Organosoluble calixarene-based quinolone carriers: syntheses, evaluation and model hydrolytic studies at the air–water interface
Guillaume
Sautrey
,
Igor
Clarot
,
Adel Ben
Salem†
,
Ewa
Rogalska
and
Jean-Bernard
Regnouf de Vains
*
SRSMC, UMR 7565 Nancy Université, CNRS; équipe GEVSM, Faculté de Pharmacie, 5, rue Albert Lebrun, 54001 Nancy cedex, France. E-mail: jean-bernard.regnouf@pharma.uhp-nancy.fr; Fax: +33 3 83 68 23 45; Tel: +33 3 83 68 23 15
First published on 24th October 2011
Abstract
Two tetra-p-tert-butyl- and two tetra-p-H-calix[4]arene species integrating one or two propylnalidixate esters at the lower rim have been synthesized as possible antibacterial prodrugs. As they display amphiphilic behavior, their properties at the air–water interface were evaluated using the Langmuir balance technique. It has been shown that the two tert-butyl analogues form stable monolayers. The two tert-butyl analogues were then studied at 20 and 37 °C on carbonate buffered subphases at pH 10.0, before being engaged in hydrolytic studies on “production” of monolayers so as to allow a simple treatment, a HPLC survey of nalidixate release. The latter was found to be effective at 37 °C, with formation of free nalidixate in 13% yield for the bisnalidixate derivative, and 10% yield for the mononalidixate after 3 days.
Introduction
Due to their intrinsic physico-chemical and chemical properties, the calixarenes have often been employed, in recent years, as carriers and spatial organisers of various kinds of active substituents, displaying properties dealing with the recognition of organic substrates or metallic cations.1–5Such organising properties could be of great interest in the building of molecular drug-organisers or dispensers, notably as medically valuable structures incorporating probes and drugs; in the last case, a biochemically controlled release should be expected, resulting in such edifices in a drug-carrier or a prodrug behaviour.
As recently reviewed by de Fatima et al.,6 Kalchenko et al.,7 and Coleman et al.,8–10 very few reports, essentially under the form of patents, describe therapeutical activities of calixarenes and derivatives; some of them, hydrophilic, have shown interesting activities against bacteria,11 fungi, cancerous cells and enveloped viruses,12–18 but also against thrombosis19 or fibrotic diseases.20 Biological studies related to plasmid DNA binding and cell transfection have been recently reported by Ungaro and co-workers.21–23 In the middle 50s, the calixarene derivative “Macrocyclon”,24 and more recently some parent structures25–27 have been studied in the treatment of tuberculosis and other mycobacterioses. Calixarene-based mimics of vancomycin have also been described.28
In this field, our contribution has been devoted to the design of new anti-HIV29 or anti-corona virus30 agents and, with regards to spreading resistances of pathogenic microorganisms against actual antibiotics,31antibacterial agents.32,33 Some of them have been conceived as prodrugs of known antibiotics, e.g.penicillins or quinolones, in which the latter are attached to the calixarene platform via an amide34,35 or a labile ester35–37 junction. A water-soluble analogue incorporating a nalidixic acid quinolone arm demonstrated the effective drug release in biological medium (rat serum), and a pronounced antibacterial activity.37 Nevertheless, the lack of solubility of organosoluble analogues34–36 in water and biological media made them until now non-compliant for in vitro standard evaluation as antimicrobial agents, as well as for chemical hydrolysis studies.
A recent study of some of these derivatives at the air–water interface and in a biological membrane model by means of Langmuir balance techniques35,38 confirmed the amphiphilic behaviour of these structures, prone to display in the aqueous layer their polar bioactive subunits and pre-organise the ester junctions for a possibly facilitated hydrolysis.
Few reports are dedicated to the study at the air–water interface of calixarenes decorated at the lower rim with bio-active substituents. In addition to our previous works,35,38 interaction of monolayers of an adenine-derivative of tetra-p-tert-butylcalix[4]arene with subphases charged in thymidine, uridine or cytidine has been described by Liet al.,39 or by Shi et al. who investigated the interactions between an amphiphilic calixarene derivative mono-substituted at the upper rim by one uracyl unit, and aqueous subphases charged in adenine, adenosine or ATP·Na2;40 very recently, Shahgaldian et al. described an important possible application of amphiphilic calixarene decorated with mimics of p-amidophenol displaying crystal nucleation properties for paracetamol.41
Here, we report on the synthesis, interfacial properties and hydrolysis at the air–water (buffer) interface of new amphiphilic calix[4]arene–quinolone adducts incorporating one or two nalidixic moieties attached as esters to the lower rim of the calixarene platform via a propyl linker, and H- or tert-butyl-groups in the para-position at the upper rim.
Experimental
Synthesis
Melting points (°C, uncorrected) were determined on an Electrothermal 9200 Capillary apparatus. 1H- and 13C-NMR spectra were recorded on a Bruker DRX 400 (chemical shifts in ppm), in CDCl3 (7.26 and 77.00 ppm). Mass spectra (electrospray-ESI) were recorded on a Micromass Platform II apparatus, at the Service Commun de Spectrométrie de Masse Organique, Nancy. Infrared analysis was performed on a Bruker Vector 22 apparatus (KBr, ν in cm−1) and UV spectra were recorded on a SAFAS UV mc2 apparatus, λmax in nm, ε in mol−1 dm3 cm−1. Elemental analyses were performed at the Service de Microanalyse, Nancy. Merck TLC plates were used for chromatography analysis (SiO2, ref. 1.05554; Al2O3, ref. 1.05581). All commercially available products were used without further purification unless otherwise specified. For NMR data of compounds 1 and 3, letter A refers to free phenols and B to substituted ones; for 2 and 4, C refers to the central free phenol ring.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Et2O (40):MeCN (20) v/v/v) to give pure 1 (0.226 g; 67%). This solid was dissolved in CH2Cl2 (1 mL) and Et2O was added until the formation of fine seeds. The mixture was let to stand at rt for 30 min, and the resulting crystals were recovered by filtration, then dried under high vacuum (0.126 g; 56% cryst.). White crystals. mp: 151–152 °C. IR (KBr): 1692 (CO); 1735 (COO). 1H NMRδ 1.00 (s, 18H, Me3C B); 1.24 (s, 18H, Me3C A); 1.38 (t, J = 7.2 Hz, 6H, NCH2CH3); 2.52 (quint, J = 6.0 Hz, 4H, OCH2CH2CH2OCO); 2.61 (s, 6H, C(7)CH3); 3.28, 4.30 (“q”, AB, J = 12.8 Hz, 8H, ArCH2Ar); 4.19 (t, J = 6.0 Hz, 4H, OCH2CH2CH2OCO); 4.32 (q, J = 7.2 Hz, 4H, NCH2CH3); 4.86 (t, J = 6.0 Hz, 4H, OCH2CH2CH2OCO); 6.82 (s, 4H, HmB); 6.97 (s, 4H, HmA); 7.17 (d, J = 8.1 Hz, 2H, H(6)); 7.78 (s, 2H, ArOH); 8.58 (d, J = 8.1 Hz, 2H, H(5)); 8.60 (s, 2H, H(2)). 13C NMRδ 15.55 (NCH2CH3); 25.38 (C(7)CH3); 29.71 (OCH2CH2CH2OOC); 31.43 (Me3C B); 32.09 (Me3C A); 32.18 (ArCH2Ar); 34.17 (Me3C A), 34.37 (Me3C B); 46.89 (NCH2CH3); 61.95 (OCH2CH2CH2OCO); 72.93 (OCH2CH2CH2OCO); 112.34 (C(3)); 121.22 (C(6)); 121.73 (C(9)); 125.43 (Cm of Ar A); 125.99 (Cm of Ar B); 127.98 (Co of Ar A); 133.15 (Co of Ar B); 137.06 (C(5)); 141.74 (Cp of Ar A); 147.32 (Cp of Ar B); 149.18 (C(10)); 148.96 (C(2)); 150.00 (Ci of Ar B); 151.15 (Ci of Ar A); 162.77 (C(7)); 165.51 (OCH2CH2CH2OCO); 175.12 (C(4)). Anal. calcd for C74H88N4O10 (1193.51): C 74. 47, H 7.43, N 4.69%; found: C 74.50, H 7.36, N 4.71%. ESI-MS (pos. mode): 1194.4 [M + H]+. λabs (CH2Cl2) 254 (29800), 290 (12900), 332 (19900).
:MeCN (15) v/v) to give pure 2 (0.335 g; 46%). This amorphous solid was treated with MeCN (1.5 mL); after a preliminary solubilisation, a voluminous precipitate was formed, and additional MeCN was added (1 mL). The solid was filtered, rinsed with MeCN (1 mL), and finally dried under high vacuum at 45 °C to give 2 (0.243 g; 73% cryst). White powder. Mp: 159–160 °C. IR (KBr): 1696 (CO); 1736 (COO). 1H NMRδ 1.18 (s, 9H, Me3C B); 1.20 (s, 18H, Me3C A); 1.22 (s, 9H, Me3C C); 1.43 (t, J = 7.2 Hz, 3H, NCH2CH3); 2.61 to 2.65 (quint + s, 5H, OCH2CH2CH2OCO + C(7)CH3); 3.40, 4.38 (AB, JAB = 13.1 Hz, 4H, Ar(A)CH2Ar(B)); 3.43, 4.27 (AB, JAB = 13.6 Hz, 4H, Ar(A)CH2Ar(C)); 4.34 to 4.42 (m, 6H, NCH2CH3 + Ar(A)CHaxHeqAr(B) + OCH2CH2CH2OCO); 4.81 (t, J = 6.2 Hz, 2H, OCH2CH2CH2OCO); 6.98 (AX, J = 2.3 Hz, 2H, ArHm(A)CH2Ar(C)); 7.03 (AX, J = 2.3 Hz; 2H, ArHm(A)CH2Ar(B)); 7.05 (s, 2H, ArHm(C)); 7.07 (s, 2H, ArHm(B)); 7.23 (d, J = 8.0 Hz, 1H, H(6)); 8.64 (d, J = 8.0 Hz, 1H, H(5)); 8.70 (s, 1H, H(2)); 9.53 (s, 2H, Ar(A)OH); 10.12 (s, 1H, Ar(C)OH). 13C NMRδ 15.15 (NCH2CH3); 25.03 (C(7)CH3); 29.26 (OCH2CH2CH2OCO); 31.20 (Me3C B); 31.44 (Me3C C); 31.47 (Me3C A); 32.10 (Ar(A)CH2Ar(B)); 32.95 (Ar(A)CH2Ar(C)); 33.87 (Me3CA); 33.98 (Me3CC), 34.19 (Me3CB); 46.54 (NCH2CH3); 61.35 (OCH2CH2CH2OCO); 73.58 (OCH2CH2CH2OCO); 111.83 (C(3)); 121.05 (C(6)); 121.51 (C(9)); 125.58 (ArCm(A)CH2Ar(C)); 125.65 (ArCm(A)CH2Ar(B) + ArCm(C)); 126.42 (ArCm(B)); 127.43 (ArCo(A)CH2Ar(B)); 128.05 (ArCo(C)); 128.32 (ArCo(A)CH2Ar(C)); 133.43 (ArCo(B)); 136.80 (C(5)); 143.05 (ArCp(A)); 143.63 (ArCp(C)); 147.65 (ArCi(C)); 148.16 (ArCp(B)); 148.45 (ArCi(A)); 148.61 (C(10)); 148.91 (C(2)); 149.09 (ArCi(B)); 162.54 (C(7)); 165.41 (OCH2CH2CH2OCO); 174.68 (C(4)). Anal. calcd for C59H72N2O7 (921.21): C 76.92, H 7.88, N 3.04%; found: C 76.57, H 7.80, N 3.54%. ESI-MS (pos. mode): 922.41 [M + H]+. λabs (CH2Cl2) 259 (27841), 281 (22170), 329 (14084); 337 (14473).
:Et2O (40):MeCN (50) v/v/v) to give pure 3 as an amorphous solid (0.232 g; 59%). This material was dissolved in a mixture of CH2Cl2 and MeCN; CH2Cl2 was evaporated under vacuum, without heating, resulting in the precipitation of pure 3 (0.193 g; 83% cryst). White solid. Mp: 224–225 °C. IR (KBr): 1638 (CO); 1695 (COO). 1H NMRδ 1.21 (t, J = 7 Hz, 6H, NCH2CH3); 2.58 (q, J = 6 Hz, 4H, OCH2CH2CH2OCO); 2.62 (s, 6H, C(7)CH3); 3.35, 4.33 (“q”, AB, JAB = 13 Hz, 8H, ArCH2Ar); 4.23 (t, J = 6 Hz, 4H, OCH2CH2CH2OCO); 4.32 (q, J = 7 Hz, 4H, NCH2CH3); 4.89 (t, J = 6 Hz, 4H, OCH2CH2CH2OCO); 6.58 (t, J = 7 Hz, 2H, HpA); 6.72 (t, J = 8 Hz, 2H, HpB); 6.88 (d, J = 8 Hz, 4H, HmB); 6.98 (d, J = 7 Hz, 4H, HmA); 7.18 (d, J = 8 Hz, 2H, H(6)); 8.16 (s, 2H, OHA); 8.57 (d, J = 8 Hz, 2H, H(5)); 8.58 (s, 2H, H(2)). 13C NMRδ 15.10 (NCH2CH3); 25.03 (C(7)CH3); 29.31 (OCH2CH2CH2OCO); 31.35 (ArCH2Ar); 46.55 (NCH2CH3); 61.52 (OCH2CH2CH2OCO); 72.82 (OCH2CH2CH2OCO); 111.78 (C(3)); 118.94 (CpA); 120.86 (C(6)); 121.32 (C(9)); 125.42 (CpB); 127.94 (CoA); 128.35 (CmA); 128.97 (CmB); 133.27 (CoB); 136.63 (C(5)); 148.52 (C(2)); 148.72 (C(10)); 151.60 (CiB); 153.28 (CiA); 162.42 (C(7)); 165.21 (OCH2CH2CH2OCO); 174.68 (C(4)). Anal. calcd for C58H56N4O10, 0.2CH2Cl2 (986.07): C 70.89, H 5.77, N 5.68%; found: C 70.66, H 5.82, N 5.72%. ESI-MS (pos. mode): 991.4 [M + Na]+. λabs (CH2Cl2) 257 (34605), 286 (13731), 336 (24264).
:Et2O:MeCN, gradient from 1/1/0.2 to 1/1/2 v/v/v) to give compound 3 and relatively impure compound 4 (0.188 g; 53%). The latter was purified again by chromatography (SiO2; CH2Cl2:CH3CN gradient from 90/10 to 80/20 v/v) giving pure 4 as an amorphous solid (0.14 g; 76%). This material was dissolved in CH2Cl2 (2 mL) and Et2O (6 mL) was added to give 4 as a white precipitate that was filtered, triturated in pentane, and dried under high vacuum (0.12 g; 84% cryst.) (total yield 34%). Mp: 204–205 °C. IR (KBr): 1691 (CO); 1734 (COO). 1H NMRδ 1.42 (t, J = 7 Hz, 3H, NCH2CH3); 2.62 to 2.66 (m, 5H, C(7)CH3 + OCH2CH2CH2OCO); 3.45 (d, 1/2AB, J = 13 Hz, 2H, Ar(A)CH2Ar(B)); 3.46, 4.27 (“q”, AB, J = 14 Hz, 4H, Ar(A)CH2Ar(C)); 4.38 to 4.41 (m, 6H, NCH2CH3 + Ar(A)CHaxHeqAr(B) + OCH2CH2CH2OCO); 4.85 (t, J = 6 Hz, 2H, OCH2CH2CH2OCO); 6.63 to 6.69 (t + t, 3H, HpC + HpA); 6.85 (t, J = 8 Hz, 1H, HpB); 6.98 to 7.07 (m, 8H, HmA + HmB + HmC); 7.24 (d, J = 8 Hz, 1H, H(6)); 8.63 (d, J = 8 Hz, 1H, H(5)); 8.68 (s, 1H, H(2)); 9.35 (s, 2H, OHA); 9.64 (s, 1H, OHC). 13C NMRδ 15.07 (NCH2CH3); 25.03 (C(7)CH3); 29.31 (OCH2CH2CH2OCO); 31.34 (Ar(A)CH2Ar(B)); 31.86 (Ar(A)CH2Ar(C)); 46.56 (NCH2CH3); 61.64 (OCH2CH2CH2OCO); 73.71 (OCH2CH2CH2OCO); 111.74 (C(3)); 120.77 (CpA); 121.10 (C(6)); 121.49 (C(9)); 121.94 (CpC); 126.18 (CpB); 128.21 (CmC); 128.33 (CmB); 128.37 (CmA); 128.72 (CoA); 129.34 (CoC); 134.10 (CoB); 136.76 (C(5)); 148.58 (C(2)); 148.91 (C(10)); 149.05 (CiC); 150.87 (CiA); 151.20 (CiB); 162.60 (C(7)); 165.64 (OCH2CH2CH2OCO); 174.62 (C(4)). Anal. calcd for C43H40N2O7 (696.79): C, 74.12; H, 5.79; N, 4.02%; found: C, 74.05; H, 5.82; N, 4.08%. ESI-MS (pos. mode): 719.3 [M − H + Na]+, 1415.5 [2M − H + Na]+. λabs (CH2Cl2) 258 (19243), 280 (10217), 336 (13432).
:Et2O (40):MeCN (20) v/v/v) to give pure 1 (0.226 g; 67%). This solid was dissolved in CH2Cl2 (1 mL) and Et2O was added until the formation of fine seeds. The mixture was let to stand at rt for 30 min, and the resulting crystals were recovered by filtration, then dried under high vacuum (0.126 g; 56% cryst.). White crystals. mp: 151–152 °C. IR (KBr): 1692 (CO); 1735 (COO). 1H NMRδ 1.00 (s, 18H, Me3C B); 1.24 (s, 18H, Me3C A); 1.38 (t, J = 7.2 Hz, 6H, NCH2CH3); 2.52 (quint, J = 6.0 Hz, 4H, OCH2CH2CH2OCO); 2.61 (s, 6H, C(7)CH3); 3.28, 4.30 (“q”, AB, J = 12.8 Hz, 8H, ArCH2Ar); 4.19 (t, J = 6.0 Hz, 4H, OCH2CH2CH2OCO); 4.32 (q, J = 7.2 Hz, 4H, NCH2CH3); 4.86 (t, J = 6.0 Hz, 4H, OCH2CH2CH2OCO); 6.82 (s, 4H, HmB); 6.97 (s, 4H, HmA); 7.17 (d, J = 8.1 Hz, 2H, H(6)); 7.78 (s, 2H, ArOH); 8.58 (d, J = 8.1 Hz, 2H, H(5)); 8.60 (s, 2H, H(2)). 13C NMRδ 15.55 (NCH2CH3); 25.38 (C(7)CH3); 29.71 (OCH2CH2CH2OOC); 31.43 (Me3C B); 32.09 (Me3C A); 32.18 (ArCH2Ar); 34.17 (Me3C A), 34.37 (Me3C B); 46.89 (NCH2CH3); 61.95 (OCH2CH2CH2OCO); 72.93 (OCH2CH2CH2OCO); 112.34 (C(3)); 121.22 (C(6)); 121.73 (C(9)); 125.43 (Cm of Ar A); 125.99 (Cm of Ar B); 127.98 (Co of Ar A); 133.15 (Co of Ar B); 137.06 (C(5)); 141.74 (Cp of Ar A); 147.32 (Cp of Ar B); 149.18 (C(10)); 148.96 (C(2)); 150.00 (Ci of Ar B); 151.15 (Ci of Ar A); 162.77 (C(7)); 165.51 (OCH2CH2CH2OCO); 175.12 (C(4)). Anal. calcd for C74H88N4O10 (1193.51): C 74. 47, H 7.43, N 4.69%; found: C 74.50, H 7.36, N 4.71%. ESI-MS (pos. mode): 1194.4 [M + H]+. λabs (CH2Cl2) 254 (29800), 290 (12900), 332 (19900).
:MeCN (15) v/v) to give pure 2 (0.335 g; 46%). This amorphous solid was treated with MeCN (1.5 mL); after a preliminary solubilisation, a voluminous precipitate was formed, and additional MeCN was added (1 mL). The solid was filtered, rinsed with MeCN (1 mL), and finally dried under high vacuum at 45 °C to give 2 (0.243 g; 73% cryst). White powder. Mp: 159–160 °C. IR (KBr): 1696 (CO); 1736 (COO). 1H NMRδ 1.18 (s, 9H, Me3C B); 1.20 (s, 18H, Me3C A); 1.22 (s, 9H, Me3C C); 1.43 (t, J = 7.2 Hz, 3H, NCH2CH3); 2.61 to 2.65 (quint + s, 5H, OCH2CH2CH2OCO + C(7)CH3); 3.40, 4.38 (AB, JAB = 13.1 Hz, 4H, Ar(A)CH2Ar(B)); 3.43, 4.27 (AB, JAB = 13.6 Hz, 4H, Ar(A)CH2Ar(C)); 4.34 to 4.42 (m, 6H, NCH2CH3 + Ar(A)CHaxHeqAr(B) + OCH2CH2CH2OCO); 4.81 (t, J = 6.2 Hz, 2H, OCH2CH2CH2OCO); 6.98 (AX, J = 2.3 Hz, 2H, ArHm(A)CH2Ar(C)); 7.03 (AX, J = 2.3 Hz; 2H, ArHm(A)CH2Ar(B)); 7.05 (s, 2H, ArHm(C)); 7.07 (s, 2H, ArHm(B)); 7.23 (d, J = 8.0 Hz, 1H, H(6)); 8.64 (d, J = 8.0 Hz, 1H, H(5)); 8.70 (s, 1H, H(2)); 9.53 (s, 2H, Ar(A)OH); 10.12 (s, 1H, Ar(C)OH). 13C NMRδ 15.15 (NCH2CH3); 25.03 (C(7)CH3); 29.26 (OCH2CH2CH2OCO); 31.20 (Me3C B); 31.44 (Me3C C); 31.47 (Me3C A); 32.10 (Ar(A)CH2Ar(B)); 32.95 (Ar(A)CH2Ar(C)); 33.87 (Me3CA); 33.98 (Me3CC), 34.19 (Me3CB); 46.54 (NCH2CH3); 61.35 (OCH2CH2CH2OCO); 73.58 (OCH2CH2CH2OCO); 111.83 (C(3)); 121.05 (C(6)); 121.51 (C(9)); 125.58 (ArCm(A)CH2Ar(C)); 125.65 (ArCm(A)CH2Ar(B) + ArCm(C)); 126.42 (ArCm(B)); 127.43 (ArCo(A)CH2Ar(B)); 128.05 (ArCo(C)); 128.32 (ArCo(A)CH2Ar(C)); 133.43 (ArCo(B)); 136.80 (C(5)); 143.05 (ArCp(A)); 143.63 (ArCp(C)); 147.65 (ArCi(C)); 148.16 (ArCp(B)); 148.45 (ArCi(A)); 148.61 (C(10)); 148.91 (C(2)); 149.09 (ArCi(B)); 162.54 (C(7)); 165.41 (OCH2CH2CH2OCO); 174.68 (C(4)). Anal. calcd for C59H72N2O7 (921.21): C 76.92, H 7.88, N 3.04%; found: C 76.57, H 7.80, N 3.54%. ESI-MS (pos. mode): 922.41 [M + H]+. λabs (CH2Cl2) 259 (27841), 281 (22170), 329 (14084); 337 (14473).
:Et2O (40):MeCN (50) v/v/v) to give pure 3 as an amorphous solid (0.232 g; 59%). This material was dissolved in a mixture of CH2Cl2 and MeCN; CH2Cl2 was evaporated under vacuum, without heating, resulting in the precipitation of pure 3 (0.193 g; 83% cryst). White solid. Mp: 224–225 °C. IR (KBr): 1638 (CO); 1695 (COO). 1H NMRδ 1.21 (t, J = 7 Hz, 6H, NCH2CH3); 2.58 (q, J = 6 Hz, 4H, OCH2CH2CH2OCO); 2.62 (s, 6H, C(7)CH3); 3.35, 4.33 (“q”, AB, JAB = 13 Hz, 8H, ArCH2Ar); 4.23 (t, J = 6 Hz, 4H, OCH2CH2CH2OCO); 4.32 (q, J = 7 Hz, 4H, NCH2CH3); 4.89 (t, J = 6 Hz, 4H, OCH2CH2CH2OCO); 6.58 (t, J = 7 Hz, 2H, HpA); 6.72 (t, J = 8 Hz, 2H, HpB); 6.88 (d, J = 8 Hz, 4H, HmB); 6.98 (d, J = 7 Hz, 4H, HmA); 7.18 (d, J = 8 Hz, 2H, H(6)); 8.16 (s, 2H, OHA); 8.57 (d, J = 8 Hz, 2H, H(5)); 8.58 (s, 2H, H(2)). 13C NMRδ 15.10 (NCH2CH3); 25.03 (C(7)CH3); 29.31 (OCH2CH2CH2OCO); 31.35 (ArCH2Ar); 46.55 (NCH2CH3); 61.52 (OCH2CH2CH2OCO); 72.82 (OCH2CH2CH2OCO); 111.78 (C(3)); 118.94 (CpA); 120.86 (C(6)); 121.32 (C(9)); 125.42 (CpB); 127.94 (CoA); 128.35 (CmA); 128.97 (CmB); 133.27 (CoB); 136.63 (C(5)); 148.52 (C(2)); 148.72 (C(10)); 151.60 (CiB); 153.28 (CiA); 162.42 (C(7)); 165.21 (OCH2CH2CH2OCO); 174.68 (C(4)). Anal. calcd for C58H56N4O10, 0.2CH2Cl2 (986.07): C 70.89, H 5.77, N 5.68%; found: C 70.66, H 5.82, N 5.72%. ESI-MS (pos. mode): 991.4 [M + Na]+. λabs (CH2Cl2) 257 (34605), 286 (13731), 336 (24264).
:Et2O:MeCN, gradient from 1/1/0.2 to 1/1/2 v/v/v) to give compound 3 and relatively impure compound 4 (0.188 g; 53%). The latter was purified again by chromatography (SiO2; CH2Cl2:CH3CN gradient from 90/10 to 80/20 v/v) giving pure 4 as an amorphous solid (0.14 g; 76%). This material was dissolved in CH2Cl2 (2 mL) and Et2O (6 mL) was added to give 4 as a white precipitate that was filtered, triturated in pentane, and dried under high vacuum (0.12 g; 84% cryst.) (total yield 34%). Mp: 204–205 °C. IR (KBr): 1691 (CO); 1734 (COO). 1H NMRδ 1.42 (t, J = 7 Hz, 3H, NCH2CH3); 2.62 to 2.66 (m, 5H, C(7)CH3 + OCH2CH2CH2OCO); 3.45 (d, 1/2AB, J = 13 Hz, 2H, Ar(A)CH2Ar(B)); 3.46, 4.27 (“q”, AB, J = 14 Hz, 4H, Ar(A)CH2Ar(C)); 4.38 to 4.41 (m, 6H, NCH2CH3 + Ar(A)CHaxHeqAr(B) + OCH2CH2CH2OCO); 4.85 (t, J = 6 Hz, 2H, OCH2CH2CH2OCO); 6.63 to 6.69 (t + t, 3H, HpC + HpA); 6.85 (t, J = 8 Hz, 1H, HpB); 6.98 to 7.07 (m, 8H, HmA + HmB + HmC); 7.24 (d, J = 8 Hz, 1H, H(6)); 8.63 (d, J = 8 Hz, 1H, H(5)); 8.68 (s, 1H, H(2)); 9.35 (s, 2H, OHA); 9.64 (s, 1H, OHC). 13C NMRδ 15.07 (NCH2CH3); 25.03 (C(7)CH3); 29.31 (OCH2CH2CH2OCO); 31.34 (Ar(A)CH2Ar(B)); 31.86 (Ar(A)CH2Ar(C)); 46.56 (NCH2CH3); 61.64 (OCH2CH2CH2OCO); 73.71 (OCH2CH2CH2OCO); 111.74 (C(3)); 120.77 (CpA); 121.10 (C(6)); 121.49 (C(9)); 121.94 (CpC); 126.18 (CpB); 128.21 (CmC); 128.33 (CmB); 128.37 (CmA); 128.72 (CoA); 129.34 (CoC); 134.10 (CoB); 136.76 (C(5)); 148.58 (C(2)); 148.91 (C(10)); 149.05 (CiC); 150.87 (CiA); 151.20 (CiB); 162.60 (C(7)); 165.64 (OCH2CH2CH2OCO); 174.62 (C(4)). Anal. calcd for C43H40N2O7 (696.79): C, 74.12; H, 5.79; N, 4.02%; found: C, 74.05; H, 5.82; N, 4.08%. ESI-MS (pos. mode): 719.3 [M − H + Na]+, 1415.5 [2M − H + Na]+. λabs (CH2Cl2) 258 (19243), 280 (10217), 336 (13432).
Interfacial studies
Chloroform used for preparing calixarene derivative solutions was from Sigma-Aldrich (ref. 288306; 100 mL; ∼99.9% pure; 0.5–1.0% EtOH), and subphases were prepared with Milli-Q water which had a surface tension of 72.8 mN m−1 at 20 °C (pH 5.6). Buffers were prepared with analytical grade NaHCO3 and NaOH. All solvents used were of analytical grade.Chromatographic procedure
The HPLC system consisted of the following Merck Hitachi modules: a model 7100 pump, a model 7200 autosampler set to inject 20 μL and a model 7450 diode array detector. The analytical column was a Nucleodur C18 Isis (150 mm × 4.6 mm id, 3 μm) reversed phase column from Macherey Nagel. The column temperature was controlled with a Merck Hitachi model T6300 external oven.Calibration curves were prepared with sodium nalidixate solutions ranging from 1.0 × 10−6 to 2.0 × 10−5 M (5 concentrations, n = 3), corresponding to the theoretical concentrations after ST. A standard curve of nalidixic acid peak area against concentration was plotted and calibration curves were fitted using least squares linear regression. A good linearity was observed (typical equation: y = 1.272 × 1010 × −990) with correlation coefficients >0.999.
The specificity of the method was investigated by injecting treated carbonate samples (without nalidixate) in order to confirm that there were no interferences in the buffer media. The sensitivity of the method was determined by means of the LOD and the LOQ (signal/noise ratio S/N of 3 and 10, respectively). The LOD and LOQ, evaluated using corresponding diluted samples, were 1.6 × 10−7 M and 5.0 × 10−7 M, respectively.
The intra-, inter-day accuracy and precision were evaluated by assaying triplicate samples of blinded concentrations representing low, middle and high analyte concentrations (1.0 × 10−6, 5.0 × 10−6 and 1.0 × 10−5 M, respectively). The intra-day variation (repeatability, n = 3) was determined on a single day whereas the inter-day variation (intermediate precision, n = 9) was assayed for three consecutive days. The precision was expressed as the relative standard deviation (RSD) and the accuracy was determined by comparing the calculated concentration of treated samples to the theoretical ones. Satisfactory results for precision (RSD < 5.0% for 5 × 10−6 and 1.0 × 10−5 M and <13.0% for 1.0 × 10−6 M) and for accuracy (from 94.9 ± 0.5 to 106.9 ± 2.0%) were obtained.
Results and discussion
Synthesis
The di-substituted calixarene 1 was previously prepared with a moderate yield of 35% by reaction of tetra-p-tert-butylcalix[4]arene with 2 equiv. of bromopropylnalidixate in MeCN, using 4 equiv. of K2CO3 as base,35 but the present procedure involving 2.2 equiv. of K2CO3 and additional KI resulted in a much better yield of 67% after chromatography.This procedure was also applied, but with ca. 1 equiv. of K2CO3, to the synthesis of the di-substituted analogue 3, that was obtained from p-H-calix[4]arene with a yield of 60%.
The mono-substituted species 2 has been previously prepared by reaction of tetra-p-tert-butylcalix[4]arene with 0.9 equiv. of bromopropylnalidixate in MeCN, using 1.3 equiv. of NaHCO3 as base and KI as catalyst, with a yield of 42%.36 In the present study, 2 was prepared by replacing NaHCO3 with 0.6 equiv. of K2CO3, with a similar yield of 46%; this procedure applied to p-H-calix[4]arene gave the derivative 4 with a yield of 53% after chromatography over NEt3-deactivated silica gel, using a CH2Cl2/Et2O/MeCN mixture (gradient from 1:1:0 to 1:1:2) as mobile phase, allowing in addition the recovery of the by-product 3 (Fig. 1).
![Synthesis of calixarene derivatives 1, 2, 3 and 4 from bromo-propyl-nalidixate (BPN), in MeCN, reflux. (i) p-tert-Butylcalix[4]arene (0.48 equiv.), K2CO3 (1.10 equiv.), KI (1.36 equiv.), 67% of 1; (ii) p-tert-butylcalix[4]arene (1.16 equiv.), K2CO3 (0.67 equiv.), KI (2.80 equiv.), 46% of 2; (iii) calix[4]arene (0.48 equiv.), K2CO3 (0.52 equiv.), KI (1.20 equiv.), 60% of 3; (iv) calix[4]arene (0.90 equiv.), K2CO3 (0.54 equiv.), KI (2.10 equiv.), 53% of 4.](/image/article/2012/NJ/c1nj20636b/c1nj20636b-f1.gif) | ||
| Fig. 1 Synthesis of calixarene derivatives 1, 2, 3 and 4 from bromo-propyl-nalidixate (BPN), in MeCN, reflux. (i) p-tert-Butylcalix[4]arene (0.48 equiv.), K2CO3 (1.10 equiv.), KI (1.36 equiv.), 67% of 1; (ii) p-tert-butylcalix[4]arene (1.16 equiv.), K2CO3 (0.67 equiv.), KI (2.80 equiv.), 46% of 2; (iii) calix[4]arene (0.48 equiv.), K2CO3 (0.52 equiv.), KI (1.20 equiv.), 60% of 3; (iv) calix[4]arene (0.90 equiv.), K2CO3 (0.54 equiv.), KI (2.10 equiv.), 53% of 4. | ||
A particular attention was brought to the purification of these compounds, in view of their use in the preparation of Langmuir monolayers. It has been previously noted that the calixarene–quinolone ester adduct could be trans-esterified into methyl nalidixate during chromatography on silica gel, using methanol as elution co-solvent.45Purification protocols preserving the ester bond and giving solid products with a high purity were explored to circumvent this fragility. The best results were obtained with crystallizations and/or chromatographic processes over silica-gel deactivated with NEt3, and non-alcoholic eluants composed of CH2Cl2, Et2O and MeCN.
All final compounds were fully characterised; NMR experiments showed notably that they were as expected in the cone conformation, as assessed by Ar–CH2–Ar 13C resonance signals located at 31–32 ppm, and their AB-like shape in 1H-NMR. Combustional analyses were found to be consistent with the proposed structures, and electrospray mass spectrometry, positive mode, showed the presence of the corresponding proto- or sodio-derivative molecular peaks.
Studies at the air–water interface
The interfacial properties of the four calixarene derivatives were studied using a Langmuir balance technique.19 The calixarene–quinolone derivatives studied here are amphiphilic. Indeed, the macrocyclic crown is apolar compared to the quinolone and hydroxyl groups; the tert-butyl groups present in the derivatives 1 and 2 increase the hydrophobicity of the upper part of the molecules compared to 3 and 4 (Fig. 1). Surface pressure–mean molecular area compression isotherms (Π–A) performed with derivatives 1–4 on pure water at 20 °C are shown in Fig. 2.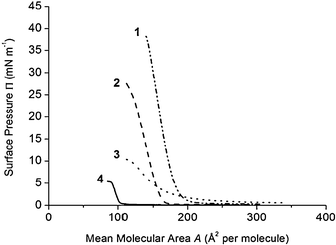 | ||
| Fig. 2 Surface pressure–mean molecular area (Π–A) compression isotherms of calixarene derivative monolayers spread on pure water (pH 5.6) at 20 °C. Results obtained with calixarene 1 (- ••), 2 (- -), 3 (••••), 4 (—). Compression rate: 5 mm min−1 (3.25 cm2 min−1). | ||
The films formed with 3 and 4 allowed compression only to low surface pressures. These results indicate that the films formed with derivatives 3 and 4 cannot be used in hydrolysis studies, as they are not stable enough. The derivatives 1 and 2 bearing tert-butyl groups on the upper rim of the calixarene crown form more stable monolayers compared to 3 and 4, as shown by the profile of the isotherms. The derivative 1 bearing two nalidixic acid arms on the lower part of the molecule forms more rigid films compared to the derivative 2 bearing one hydrophilic arm, as indicated by the compressibility of the monolayers. Indeed, the compressibility modulus (Cs−1max)42,43 values calculated at the most condensed state of the monolayers were 159 mN m−1 (Π = 30 mN m−1) and 98 mN m−1 (Π = 20 mN m−1) with 1 and 2, respectively.
The isotherms show that the presence of tert-butyl moieties on the upper rim of the calixarene crown increases the stability of the films formed with 1 and 2 compared to 3 or 4. On the other hand, the presence of the second nalidixic acid arm on the lower rim is less important relative to film stability, as judged by the profiles of the isotherms 1 and 2. Isotherms obtained at higher compression rates (results not shown) were superposed with those presented in Fig. 2, indicating that the derivatives studied form stable films at the experiment time scale.
To get more information on film stability, evolution of film properties of 1 and 2 was monitored as a function of time (Fig. 3). To this aim, mean molecular area variation at a constant surface pressure was measured.46,47 The films were compressed to an initial value of 25.0 mN m−1 for 1 and 17.0 mN m−1 for 2 that is below the corresponding collapse points.
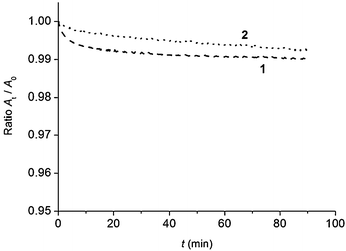 | ||
| Fig. 3 Evolution of the mean molecular area at constant surface pressure of the monolayers formed with 1 (dashed line), and 2 (dotted line), spread on pure water (pH 5.6) at 20 °C. | ||
The results are presented as At/A0, where A0 and At represent values obtained at the beginning of the experiment, and at a given time, respectively. It can be observed that the decrease in the molecular area, which can be due to a slight dissolution and/or aggregation, is lower than 1%.
The stability of the films formed with 1 and 2 was further checked in compression–expansion experiments.46–48 As shown in Fig. 4, successive compression–expansion cycles of the calixarene films were performed from the gas phase to a mean molecular area around the more condensed state before collapse. The compression isotherms obtained with 1 and 2 in the consecutive cycles are almost superposed, except the parts of the isotherms corresponding to the lowest surface pressures. The latter effect can be due to some minor aggregation of the molecules in the film upon compression.48
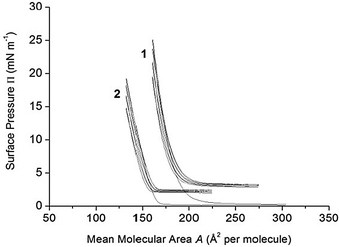 | ||
| Fig. 4 Four successive compression–expansion P–A cycle isotherms of calixarene derivatives 1 and 2 spread on pure water, pH 5.6, at 20 °C. Compression rate: 5 mm min−1 (3.25 cm2 min−1). | ||
It was concluded that the films formed with 1 and 2 are stable enough to be used in the studies of the reactivity of the ester bonds present in the two molecules. Indeed, in the absence of calixarene derivatives dissolved in the subphase, the hydrolysis reaction, if any, would take place only at the film–subphase interface.
Drug-release at the air–water interface
The calixarene derivatives 1–4 were conceived as prodrugs of the nalidixic acid, using the carboxylate moiety of the nalidixic arm as a linker via an in vivo labile ester function. The drug-release ability and the antibacterial activity of the water-soluble tetra-para-aminoethyl analogue of 2 or 4 were recently evaluated.37In order to assess the drug-release ability at the air–water (buffer) interface of the amphiphilic-water insoluble derivatives 1 and 2, an analytical procedure using HPLC was adapted from a previous work,37 and developed so as to detect and quantify the nalidixate released from the monolayer into the subphase.
For this purpose, and as shown in Fig. 5, the behavior of the monolayers of calixarenes 1 and 2 on a buffered subphase was first characterized at 20 °C and 37 °C by measurement of the Π–A isotherms, using a carbonate buffer (NaHCO3/Na2CO3 50 mM) at pH 10.0. Similar experiments were carried out with buffers at pH 7.0, 8.0 and 9.0.
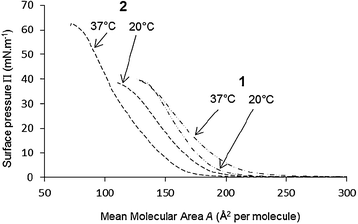 | ||
| Fig. 5 Π–A compression isotherms of calixarenes 1 and 2 spread on carbonate buffer 50 mM pH 10.0, at 20 and 37 °C. | ||
Suspecting a long-term hydrolysis process (several days), these experiments were performed in covered Petri dishes (see experimental part) in order to limit evaporation of the subphase.
The quantity of calixarene derivatives needed to form a condensed monolayer at the air–buffer interface in the Petri dishes was calculated at mean molecular areas of 155 and 130 Å2 for 1 and 2, respectively. The Petri dishes were half-filled with 120 mL of freshly prepared carbonate buffer, and the needed volume of a chloroformic solution of calixarene derivatives was spread at the buffer surface.
The drug-release kinetic of 1 and 2 was evaluated by the validated HPLC method described in the Experimental section, with buffers at pH 7.0, 8.0, 9.0 and 10.0, from 0 to 3 days, at 20 °C and 37 °C.
The limit of 3 days was imposed by the condensation of water on the internal face of the lid of Petri dishes at 37 °C. Due to this time limitation, experiments done with buffers at pH 7.0, 8.0 and 9.0 were not pursued, as no clearly detectable nalidixic salt was formed within this period.
The nalidixic acid quantity (drug release percentage) was calculated versus the maximal theoretical quantity that could be obtained after total hydrolysis from 1 (bearing two nalidixic acid moieties) or 2 (bearing one nalidixic acid moiety). Chromatograms are given in Fig. 6, and the kinetic survey of nalidixate release is resumed in Fig. 7.
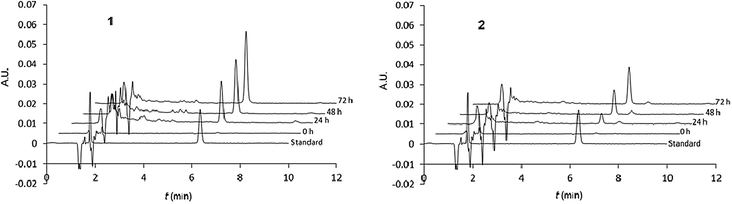 | ||
| Fig. 6 Hydrolysis survey of calixarenes 1 (left) and 2 (right). Typical chromatograms (λ = 254 nm) of, from bottom to top, standard (nalidixic acid 5 × 10−6 M), subphase after ST at t = 0, 24, 48 and 72 h. | ||
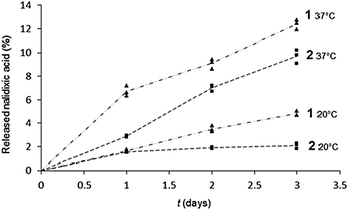 | ||
| Fig. 7 Kinetic of the nalidixic acid release from 1 (▲) and 2 (■) monolayers spread on carbonate buffer NaHCO3/Na2CO3 50 mM pH 10.0, at 20 and 37 °C, in the Petri dishes. | ||
It appears that the release of nalidixic acid into the subphase is effective and regular. After 3 days at 37 °C, we observe that 13% and 10% of nalidixate was released from monolayers of 1 and 2, respectively, against only 5% and 2% at 20 °C.
In parallel to these studies, other experiments were conducted with compound 2, at the liquid–liquid interface (stirred solution of 2 in CH2Cl2 against buffer at pH 10.0). In the time scale of 3 days, no release of nalidixate was observed, and only some traces of this salt were detected after 7 days. This observation indicates that the organization of substrate molecules at the air–aqueous interface may be important for the advancement of the reaction. The latter may be due to easier intermolecular interactions compared to bulk phases.
Conclusions
In order to circumvent strong difficulties encountered in the evaluation of their antibacterial activities, four amphiphilic calix[4]arene derivatives, integrating for two of them four tert-butyl-groups at the upper rim, and one or two propylnalidixate esters at the lower rim, have been prepared and fully characterized.Their amphiphilic behavior has been studied at the air–water interface on a Langmuir balance, allowing the selection of the two p-tert-butylated derivatives that gave stable monolayers suitable for further investigations.
These two derivatives were then studied at the air–carbonate buffer (pH 10.0) interface, at 20 and 37 °C, here also showing that they form stable monolayers during the measurement of compression isotherms. Monolayers of these two compounds were prepared on the same buffer, in large Petri dish reactors, and nalidixate salt release was followed by HPLC at 20 and 37 °C, according to a specific sample treatment developed for this purpose. This release is effective, and, even if hydrolysis yields are low, much better than with a liquid–liquid interface, indicating that by engineering the interfacial processes, the hydrolysis yield could be enhanced. As expected initially, these results converge towards a possible prodrug behavior for these compounds. Further experiments implying other antibacterial or antiviral agents, as well as much complex aqueous subphases are on the way, with the aim of developing an alternative method of pre-biological screening for such compounds.
Acknowledgements
The authors thank the French Ministère de la Recherche et de l'Enseignement Supérieur, particularly G. Sautrey for a PhD grant, the CNRS and the Région Lorraine for financial support. We also thank Mr Eric Dubs for synthesis of starting materials, Mrs B. Fernette for NMR experiments, as well as Mrs S. Adach for elemental analyses and Mr F. Dupire for mass measurements.Notes and references
- C. D. Gutsche, Monographs in Supramolecular Chemistry, Calixarenes, ed. J. F. Stoddart, Royal Society of Chemistry, Cambridge, 1989 Search PubMed.
- Calixarenes. A Versatile Class of Macrocyclic Compounds, ed. J. Vicens and V. Böhmer, Kluwer Academic Publishers, Dordrecht, 1991 Search PubMed.
- C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, 1998 Search PubMed.
- Calixarenes 2001, ed. J. Asfari, Z. Böhmer, V. Harrowfield and J. Vicens, Kluwer, Dordrecht, The Netherlands, 2001 Search PubMed.
- C. Wieser, D. B. Dieleman and D. Matt, Coord. Chem. Rev., 1997, 165, 93 CAS.
- A. de Fatima, S. A. Fernandes and A. A. Sabino, Curr. Drug Discovery Technol., 2009, 6, 151 CrossRef CAS.
- R. V. Rodik, V. I. Boyko and V. I. Kalchenko, Curr. Med. Chem., 2009, 16, 1630 CrossRef CAS.
- E. da Silva and A. W. Coleman, J. Drug Delivery Sci. Technol., 2004, 14, 3 CAS.
- F. Perret, A. N. Lazar and A. W. Coleman, Chem. Commun., 2006, 2425 RSC.
- F. Perret and A. W. Coleman, Chem. Commun., 2011, 47, 7303 RSC.
- T. Yo, K. Fujiwara and M. Otsuka, JP 10203906, 1998 Search PubMed.
- K. M. Hwang, Y. M. Qi, S. Y. Liu, W. Choy and J. Chen, WO Patent 9403164, 1994 Search PubMed.
- S. J. Harris, WO Patent 9519974, 1995 Search PubMed.
- S. J. Harris, WO Patent 0244121, 2002 Search PubMed.
- D. Coveney and B. Costello, US 2005113454, 2005 Search PubMed.
- V. Kral, P. Cigler, J. Konvalinka, M. Kozisek, J. Prejdova, B. Gruener, J. Plesek, M. Lepsik, J. Pokorna, H.-G. Kraeusslich and J. Bodem, WO 2005073240, 2005 Search PubMed.
- R. P. M. Dings, X. Chen, D. M. E. I. Hellebrekers, L. I. van Eijk, Y. Zhang, T. R. Hoye, A. W. Griffioen and K. H. Mayo, J. Natl. Cancer Inst., 2006, 98, 932 CrossRef CAS.
- A. E. Motornaya, L. M. Alimbarova, E. A. Shokova and V. V. Kovalev, Pharm. Chem. J., 2006, 40, 68 CrossRef CAS.
- K. M. Hwang, Y. M. Qi, S. Y. Liu, T. C. Lee, W. Choy and J. Chen, WO Patent 9403165, 1994 Search PubMed.
- D. Hulmes, A. Coleman and E. Aubert-Foucher, (CNRS, Fr) WO Patent 0007585, 2000 Search PubMed.
- M. Dudic, A. Colombo, F. Sansone, A. Casnati, G. Donofrio and R. Ungaro, Tetrahedron, 2004, 60, 11613 CrossRef CAS.
- F. Sansone, M. Dudic, G. Donofrio, C. Rivetti, L. Baldini, A. Casnati, S. Cellai and R. Ungaro, J. Am. Chem. Soc., 2006, 128, 14528 CrossRef CAS.
- V. Bagnacani, F. Sansone, G. Donofrio, L. Baldini, A. Casnati and R. Ungaro, Org. Lett., 2008, 10, 3953 CrossRef CAS.
- J. W. Cornforth, P. D'Arcy Hart, G. A. Nicholls, R. J. W. Rees and J. A. Stock, Br. J. Pharmacol., 1955, 10, 73 CAS.
- P. D'Arcy Hart, A. J. Armstrong and E. Brodaty, Infect. Immun., 1996, 64, 1491 Search PubMed.
- M. J. Colston, H. C. Hailes, E. Stavropoulos, A. C. Herve, G. Herve, K. J. Goodworth, A. M. Hill, P. Jenner, P. D. Hart and R. E. Tascon, Infect. Immun., 2004, 72, 6318 CrossRef CAS.
- F. A. Loiseau, A. M. Hill and K. K. Hii, Tetrahedron, 2007, 63, 9947 CrossRef CAS.
- A. Casnati, M. Fabbi, N. Pelizzi, A. Pochini, F. Sansone and R. Ungaro, Bioorg. Med. Chem. Lett., 1996, 6, 2699 CrossRef CAS.
- M. Mourer, N. Psychogios, G. Laumond, A.-M. Aubertin and J.-B. Regnouf-de-Vains, Bioorg. Med. Chem., 2010, 18, 36 CrossRef CAS.
- C. Geller, S. Fontanay, M. Mourer, H. M. Dibama, J.-B. Regnouf-de-Vains, C. Finance and R. E. Duval, Antiviral Res., 2010, 88, 343 CrossRef CAS.
- M. Leeb, Nature, 2004, 431(7011), 892 CrossRef CAS.
- M. Mourer, H. M. Dibama, S. Fontanay, M. Grare, R. E. Duval, C. Finance and J.-B. Regnouf-de-Vains, Bioorg. Med. Chem., 2009, 17, 5496 CrossRef CAS.
- M. Grare, H. M. Dibama, S. Lafosse, A. Ribon, M. Mourer, S. Fontanay, C. Finance, J.-B. Regnouf-de-Vains and R. E. Duval, Clin. Microbiol. Infect., 2010, 16, 432 CrossRef CAS.
- A. Ben Salem and J.-B. Regnouf-de-Vains, Tetrahedron Lett., 2001, 42, 7033 CrossRef CAS.
- B. Korchowiec, A. B. Salem, Y. Corvis, J.-B. Regnouf-de-Vains, J. Korchowiec and E. Rogalska, J. Phys. Chem. B, 2007, 111, 13231 CrossRef CAS.
- A. B. Salem and J.-B. Regnouf-de-Vains, Tetrahedron Lett., 2003, 44, 6769 CrossRef.
- H. M. Dibama, I. Clarot, S. Fontanay, A. B. Salem, M. Mourer, C. Finance, R. E. Duval and J.-B. Regnouf-de-Vains, Bioorg. Med. Chem. Lett., 2009, 19, 2679 CrossRef CAS.
- B. Korchowiec, M. Orlof, G. Sautrey, A. B. Salem, J. Korchowiec, J.-B. Regnouf de Vains and E. Rogalska, J. Phys. Chem. B, 2010, 114, 10427 CrossRef CAS.
- X. Guo, G.-Y. Lu and Y. Li, Thin Solid Films, 2004, 460, 264 CrossRef CAS.
- H.-J. Shi, X.-F. Shi, T.-M. Yao and L.-N. Ji, Chin. J. Chem., 2008, 26, 170 CrossRef CAS.
- N. Moridi, D. Elend, O. Danylyuk, K. Suwinska and P. Shahgaldian, Langmuir, 2011, 27, 9116 CAS.
- J. T. Davies and E. K. Rideal, Interfacial Phenomena, Academic Press, New York, USA, 2nd edn, 1963 Search PubMed.
- G. L. Gaines, Insoluble monolayers at liquid–gas interfaces, Wiley Interscience, New York, USA, 1966 Search PubMed.
- Synthesized as follows: nalidixic acid suspension stirred in water. Addition of 1.0 equiv. of calibrated NaOH solution. Solution stirred 30 min and lyophilized. 100%. White powder.
- A. B. Salem, H. M. Dibama and J.-B. Regnouf-de-Vains, unpublished results.
- C. Wang, M. Micic, M. Ensor, S. Daunert and R. M. Leblanc, Langmuir, 2007, 23, 7602 CrossRef CAS.
- R. D. Smith, Stability of sparingly soluble monolayers at the air–water interface, PhD thesis, Univ. Washington, Seattle, USA, 1977 Search PubMed.
- I. Capan, R. Capan, T. Tanrisever and S. Can, Mater. Lett., 2005, 59, 2468 CrossRef CAS.
Footnote |
| † Present address: Institut Préparatoire aux Etudes d'Ingénieur de Sfax, Route Menzel Chaker Km 0.5, BP 1172, 3018 Sfax, Tunisie. E-mail: adel_bs2002@yahoo.fr; Fax: 00216 7449 0320; Tel: 00216 2427 5642. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2012 |