
Optimized Li and Fe recovery from spent lithium-ion batteries via a solution-precipitation method
Rujuan Zhengab,
Li Zhaoa,
Wenhui Wanga,
Yuanlong Liua,
Quanxin Maa,
Deying Mua,
Ruhong Lia and
Changsong Dai*a
aSchool of Chemical Engineering and Technology, Harbin Institute of Technology, Harbin 150001, China. E-mail: changsd@hit.edu.cn
bCollege of Chemistry and Chemical Engineering, Qiqihar University, Qiqihar 161006, China
First published on 19th April 2016
Abstract
A new process is optimized and presented for the recovery and regeneration of LiFePO4 from spent lithium-ion batteries (LIBs). The recycling process reduces the cost and secondary pollution caused by complicated separation and purification processes in spent LIB recycling. Amorphous FePO4·2H2O was recovered by a dissolution-precipitation method from spent LiFePO4 batteries. The effects of different surfactants (i.e. CTAB, SDS and PEG), which were added to the solution on the recovered FePO4·2H2O, were investigated. Li2CO3 was precipitated by adding Na2CO3 to the filtrate. Then the LiFePO4/C material was synthesized by a carbon thermal reduction method using recycled FePO4·2H2O and Li2CO3 as the Fe, P, and Li sources. The as-prepared LiFePO4/C shows comparable electrochemical performance to that of commercial equivalents.
Introduction
With the rapid development of global economy, energy demand and consumption grows every day. There are many advantages of lithium-ion batteries (LIBs) due to their high working voltage, high energy density, small self discharge, long cycle life, convenient use and no memory effect reservoir.1–3 LIBs have been widely used in consumer electronics and related electrical vehicles, and their production and consumption is increasing year by year.4 The rapid expansion of hybrid electric vehicles (EVs), plug-in hybrid electric vehicles (PHEVs) and pure EVs is now greatly increasing the consumption of LIBs. Large quantities of spent LIBs along with scrap will be generated due to their limited life spans and the rapid updating of electronic products. Disposed LIBs in the environment are a waste of resources (e.g. Li salts), lead to environmental pollution, and have a negative impact on the energy crisis. Therefore, the recovery of spent LIBs is necessary to prevent environmental pollution and resource depletion.As for battery recovery, many efforts have been focused on the expensive and toxic metal containing cathodes, for example LiCoO2 5–10 and LiNixCoyMnzO2.11–15 However, little attention has been paid to the recovery of LiFePO4, which has been widely employed as a cathode of high-power LIBs. For example, in November 2015, LiFePO4-based high-power LIBs shared 20.08% of Chinese market of pure electric passenger vehicles (i.e. 6230 cars have been selected to employ LiFePO4-based high-power LIBs). More surprisingly, it shares 64.9% of the Chinese market of electric buses, that is, 14![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 143 electric buses employ LiFePO4-based high-power LIBs.16
143 electric buses employ LiFePO4-based high-power LIBs.16
Under the circumstances, developing a LiFePO4 recovery technique is undoubtedly necessary, which is the focus of this work. This paper presents the whole diagram for recovering a LiFePO4 cathode, including the Li salt, iron salt and current collector. In addition, the high quality of the recovered materials was verified by using them as raw materials to synthesize high performance LiFePO4/C (comparable to the commercial one). The optimized method presented in this work is environmental friendly, economically feasible, and scalable.
Experimental
Recovery of “cathode powder” from spent batteries
Prior to the recovery process, the spent LIBs were fully discharged and then disassembled. While the electrolyte was collected and recycled by a supercritical method, the diaphragm was recycled by wind sieving, which takes advantage of the weight difference between the electrodes and diaphragm. Then, LiFePO4 cathode sheets were crushed into about 2 cm2 and heated at different temperatures ranging from 450–650 °C for 1 h, which not only removes the binders and the carbon in the electrode, but also oxidized Fe2+ to Fe3+ (which favors the subsequent recovery of FePO4). Powder can be separated from the aluminum current collector via oscillation sieving.Recovery of FePO4
The mixed powder was dissolved in sulphuric acid in a reactor with continuous stirring at a speed of 500 rpm. The dissolution efficiency was optimized via removing insoluble impurities (e.g. residual graphite) and tuning the liquid–solid concentration acid ratio, reaction time, and temperature.Ammonia was added to the precursor solution in order to manipulate the pH value at 2. After filtration, the filter residue was washed with deionized water and then dried at 80 °C until it reached a constant weight. Then the amorphous hydrated FePO4 (sample A, without adding any surfactant to the precursor solution) was obtained.
The surfactant effect on the phase and morphology of precipitated FePO4·2H2O was investigated by adding 1 wt% PEG-6000, CTAB and SDS in the precursor solution before adding ammonia, and the obtained amorphous hydrated FePO4 was named as samples B, C, and D.
To form FePO4 with an alpha quartz structure, the amorphous hydrated FePO4 was then annealed at 700 °C for 5 h.
Recovery of Li2CO3
Li2CO3 was recovered according to the following steps: the filtrate was concentrated, brought to a boil, Na2CO3 was added, it was filtered, and the filter cake was washed thoroughly with deionized water.17 Finally, the filter cake was dried at 80 °C until a constant weight, and Li2CO3 was then obtained.Re-synthesis of LiFePO4 via a carbon thermal reduction method using recycled FePO4 and Li2CO3 as raw materials
To re-synthesize LiFePO4, the recycled Li2CO3 (Li source) and FePO4 (both Fe source and P source) with a molar ratio of Li:Fe:P = 1.05:1:1 were used as raw materials, and an additional 20 wt% of sucrose (relative to the weight of the raw materials) was used as a carbon source, which not only reduces the Fe3+ to Fe2+, but also decomposes to a continuous carbon network for electrical conduction. These raw materials were first ball-milled together for 7 h with ethanol as a dispersant. The obtained mixture was heated to 300 °C under an argon atmosphere for 4 h. The mixture was then ground and heated to 700 °C under a reducing atmosphere for 10 h. The product (i.e. LiFePO4/C) was ground in an agate mortar for later use.
Characterization of materials
The concentration of P, Fe and Li was determined using an inductively coupled plasma emission spectrometer (ICP-AES, American Perkin-Elmer Company Optima 5300 DV). X-ray diffraction (XRD) patterns were collected using a D/max-gamma B X-ray diffractometer (Rigaku, Japan) with Cu Kα radiation (λ = 0.15405 nm), a voltage of 45 kV, and a current of 50 mA. The patterns with 2θ ranging from 10 to 90° were collected using a scan rate and step of 10° min−1 and 0.02°, respectively. The FePO4 and LiFePO4 products were characterized using an infrared spectrometer (IR Magna 560 Nicolet companies in the United States). The morphology was examined using a scanning electron microscope (QUANTA-200F American FEI Company) with a working voltage of 10 kV. To study the effect of the annealing temperature on the LiFePO4 cathode electrode, TG-DSC analysis (STA449F3, NETZSCH) was carried out with temperatures ranging from room temperature to 800 °C at a heating rate of 5 °C min−1 under a flow of air. XPS measurement was conducted using a PHI 5700 ESCA System (USA), using a monochromatised Mg-Ka radiation source. X-ray absorption near edge structure (XANES) spectroscopy of the Fe K-edge was conducted using the Soft X-ray beamlines and Variable Line Spacing Plane Grating Monochromator (VLS-PGM) beamlines at the Canadian Light Source (CLS). XANES was recorded in the surface sensitive Total Electron Yield (TEY) or Fluorescence Yield (FY) using a Silicon Drift Detector (SDD) at SXRMB and a Micro-Channel Plate (MCP) detector at PGM.Battery assembly and electrochemical test
The as-synthesized LiFePO4/C was mixed with acetylene black and polyvinylidene fluoride (PVDF) in a mass ratio of 8:1:1 to form a cathode slurry with N-methyl-2-pyrrolidone (NMP) as solvent. The slurry was coated on aluminum foil and vacuum dried at 100 °C for 10 h to obtain a cathode sheet. The dry cathode sheet was then punched into 16 mm wafers, and pressed at 1 MPa of pressure for 3–5 min. The pressed wafers were vacuum dried at 120 °C for 10 h, and transferred to a glove box for later use. The cathode sheet, lithium foil anode, and Celgard2300 microporous polypropylene diaphragm were assembled into CR2025-type coin cells in a glove box filled with high-purity argon. 1 mol L−1 LiPF6 in a mixture of ethylene carbonate (EC) and dimethyl carbonate (DMC) (with volume ratio of 1:1) solution was used as the electrolyte. The charge–discharge test was carried out using Neware Battery under different current densities (1C = 170 mA g−1) in a voltage range of 2.5–4.2 V.
Results and discussion
Effect of heat treatment on the spent LiFePO4 electrode
In order to find an appropriate temperature for annealing, TG-DSC analysis of the LiFePO4 electrode was performed from room temperature to 800 °C under air flow at a heating rate of 5 °C min. As shown in Fig. 1(a), the TG curve shows a weight loss of about 6.68% as the temperature increases from room temperature to 600 °C. The DSC curves show several exothermic peaks, which are around 421.8 °C, 475.5 °C, and 579.2 °C, which can be due to decomposition of the binder and carbon in the electrode. According to the previous report,18 the decomposition of the PVDF begins at 350 °C under an oxygen atmosphere and 600 °C is ideal for PVDF decomposition. The weight of the electrode shows little change at temperatures above 600 °C, indicating the completion of pyrolysis of the binder and carbon. There is an obvious endothermic peak around 658.6 °C ascribed to the oxidation of aluminum foil, which is confirmed by the DSC/TG test of Al foil (shown in Fig. 1(b)). Thus, we conclude that the optimum temperature of the heat treatment is 600 °C. | ||
| Fig. 1 TG-DSC curves of waste LiFePO4 cathode sheet (a) and aluminum foil (b). | ||
Table 1 shows the effect of annealing temperature on the color of the electrode material and the degree of difficulty in separating the materials from the current collector via oscillating sieve separation. The separation effect of the active material in the collection fluid demonstrates that the decomposition of the binder is not complete when the annealing temperature is lower than 550 °C. Meanwhile, when the temperature reaches 600 °C, the binder is completely decomposed and the active material can be easily separated from the current collector. The color change is due to the pyrolysis of the binder and the oxidation of Fe2+ to Fe3+ during the annealing process in air. Fig. 2 shows XRD patterns of the electrode after the 1 h annealing process at different temperatures. Despite the annealing temperature, the major phases of the material after heat treatment are monoclinic Li3Fe2(PO4)3 and Fe2O3, thus the oxidation of the LFP was expected to follow eqn (1). When the temperature reaches 650 °C, diffraction peaks related to Al2O3 appear, indicating the oxidation of the Al current collector (which is consistent with the analysis of the TG-DSC curves) and exfoliation surface oxidized layer in the fluid flow. These results clearly demonstrate that the best annealing temperature should be 600 °C.
| 12LiFePO4 + 3O2 → 4Li3Fe(PO4)3 + 2Fe2O3 | (1) |
| Temperature (°C) | 450 | 500 | 550 | 600 | 650 |
|---|---|---|---|---|---|
| Color | Dark brown | Light brown | Maroon | Brick red | Brick red |
| Amount of active materials separated from current collector | 72% | 84% | 96% | 100% | 100% |
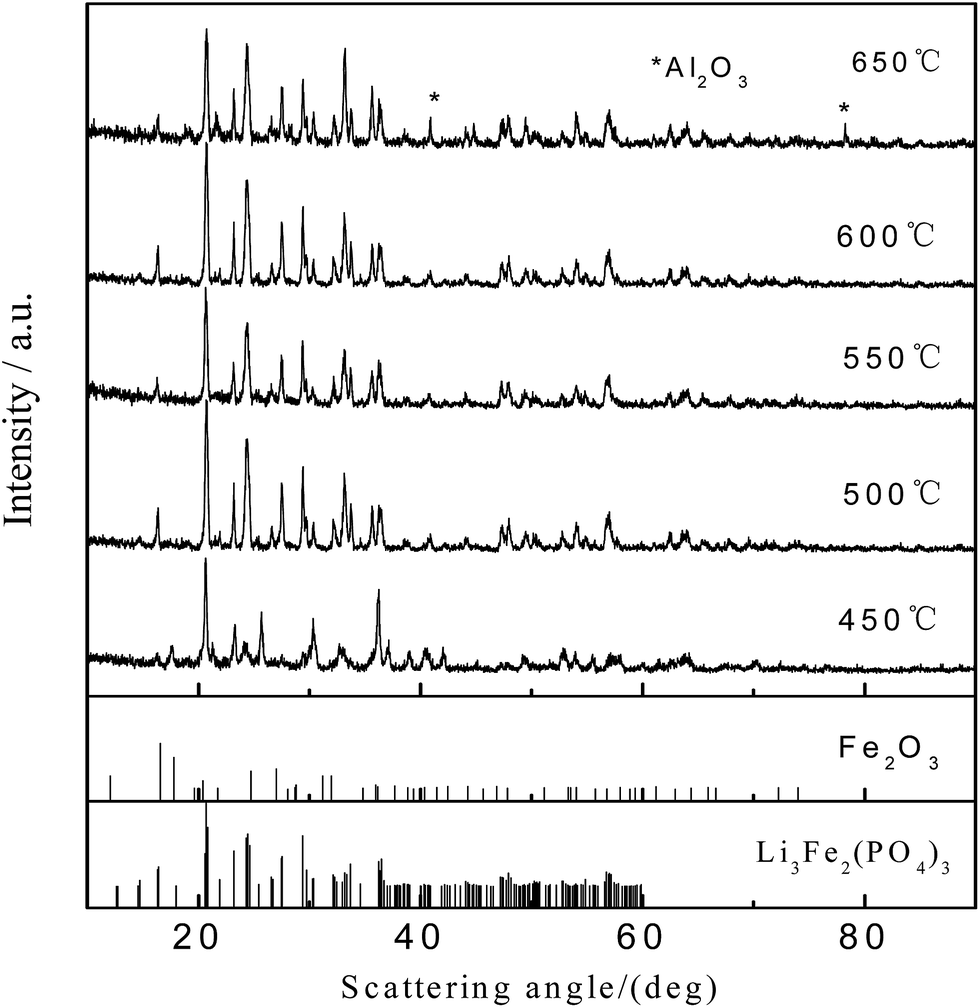 | ||
| Fig. 2 XRD patterns of the mixed powder after annealing at different temperatures for 1 h. | ||
The XPS spectrum (Fig. 3) of the mixed powder both before and after annealing at 600 °C for 1 h shows the existence of O, C, Li, Fe, and P. The Li curve peaks are at about 60 eV. The XPS Fe 2p3/2 spectra (Fig. 3(b)) of the mixed powder before and after annealing show binding energies of 709.7 and 711.3 eV, which correspond to valence numbers of +2 and +3, respectively.19 Therefore it can be assumed that Fe has been completely oxidized to Fe3+. Thus, it can concluded that the Fe2+ ion within LiFePO4 can be completely oxidized to Fe3+ via annealing at 600 °C for 1 h, which is consistent with analysis of the XRD patterns before and after heat treatment.
 | ||
| Fig. 3 XPS spectra of the mixed powder before and after annealing at 600 °C for 1 h. | ||
Optimization of the acid leaching process
In order to optimize the conditions for acid leaching of the mixed powder, the effects of sulfuric acid concentration, liquid to solid ratio, reaction time, and reaction temperature on the dissolution efficiency were investigated systematically.With a temperature of 60 °C, a liquid to solid ratio of 10:1, and a reaction time of 4 h, the concentration of sulfuric acid was varied from 0.5 mol L−1 to 5 mol L−1. As shown in Fig. 4(a), with the increase in concentration of sulfuric acid from 0.5 to 2.5 mol L−1, the leaching efficiency of Li and Fe increased from 25% to 97.2% and from 46% to 98.5%, respectively, while further increasing the concentration of sulfuric acid above 2.5 mol L−1 shows a negligible effect on the leaching rate. Therefore, the best concentration of sulfuric acid for leaching is 2.5 mol L−1.
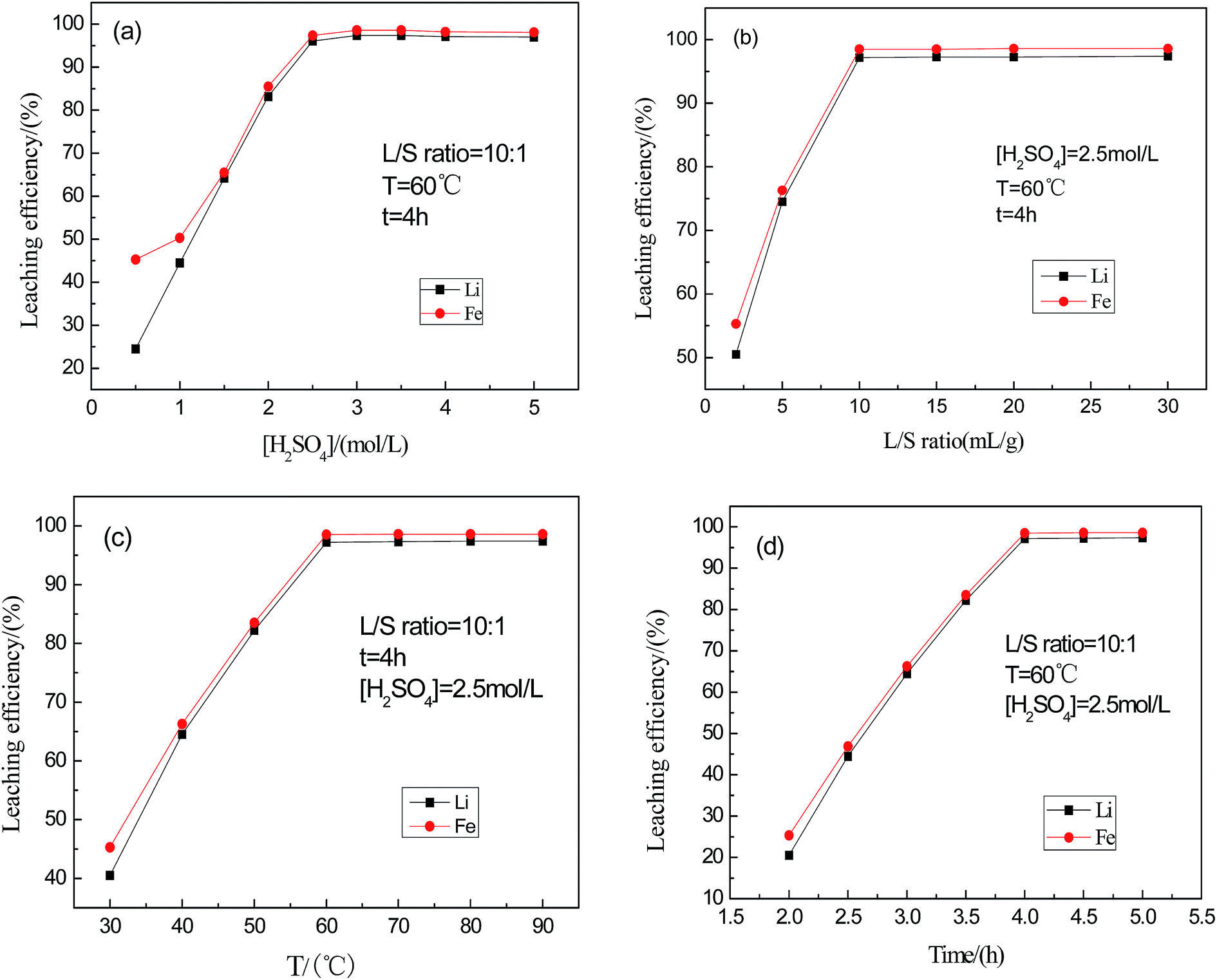 | ||
| Fig. 4 Effect of sulfuric acid concentration, L/S ratio, temperature and time on the leaching of Fe and Li from the battery powder. | ||
Fig. 4(b) shows the effect of the L/S ratio on the leaching rate when the concentration of sulfuric acid is 2.5 mol L−1, the temperature is 60 °C, and the reaction time is 4 h. When the ratio of L/S reached 10, the leaching rates of Fe and Li were 97.4% and 96.1%, respectively. Since the leaching rate shows little increase with further increases of L/S, the optimal L/S (mL g−1) ratio was determined to be 10.
Fig. 4(c) shows the effect of temperature on the leaching rate when the concentration of sulfuric acid is 2.5 mol L−1, the L/S ratio is 10:1, and the reaction time is 4 h. When the temperature is lower than 60 °C, the leaching rate increases when the reaction temperature increases, while when the temperature reaches above 60 °C the leaching rate shows little change as the temperature increases. Therefore, the optimum leaching temperature is 60 °C.
Fig. 4(d) shows the effect of the reaction time on the leaching rate when the concentration of sulfuric acid is 2.5 mol L−1, the temperature is 60 °C, and the L/S ratio is 10:1. Similar to previous discussions, the optimal extraction time was determined to be 4 h.
It can be concluded that the optimal leaching efficiency (i.e. 97% and 98% for Li and Fe, respectively) can be achieved when the sulfuric acid concentration, L/S, temperature and time are 2.5 mol L−1, 10 mL g−1, 60 °C and 4 h, respectively.
Compositional and structural analysis of the “FePO4”
The pH value is known to be the key factor that controls the precipitation of FePO4, and thus will be investigated in the following part. When the pH > 1, the solution begins to precipitate. As shown in Fig. 5, the Fe/P molar ratio (analyzed by ICP-AES) in the precipitation increases gradually accompanied with the increase of pH value. When the pH value was adjusted to between 1.9 and 2.1, the Fe/P molar ratio was analyzed as 0.961–1.008, which is consistent with the molar ratio in FePO4. Therefore, the best pH value for “FePO4” precipitation is tentatively set to be 2 ± 0.5 in the control experiment. | ||
| Fig. 5 Effect of pH value on the Fe/P molar ratio of recovered “FePO4”. | ||
Table 2 shows the products analyzed by detailed ICP-AES analysis of the precipitate obtained under a pH value of 2. The results show that the Fe/P molar ratio of the precipitate is 1.01, and the overall mass fraction of metal impurities in the products is less than 0.005%, and the sulfur content is less than 0.018%, which is in the standard range of values, indicating the high quality of the recovered “FePO4”.
| Fe (wt%) | Mg (wt%) | Na (wt%) | K (wt%) | Cu (wt%) | Al (wt%) | Pb (wt%) | S (wt%) | P (wt%) | P/Fe | |
|---|---|---|---|---|---|---|---|---|---|---|
| Standard values20 | 28–30 | ≤0.005 | ≤0.005 | ≤0.005 | ≤0.001 | ≤0.005 | ≤0.0015 | ≤0.05 | 15.5–17.0 | 0.99–1.03 |
| Measured values | 29.5 | 0.003 | 0.0035 | 0.0025 | 0.0006 | 0.0031 | 0.0010 | 0.018 | 16.6 | 1.01 |
Fig. 6 shows the XRD patterns of the products recovered from the solution with/without surfactants. Despite the addition of surfactants (including the one recovered without adding surfactant), the products show an amorphous nature, which is consistent with the literature when the pH value of the solution was adjusted to 2.21 The XRD result has hindered us in determining what the product is. Therefore, to clarify this point, infrared spectroscopy and DSC-TG of the as-precipitated products, and XRD data of the annealed products was collected and analyzed.
 | ||
| Fig. 6 XRD patterns of the recovered FePO4 xH2O with and without surfactants. | ||
Infrared spectroscopy is a method to identify molecular structure and compounds according to the relative vibration and rotation of the atoms and molecules.22 Fig. 7 shows the infrared spectrum of the recycled FePO4. The absorption peaks at 3389 and 1635 cm−1 correspond to the stretching vibration and bending vibration of O–H in the H2O molecules, which indicated the existence of crystallized water in the recovered sample. Meanwhile, the absorption peaks at 1050 and 600 cm−1 correspond to telescopic vibration and symmetric bending vibration of P–O in PO43−, respectively. The results indicate that the recovered sample may be FePO4·xH2O.
 | ||
| Fig. 7 IR spectra of recovered FePO4 xH2O with and without surfactants. | ||
Fig. 8 shows the DSC-TG curve of FePO4·xH2O. From the DSC curve we can see that there is an obvious endothermic peak at about 152.45 °C. The TG curve showed that there are obvious changes in this range of temperature between the slope curves of 90–200 °C, indicating that the loss of crystal water changes the mass percentage. From room temperature to 300 °C, the TG curve measured a weight loss of about 19.24%. Complimented by the Fe content from ICP (i.e. 29.9 wt%), the recovered sample can be identified as FePO4·2H2O. There appears to be an exothermic peak at 670.36 °C in the DSC curve, which may indicate a phase transition in the material (e.g. from amorphous to an alpha quartz structure).
 | ||
| Fig. 8 TG-DSC curves of the recycled FePO4·xH2O. | ||
To further support the analysis from Fig. 8, XRD patterns of the recycled amorphous FePO4·2H2O after being annealed at 700 °C for 5 h were collected and are shown in Fig. 9. The diffraction patterns of the products match well with FePO4 with an alpha quartz structure (PDF#29-0715); the phase transition is consistent with the expectation from the DSC-TG analysis. The unit cell parameters were calculated to be a = 0.50330 nm, b = 0.50330 nm, and c = 1.12470 nm, via a least squares fit. The complementary results presented above clearly prove that the precipitated products are amorphous FePO4·2H2O.
 | ||
| Fig. 9 XRD spectra of FePO4 after heat treatment: (A) without surfactant, (B) with PEG-6000, (C) with CTAB, (D) with SDS. | ||
The SEM images of the as-precipitated FePO4·2H2O are shown in Fig. 10. A severe degree of agglomeration occurred when there was no surfactant added during the phase precipitation process. Image C shows that CTAB is the surfactant for large particle size but agglomeration also exists. Image (D) shows when SDS was used as the surfactant, the precipitates tends to reunion although the primary particle size is small. Image (B) shows that with PEG-6000 as the surfactant, the particle size is smaller and distribution is more uniform.
 | ||
| Fig. 10 SEM images of the recovery of FePO4·2H2O: (A) without surfactant, (B) PEG-6000, (C) CTAB, and (D) SDS. | ||
Fig. 11 shows the magnitudes of the Fourier transforms of the k2-weighted EXAFS spectra of Fe K-edge in both TEY and FY mode. While TEY shows information about the surface of the materials (i.e. 5–10 nm), FY depicts structural information of the bulk material (60 nm under the surface).23 Clearly, when PEG-6000 surfactant was adding during phase precipitation, the recovered FePO4·2H2O is much more similar to the commercial one compared to the one precipitated without adding surfactant. This indicates the higher quality of the recovered FePO4 when PEG-6000 surfactant was added.
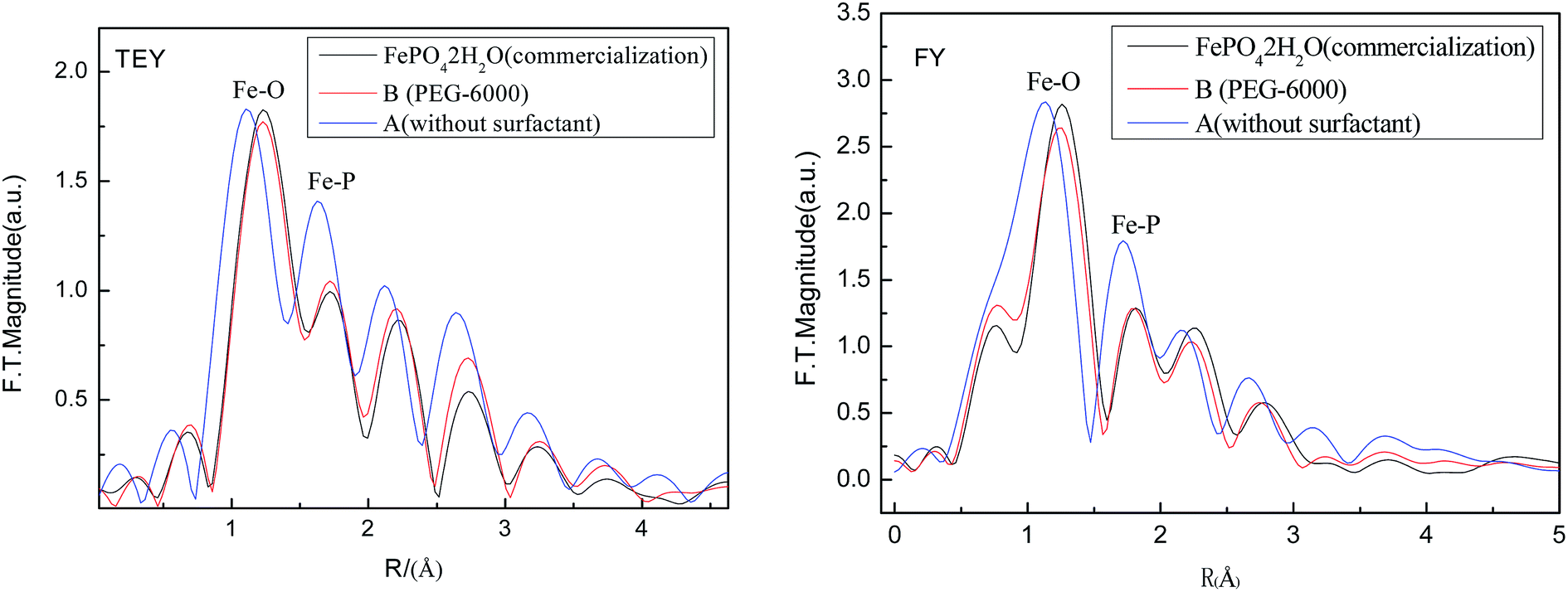 | ||
| Fig. 11 EXAFS analysis of the FePO4·2H2O material. | ||
Fig. 12 illustrates the Fe K-edge XANES spectra within FePO4·2H2O. EXAFS curves of the recovered sample show similar characteristics to that of the commercial one. When PEG-6000 surfactant is added during phase precipitation, the recovered sample and the commercial FePO4·2H2O share almost the same spectrum.
 | ||
| Fig. 12 K-edge XANES spectrum for Fe in the FePO4·2H2O material. (a) is an enlargement of the first rectangle; (b) is the enlargement of the second rectangle. | ||
The locations on the graph of XANES edge absorption and oxidation state are closely related. This relationship can be used to determine the valence number of the elements because an increase in oxidation state corresponds to a movement of higher energy in absorption. From Fig. 12, the absorption graph location of FePO4·2H2O and B are the same. This means that the iron element has the same valence number.
The recovered Li2CO3
Fig. 13 clearly shows that the recovered Li2CO3 is pure and highly crystalline, as indicated by the well matched patterns to PDF#22-1141 and sharp diffraction peaks, respectively. The particle size ranges from several microns to about twenty microns. | ||
| Fig. 13 XRD pattern and SEM images of the recovered Li2CO3. | ||
Synthesis of LiFePO4/C via carbon thermal reduction using the recycled products as raw materials
The recycled FePO4·2H2O (A, B, C, D) and Li2CO3 are used as the raw material to synthesize LiFePO4/C. The obtained LiFePO4/C products were named A1, B1, C1, and D1, which correspond to using FePO4·2H2O precipitated under different conditions (i.e. A, B, C, and D). As shown in Fig. 14, all the synthesized samples can be indexed as LiFePO4 with olivine structure (PDF# 40-1499). We cannot find the diffraction peaks of carbon in the XRD diagram, which may be associated with low carbon content and/or its amorphous nature. | ||
| Fig. 14 XRD spectra of the synthesized LiFePO4/C. | ||
Fig. 15 shows that the spectra of all the synthesized samples of LiFePO4/C. These spectra are mainly distributed in two spectral bands, namely the strong absorption of 1120–940 cm−1 and that of 650–540 cm−1. We can see from Fig. 15 that the group of the PO2 at 1140 cm−1 is the stretching vibration and those at 1096 cm−1 and 1054 cm−1 are the PO anti-symmetric stretching vibration. Wavenumbers 968 cm−1 and 638 cm−1 are symmetric stretching vibrations in LiFePO4 PO. 577 cm−1 is the anti-symmetric bending vibration in PO2, and 551 cm−1 and 473 cm−1 are symmetric bending vibrations in PO. Wavenumber 505 cm−1 is the swing vibration peak in PO2.24 The existing carbon particles do not affect the spectral structure of LiFePO4/C.
 | ||
| Fig. 15 IR spectra of the synthesized LiFePO4/C. | ||
As can be seen from Fig. 16, the morphology of the synthesized LiFePO4/C can be affected by the different raw materials of FePO4·2H2O (i.e. A, B, C, and D), which are recovered with/without different surfactants. The synthesized LiFePO4/C using FePO4 B has the smallest particle sizes when compared to LiFePO4/C synthesized using other FePO4 samples, which is believed to be beneficial for the electrochemical performance due to the shorter diffusion length of Li+.
 | ||
| Fig. 16 SEM images of the synthesized FePO4/C. | ||
Fig. 17 compares the EXAFS graph of LiFePO4/C resynthesized with the addition of surfactant (B1) and commercial LiFePO4/C. From the perspective of fingerprint analysis, the oscillation peaks and intensity of B1 and LiFePO4/C are very similar.
 | ||
| Fig. 17 EXAFS analysis of the LiFePO4 material. | ||
Fig. 18 shows that the samples have a weak inclination. This means that the 3d orbital of Fe and the 3p orbital or the p orbital of oxygen form a hybrid.25 Thus, Fe atoms are from the distorted FeO6 octahedron. The XANES absorption position and oxidation state are closely related. This information can be used to determine the valence number of elements. As the oxidation state increases, the absorption energy will also increase. From Fig. 18, the absorption position of LiFePO4/C and B1 are the same. This means that the Fe element has the same valence number. XANES can offer qualitative information regarding the three-dimensional structures around the atomic absorption. Because XANES can only detect absorption to 200 eV, there is a better signal-to-noise ratio. Within the appropriate parameters of signal-to-noise ratio, as shown in Fig. 18, the LiFePO4/C XANES data was consistent. This indicates that the Fe within the sample has the same structure, which all exist in the form of LiFePO4/C.
 | ||
| Fig. 18 The K-edge XANES spectrum for Fe in the LiFePO4/C material. ((a) is an enlargement of the first rectangle from the figure; (b) is the enlargement of the second rectangle from the figure.) | ||
The electrochemical performance test of LiFePO4/C
Fig. 19 shows the first charge/discharge curve of LiFePO4/C (A1–D1) synthesized from the recycled FePO4 precursors (A–D).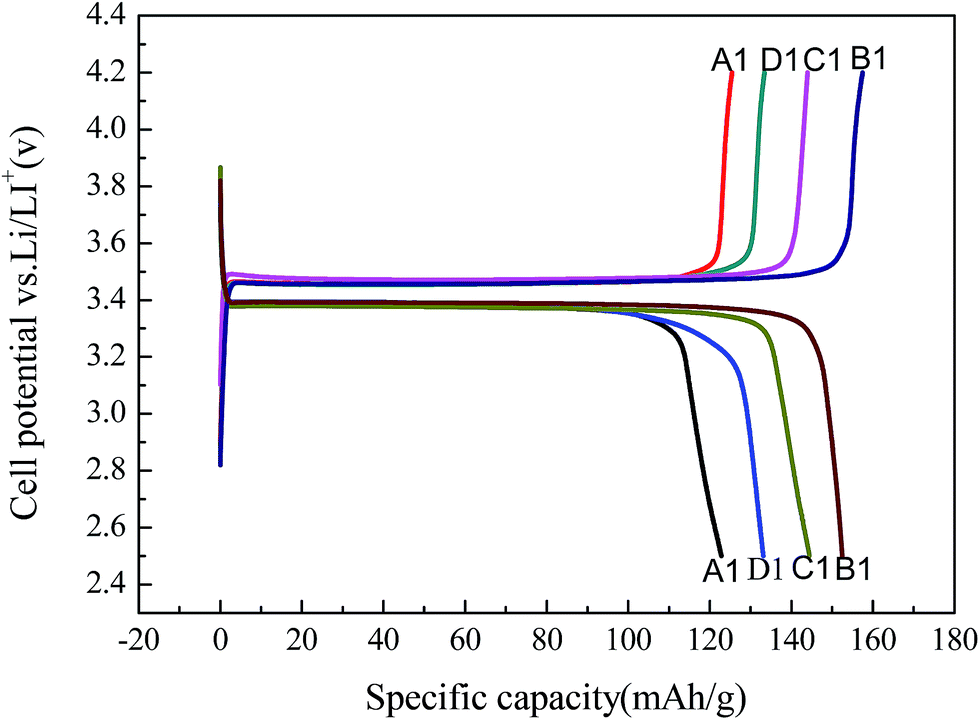 | ||
| Fig. 19 Charge/discharge curves of LiFePO4/C synthesized using different FePO4·2H2O samples: (A1) without surfactant, (B1) PEG-6000, (C1) CTAB, and (D1) SDS. | ||
The first charge/discharge capacities of B1 are 155.4 mA h g−1 and 153.3 mA h g−1, respectively, which corresponds to an initial coulombic efficiency of 98.6%. These results are comparable to the performance of LiFePO4 prepared via a carbon thermal reduction method using FePO4 2H2O as the iron source from the previous literature.26 With SDS and CTAB as the surfactants, the first discharge capacities are 147.5 mA h g−1 and 133.4 mA h g−1, respectively. However, without a surfactant the first discharge capacity was 122.8 mA h g−1. These electrochemical results are consistent with the particle size analysis of the samples (shown in Fig. 16), that is, a smaller particle size leads to better Li+ diffusion kinetics, and thus a higher discharge capacity. To demonstrate the high quality of the regenerated LiFePO4/C, B1 was selected as a example to compare with the commercial LiFePO4/C in the following parts.
Fig. 20(a) presents the charge/discharge curve of B1 under different current densities. The discharge capacity under rates of 0.2, 0.5, 1, 2, and 5C are 152.2 mA h g−1, 150.1 mA h g−1, 137.8 mA h g−1, 120.4 mA h g−1 and 99.8 mA h g−1, respectively. Fig. 20(b) presents the charge/discharge curve of a commercial sample under different current densities. The discharge capacity under rates of 0.2, 0.5, 1, 2, and 5C are 156.8 mA h g−1, 151.3 mA h g−1, 138.2 mA h g−1, 121.5 mA h g−1 and 100.6 mA h g−1, respectively.
 | ||
| Fig. 20 The charge–discharge curves of (a) the regenerated LiFePO4/C (B1) sample and (b) the commercial sample, between 2.5 and 4.2 V at rates of 0.2, 0.5, 1, 2 and 5C. | ||
The capacity evolution during the whole rate test is shown in Fig. 21. It is observed that at 0.1C, 0.2C, and 0.5C charge/discharge rates, there is no capacity decay among the 20 cycles, while at 5C charge/discharge rate, the rate of capacity retention over the 20 cycles was 91.6% and 92.3%, respectively. One reason for this is the significant polarization effects. A larger current electrode also distorts the host structure and limits the extraction and insertion of Li+. Another noteworthy point is that when the current density was reset to 0.1C, the capacity was almost recovered, indicating the good electrochemical reversibility of the material.
 | ||
| Fig. 21 Rate capability comparison of the regenerated LiFePO4/C (B1) sample and the commercial sample from the pure compound cycled between 2.5 and 4.2 V. | ||
Both the capacity and cyclability of the regenerated sample are comparable to the commercial sample, while the cost is competitive with the market price for the same chemicals prepared from primary resources.
Conclusion
This paper presents a relatively simple recovery process for FePO4 and Li2CO3 from waste lithium iron phosphate batteries and a preparation process for lithium iron phosphate battery cathode materials from the recycled material, as shown in Fig. 22. This process includes the following steps: (1) after crushing the discharged battery, recycle the membrane, battery shell and copper content, (2) heat treatment of the anode of 600 °C was conducted to remove the coating binder, surface active substances, and carbon. This also oxidizes Fe2+ to Fe3+ and aluminum foil is recycled using 0.5 mm sieve oscillation sieving, and the powders are mixed. (3) The mixed powder was then dissolved in 2.5 mol L−1 sulfuric acid, with the L/S equal to 10, a temperature of 60 °C, and a time of 4 h. As a result, 98% of the iron and 97% of the lithium are leached from the mixed powder. (4) With PEG-6000 as the surfactant, the pH value was adjusted to 2, causing precipitation of FePO4, and (5) the filtrate is concentrated and heated to boiling point. This results in the precipitation of Na2CO3 and Li2CO3. (6) The recycled FePO4 is used as the iron source and phosphorus source. Li2CO3 is used as the lithium source, and sucrose was used as the carbon source. The molar ratio of lithium, iron and phosphorus was set at 1.05:1:1. Then, the ingredients were ball-milled, dried, and the resulting mixture was obtained. (7) The mixture was then pre-sintered under argon, at a temperature of 350 °C, and calcined at a temperature of 750 °C for 10 h to obtain the LiFePO4/C cathode material.
 | ||
| Fig. 22 Proposed flow-sheet for the spent LIB recycling process. | ||
Under the optimized conditions, the recovered FePO4 and Li2CO3 crystals are high purity products, successfully reaching the standard of battery level.
The battery using regenerated LiFePO4/C as cathode was then subjected to an electrochemical performance test, and its resulting electrochemical performance was superb.
Acknowledgements
This research was supported by the National Natural Science Foundation of China (no. 51274075), the National Environmental Technology Special Project (no. 201009028), and Guangdong Province-Department University-Industry Collaboration Project (grant no. 2012B091100315).Notes and references
- M. J. Lain, J. Power Sources, 2001, 97–98, 736–738 CrossRef CAS.
- B. Scrosati and J. Garche, J. Power Sources, 2010, 195, 2419–2430 CrossRef CAS.
- D. Ra and K. S. Han, J. Power Sources, 2006, 163(1), 284–288 CrossRef CAS.
- J. Li, P. Shi, Z. Wang, Y. Chena and C. C Chang, Chemosphere, 2009, 77, 1132–1136 CrossRef CAS PubMed.
- C. K. Lee and K. I. Rhee, J. Power Sources, 2002, 109(1), 17–21 CrossRef CAS.
- L. Chen, X. Tang, Y. Zhang, L. Li, Z. Zeng and Y. Zhang, Hydrometallurgy, 2011, 108(1), 80–86 CrossRef CAS.
- D. M. Robinson, Y. B. Goreenblatt and G. C. Dismukes, J. Am. Chem. Soc., 2010, 132(33), 11467–11469 CrossRef CAS PubMed.
- D. Song, X. Wang, H. Nie, H. Shi, D. Wang, F. Guo, X. Shi and L. Zhang, J. Power Sources, 2014, 249, 137–141 CrossRef CAS.
- T. Zhang, Y. He, F. Wang, L. Ge, X. Zhu and H. Li, Waste Manag., 2014, 34, 1051–1058 CrossRef CAS PubMed.
- X. Zeng, J. Li and B. Shen, J. Hazard. Mater., 2015, 295, 112–118 CrossRef CAS PubMed.
- E. Gratz, Q. Sa, D. Apelian and Y. Wang, J. Power Sources, 2014, 262, 255–262 CrossRef CAS.
- Y. Weng, S. Xu, G. Huang and C. Jiang, J. Hazard. Mater., 2013, 246–247, 163–172 CrossRef CAS PubMed.
- H. Zou, E. Gratz, D. Apelian and Y. Wang, Green Chem., 2013, 15, 1183–1191 RSC.
- L. Li, X. Zhang, R. Chen, T. Zhao, J. Lu, F. Wu and K. Amine, J. Power Sources, 2014, 249, 28–34 CrossRef CAS.
- X. Liu, H. Li, D. Li, M. Ishida and H. Zhou, J. Power Sources, 2013, 243, 374–380 CrossRef CAS.
- http://www.dvdc100.com/v-auto-d-20160112-n-434339829/.
- P. Zhang, T. Yokoyama, O. Itabashi, T. M. Suzuki and I. Katsutoshi, Hydrometallurgy, 1998, 47, 259–271 CrossRef CAS.
- C. Hanisch, T. Loellhoeffel, J. Diekmann, K. J. Markley, W. Haselrieder and A. Kwade, J. Cleaner Prod., 2015, 108, 301–311 CrossRef CAS.
- S. Praneetha and A. Vadivel Murugan, RSC Adv., 2013, 3, 25403–25409 RSC.
- A. G. Peng, Z. C. He and C. Y. Yu, J. Wuhan Inst. Technol., 2013, 35(7), 1–5 CAS.
- Y. Huang, H. Ren, Z. Peng and Y. Zhou, Electrochim. Acta, 2009, 55, 311–315 CrossRef CAS.
- R. A. Nyquist and R. O. Kagel, Infrated spectra of inorganic compounds (3800–45 cm−1) [M], Academic Press, New York, 1997 Search PubMed.
- J. R. Croy, M. Balasubramanian, D. Kim, S. H. Kang and M. M. Thackeray, Chem. Mater., 2011, 23, 5415–5424 CrossRef CAS.
- Y. Zhao, L. Peng, B. Liu and G. Yu, Nano Lett., 2014, 14, 2849–2853 CrossRef CAS PubMed.
- M. S. Chen, S. H. Wu and W. K. Pang, J. Power Sources, 2013, 241, 690–695 CrossRef CAS.
- W. Zheng, H. Yu, G. Cao and X. Zhao, Chin. J. of Nonferrous Met., 2010, 20(3), 572–577 CAS.
| This journal is © The Royal Society of Chemistry 2016 |