Am. Chem. Soc., 2004, 126, 6193–6199 CrossRef CAS.
- R. Guo and R. W. Murray, J. Am. Chem. Soc., 2005, 127, 12140–12143 CrossRef CAS.
- G. L. Wang, T. Huang, R. W. Murray, L. Menard and R. G. Nuzzo, J. Am. Chem. Soc., 2005, 127, 812–813 CrossRef CAS.
- Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2003, 125, 4046–4047 CrossRef CAS.
- H. Murayama, T. Narushima, Y. Negishi and T. Tsukuda, J. Phys. Chem. B, 2004, 108, 3496–3503 CrossRef CAS.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- Y. Shichibu, Y. Negishi, T. Tsukuda and T. Teranishi, J. Am. Chem. Soc., 2005, 127, 13464–13465 CrossRef CAS.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Phys. Chem. B, 2006, 110, 12218–12221 CrossRef CAS.
- Y. Negishi, H. Tsunoyama, M. Suzuki, N. Kawamura, M. M. Matsushita, K. Maruyama, T. Sugawara, T. Yokoyama and T. Tsukuda, J. Am. Chem. Soc., 2006, 128, 12034–12035 CrossRef CAS.
- H. Tsunoyama, Y. Negishi and T. Tsukuda, J. Am. Chem. Soc., 2006, 128, 6036–6037 CrossRef CAS.
- K. Ikeda, Y. Kobayashi, Y. Negishi, M. Seto, T. Iwasa, K. Nobusada, T. Tsukuda and N. Kojima, J. Am. Chem. Soc., 2007, 129, 7230–7231 CrossRef CAS.
- E. S. Shibu, M. A. H. Muhammed, T. Tsukuda and T. Pradeep, J. Phys. Chem. C, 2008, 112, 12168–12176 CAS.
- K. Nobusada, T. Tsukuda and N. Kojima, J. Am. Chem. Soc., 2007, 129, 7230–7231 CrossRef.
- Y. Negishi, N. K. Chaki, Y. Shichibu, R. L. Whetten and T. Tsukuda, J. Am. Chem. Soc., 2007, 129, 11322–11323 CrossRef CAS.
- N. K. Chaki, Y. Negishi, H. Tsunoyama, Y. Shichibu and T. Tsukuda, J. Am. Chem. Soc., 2008, 130, 8608–8610 CrossRef CAS.
- H. Qian and R. Jin, Nano Lett., 2009, 9, 4083–4087 CrossRef CAS.
- M. Zhu, H. Qian and R. Jin, J. Am. Chem. Soc., 2009, 131, 7220–7221 CrossRef CAS.
- R. Jin, H. Qian, Z. Wu, Y. Zhu, M. Zhu, A. Mohanty and N. Garg, J. Phys. Chem. Lett., 2010, 1, 2903–2910 CrossRef CAS.
- H. Qian and R. Jin, Chem. Mater., 2011, 23, 2209–2217 CrossRef CAS.
- H. Qian, Y. Zhu and R. Jin, J. Am. Chem. Soc., 2010, 132, 4583–4585 CrossRef CAS.
- R. Jin, G. Wu, Z. Li, C. A. Mirkin and G. C. Schatz, J. Am. Chem. Soc., 2003, 125, 1643–1654 CrossRef CAS.
- M. Zhu, H. Qian, X. Meng, S. Jin, Z. Wu and R. Jin, Nano Lett., 2011, 11, 3963–3969 CrossRef CAS.
- G. Schmid, Chem. Soc. Rev., 2008, 37, 1909–1930 RSC.
- C. E. Briant, R. C. Theobald, J. W. White, L. K. Bell and D. M. P. Mingos, J. Chem. Soc., Chem. Commun., 1981, 201–206 RSC.
- B. K. Teo, X. Shi and H. Zhang, J. Am. Chem. Soc., 1992, 114, 2743–2744 CrossRef CAS.
- M. C. Fairbanks, R. E. Benfield, R. J. Newport and G. Schmid, Solid State Commun., 1990, 73, 431–436 CrossRef CAS.
- D. Micheal and P. Mingos, J. Chem. Soc., Dalton Trans., 1976, 1163–1169 Search PubMed.
- H. Häkkinen, R. N. Barnett and U. Landman, Phys. Rev. Lett., 1999, 82, 3264–3267 CrossRef.
- A. Ulman, Chem. Rev., 1996, 96, 1533–1554 CrossRef CAS.
- L. Garzón, C. Rovira, K. Michaelian, M. R. Beltran, P. Ordejorn, J. Junquera, D. Sanchez-Portal, E. Artacho and J. M. Soler, Phys. Rev. Lett., 2000, 85, 5250–5251 CrossRef.
- H. Häkkinen, M. Walter and H. Grönbeck, J. Phys. Chem. B, 2006, 110, 9927–9931 CrossRef.
- P. D. Jadzinsky, G. Calero, C. J. Ackerson, D. A. Bushnell and R. D. Kornberg, Science, 2007, 318, 430–433 CrossRef CAS.
- R. L. Whetten and R. C. Price, Science, 2007, 318, 407–408 CrossRef CAS.
- M. W. Heaven, A. Dass, P. S. White, K. M. Holt and R. W. Murray, J. Am. Chem. Soc., 2008, 130, 3754–3755 CrossRef CAS.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 99–104 CrossRef CAS.
- H. Häkkinen, Chem. Soc. Rev., 2008, 37, 1847–1859 RSC.
- D. Jiang, Acta Phys.-Chim. Sin., 2010, 26, 999–1016 CAS.
- A. P. Sutton, Philos. Mag. Lett., 1990, 61, 139–164 CrossRef.
- R. P. Gupta, Phys. Rev. B, 1981, 23, 6265–6270 CrossRef CAS.
- N. T. Wilson and R. L. Johnston, Eur. Phys. J. D, 2000, 12, 161–169 CrossRef CAS.
- L. Garzön, K. Michaelian, M. R. Beltrán, A. Posada-Amarillas, P. Ordejón, E. Artacho, D. Sanchez-Portal and J. M. Soler, Eur. Phys. J. D, 1999, 9, 211–215 CrossRef.
- M. Born and J. R. Oppenheimer, Ann. Phys., 1927, 389, 457–484 CrossRef.
- B. Hartke, J. Phys. Chem., 1993, 97, 9973–9976 CrossRef CAS.
- S. Kirkpatrick, C. D. Gelatt and M. P. Vecchi, Science, 1983, 220, 671–680 CAS.
- D. J. Wales and J. P. K. Doye, J. Phys. Chem. A, 1997, 101, 5111–5116 CrossRef CAS.
- E. Runge and E. K. U. Gross, Phys. Rev. Lett., 1984, 52, 997–1000 CrossRef CAS.
- W. D. Luedtke and U. Landman, J. Phys. Chem. A, 1996, 100, 13323–13329 CAS.
- N. T. Wilson and R. L. Johnston, Phys. Chem. Chem. Phys., 2002, 4, 4168–4171 RSC.
- C. Majumder, T. M. Briere, H. Mizuseki and Y. Kawazoe, J. Chem. Phys., 2002, 117, 2819–2822 CrossRef CAS.
- A. Larsson, M. Nolan and J. C. Greer, J. Phys. Chem. B, 2002, 106, 5931–5937 CrossRef.
- P. Maksymovych, D. C. Sorescu and J. T. Yates, Phys. Rev. Lett., 2006, 97, 146103–146106 CrossRef.
- T. Iwasa and K. Nobusada, J. Phys. Chem. C, 2007, 111, 45–49 CAS.
- Y. Gao, N. Shao, X. C. Zeng and X. C., ACS Nano, 2008, 2, 1497–1503 CrossRef CAS.
- Y.-K. Han, H. Kim, J. Jung and Y. C. Choi, J. Phys. Chem. C, 2010, 114, 7548–7552 CAS.
- T. Li, G. Galli and F. Gygi, ACS Nano, 2008, 2, 1896–1902 CrossRef.
- R. L. Donkers and R. W. Murray, Langmuir, 2004, 20, 1945–1952 CrossRef CAS.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS.
- Y. Schichibu, Y. Negishi, T. Watanabe, N. K. Chaki, H. Kawaguchi and T. Tsukuda, J. Phys. Chem. C, 2007, 111, 7845–7847 Search PubMed.
- J. Akola, M. Walter, R. L. Whetten, H. Hakkinen and H. Grönbeck, J. Am. Chem. Soc., 2008, 130, 3756–3757 CrossRef CAS.
- D. Jiang, M. L. Tiago, W. D. Luo and S. Dai, J. Am. Chem. Soc., 2008, 130, 2777–2779 CrossRef CAS.
- D. Jiang, W. Luo, M. L. Tiago and S. Dai, J. Phys. Chem. C, 2008, 112, 13905–13910 CAS.
- Y. Pei, Y. Gao and X. C. Zeng, J. Am. Chem. Soc., 2008, 130, 7830–7832 CrossRef CAS.
- O. Lopez-Acevedo, H. Tsunoyama, T. Tsukuda, H. Häkkinen and C. M. Aikens, J. Am. Chem. Soc., 2010, 132, 8210–8218 CrossRef CAS.
- T. G. Schaaff and R. L. Whetten, J. Phys. Chem. B, 2000, 104, 2630–2641 CrossRef CAS.
- H. Qian, W. T. Eckenhoff, Y. Zhu, T. Pintauer and R. Jin, J. Am. Chem. Soc., 2010, 132, 8280–8281 CrossRef CAS.
- T. G. Schaaff, M. N. Shafigullin, J. T. Khoury, I. Vezmar and R. L. Whetten, J. Phys. Chem. B, 2001, 105, 8785–8796 CrossRef CAS.
- O. Lopez-Acevedo, J. Akola, R. L. Whetten, H. Grönbeck and H. Häkkinen, J. Phys. Chem. C, 2009, 113, 5035–5038 CAS.
- D. Jiang, W. Chen, R. L. Whetten and Z. Chen, J. Phys. Chem. C, 2009, 113, 16983–16987 CAS.
- D. Jiang, R. L. Whetten, W. Luo and S. Dai, J. Phys. Chem. C, 2009, 113, 17291–17295 CAS.
- Y. Zhang, S. Shuang, C. Dong, C. K. Lo, M. C. Paau and M. M. F. Choi, Anal. Chem., 2009, 81, 1676–1685 CrossRef CAS.
- R. C. Price and R. L. Whetten, J. Am. Chem. Soc., 2005, 127, 13750–13751 CrossRef CAS.
- D. Jiang, M. Walter and J. Akola, J. Phys. Chem. C, 2010, 114, 15883–15889 CAS.
- P. R. Nimmala and A. Dass, J. Am. Chem. Soc., 2011, 133, 9175–9177 CrossRef CAS.
- A. C. Dharmaratne, T. Krick and A. Dass, J. Am. Chem. Soc., 2009, 131, 13604–13605 CrossRef CAS.
- Y. Pei, Y. Gao and X. C. Zeng, J. Am. Chem. Soc., 2009, 131, 13619–13621 CrossRef CAS.
- S. M. Reilly, T. Krick and A. Dass, J. Phys. Chem. C, 2010, 114, 741–745 CAS.
- A. Tlahuice and I. L. Garzon, Phys. Chem. Chem. Phys., 2012, 14, 3737–3740 RSC.
- T. Tsukuda, H. Tsunoyama and Y. Negishi, in Metal Nanoclusters in Catalysis and Materials Science: The Issue of Size Control, ed. B. Corain, G. Schmid and M. Toshima, Elsevier, Amsterdam, 2008, p. 373 Search PubMed.
- D. Jiang, M. Walter and J. Akola, Chem.–Eur. J., 2011, 17, 12289–12293 CrossRef CAS.
- Z. Wu, M. A. MacDonald, J. Chen, P. Zhang and R. Jin, J. Am. Chem. Soc., 2011, 133, 9670–9673 CrossRef CAS.
- Y. Pei, R. Pal, C. Liu, Y. Gao, Z. Zhang and X. C. Zeng, J. Am. Chem. Soc., 2012, 134, 3015–3024 CrossRef CAS.
- V. N. Staroverov, G. E. Scuseria, J. Tao and J. P. Perdew, J. Chem. Phys., 2003, 119, 12129–12137 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, J. Chem. Phys., 2006, 125, 194101–194118 CrossRef.
- B. M. Barngrover and C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 990–994 CrossRef CAS.
- R. Wiseman, P. A. Marsh, P. T. Bishop, B. J. Brisdon and M. F. Mahon, J. Am. Chem. Soc., 2000, 122, 12598–12599 CrossRef.
- N. Shao, Y. Pei, Y. Gao and X. C. Zeng, J. Phys. Chem. A, 2009, 113, 629–632 CrossRef CAS.
- H. Grönbeck, M. Walter and H. Häkkinen, J. Am. Chem. Soc., 2006, 128, 10268–10275 CrossRef.
- E. Zeller, H. Beruda and H. Schmidbaur, Inorg. Chem., 1993, 32, 3203–3204 CrossRef CAS.
- C. E. Briant, K. P. Hall, D. M. P. Mingos and A. C. Wheeler, J. Chem. Soc., Dalton Trans., 1986, 687–692 RSC.
- Y. Gao, S. Bulusu and X. C. Zeng, J. Am. Chem. Soc., 2005, 127, 15680–15681 CrossRef CAS.
- L.-M. Wang, S. Bulusu, H.-J. Zhai, X. C. Zeng and L. S. Wang, Angew. Chem., Int. Ed., 2007, 46, 2915–2918 CrossRef CAS.
- D. Jiang and S. Dai, Inorg. Chem., 2009, 48, 2720–2722 CrossRef CAS.
- D. Jiang and R. L. Whetten, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 115402–115406 CrossRef.
- H. Qian, E. Barry, Y. Zhu and R. Jin, Acta Phys.-Chim. Sin., 2011, 27, 513–519 CAS.
- Y. Negishi, K. Igarashi, K. Munakata, W. Ohgake and K. Nobusada, Chem. Commun., 2012, 48, 660–662 RSC.
- Y. Negishi, T. Iwai and M. Ide, Chem. Commun., 2010, 46, 4713–4715 RSC.
- Y. Negishi, W. Kurashige, Y. Niihori, T. Iwasa and K. Nobusada, Phys. Chem. Chem. Phys., 2010, 12, 6219–6225 RSC.
- F. Parker, J. E. F. Weaver, F. McCallum, C. A. Fields-Zinna and R. W. Murray, Langmuir, 2010, 26, 13650–13654 CrossRef.
- A. Kacprzak, L. Lehtovaara, J. Akola, O. Lopez-Acevedoa and H. Häkkinen, Phys. Chem. Chem. Phys., 2009, 11, 7123–7129 RSC.
- M. Walter and M. Moseler, J. Phys. Chem. C, 2009, 113, 15834–15837 CAS.
- Z. Wu and R. Jin, ACS Nano, 2009, 3, 2036–2042 CrossRef CAS.
- A. Dass, G. B. Dubay, C. A. Fields-Zinna and R. W. Murray, Anal. Chem., 2008, 80, 6845–6851 CrossRef CAS.
- A. Angel, L. T. Majors, A. C. Dharmaratne and A. Dass, ACS Nano, 2010, 4, 4691–4700 CrossRef.
- Z. H. Tang, B. Xu, B. H. Wu, D. Robinson, N. Bokossa and G. L. Wang, Langmuir, 2011, 27, 2989–2996 CrossRef CAS.
- D. Jiang, S. Walter and S. Dai, Chem.–Eur. J., 2010, 16, 4999–5003 CrossRef CAS.
- Y. Pei, N. Shao, H. Li, D. Jiang and X. C. Zeng, ACS Nano, 2011, 5, 1441–1449 CrossRef CAS.
- O. Lopez-Acevedo and H. Häkkinen, Eur. Phys. J. D, 2011, 63, 311–314 CrossRef.
- R. I. G. Hughes, Perspect. Sci., 2006, 14, 457–524 CrossRef.
- M. Walter, J. Akola, O. Lopez-Acevedo, P. D. Jadzinsky, G. Calero, C. J. Ackerson, R. L. Whetten, H. Gronbeck and H. Hakkinen, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 9157–9162 CrossRef CAS.
- A. Dass, J. Am. Chem. Soc., 2009, 131, 11666–11667 CrossRef CAS.
- K. Clemenger, Phys. Rev. B, 1985, 32, 1359–1362 CrossRef CAS.
- C. M. Aikens, J. Phys. Chem. C, 2008, 112, 19797–19800 CAS.
- J. R. Reimers, Y. Wang, B. O. Cankurtaran and M. J. Ford, J. Am. Chem. Soc., 2010, 132, 8378–8384 CrossRef CAS.
- Y.-K. Han, H. Kim, J. Jung and Y. C. Choi, J. Phys. Chem. C, 2010, 114, 7548–7552 CAS.
- B. Yoon, H. Häkkinen, U. Landman, A. S. Wörz, J.-M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403–407 CrossRef CAS.
- Y. Zhu, H. Qian and R. Jin, Chem.–Eur. J., 2010, 16, 11455–11462 CrossRef CAS.
- Y. Zhu, H. Qian, M. Zhu and R. Jin, Adv. Mater., 2010, 22, 1915–1920 CrossRef CAS.
- M. Zhu, W. T. Eckenhoff, T. Pintauer and R. Jin, J. Phys. Chem. C, 2008, 112, 14221–14224 CAS.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Häkkinen, Nat. Chem., 2010, 2, 329 CrossRef CAS.
- A. M. Barngrover and C. M. Aikens, J. Phys. Chem. Lett., 2011, 2, 990–994 CrossRef.
- H. Qian, Y. Zhu and R. Jin, J. Am. Chem. Soc., 2010, 132, 4583–4585 CrossRef CAS.
- H. Qian, Y. Zhu and R. Jin, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 696–700 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |