Crystal structure of the multicopper oxidase from the pathogenic bacterium Campylobacter jejuni CGUG11284: characterization of a metallo-oxidase†
Catarina S.
Silva
,
Paulo
Durão
,
Amanda
Fillat‡
,
Peter F.
Lindley
,
Lígia O.
Martins
* and
Isabel
Bento
*
Instituto de Tecnologia Química e Biológica, Universidade Nova de Lisboa, Av. da República, 2781-901 Oeiras, Portugal. E-mail: bento@itqb.unl.pt; lmartins@itqb.unl.pt; Web: http://www.itqb.unl.pt/researchers/bento Web: http://www.itqb.unl.pt/martins Fax: +351-214433644; Tel: +351-214469662
First published on 29th November 2011
Abstract
Multicopper oxidases are a multi-domain family of enzymes that are able to couple oxidation of substrates with reduction of dioxygen to water. These enzymes are capable of oxidizing a vast range of substrates, varying from aromatic to inorganic compounds such as metals. This metallo-oxidase activity observed in several members of this family has been linked to mechanisms of homeostasis in different organisms. Recently, a periplasmic multicopper oxidase, encoded by Campylobacter jejuni, has been characterised and associated with copper homeostasis and with the protection against oxidative stress as it may scavenge metallic ions into their less toxic form and also inhibit the formation of radical oxygen species. In order to contribute to the understanding of its functional role, the crystal structure of the recombinant McoC (Campylobacter jejuni CGUG11284) has been determined at 1.95 Å resolution and its structural and biochemical characterizations undertaken. The results obtained indicate that McoC has the characteristic fold of a laccase having, besides the catalytic centres, another putative binding site for metals. Indeed, its biochemical and enzymatic characterization shows that McoC is essentially a metallo-oxidase, showing low enzymatic efficiency towards phenolic substrates.
Introduction
Laccases (p-diphenol: dioxygen oxidoreductase, EC 1.10.3.2) are the simplest members of the multicopper oxidase (MCO) family of enzymes (ref. 1–4 and references therein) exerting a wide range of physiological functions. Overall, these enzymes are capable of coupling the oxidation of a vast range of substrates, from organic aromatic amines, non-phenols and phenols to inorganic compounds (ref. 5, 6 and references therein), with reduction of dioxygen to water. Such an asset makes these enzymes interesting targets for biotechnological purposes.7 To date, the vast majority of laccases whose structures have been reported are derived from fungi, whereas bacterial laccases remain far less well characterized (ref. 8 and references therein). Nevertheless, recent progress in whole genome analysis has provided evidence for novel bacterial laccases or laccase-like proteins, identified in many gram-negative and gram-positive bacteria.4,8,9 Even in thermophilic organisms, for which such proteins are rare, corresponding laccase-like proteins have been identified.10–12The overall laccase structural fold comprises three cupredoxin-type domains, characterized by a Greek key β-barrel topology (ref. 5 and references therein). In addition, two copper centres, a mononuclear type 1 blue copper centre (T1) and a trinuclear cluster, consisting of two type 3 (T3) and one type 2 (T2) copper atoms, mediate the oxidation of substrates and the concomitant reduction of dioxygen to water, respectively. The oxidation of four substrate molecules at the T1 copper centre (the primary electron acceptor site) leads to the reductive cleavage of dioxygen to two molecules of water at the trinuclear site.1–3,13 The first three-dimensional structure of a bacterial laccase was reported for the CotA laccase,14–16 a laccase with oxidase activity towards aromatic compounds, from the outer layer of the spore coat of Bacillus subtilis.17 Another structure of a bacterial MCO is that of the Escherichia coli CueO protein.18,19 This enzyme, contrary to CotA laccase, is able to oxidize lower valence metal ions (Cu1+ and Fe2+) more efficiently than aromatic substrates.20 As such, this enzyme is more specifically a metallo-oxidase.21 The presence of a methionine-rich (Met-rich) region near the T1 Cu site has been suggested to provide additional binding sites for exogenous Cu, playing a role in Cu-dependent activity.19,20 Just recently, Singh et al. have hypothesized that this region may in fact be used for copper binding during CueO's response to copper toxicity.22 Such a region is also present in other proteins implicated in Cu homeostasis, additionally supporting its participation in Cu tolerance.23–25 Furthermore, CueO shows ferroxidase activity in a similar manner to its mammalian counterpart human ceruloplasmin (hCp), the major Cu-containing protein in plasma, which plays a vital role in iron homeostasis in vertebrates.26 Similarly, the yeast MCO Fet3 protein (Fet3p) catalyses the conversion of ferrous to ferric iron, allowing its uptake across the yeast plasma membrane into the cytosol. Hence, a correlation between both copper and iron metabolism in these organisms has been suggested.27
Campylobacter jejuni, a pathogenic, Gram-negative, microaerobic, flagellated, spiral bacterium, is one of the most significant causes of human enteric diseases worldwide.28,29 Included in C. jejuni's virulence factors is its ability to acquire iron, a homeostasis system known in some detail.30,31 However, the way that other essential metals, such as copper, are acquired, processed and regulated in C. jejuni remains to be fully clarified. From its genome sequence (strain NCTC 11168),28 a gene (Cj1516) was identified as encoding for a periplasmic oxidoreductase, sharing 28% and 24% sequence identity with CueO and CotA laccase, respectively. Hall et al.32 confirmed that the Cj1516 protein is a MCO as its spectroscopic properties matched those of typical MCOs. Moreover, and similar to its homologue CueO, C. jejuni's MCO showed evidence of metallo-oxidase activity towards Cu1+ and Fe2+. Further studies conducted by Hall et al.32 led these authors to propose that the major physiological role of the identified MCO seems to be copper detoxification in the periplasm.
In this paper the crystal structure of the recombinant multicopper oxidase from Campylobacter jejuni (McoC), obtained from the type strain CGUG11284, is reported as well as its biochemical characterization. The results clearly provide evidence for the enzyme's function as a metallo-oxidase. The presence of a Met-rich region (resembling its homologue CueO) and other secondary structure elements near the mononuclear T1 Cu site occludes a substantial portion of the highly exposed substrate binding cavity identified for CotA laccase, functioning as a barrier for the access of bulkier substrates to this site. McoC mainly shows specificity as a cuprous oxidase, possibly being involved in the detoxification of Cu1+ exported from the cytoplasm to the periplasm by converting it to the less toxic Cu2+ form. Moreover, it also exhibits slight ferroxidase activity.
Materials and methods
Construction of a strain containing the mcoc gene
The mcoC gene was amplified from the genome of C. jejuni CGUG11284 by PCR using oligonucleotides mcoC-181D (5′-CTATAATACAAAAAAGCTAGCAAAAGG-3′) and mcoC-1756R (5′-CCTAAGAATTCCACTTTTACCATATTATTCC-3′). The primers were designed taking into account the Cj1516 gene sequence of C. jejuni NCTC11168. The PCR product was digested with NheI and EcoRI and inserted between the same restriction sites of plasmid pET-21a(+) (Novagen) to yield pATF-5. A codon usage analysis of mcoC revealed that it contains codons rarely used by E. coli (25 arginine tRNAArg(AGG/AGA/CGG/CGA), 18 lysine tRNALys(CTC) and 22 isoleucine tRNAIso(ATA)), a limiting expression factor by common E. coli strains. Therefore, the host strain Rosetta (DE3) pLysS (Novagen), expressing rare tRNAs, was used. Introduction of pATF-5 into Rosetta E. coli created strain LOM 418, in which the protein was produced under the control of the T7lac promoter.Overproduction and purification of McoC
Strain LOM 418 was grown in Luria-Bertani (LB) culture medium (10 g L−1 tryptone; 5 g L−1 yeast; 5 g L−1 NaCl) supplemented with ampicillin (100 μg mL−1) and chloramphenicol (34 μg mL−1). The cells were grown at 30 °C until reaching an OD600 of 0.6. At this time 0.1 mM isopropyl-β-D-thiogalactopyranoside (IPTG) and 0.25 mM CuCl2 were added and the temperature reduced to 25 °C. Incubation was continued for further 4 h, when a change to microaerobic growth conditions was achieved.33 Cells were harvested by centrifugation (8000 × g, 10 min, 4 °C) and the cell sediment suspended in 20 mM Tris–HCl buffer, pH 7.6, with a mixture of protease inhibitors, DNase I and MgCl2 (5 mM). Cells were disrupted in a French pressure cell (at 900 psi), followed by centrifugation (18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 × g, 60 min, 4 °C) to remove cell debris. The cell lysate was then loaded onto a SP-Sepharose column equilibrated with 20 mM Tris–HCl, pH 7.6, and the protein was eluted using a salt gradient up to 1 M NaCl. The active fractions were pooled, concentrated and applied onto a Superdex 200 HR 10/300 column equilibrated with 20 mM Tris–HCl, pH 7.6, with 0.2 M NaCl. All purification steps were carried out at room temperature in ÅktaPurifier (GE Healthcare).
000 × g, 60 min, 4 °C) to remove cell debris. The cell lysate was then loaded onto a SP-Sepharose column equilibrated with 20 mM Tris–HCl, pH 7.6, and the protein was eluted using a salt gradient up to 1 M NaCl. The active fractions were pooled, concentrated and applied onto a Superdex 200 HR 10/300 column equilibrated with 20 mM Tris–HCl, pH 7.6, with 0.2 M NaCl. All purification steps were carried out at room temperature in ÅktaPurifier (GE Healthcare).
UV-visible, EPR and RR spectra and redox titration
UV-Visible, electron paramagnetic resonance (EPR) and resonance Raman (RR) spectra were acquired, as described before,11,33 using a Nicolet Evolution 300 spectrophotometer from Thermo Industries, a Bruker EMX spectrometer equipped with an Oxford Instruments ESR-900 continuous-flow helium cryostat and a confocal spectrograph (Jobin Yvon, XY) equipped with grating of 1800 lines per millimetre and a liquid nitrogen cooled back-illuminated CCD camera, respectively. Redox titrations were performed at 25 °C and pH 7.6, under an argon atmosphere, and were monitored by visible spectroscopy (300–900 nm) in a Shimadzu Multispec-1501 spectrophotometer as described by Durão et al.34Enzyme activities
The McoC oxidation reactions of ABTS (2,2′-azinobis-(3-ethylbenzothiazoline-6-sulfonic acid)), SGZ (syringaldazine), 2,6-DMP (2,6-dimethoxyphenol) and ferrous sulfate (FeSO4) were monitored with either a Nicolet Evolution 300 spectrophotometer from Thermo Industries or a Molecular Devices Spectra Max 340 microplate reader with a 96-well plate. The oxidation reactions of ABTS, SGZ, 2,6-DMP and FeSO4 were followed at 420 nm (ε = 36000 M−1 cm−1), 530 nm (ε = 65000 M−1 cm−1), 468 nm (ε = 49600 M−1 cm−1) and 315 nm (ε = 2200 M−1 cm−1), respectively. Oxidations were determined by using Britton-Robinson (BR) buffer (100 mM phosphoric acid, 100 mM boric acid and 100 mM acetic mixture with 0.5 M NaOH to the desired pH). Ferrous sulfate oxidation was performed in 100 mM Mes buffer, pH 5. The effect of pH on the enzyme activity was determined at 37 °C for the different substrates, in BR buffer (pH 3–9). The reaction mixtures contained 1 mM of ABTS, 0.1 mM SGZ, 1 mM 2,6-DMP and 300 μM of FeSO4. Kinetic parameters were determined at 37 °C. Reaction mixtures contained ABTS (100–10000 μM, pH 4), SGZ (1–100 μM, pH 8), 2,6-DMP (40–400 μM, pH 8) and FeSO4 (10–300 μM, pH 5). The initial reaction rates were obtained from the linear portion of the progress curve. Kinetic constants Km and kcat were fitted directly to the Michaelis–Menten equation (OriginLab, Northampton, MA, USA). All enzymatic assays were performed at least in triplicate. Cuprous oxidase activity was measured in terms of rate of oxygen consumption by using an oxygen electrode (Oxygraph; Hansatech, Cambridge, UK) at 37 °C, following the method described by Singh et al.35 Stock solutions of [Cu(I)(MeCN)4]PF6 (Sigma-Aldrich, St Louis, MO, USA) were freshly prepared in argon-purged acetonitrile and subsequently diluted anaerobically by using gas-tight syringes. Reactions were initiated by adding the enzyme to an air-saturated mixture containing substrate (1 mM), 100 mM MES buffer, pH 5 and 5% acetonitrile. The buffer was chosen to provide the best stability for the substrate used, and all reactions were corrected for background auto-oxidation rates of Cu(I). The protein concentration was measured by using the absorption band at 280 nm (ε280 = 40680 M−1 cm−1) or the Bradford assay using bovine serum albumin as standard.
Crystallisation and data collection
Crystallisation experiments were carried out by the vapour diffusion method at 298 K. Sitting-drops were set in a 1:1 ratio, using 1.5 μL of protein sample (at concentrations of 7–8 mg mL−1, in 20 mM Tris–HCl, pH 7.6 with 200 mM NaCl) and precipitant solutions. Suitable well diffracting crystals were grown after 4–10 days, within the following range of conditions: 25–32% PEG 3350 with 0.15/0.2 M MgCl2 in 0.1 M Bis-Tris Propane, pH 5.5, containing 3% dioxane. The crystals used in X-ray analysis were grown in a drop containing 32% PEG 3350 with 0.2 M MgCl2 in 0.1M Bis-Tris Propane, pH 5.5, with 3% dioxane. McoC crystals appeared as blue rectangular prisms of variable size (ca. 100–250 μm). Cryo conditions were provided by adding 22% ethylene glycol to the crystallisation condition, prior to data collection.
Diffraction data were collected under a cold nitrogen stream at 100 K, on ID14-EH4 at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. Crystals diffracted to 1.95 Å resolution and belong to the monoclinic space group P21, containing one single molecule in the asymmetric unit and a solvent content of around 37% (as estimated by the Matthews coefficient).36 Data sets were processed with iMOSFLM v1.0.437,38 and scaled using SCALA from the CCP4 programs suite.39 Data collection details and processing statistics are listed in Table 1.
| Values in parentheses refer to the highest-resolution shell (2.06–1.95 Å). Ramachandran analysis was determined by RAMPAGE.39 ESRF, European Synchrotron Radiation Facility; EDO ethylene glycol.a Based on maximum likelihood.b Rms deviations from standard values. | |
|---|---|
| Data collection statistics | |
| Synchrotron beam line | ESRF ID14-EH4 |
| Wavelength/Å | 0.947 |
| Detector distance/mm | 301.8 |
| Detector | ADSC Q315r |
| Resolution/Å | 1.95 |
| Space group | P21 |
| Cell parameters a, b, c/Å | 48.8, 94.7, 50.4 |
| α = γ, β/° | 90, 100.6 |
| Mosaicity/° | 0.63 |
| Oscillation angle/° | 1.45 |
| Oscillation range/° | 217.5 |
| No. of observed hkl reflections | 141798 (19386) |
| No. of unique hkl reflections | 32673 (4624) |
| Completeness (%) | 99.4 (97.3) |
| Mn(I/σ(I)) | 14.9 (3.5) |
| I/σ(I) | 6.6 (2.0) |
| R merge | 0.073 (0.369) |
| Multiplicity | 4.3 (4.2) |
| Refinement statistics | |
| No. of protein atoms | 3836 |
| No. of solvent atoms | 332 |
| No. of hetero atoms | 4Cu + 2O + 3EDO |
| Final R-factor | 0.160 |
| Final free R-factor | 0.207 |
| FOM | 0.89 |
| Mean B values/Å2 | |
| Protein | 16.080 |
| Solvent | 21.475 |
| Overall | 16.514 |
| Estimated overall coordinate uncertaintya/Å | 0.097 |
| Distance deviationsb | |
| Bond distances/Å | 0.015 |
| Bond angles/° | 1.534 |
| Planar groups/Å | 0.007 |
| Chiral volume deviation/Å3 | 0.110 |
| Quality of models | |
| Ramachandran analysis (%) | |
| Favourable | 98.1 |
| Allowed | 1.7 |
| Disallowed | 0.2 |
Structure determination and refinement
The three-dimensional structure of McoC was solved by the molecular replacement (MR) method. At a first stage, the molecular replacement software MrBUMP40 was used based on the protein sequence. The best search model found was the Met-rich region deleted form of CueO (PDB idcode 2YXW), which was identified only as a marginal solution (Rfreeca. 48%,) by both molecular replacement programs, MOLREP41 and PHASER.42 At a second stage, the output model obtained from MrBUMP was used as a search model in PHENIX Auto-MR,43 using data up to 2.5 Å resolution, and a correct structure solution was found with rotation and translation function Z-score values of RFZ = 18.1 and TFZ = 13.5. The structural model obtained was then submitted to 10 cycles of rigid body refinement using REFMAC44 and with all the data up to 1.95 Å, yielding the values for R and Rfree factors of 42.4% and 48.0%, respectively. Before the initial model building and correction ARP/wARP45,46 was used to improve the quality of the electron density maps and minimise model bias. Subsequent iterative cycles of refinement and manual model building were performed using the maximum-likelihood functions enclosed in REFMAC and the program COOT,47 respectively. Some 5% of the data was excluded for Rfree calculation. Close to the end of the refinement procedure, BUSTER-TNT48 was additionally used for map improvement on the poorly defined region that comprises residues Met384 to Ser393. Based on the output maps, this region was manually built into the 2|Fo| – |Fc| electron density maps using COOT. For the very last stages of refinement REFMAC was run including translation/libration/screw (TLS) refinement.49 The positions of the T1 Cu and T3 Cu2 ions became evident after the first cycles of refinement, based on electron density synthesis maps, but the positions of the remaining ions in the trinuclear cluster, Cu3 and Cu4, took longer to become apparent because of reduced occupancies. Solvent molecules were placed in the model after a few rounds of refinement corresponding to standard geometrical and stereochemical restraints. Three molecules of ethylene glycol were also modelled in the structure. Isotropic refinement of the atomic displacement parameters was performed for all the atoms present in the model. Copper ion occupancy was assessed by adjusting their value so that their isotropic thermal vibration parameters refined approximately to those observed for their neighbouring atoms. The determination of the species located in between both T3 Cu atoms, Cu2 and Cu3, was achieved by combination of a cautious use of omit and standard difference Fourier synthesis, and inspection of thermal vibration coefficients throughout refinement, as previously described.16 The positive ovoid electron density was best modelled as a dioxygen species rather than an hydroxide or chlorine ion. However, the resolution of the data does not permit a distinction between, for example, O22− and H2O2. Model validation was monitored using RAMPAGE from the CCP4 programs suite39 and MOLPROBITY.50,51 The coordinates and structure factors have been deposited in the PDB as entry 3ZX1.Refinement statistics and details of the final quality of the model are listed in Table 1. PyMOL52 was used to prepare the figures of the McoC and related molecules.
Other experimental methods
McoC peptides after tryptic digestion were analysed by MALDI-TOF-MS using a mass spectrophotometer Voyager STR (Applied Biosystems). Protein identification was performed using the peptide mass fingerprint as a query for the MASCOT-PMF software using the Swiss-Prot database. The N-terminal amino acid sequence was determined from protein transferred to PVDF using stepwise Edman degradation on an Applied Biosystems Procise 491HT protein sequencer. The overall amount of copper was determined with the trichloroacetic acid/bicinchoninic acid (BCA) method of Brenner and Harris.53Results and discussion
Overproduction, purification and biochemical characterization of recombinant McoC
The mcoC gene was amplified by PCR and inserted into the pET-21a(+) expression vector resulting in pATF-5. Expression of the protein using E. coli Rosetta (DE3) pLysS (coding for rare tRNA's) revealed that upon induction with IPTG, a major band of around 59 kDa, that was absent in extracts prepared from non-induced LOM 418, appeared in the soluble and insoluble fraction of cell lysates, as visualised by SDS-PAGE gels (results not shown). Two chromatographic steps purified the protein close to homogeneity (Fig. 1a) and an enzyme solution with blue colour (typical of blue copper oxidases) with a copper per protein ratio close to four was obtained. The protein was identified by MALDI-TOF-MS with a score of 95 (31% coverage, 10 peptides identified). Interestingly, the N-terminal amino acid sequence determined was KNHSIN, eleven amino acids displaced from the most expected (theoretical) signal peptide incision (AYA-NP), as determined by SignalP.54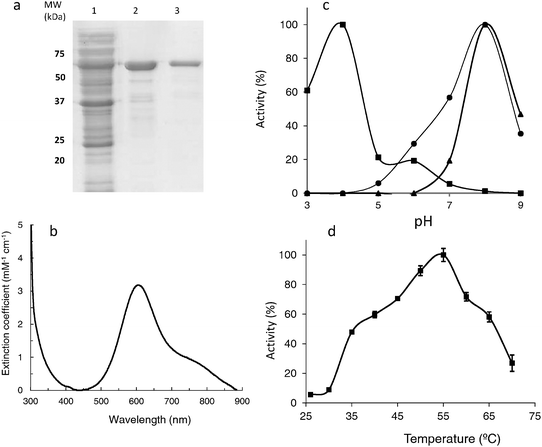 | ||
| Fig. 1 (a) SDS-PAGE analysis of McoC purified from recombinant E. coli LOM418. Lane 1, protein mixture before purification, Lane 2, McoC after the first step of purification (SP-sepharose pH 7.6), Lane 3, McoC after the second purification step (Superdex 200 pH 7.6). (b) UV-visible spectra of purified McoC. (c) pH profile for catalytic activities using as substrates ABTS (full squares), 2,6-DMP (full circles) and SGZ (full triangles) of recombinant McoC. (d) Effect of temperature on the activity of recombinant purified McoC. The enzyme activity was assayed at each temperature in 100 mM Briton Robinson buffer (pH 4.0) in the presence of 2 mM ABTS. Activity was monitored at 420 nm. | ||
The UV-visible, electron paramagnetic resonances (EPR) and Resonance Raman (RR) spectra obtained for McoC showed all the features that are characteristic of MCOs namely: a band at ∼600 nm that corresponds to the Cu–Cys interaction at the T1 Cu centre, with a ε = 3.2 mM−1 cm−1;55 resonances characteristic of the T1 and T2 copper centres; the intensity weighted frequency (νCu–S = 417 ± 1 cm−1) obtained for the Cu–S bond length in the T1 site was within the range reported for other well-studied MCOs (Fig. 1b and data not shown).56 The T1 site redox potential was measured by monitoring the absorbance decrease at 600 nm in the visible spectra. McoC displayed a redox potential of 422 ± 20 mV, a relatively low value within usual MCO range, from 340 to 790 mV for some fungal laccases.55
Catalytic properties of recombinant McoC
Compared to CotA, McoC showed little activity towards aromatic substrates (Tables 2 and 3), but it has maximal activity at pH 4 for ABTS and at pH 8 for SGZ and 2,6-DMP (Fig. 1c), pH values consistent with those exhibited by other bacterial MCOs such as CotA laccase. The temperature for this catalytic activity of McoC was found to be maximal at 55 °C (Fig. 1d), identical to that observed for the CueO of E. coli,18 but lower than CotA laccase or McoA that show an optimal temperature for activity around 75 °C.12,17 The steady state kinetic parameters of recombinant McoC were obtained from substrate saturation curves for the metal ion Cu(I), for ferrous sulfate Fe(II)SO4 and also for the typical laccase substrates ABTS (non-phenolic), SGZ and 2,6-DMP (phenolics) in air saturated solutions, at 37 °C. The dependence of the enzymatic rate on the substrate concentration followed Michaelis–Menten kinetics (Table 2). These results showed that recombinant McoC is clearly a metallo-oxidase, exhibiting higher enzyme efficiency (kcat/Km) towards the oxidation of the metal ions than for the oxidation of aromatic substrates. The kinetic constants of McoC for Cu(I) are in the same range as those obtained for E. coli CueO, but quite different than the ones obtained for the yeast ferroxidase Fet3p, human ceruloplasmin (hCp) or McoA12,35,57 indicating that McoC is most likely a cuprous oxidase enzyme. Although the efficiency (kcat/Km) of McoC towards Fe(II) is 8 times lower than the one obtained for Cu(I), it is still higher than the efficiency observed towards all of the aromatic substrates tested, and in the same range as exhibited by CueO (Tables 2 and 3). The greatly diminished activity of McoC towards aromatic substrates compared to its bacterial homologue CotA laccase (Tables 2 and 3) strengthens the fact that McoC does not function as a phenoloxidase but instead as a metallo-oxidase.| Substrate | K m/μM | k cat/min−1 | k cat/Km/min−1 μM−1 |
|---|---|---|---|
| Cu(I) | 128 ± 25 | 1038 ± 89 | 8.1 |
| Fe(II) | 30 ± 4 | 34 ± 1.3 | 1.1 |
| ABTS | 791 ± 106 | 466 ± 22 | 0.59 |
| SGZ | 35 ± 2 | 10 ± 0.1 | 0.29 |
| 2,6-DMP | 227 ± 36 | 0.70 ± 0.02 | 0.003 |
| O2 (ABTS) | 31 ± 4 | 247 ± 10 | 8.0 |
| Substrate | k cat/Km/min−1 μM−1 | |
|---|---|---|
| a Only measurable when in the presence of endogenous Cu(II). b Values calculated based in the referred references. | ||
| CotA laccase | ABTS | 156.0 |
| SGZ | 267.0 | |
| 2,6-DMP | 8.1 | |
| CueO | Cu(I) | 5.5 |
| Fe(II) | 1.7a | |
| McoA | Cu(I) | 1.6 |
| Fe(II) | 3.4 | |
| Fet3p | Cu(I) | 2.1b |
| Fe(II) | 11.8b | |
| hCp | Cu(I) | 0.6b |
| Fe(II) | 3.6b |
The dioxygen-binding affinity, determined by measuring the initial rate of oxygen consumption, has also been investigated as a function of varying concentrations of dioxygen. The result obtained for the recombinant McoC (Km (O2) of 31 ± 4 μM) is within the same range of values reported for other MCOs.58–60
The overall structure of McoC
McoC has a typical laccase fold with three cupredoxin-like domains. However, there are some slight differences from the customary conformation (Fig. 2a) with domain 1 (residues 43–203) consisting of eight β-strands, while domains 2 (residues 212–353) and 3 (residues 373–513) comprise eleven β-strands each. Fig. 2b and c enable a comparison of McoC with its bacterial homologues CotA laccase (36% sequence similarity, PDB:1W6L)15 and CueO (42% sequence similarity, PDB:1N68),20 respectively. Major differences were observed mainly at the protein surface in close proximity to the T1 copper centre, in the region of the substrate binding site (vide infra). Superposition of the McoC structure onto the CueO and CotA laccase structures, performed by Secondary Structure Matching (SSM),39 gave root mean square deviations of only 1.712 Å and 1.670 Å, respectively. Besides differing in β-strand lengths and disposition within the same β-barrel, in loop sizes and in the number of α-helices and 310-helices, all three MCOs retain a similar location for the C-terminus whereas the position of the N-terminus varies. In the McoC structure, in addition to the 22 amino acid residues corresponding to the putative signal peptide, 20 further residues in the protein N-terminus were not visible in the structure. Ten of these are not identifiable by MALDI-TOF-MS and the remaining 10 are apparently highly disordered in the structure. However, the McoC N-terminal domain still contains a long starting loop that spans over the top of the molecule and extends all the way towards domain 3; in CotA and CueO the equivalent loop is smaller starting on the proximity of the interface between domains 1 and 2, and not crossing the molecule over the top in either case (Fig. 2a–c). In all three laccases, the linker peptides connecting the domains are placed at the bottom of the structure, constituting an external feature (Fig. 2a–c, in grey).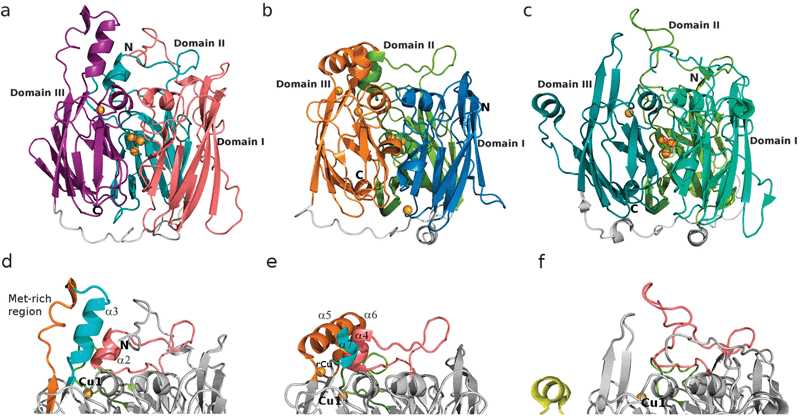 | ||
| Fig. 2 Overall three-dimensional structure of McoC (a, d), CueO (PDB:1N68)19 (b, e) and CotA laccase (PDB:1W6L)15 (c, f). Upper panel (a–c): Tripartite cupredoxin-domain organization with the mononuclear T1 Cu site located in domain 3 and the trinuclear cluster placed at the interface between domains 1 and 3. Lower panel (d–f): Major structural differences are observed at the protein surface, near the T1 Cu site. Highlighted are secondary structure elements interfering with the access to this site: N-terminal loop and other loops; α-helices; and Met-rich regions. The same colour code is used for corresponding regions between structures. Cu ions are represented as dark-yellow spheres, with a fifth regulatory Cu ion (rCu) identified in the CueO structure, near the T1 Cu site, and a sixth one near the exit channel with no apparent functional importance and only partially occupied.19 | ||
The copper binding sites
The crystal structure of McoC contains four copper atoms with the same spatial arrangement, relative to the polypeptide chains, as other members of the MCO family of enzymes (ascorbate oxidase, Fet3p and laccases in general). A mononuclear blue copper centre or T1 site is placed in domain 3 and a trinuclear cluster (T2/T3 site) is embedded in the interface between domains 1 and 3 (Fig. 2a and 3). The T1 Cu is coordinated by two histidines, a cysteine and a fourth axial “soft” ligand methionine, which confers distorted tetrahedral coordination geometry at this centre (Fig. 3a and b and Table S1, ESI†). The trinuclear site (Fig. 3a and c), composed of two T3 and one T2 copper ions, is coordinated by four HXH structural motifs, 2 pairs coming from domain 1 and two other from domain 3. Each T3 Cu binds to three histidine residues whereas T2 Cu (Cu4) binds to two histidine residues (Fig. 3 and Table S1, ESI†). In a similar manner to other MCOs, the mononuclear Cu site interacts with the trinuclear cluster via the highly conserved HCH motif, where the cysteine binding to the T1 Cu shuttles electrons via the intramolecular pathway (13.0/12.3 Å for Cu2/Cu3), to each of the adjacent histidines binding to one of the T3 copper ions (His496 for Cu2 and His494 for Cu3).55 Refinement of the occupancies of the copper ions in the McoC structure showed full occupancy for the T1 Cu centre, but substantial Cu depletion for the trinuclear centre. Occupancies of 60% and 30% were obtained for the T3 copper ions, Cu2 and Cu3, respectively, but only 20% for the T2 ion, Cu4 (Fig. 3c and Table S1, ESI†). As observed for other MCOs the T2 copper appears to be the most labile. A protein copper content of 6.4 atoms per polypeptide chain is reported by Hall et al.32 for C. jejuni Cj1516 (strain NCTC 11168) and such a contrast with the present crystal structure presumably results from the different expression protocols and strains used and possibly the crystallisation conditions. In between the type 3 copper ions the dioxygen species was refined with occupancy of 60%, corresponding to the occupancy for Cu2 (Fig. 3c). | ||
| Fig. 3 (a) Overall organization of the catalytic motif in the MCO family of enzymes, comprising a mononuclear copper site and a trinuclear cluster. (b) Superposition of the mononuclear copper centres of McoC (blue), CueO (purple) and CotA laccase (green). (c–e) Structural detail of the trinuclear centre and its neighbourhood in McoC (c), CueO (d, PDB:1KV7) and CotA laccase (e, PDB:1W6L). Important acidic residues placed in the entrance and exit channels are identified (Glu and Asp residues, respectively). Cu ions are represented as spheres. In panel (c) the electron density for the molecular oxygen is derived from omit Fourier synthesis computed with Sigma A weighted coefficients |Fo|–|Fc|, where the dioxygen was not included in the structure factor calculations; the contour level is 5.0 rms. | ||
In a similar way to its bacterial homologues, McoC also has two well-defined solvent channels formed mainly by polar and neutral residues, giving clear access to the trinuclear cluster. McoC maintains a carboxylate moiety, Glu501 (equivalent residues in CotA,15,58 CueO18,64 and Fet3p64–67 are Glu498, Glu506 and Glu487, respectively) strategically sited in the entrance channel to the trinuclear cluster8,58 and a second acidic residue, Asp150 (equivalent residues Asp116, Asp112 and Asp94 in CotA, CueO and Fet3p, respectively) placed within the exit channel (Fig. 3c). These acidic residues are believed to participate in the protonation processes required to convert dioxygen to two molecules of water.15 However, unlike the CotA structure, no well-defined water molecule bridging Glu501 and the dioxygen moiety in the access channel appears to be present (Fig. 3c).
The substrate binding site
In general, MCOs are able to oxidize a wide range of aromatic compounds of different nature and size, as well as inorganic ones, such as lower valence metal ions. Thus, these enzymes tend to lack a unique way of substrate binding, having broad binding pockets, often able to accommodate more than one type of substrate. Some, as is the case of human ceruloplasmin, appear to possess more than one binding site.68 For the reported structures of substrate complexed MCOs, the T1 Cu centre is sited at the bottom of the substrate binding region, relatively exposed to the solvent and interacting with the substrate molecule through the imidazole ring of one of its His ligands (His497 in CotA).69 However, in McoC and CueO the equivalent residues, His500 and His505, are virtually buried and this constitutes a major difference between these two laccases and the remainder of the laccase family whose structures have been reported. Thus, in McoC, the T1 site becomes occluded by several secondary structure elements (Fig. 2d). Firstly, a Met-rich region (Fig. 2d, in orange), comprising residues Met379–Gly391, blocks the site entrance and secondly two extra α-helixes, α2 and α3, placed not very far from the mononuclear Cu centre (Fig. 2d, in salmon and cyan, respectively), partially hinder this substrate binding region. In this respect, the region containing the residues 383–396, and enclosing most of the Met-rich region, is somewhat disordered showing discontinuous electron density, except for His387 which is clearly defined. This is the only residue in the entire Met-rich region that establishes crystal contacts, whereas all the remaining residues are more loose and hence poorly defined in the structure. However, such crystal contacts were enough to provide some packing constraints to this loop, making it possible to be modelled. Within the Met-rich region it is likely that residues His387 to Gly391, shown as a random coil due to local disorder, comprise in fact a helix (as it resembles in Fig. 2d). Finally, the domain 2 loop (residues 327–344; Fig. 2d, in green) stretches over the substrate binding cavity, closely to the T1 Cu, and covers part of the access to this site (Fig. 2d). Thus, it is difficult to see how bulky aromatic substrates such as ABTS, which readily binds to CotA, can be easily accommodated by McoC (Fig. 4b). In CotA this region is mainly composed by apolar residues, in agreement with the enzymes' lack of specificity towards substrates.69 | ||
| Fig. 4 Molecular surface representation and putative substrate binding sites identified for CotA laccase (PDB:1UVW),69 McoC and CueO (PDB:1KV7), near the T1 Cu. All molecular surface representations are in the same orientation. Cavity analysis was performed using the CASTp server.70 (a) CotA laccase shows a widely exposed substrate binding cavity (in green), where the bulky organic substrate ABTS has been shown to bind.69 (b, c) Superposition of the three-dimensional structures of McoC and CueO, respectively, onto the molecular surface and substrate binding pocket identified for CotA laccase. In both cases, secondary structure elements obstruct to some extent the binding cavity. (d–f) Overall molecular surface representation of CotA, McoC and CueO, with respective putative substrate binding cavities highlighted: green for CotA, cyan for McoC; for CueO the site is not visible in the orientation shown. For the latter two enzymes, a reduced access to the T1 Cu is observed, diminishing the accessibility of bulkier substrates to this site. | ||
For CueO, the other bacterial MCO, whose structure has been reported (1N68),19 the access to the T1 Cu site is also greatly reduced due to a large Met-rich helical region (Fig. 2e, in orange). In CueO, this structural feature is shown to interfere with the access of organic substrates to the T1 Cu. Studies conducted by Kataoka et al.,20 where the entire Met-rich helical region was deleted, together with α-helix 7 (Δα5–7 CueO) yield higher specificity of the truncated enzyme for bulky substrates (ABTS and p-PD) than wt CueO. Nevertheless, these became comparable when in the presence of excess Cu2+, and newly emerged activities are verified for some phenolic substrates (2,6-DMP, catechol, guaiacol and SGZ), with overall higher values for Δα5–7 CueO. A severe cuprous oxidase catalytic impairment is also attained for Δα5–7 CueO, while its ferroxidase activity is not affected by the truncation, becoming enhanced for both wt and truncated forms when in the presence of exogenous Cu2+. For this enzyme, the presence of charged and polar residues in the active centre comes as a consequence of the higher substrate specificity (in Δα5–7 CueO a decrease of the negative potential area was observed).20 Similarly, in McoA when the equivalent Met-rich region (residues 321–363) was deleted higher specificity for larger aromatic substrates is observed, while it remains unchanged for the metal substrates tested, Cu(I) and Fe(II), suggesting that this region occludes the substrate binding site.12
To identify possible substrate binding pockets in McoC and respective accessibility, calculations on the enzyme's solvent accessible surface near the T1 Cu were undertaken. A cavity analysis was performed on all three bacterial MCOs, using the CASTp server70 and Areamol from CCP4 programs suite.39 As expected, CotA shows a higher accessible area in the proximity of the T1 Cu centre and hence higher solvent accessibility, favouring substrate binding (Fig. 4a and d). At the other extreme, CueO shows a very limited access to the T1 Cu with a considerably low accessible area for substrates, in particular bulky ones (Fig. 4c,f). For McoC (Fig. 4b,e), half of the substrate binding region, identified for the binding of ABTS to CotA and located above the T1 copper site, is occluded. However, there is still some solvent accessibility suggesting substrates can bind, but less easily than for CotA. For both McoC and CueO the decrease in accessibility of the T1 Cu site becomes more evident when considering the accessibility of the histidine ligand (His500 and His505 in McoC and CueO, respectively) thought to be the initial electron acceptor (Table 4). This residue becomes non-accessible in these two MCOs. Identical results are observed for other residues placed at the bottom of CotA laccase binding pocket, in close proximity to the type 1 copper ion. This is also true for the counterparts of CotA laccase residues Ile494, His419 and even for Leu386 (in this latter case spatially equivalent to Phe405 in McoC and Asn408 in CueO), which again become non-accessible (Table 4). Other residues sited at the entrance site for ABTS, such as Pro384 in the CotA structure, also become highly concealed in McoC and CueO structures, as was seen from the accessibility of the spatially equivalent residues Met404 and Leu359, respectively (Table 4). Therefore, in accordance with the measured kinetic parameters, McoC should be considered predominantly as a metallo-oxidase, able to accommodate and oxidise smaller compounds such as those containing the lower valence metal ions Cu1+ and Fe2+, but showing radically reduced or non-existent affinity for aromatic substrates (Table 4). However, the possibility that McoC is able to oxidise smaller phenolic or non-phenolic substrates cannot be completely eliminated purely from structural considerations.
For CueO a fifth Cu ion, buried just under the Met-rich region and close to the T1 copper site, is reported in the structure of CueO soaked with CuCl2 (PDB:1N68) (Fig. 5a). A regulatory role is proposed for this additional Cu ion (rCu).19 Just recently Singh et al. have determined other CueO's structures, namely those of the C500S mutant bound to Cu(I) or Cu(II).22 In the former structure, besides the fifth Cu ion, two additional coppers are found binding along the Met-rich region.22 Removal of those two coppers by mutation of the binding methionines into serines results in a four- and two-fold decrease of the Cu(I) and Fe(II) oxidation catalytic rates, respectively. Additionally, these sites appear to be specific for cuprous ions as neither of them was observed on the Cu(II) soaked structure of CueO. For this reason, Singh et al. have renamed this site from regulatory Cu site to substrate copper site, and suggest that the Met-rich loop plays a role in the binding and oxidation of Cu(I).22
 | ||
| Fig. 5 (a) Representation of the regulatory copper ion site (rCu) region in the CueO structure (PDB:1N68). This site is directly linked to the T1 Cu site through a H-bond with one of its His ligands, assisting in the mediation of electrons from the substrates to the T1 Cu.19 (b) Structurally equivalent region in McoC. Cu ions are represented as spheres. | ||
A structural comparison showed the presence of spatially equivalent residues at the rCu site in McoC (Glu381, His383, Ser435, Met437) (Fig. 5b). Furthermore, residues Met335 and His436 are also placed within very close proximity. Interestingly, in McoC neither of the two methionines constituting this ‘site’ take part in the Met-rich region, whereas Glu381 and His383 do. However, in the McoC structure no additional Cu ions were observed. This is not too surprising, since copper depletion was observed in the structure even for the catalytic copper centres, as no additional copper was added either to the protein solution or to the protein crystals. Attempts are being made in order to obtain a full copper content of the McoC.
Concluding remarks
The structure of a multicopper oxidase present in the pathogenic bacterium Campylobacter jejuni has been solved at 1.95 Å resolution. Spectroscopic, biochemical and functional studies showed that McoC is a typical MCO, with higher efficiency (kcat/Km) towards metal ions, such as Cu(I) and Fe(II), than for the aromatic substrates ABTS, SGZ and 2,6-DMP. Similarly to its homologue CueO, McoC acts predominantly as a cuprous oxidase, although it also showed activity as a ferroxidase. The three-dimensional structure determined by X-ray studies showed that this enzyme is a three-domain laccase-like MCO, with a significantly smaller substrate binding pocket near the T1 copper site, when compared with its homologue CotA laccase. The presence of secondary structure elements partially occluding this site is likely to function as a barrier to the access of bulkier substrates. Such a site much resembles CueO from E. coli, for which the site cavity is even smaller. One of the secondary structure elements hindering this site corresponds to a Met-rich region followed by an α-helix, also observed for CueO. For the latter enzyme, this region has been reported19,22 to bind additional Cu, constituting an extra Cu site proposed to have regulatory functions in electron transfer mediation during substrate oxidation, by facilitating Cu(I) binding and perhaps accelerating the oxidation rate. Such a binding site is not apparent in McoC, although residues seem to exist that could bind such copper ion. Examples of additional copper centres have also been reported for other MCOs of known structure, as is the case of human ceruloplasmin,71 phenoxazinone synthase laccase72 and holoCotA laccase.16Altogether, these results indicate that McoC may act as a metallo-oxidase also in vivo, thus playing a protective role while converting Cu(I) and/or Fe(II) in C. jejuni towards the less toxic forms of copper and iron, Cu(II) and Fe(III). A role in copper homeostasis has also been proposed by Hall et al. for the recombinant MCO Cj1516 obtained from a different strain of C. jejuni (NCTC11168).32
Acknowledgements
André T. Fernandes is acknowledged for assistance with gene cloning, Manuela M. Pereira with the EPR spectrum and Smilja Todorovic with the RR spectrum of McoC. Maria Arménia Carrondo is gratefully acknowledged for support. The European Synchrotron Radiation Facility in Grenoble, France, and the Macromolecular Crystallography staff are sincerely acknowledged for provision of synchrotron radiation facilities and support. This work was supported by a project grant from the European Union (BIORENEW, FP6-2004-NMP-NI-4/026456). C.S.S. and P.D. hold PhD fellowships from FCT, Portugal (SFRH/BD/40586/2007 and SFRH/BD/40696/2007, respectively).References
- A. Messerschmidt, Multi-copper Oxidases, World Science Press, Singapore, 1997 Search PubMed.
- P. F. Lindley, Multi-copper oxidases, Marcel Dekker, Inc., Basel, New York, 2001 Search PubMed.
- P. J. Hoegger, S. Kilaru, T. Y. James, J. R. Thacker and U. Kues, FEBS J., 2006, 273, 2308–2326 CrossRef CAS.
- P. Giardina, V. Faraco, C. Pezzella, A. Piscitelli, S. Vanhulle and G. Sannia, Cell. Mol. Life Sci., 2010, 67, 369–385 CrossRef CAS.
- K. Nakamura and N. Go, Cell. Mol. Life Sci., 2005, 62, 2050–2066 CrossRef CAS.
- D. J. Kosman, JBIC, J. Biol. Inorg. Chem., 2010, 15, 15–28 CrossRef CAS.
- S. Rodríguez Couto and J. L. Toca Herrera, Biotechnol. Adv., 2006, 24, 500–513 CrossRef.
- P. Sharma, R. Goel and N. Capalash, World J. Microbiol. Biotechnol., 2007, 23, 823–832 CrossRef CAS.
- H. Claus, Micron, 2004, 35, 93–96 CrossRef CAS.
- K. Miyazaki, Extremophiles, 2005, 9, 415–425 CrossRef CAS.
- A. T. Fernandes, J. M. Damas, S. Todorovic, R. Huber, M. C. Baratto, R. Pogni, C. M. Soares and L. O. Martins, FEBS J., 2010, 277, 3176–3189 CrossRef CAS.
- A. T. Fernandes, C. M. Soares, M. M. Pereira, R. Huber, G. Grass and L. O. Martins, FEBS J., 2007, 274, 2683–2694 CrossRef CAS.
- E. I. Solomon, P. Chen, M. Metz, S. K. Lee and A. E. Palmer, Angew. Chem., Int. Ed., 2001, 40, 4570–4590 CrossRef CAS.
- F. J. Enguita, L. O. Martins, A. O. Henriques and M. A. Carrondo, J. Biol. Chem., 2003, 278, 19416–19425 CrossRef CAS.
- I. Bento, L. O. Martins, G. Gato Lopes, M. Armenia Carrondo and P. F. Lindley, Dalton Trans., 2005, 3507–3513 RSC.
- I. Bento, C. S. Silva, Z. Chen, L. O. Martins, P. F. Lindley and C. M. Soares, BMC Struct. Biol., 2010, 10, 28 CrossRef.
- L. O. Martins, C. M. Soares, M. M. Pereira, M. Teixeira, T. Costa, G. H. Jones and A. O. Henriques, J. Biol. Chem., 2002, 277, 18849–18859 CrossRef CAS.
- S. A. Roberts, A. Weichsel, G. Grass, K. Thakali, J. T. Hazzard, G. Tollin, C. Rensing and W. R. Montfort, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 2766–2771 CrossRef CAS.
- S. A. Roberts, G. F. Wildner, G. Grass, A. Weichsel, A. Ambrus, C. Rensing and W. R. Montfort, J. Biol. Chem., 2003, 278, 31958–31963 CrossRef CAS.
- K. Kataoka, H. Komori, Y. Ueki, Y. Konno, Y. Kamitaka, S. Kurose, S. Tsujimura, Y. Higuchi, K. Kano, D. Seo and T. Sakurai, J. Mol. Biol., 2007, 373, 141–152 CrossRef CAS.
- C. S. Stoj and D. J. Kosman, Copper proteins: oxidases, New York, 2005 Search PubMed.
- S. K. Singh, S. A. Roberts, S. F. McDevitt, A. Weichsel, G. F. Wildner, G. B. Grass, C. Rensing and W. R. Montfort, J. Biol. Chem., 2011, 286, 37849–37857 CrossRef CAS.
- F. Arnesano, L. Banci, I. Bertini and A. R. Thompsett, Structure, 2002, 10, 1337–1347 CrossRef CAS.
- D. L. Huffman, J. Huyett, F. W. Outten, P. E. Doan, L. A. Finney, B. M. Hoffman and T. V. O'Halloran, Biochemistry, 2002, 41, 10046–10055 CrossRef CAS.
- F. W. Outten, D. L. Huffman, J. A. Hale and T. V. O'Halloran, J. Biol. Chem., 2001, 276, 30670–30677 CrossRef CAS.
- Z. L. Harris, A. P. Durley, T. K. Man and J. D. Gitlin, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 10812–10817 CrossRef CAS.
- A. B. Taylor, C. S. Stoj, L. Ziegler, D. J. Kosman and P. J. Hart, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 15459–15464 CrossRef CAS.
- J. Parkhill, B. W. Wren, K. Mungall, J. M. Ketley, C. Churcher, D. Basham, T. Chillingworth, R. M. Davies, T. Feltwell, S. Holroyd, K. Jagels, A. V. Karlyshev, S. Moule, M. J. Pallen, C. W. Penn, M. A. Quail, M. A. Rajandream, K. M. Rutherford, A. H. van Vliet, S. Whitehead and B. G. Barrell, Nature, 2000, 403, 665–668 CrossRef CAS.
- M. C. Peterson, West J. Med., 1994, 161, 148–152 CAS.
- K. Palyada, D. Threadgill and A. Stintzi, J. Bacteriol., 2004, 186, 4714–4729 CrossRef CAS.
- K. Holmes, F. Mulholland, B. M. Pearson, C. Pin, J. McNicholl-Kennedy, J. M. Ketley and J. M. Wells, Microbiology, 2005, 151, 243–257 CrossRef CAS.
- S. J. Hall, A. Hitchcock, C. S. Butler and D. J. Kelly, J. Bacteriol., 2008, 190, 8075–8085 CrossRef CAS.
- P. Durão, Z. Chen, A. T. Fernandes, P. Hildebrandt, D. H. Murgida, S. Todorovic, M. M. Pereira, E. P. Melo and L. O. Martins, JBIC, J. Biol. Inorg. Chem., 2008, 13, 183–193 CrossRef.
- P. Durão, I. Bento, A. T. Fernandes, E. P. Melo, P. F. Lindley and L. O. Martins, JBIC, J. Biol. Inorg. Chem., 2006, 11, 514–526 CrossRef.
- S. K. Singh, G. Grass, C. Rensing and W. R. Montfort, J. Bacteriol., 2004, 186, 7815–7817 CrossRef CAS.
- K. A. Kantardjieff and B. Rupp, Protein Sci., 2003, 12, 1865–1871 CrossRef CAS.
- A. Leslie, Newsletter on Protein Crystallography, 1992, No. 26.
- A. G. Leslie, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2006, 62, 48–57 CrossRef.
- C. C. P. CCP4, Number 4, Acta Cryst., 1994, D50, 760–763.
- R. M. Keegan and M. D. Winn, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2007, 63, 447–457 CrossRef.
- A. Vagin and A. Teplyakov, J. Appl. Crystallogr., 1997, 30, 1022–1025 CrossRef CAS.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, J. Appl. Crystallogr., 2007, 40, 658–674 CrossRef CAS.
- P. D. Adams, P. V. Afonine, G. Bunkóczi, V. B. Chen, I. W. Davis, N. Echols, J. J. Headd, L. W. Hung, G. J. Kapral, R. W. Grosse-Kunstleve, A. J. McCoy, N. W. Moriarty, R. Oeffner, R. J. Read, D. C. Richardson, J. S. Richardson, T. C. Terwilliger and P. H. Zwart, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2010, 66, 213–221 CrossRef.
- G. N. Murshudov, A. A. Vagin and E. J. Dodson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1997, 53, 240–255 CrossRef CAS.
- V. S. Lamzin and K. S. Wilson, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1993, 49, 129–147 CrossRef CAS.
- W. T. Mooij, S. X. Cohen, K. Joosten, G. N. Murshudov and A. Perrakis, Structure, 2009, 17, 183–189 CrossRef CAS.
- P. Emsley and K. Cowtan, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- G. Bricogne, E. Blanc, M. Brandl, C. Flensburg, P. Keller, W. Paciorek, P. Roversi, O. S. Smart, C. Vonrhein and T. O. Womack, Global Phasing Ltd., Cambridge, UK, version 2.8.0 edn, 2009.
- M. D. Winn, M. N. Isupov and G. N. Murshudov, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2001, 57, 122–133 CrossRef CAS.
- S. C. Lovell, I. W. Davis, W. B. Arendall 3rd, P. I. de Bakker, J. M. Word, M. G. Prisant, J. S. Richardson and D. C. Richardson, Proteins: Struct., Funct., Genet., 2003, 50, 437–450 CrossRef CAS.
- I. W. Davis, A. Leaver-Fay, V. B. Chen, J. N. Block, G. J. Kapral, X. Wang, L. W. Murray, W. B. Arendall, J. Snoeyink, J. S. Richardson and D. C. Richardson, Nucleic Acids Res., 2007, 35, W375–W383 CrossRef.
- W. L. DeLano, DeLano Scientific, San Carlos, CA, USA, 2002.
- A. J. Brenner and E. D. Harris, Anal. Biochem., 1995, 226, 80–84 CrossRef CAS.
- J. D. Bendtsen, H. Nielsen, G. von Heijne and S. Brunak, J. Mol. Biol., 2004, 340, 783–795 CrossRef.
- E. I. Solomon, U. M. Sundaram and T. E. Machonkin, Chem. Rev., 1996, 96, 2563–2605 CrossRef CAS.
- D. F. Blair, G. W. Campbell, W. K. Cho, A. M. English, H. A. Fry, V. Lum, K. A. Norton, J. R. Schoonover and S. I. Chan, J. Am. Chem. Soc., 1985, 5755–5766 CrossRef CAS.
- C. Stoj and D. J. Kosman, FEBS Lett., 2003, 554, 422–426 CrossRef CAS.
- Z. Chen, P. Durão, C. S. Silva, M. M. Pereira, S. Todorovic, P. Hildebrandt, I. Bento, P. F. Lindley and L. O. Martins, Dalton Trans., 2010, 2875–2882 RSC.
- Y. Ueki, M. Inoue, S. Kurose, K. Kataoka and T. Sakurai, FEBS Lett., 2006, 580, 4069–4072 CrossRef CAS.
- F. Xu, Appl. Biochem. Biotechnol., 2001, 95, 125–133 CrossRef CAS.
- A. Messerschmidt, R. Ladenstein, R. Huber, M. Bolognesi, L. Avigliano, R. Petruzzelli, A. Rossi and A. Finazzi-Agro, J. Mol. Biol., 1992, 224, 179–205 CrossRef CAS.
- N. Hakulinen, L. L. Kiiskinen, K. Kruus, M. Saloheimo, A. Paananen, A. Koivula and J. Rouvinen, Nat. Struct. Biol., 2002, 9, 601–605 CAS.
- K. Piontek, M. Antorini and T. Choinowski, J. Biol. Chem., 2002, 277, 37663–37669 CrossRef CAS.
- K. Kataoka, R. Sugiyama, S. Hirota, M. Inoue, K. Urata, Y. Minagawa, D. Seo and T. Sakurai, J. Biol. Chem., 2009, 284, 14405–14413 CrossRef CAS.
- E. I. Solomon, A. J. Augustine and J. Yoon, Dalton Trans., 2008, 3921–3932 RSC.
- A. J. Augustine, L. Quintanar, C. S. Stoj, D. J. Kosman and E. I. Solomon, J. Am. Chem. Soc., 2007, 129, 13118–13126 CrossRef CAS.
- L. Quintanar, C. Stoj, T. P. Wang, D. J. Kosman and E. I. Solomon, Biochemistry, 2005, 44, 6081–6091 CrossRef CAS.
- V. N. Zaitsev, I. Zaitseva, M. Papiz and P. F. Lindley, JBIC, J. Biol. Inorg. Chem., 1999, 4, 579–587 CrossRef CAS.
- F. J. Enguita, D. Marcal, L. O. Martins, R. Grenha, A. O. Henriques, P. F. Lindley and M. A. Carrondo, J. Biol. Chem., 2004, 279, 23472–23476 CrossRef CAS.
- J. Liang, H. Edelsbrunner and C. Woodward, Protein Sci., 1998, 7, 1884–1897 CrossRef CAS.
- P. F. Lindley, G. Card, I. Zaitseva, V. Zaitsev, B. Reinhammar, E. Selin-Lindgren and K. Yoshida, JBIC, J. Biol. Inorg. Chem., 1997, 2, 454–463 CrossRef CAS.
- A. W. Smith, A. Camara-Artigas, M. Wang, J. P. Allen and W. A. Francisco, Biochemistry, 2006, 45, 4378–4387 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c1mt00156f |
| ‡ Present address: Textile and Paper Engineering Department, ETSEIAT, Universitat Politècnica de Catalunya, Colom 11, E-08222 Terrassa, Spain |
| This journal is © The Royal Society of Chemistry 2012 |