Advances in metal–carbene complexes as potent anti-cancer agents
Arnaud
Gautier
*ab and
Federico
Cisnetti
*b
aCNRS, UMR 6504, F-63177 Aubière, CEDEX, France. E-mail: Arnaud.Gautier@univ-bpclermont.fr; Federico.Cisnetti@univ-bpclermont.fr; Fax: +33 473-40-77-17; Tel: +33 473-40-76-46
bClermont Université, Université Blaise Pascal, Laboratoire SEESIB, F-63000 Clermont-Ferrand, France
First published on 25th October 2011
Abstract
This critical review is an updated survey of metal–carbenes as potential anticancer chemotherapeutics. We report on the recent advances in the discovery of N-heterocyclic carbenes, acyclic diamino carbenes and abnormal NHCs associated with metals from groups 10 and 11 that displayed antiproliferative activity and emphasize, when possible, their molecular target(s) and their mechanism of action.
 Arnaud Gautier | Arnaud Gautier obtained his PhD from the Université d'Auvergne in 1995 and undertook postdoctoral training at Stanford University (Professors B. M. Trost and W. S. Johnson). In 1997, he moved to the Professor István E. Markó's team at the Université Catholique de Louvain. He was appointed as a CNRS member (IRCOF, Rouen, France, 1999) in Pr. Serge Piettre's group. In 2005, he moved to the Université Blaise Pascal. His current research interests include platinum complexes obtained by click chemistry, the synthesis of metal–carbenes and their anti-cancer properties. |
 Federico Cisnetti | Federico Cisnetti studied in the University of Geneva, ENS and Paris-XI University where he obtained his PhD in inorganic chemistry in 2007 (Pr. Policar). After a post-doctoral stay in CEA-Grenoble with Dr Pascale Delangle, he was appointed in 2009 as “Maître de Conférences” (lecturer) in Blaise Pascal University (Clermont-Ferrand, France) and works currently in Arnaud Gautier's group. At the present time, his research interests are mainly focused on (bio)organometallic chemistry. |
Introduction
Chemotherapy, along with surgery and radiotherapy, is widely used in treatments for cancers. Since its initial discovery by Rosenberg et al.,1 cisplatin or cis-diamminedichloroplatinum(II) (DDP) is considered as the successful historical example of a metal-based drug. However, severe side effects—nephrotoxicity, neurotoxicity and ototoxicity—are displayed by this platinum complex and sometimes limit its applications. In addition, DDP presents also a limited aqueous solubility, and is chemically incompatible with thiols. Finally, some tumours are naturally cisplatin-resistant or develop resistances during DDP treatment.2 Many DDP analogues have been biologically evaluated but only few possess pharmacological advantages relative to cisplatin. Only three platinum-based anticancer drugs have been worldwide approved (Scheme 1), and a few others are approved in some countries.3 The global market of platinum-based drugs represents several billion €—oxaliplatin alone is expected to reach more than 2 billion € in the next two years.4 This figure illustrates well their impact within the global pharmaceutical market.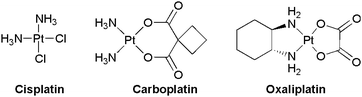
Consequently, circumvolution of the aforementioned limitations of DDP is an actual challenge in pharmaceutical research. It is thus not surprising that a wide range of transition-metal drug candidates are currently under consideration for future development.5 For example, NAMI-A (ruthenium, phase I/II: 2008, undergoing),6 KP1019 (ruthenium, phase I: 2004),7 Ferrocifen8 (iron, preclinical formulation studies), Budotitane (titanium, phase I: 1986), titanocene dichloride (titanium, phase II: 1993)9 and [Au(dppe)]+ (gold, preclinical studies in the late 1980's) were subjected to preclinical or clinical studies (Scheme 2). However, until today, no non-platinum anticancer drug has successfully passed all stages of clinical development.
 | ||
| Scheme 2 Examples of anti-cancer transition metal complexes. | ||
As potential anticancer drugs, metal–NHCs (NHC: N-Heterocyclic Carbene) constitute a recent and very rapidly growing field of research. Öfele and Wanzlick reported in 1968 the first synthesis of stable metal–N-heterocyclic carbenes.10 Since then, metal–NHCs have become systems very well-known to the organometallic chemists as they have found a plethora of applications in catalysis, thanks to their strong and stable metal–ligand bond (compared to phosphines).11 The same feature has recently allowed therapeutic investigations of NHC–metal complexes, at first in the field of antimicrobials12 and more recently as anti-cancer agents. Importantly, the great potential of metal–NHCs in the pharmaceutical domain is highlighted by several recent patents taken by universities and “big Pharma”.13,14 We have already reviewed in 2009 the growing field of metal–NHC complexes as anti-cancer agents;15 this article presents an update of this subject—as of July 2011—as new compounds and metals appeared since, broadening considerably the scope and applicability of metal–NHCs as novel anticancer agents. Other biological applications of metal–NHCs—especially in the field of antibacterials—are out of the scope of this review. The reader may refer to recent reviews dealing mainly with this subject.16
Group 10
Group 10 comprises nickel, palladium and platinum. The latter has attracted considerable interest since the discovery of cisplatin. DNA is considered to be the main target of group 10 metal-based drugs via the formation of intra- and inter-stand adducts, although metal-bonding to sulfur-containing groups in proteins was also reported.17 The strong metal–chloride bond in platinum complexes explains their therapeutic success: these complexes remain intact as neutral species in blood in which a high chloride concentration is present. Hydrolysis can occur after diffusion or active transport to the cell where the chloride concentration is lower. The released aqua complex is the actual active species.2 This also explains that nickel and palladium complexes—in which the metal–Cl bonds are weaker—have intrinsic limitations as cisplatin equivalents. Moreover, free nickel ions present a high toxicity. However, recently it was reported that tetradentate Ni(II)–NHCs complexes displayed poor toxicity toward Hela, HL60 (cancerous) and CHO (non-cancerous) cell lines.18Palladium(II)–NHCs
The antiproliferative potential of two neutral trans-Pd(II)–NHC complexes was explored by Panda and Ghosh et al.: the mono-NHC complex 1, and the bis-NHC complex 2 (Scheme 3).19 Complex 2 was an efficient inhibitor of the proliferation of cancerous cells with 2- to 20-fold increased activity in comparison with DDP: IC50s were 4 μM (HeLa), 0.8 μM (HCT 116: colon carcinoma) and 1 μM (MCF-7). | ||
| Scheme 3 Pd(II)–bis-NHC complex evaluated for their biological properties. | ||
Despite its trans configuration, the authors report biological evidence indicative that complex 2 follows the same cellular pathway as DDP with cell cycle arrest in G2/M transition, the activation of the cyclin B1, the phosphorylation of cdc2 (a M phase promoting factor) and the activation of a p53-dependent pathway in cell death. Cytotoxicity is not limited to bis-NHCs as Ong, Li et al. reported that the heteroleptic palladium–NHC–π-allyl complex 3 (Scheme 4) containing a pending amine group displayed up to a 10-fold increased cytotoxicity compared to DDP: IC50 = 6.5 μM, 4.5 μM and 10.25 μM for MCF-7, MDA-MB-231 and U-87 MG cell-lines, respectively.20

Platinum NHCs and ADC
Mailliet, Marinetti and co-workers have published21 and patented with Sanofi-aventis14 the use of trans-[Pt(NHC)(amine)] complexes 4 (Scheme 5). This patent could be understood as a testimony of the interest of big Pharma for the development of new cytotoxic compounds. Their synthetic methodology relies on the oxidative addition of iodine on Markó's Pt(0) complex, [Pt(NHC)(dvtms)]22 followed by the introduction of the amine group (NHCs derived from imidazole, benzimidazole or 1,2,4-triazole with symmetric or dissymmetric substitution). The amine substitution of dvtms results in trans geometry, in contrast to phosphines which yield cis complexes. Nearly all complexes showed micromolar cytotoxicity towards cancer cell lines such as CCRF-CEM (T leukemia) and NCI-H460 (lung cancer). Interestingly, relatively high cytotoxicities were determined against DDP-resistant cells (ovarian cells A2780/DDP, CH1/DDP and SKOV3). To exemplify the above-mentioned results, IC50s for 4a and 4b, the most active complexes on non-DDP-resistant and DDP-resistant cancer cells, respectively, are reported in Table 1. The initial choice of iodide as an ancillary ligand proved to be optimal since bromide or nitrate ligands resulted in decreased activity. NHC ligands derived from caffeine (5) or containing protected monosaccharide pendants were also considered although these were not the most active examples. Nevertheless, the authors point out that biologically non-innocent ligands—i.e. including cancer cell targeting groups—may be considered in future works as extensions of their strategy.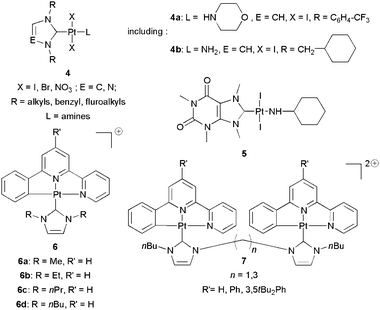 | ||
| Scheme 5 Pt(II)–NHCs considered as anticancer agents. | ||
| CCRF-CEM | NCI-H460 | A2780/DDP | CH1/DDP | SKOV3 | |
|---|---|---|---|---|---|
| a ND: not determined. | |||||
| 4a | 0.6 | 0.9 | NDa | ND | ND |
| 4b | 2.0 | 3.8 | 1.2 | 2.0 | 5.3 |
| Cisplatin | 3 | 2.4 | >10 | >10 | 6.1 |
| Oxaliplatin | 0.9 | 4 | 17.3 | 6.2 | >10 |
Che et al.23 investigated the anticancer potential of cyclometalated [Pt(II)(6-phenyl-2,2′-bipyridine)(NHC)] complexes. A range of complexes of variable lipophilicity (NHC N-substituents) and nuclearity (mono- or bis-) was subjected to cytotoxicity assays (6 and 7, Scheme 5). The most favorable case was the simplest mononuclear complex with methyl NHC substituents: submicromolar cytotoxicity toward three different cell lines was outlined as well as a significantly lower effect on healthy CCD-19Lu lung fibroblast cells (Table 2). The length of alkyl groups seems to modulate activity and selectivity, the most interesting example being 6a (R = Me), that was subsequently studied in vivo.6a significantly inhibited the NCI-H460 (non-small cells lung carcinoma) tumour growth by 55 ± 11% in nude mice when injected at 3 mg kg−1. No toxic effects were observed on healthy tissues. Cellular localisation studies were conducted taking advantage of the photoluminescent nature of the complexes, which were shown to accumulate in mitochondria and cytoplasm. DNA was not the primary target of the complexes. The cytotoxic effects towards cancer cells were ascribed to inhibition of survivin—an apoptosis inhibitor—but the authors did not rule out a mitochondria-based mechanism of action: unlike Che's gold complexes (see below), Topoisomerase I is not inhibited by 6 complexes.
| HeLa | HepG2 | SUNE1 | CCD-19Lu | |
|---|---|---|---|---|
| 6a | 0.057 | 0.77 | 0.14 | 11.6 |
| 6b | 0.052 | 1.1 | 0.16 | 4.3 |
| 6c | 0.48 | 1.3 | 0.32 | 2.1 |
| 6d | 0.33 | 0.31 | 0.51 | 5.7 |
| Cisplatin | 15 | 15 | 2.4 | >100 |
Bellemin-Laponnaz, Guichard and co-workers described recently a strategy in which platinum and palladium trans-NHCs sharing the [M(NHC)2X2] formula (M = Pd, Pt; X = Br, I) were functionalised by ruthenium catalysed azide–alkyne cycloaddition (RuAAC).24 One of the usual drawbacks in the use of organometallic complexes as therapeutic agents is the introduction of the metal, which is usually performed at the end of the synthetic sequence. The authors point out that this step often requires stringent conditions which are incompatible with a variety of functional groups. Moreover, a strategy based upon direct functionalisation of a preformed organometallic complex is certainly an important asset with regard to modularity. The failure of copper-catalysed azide–alkyne cycloaddition (CuAAC)—frequently employed in ligand design25—on their complexes was also reported. The starting complexes bore alkyne-containing groups either as N- or C-substituents on the imidazol-2-ylidene ring. Among the moieties grafted by RuAAC, biologically relevant groups were chosen: PEG groups for water solubility, an amino acid (albeit in the Fmoc protected form), and most interestingly an estrogen derivative (8, Scheme 6) to introduce selectivity in the mode of action of the complexes. However, the biological activity of the complexes was not yet reported.
 | ||
| Scheme 6 Estrogen–Pt–NHC conjugate obtained by RuAAC. | ||
Until this year, all the platinum and palladium complexes considered in the biological context present a trans geometry due to the strong σ-donation of the carbene ligand that dictates the configuration along the synthetic path. We believe therefore that these intrinsically trans metal carbenes cannot constitute true DDP equivalents and that a platinum carbene featuring a cis geometry would be highly preferable. In addition, the steric hindrance around the metal should be low and the complex should be able to establish H-bonds with biomolecules. Indeed, platinum Chugaev-type carbenes such as 9, Scheme 7, which were initially synthesized in 1915—of course, without the knowledge, at that time, of their real molecular structure—and are therefore the historical example of metal–carbene complexes, possess all of the requirements mentioned above.

The chemistry of such ADCs (Acyclic Diamino Carbene) has been well established in the 1960's and the investigation in the palladium series was reported recently by Slaughter's group since 2005.26 In addition, X-ray structural data highlight the capacity of their hydrazine moiety to behave as a H-bond donor. Therefore, we reported recently the cytotoxicity for cancerous cells of the complex 9 and its ability to interact with both supercoiled DNA and thiols (Table 3).27
| MCF-7 | MCF7-R | PC3 | |
|---|---|---|---|
| 9 | 4.5 | 14.6 | 5.1 |
| Cisplatin | 4.9 | 3.1 | 2.9 |
The reaction of DDP with cysteine (Cys) and glutathione (GSH) is implicated in the detoxification path for resistant cell lines. With the former, N2S2 adducts are obtained after a rapid expulsion of NH3. With glutathione, cis-[PtCl(GS)(NH3)2] is rapidly formed, also followed by a fast loss of NH3. The Pt–ADC 9 experiences similar complexation to Cys and GSH, but as the metal–carbene bonds are stronger than the Pt–N bonds in DDP, expulsion of the carbene ligand does not occur. Thus, with Cys a C2NS complex is the only species formed and in the presence of glutathione, a bis-μ-sulfido dimer is observed (Scheme 8).
 | ||
| Scheme 8 Reactions of thiols with the platinum ADC 9. | ||
Group 11
Group 11 comprises the coinage metals copper, silver and gold. The contribution of group 11 metals to the pool of metal-based drugs is diverse for each element. Studies indicating that copper complexes behaved as potent anticancer agents have been, up to now, limited to the academic community. Silver salts possess a long history as antimicrobial agents and proved generally to exhibit low toxicity for humans.16 Gold species might display non-cisplatin like pharmacodynamic and pharmacokinetic properties and thus have attracted a great interest because they may give rise to effective treatments for DDP-resistant cells. The first Au complex for which anticancer activity was evidenced—Auranofin (Scheme 14)—was taken from the pool of antiarthritic gold drugs. Nowadays, several gold(I) and more recently gold(III) complexes based on different ligand families have attracted interest as potential antitumour agents. Their mechanisms of action are mainly focused at the mitochondrion—even though other sites of activity including DNA cannot be completely excluded.Copper(I)–NHCs
Most of the reports regarding copper complexes as anti-cancer agents28 have focused on copper(II) precursors in the presence of reducing agents that yield a copper(I) active species which acts as a chemical nuclease.29,30 For a review on copper- and other metal-based nucleases see the review by Sigman et al.31 Despite that Cu(II) complexes are active via the generation of Cu(I), few investigations were conducted on stable copper(I) complexes.32 We hypothesized that copper(I)–NHCs—thanks to their intrinsic stability under numerous conditions33—would be able to reach biological targets inside the cell. Subsequently, Cu(I)–NHC complexes could react with intracellular oxygen or hydrogen peroxide, producing in situ Reactive Oxygen Species (ROS) ultimately leading to cellular damages.34 Therefore, we first evaluated the effect of the well-known copper(I)–NHCs 10–14, Scheme 9 on the MCF-7 cell line (Table 4).35 | ||
| Scheme 9 Selected Cu(I)–NHCs for biological studies. | ||
| Cisplatin | 10 | 11 | 12 | 13 | 14 |
|---|---|---|---|---|---|
| 10.4 | 0.075 | 0.13 | 0.04 | 0.075 | 4.4 |
All candidates—except the hindered complex 14 bearing bulky adamantyl groups—exhibited submicromolar IC50s significantly lower than that of the reference drug cisplatin. The biological action of [CuCl(SIMes)] 10 was studied from the point of view of the effect on the cell cycle arrest. A G1-phase stop was induced at concentrations at least ten times lower than those of DDP. Furthermore, an in vitro comparative evaluation with silver and palladium of genotoxicity was performed using supercoiled plasmids. As expected, the copper(I)–NHCs are unique in being able to act as chemical nucleases. This action follows a Fenton-type reaction path similar to other copper complexes such as [Cu(Phen)2]+31 or Clip-Phen.30,36 The mechanism of 10 is believed to imply the metal as a direct dioxygen activator in the presence of DNA as the reaction is inhibited by 1O2 scavengers.
Silver(I)–NHCs
In their series of papers dedicated to the biological activities of Ag–NHCs, Youngs et al. have reported the anticancer activities of silver acetate NHCs, some of them bearing chlorine atoms on the imidazol-2-ylidene skeleton37 (15–20, Scheme 10). The electron-withdrawing chlorines are not innocent because the stability of Ag–NHC complexes is greatly enhanced in comparison with non-chlorinated analogues (in D2O, an increase of the complex half-life was observed from 2 h for 15 to more than 17 weeks for 16).38 | ||
| Scheme 10 Ag(I)–NHCs for biological studies by Youngs et al. | ||
IC50s were determined and compared with those of DDP. The heteroleptic complexes 16–18 proved efficiency similar to that of DDP on OVCAR-3 (ovarian) and MB157 (breast) cells (Table 5). In contrast, the homoleptic complexes 19, 20 are 10 fold less active than DDP toward H460 (lung cancer), no significant activity was observed on HeLa cells (cervical cancer).
| OVCAR-3 | MB157 | HeLa | H460 | |
|---|---|---|---|---|
| Cisplatin | 12 | 25 | 25 | 2 |
| 16 | 35 | 8 | >200 | ND |
| 17 | 30 | 20 | >200 | ND |
| 18 | 20 | 10 | >200 | ND |
| 19 | ND | ND | ND | 19 |
| 20 | ND | ND | ND | 18 |
Importantly, in addition to in vitro results, complex 16 is efficient in vivo on the ovarian cancer xenograft model OVCAR-3. Nude mice were subjected to a high dose of 333 mg kg−1, causing major cell death in the tumours while provoking no toxic effects to the major organs. A patent from the same team evoked the possibility of incorporating these complexes in biodegradable nanoparticles.13c Still in the silver acetate series, the Tacke group published several papers39 demonstrating the cytotoxicity of symmetrical and unsymmetrical silver N-heterocyclic carbenes toward the renal cancerous Caki-1 cell line (Scheme 11 and Table 6). The results are consistent with what is reported by the Youngs group: the majority of the acetate complexes are active but in less extent than DDP, with few exceptions.
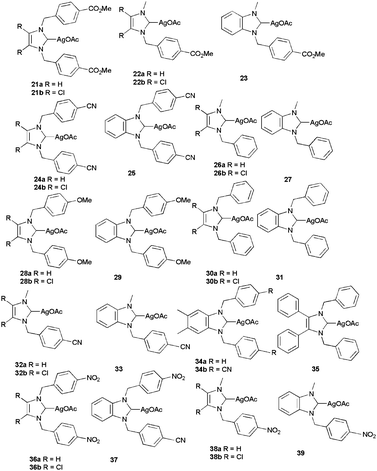 | ||
| Scheme 11 Anticancer Ag(I)–NHCs by Tacke et al. | ||
| Cisplatin | 21a | 21b | 22a | 22b | 23 |
|---|---|---|---|---|---|
| 3.3 | 13.5 | 68.5 | 3.3 | 13.2 | 9.4 |
| 29 | 30 | 53 | 3.2 | 24 | 34 |
| 28a | 28b | 29 | 30a | 30b | 31 |
| 7.6 | 12.7 | 25.2 | 2.5 | 10.8 | 12.5 |
| 32a | 32b | 33 | 34a | 34b | 35 |
| 6.2 | 7.7 | 1.2 | 13.6 | 10.8 | 24.2 |
| 36a | 36b | 37 | 38a | 38b | 39 |
| 27 | 15 | 22 | 20 | 17 | 16 |
The IC50 values for heteroleptic silver chloride NHCs complexes 40–43 have been reported by us15 on the cancer cell line MCF-7 (breast) and by Roland et al. on both MCR5, a non-cancerous cell line in rapid proliferation and EPC, a quiescent one.40 We decided to select chloride rather than acetate to mimic cisplatin's fate in blood where chloride concentration is high. Moreover, although satisfying cytotoxicities were reported for silver acetate NHCs toward cancerous cell lines, no comparison with non-cancerous cells, in rapid or slow proliferation was reported (Scheme 12, Table 7).
 | ||
| Scheme 12 Biologically evaluated silver chloride NHCs. | ||
In all cases, a high cytotoxicity is observed against MCF-7, exemplified with a 350-fold increased activity for complex 43 compared to that of DDP. 43 is until today the most active silver–NHC. However, no discrimination between the cancer cell line and the non-cancerous one in rapid proliferation (MCR5) is notable. Moreover, Roland demonstrated that Ag–NHCs displayed a 2- to 18-fold lower cytotoxicity for the quiescent EPC cell line, indicating that the activity is directly correlated with the proliferation rate.
Finally, the silver NHC 44 (Scheme 13) with an amino-pendant previously reported in the palladium series was also tested against breast adenocarcinoma and glioblastoma; the cytotoxicities were found in the range of that of cisplatin: IC50 = 28.7, 46.6 μM and 25.2 μM for MCF-7, MDA-MB-231 and U-87 MG, respectively.20
 | ||
| Scheme 13 Silver bromide NHC with an amine pending group. | ||
Gold(I)–NHCs
Nowadays, gold organometallics are recognized as promising species for future treatments for cancer (and other diseases).41 For gold, starting from Auranofin,42 the most numerous ligand family that was considered in the biological context was that of phosphines. NHCs were subsequently studied as more σ-donor phosphine analogues. This may be correlated to enhanced stability under biological conditions of Au–NHC species in comparison with Au–phosphine species. The gold–NHC bond is the most stable in the coinage metal family.43 A series of NHC analogues of Auranofin (45a–e) was first reported by Baker et al. (Scheme 14).44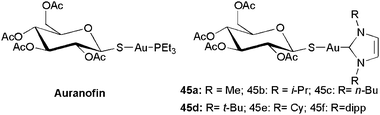 | ||
| Scheme 14 Auranofin and NHC analogues. | ||
Cationic and neutral gold–NHCs for which an anticancer activity was reported are depicted in Schemes 15 and 16, respectively.
 | ||
| Scheme 15 Cationic anticancer Au(I)–NHC complexes. | ||
 | ||
| Scheme 16 Neutral Au(I)–NHC complexes displaying anti-cancer activity. | ||
The mitochondrion plays a central role in the onset of apoptosis, thus mitochondrial targeting is highly desirable in the design of novel pro-apoptotic anticancer drugs.45 To this end, various strategies have been recently described in the literature. Gold complexes antimitochondrial properties may exploit—depending on the complex—metal-based reactivity with the selenoenzyme Thioredoxin Reductase (TrxR)46 and/or mitochondrial swelling induced by their delocalized lipophilic cation (DLC) behaviour. Reaction with TrxR is likely to imply complete ligand loss, by analogy as what established for the interaction of GoPI—a gold-phosphole complex—with TrxR.47 Both mechanisms of action may imply some degree of selectivity for tumour cells because of their abnormally high mitochondrial Δϕms and increased use of antioxidant TrxR. High activities for the latter enzyme have been correlated with tumour growth and evasion of apoptosis. Conversely, the inhibition of TrxR can lead to apoptosis of cancer cells via mitochondrial membrane permeabilisation (MMP), followed by release of cytochrome c. While DLC behaviour is obviously not restricted to gold species, TrxR inhibition takes advantage of the very high affinity of gold(I) to selenium, even in comparison with sulfur. The selenocysteine-containing enzyme TrxR has thus emerged as an important new drug target for gold-based metallodrugs.
Besides the analogy between strong σ-donor phosphines and NHCs, synthetic tuning of NHC precursors is believed to be easier. Indeed, tuning the lipophilicity of the metallo-drugs is of importance when targeting specific organelles in the cell. The Berners-Price research group reported seminal work on the rational design of mitochondrial targeting gold(I) N-heterocyclic carbenes.48,49 The potency of homoleptic Au(I)–NHC as inducers of MMP on isolated rat liver mitochondria was demonstrated, although in a lesser extent than Auranofin. Interestingly, the lipophilicity of the gold–NHCs could be fine-tuned by modulating the nature of the substituents of the aromatic wings, and correlated well with the antimitochondrial activity. The mechanism of inhibition has been demonstrated to rely on the reaction between the homoleptic gold carbenes and the selenocysteine residue of TrxR. The rate constants for the NHC–thiolate/selenolate exchange have proven to be 1–2 orders of magnitude in favour of the selenolate attack. Under the biological conditions, the formation of a (Sec)Se-Au-S(Cys) has been assumed to be the final step of the irreversible TrxR inhibition. As an example, compound 46 inhibited TrxR activity by 50% in MDA-MB-231 cells (IC50 = 12.5 μM, breast cancer). Mitochondria-induced apoptosis was supported by a reported rise of caspases 3 and 9 levels. Noteworthy was the selectivity of the complexes (cancerous cells vs. healthy human mammary epithelial cells). The luminescence associated with aurophilic interactions was advantageously exploited to design structures such as 47.49,50 Confocal fluorescence microscopy images of murine macrophage cancer cells with complex 47b showed a lysosomal intracellular localization along with the preservation of cell morphology.49
Horvath, Raubenheimer et al. designed an homoleptic gold(I) complex 48 bearing ferrocenyl groups as N-substituents.51 The authors also assumed an antimitochondrial activity for their complex, by analogy with other gold NHC complexes. The ferrocenyl groups were introduced for their electrophoric properties (as for the ferrocifen strategy8)—they are supposed to act as cytotoxic radical generators while cycling between +2 and +3 redox states. Cytotoxicities were reported on three cancerous cell lines (HeLa, CoLo 320 DM: colon adenocarcinoma, Jurkat: leukemia). Values were in the same range as DDP (Table 8). Interestingly, a 7-fold selectivity for cancerous cells versus human lymphocytes was demonstrated.
| HeLa | CoLo 320 DM | Jurkat | |
|---|---|---|---|
| 48 | 0.57 | 1.01 | 0.25 |
| Cisplatin | 0.638 | 0.41 | 0.78 |
In the field of homoleptic gold NHCs, an original piece of work is constituted by the very recent report by Kunz et al.52 These authors developed a 3-step high yielding synthetic procedure to obtain the bis-protic homoleptic gold-bis(imidazol-2-ylidene) complex 49. The complex was stable even under harsh acidic conditions and its solubility in water was found to be very high, an important parameter for biology. Preliminary biological studies showed a modest cytotoxicity at relatively high (10−4 M) concentrations. Interestingly, the complex seemed more active on cells overexpressing the CTR1 copper transporter.
Nolan, Riches et al. published two articles dealing with a family of complexes of general formula Au(NHC)L (50 and 51).53,54 These were obtained in a straightforward manner using the facile functionalisation of [Au(IPr)(OH)].55 Cationic 50 [Au(NHC)L]+ complexes with L = NHC′ (BMIM, ICy, ItBu, IMes, SIMes, IPr) or L = phosphine (PPh3, PtBu3, PnBu3) were shown to be potent cytotoxic agents with IC50 in the 0.38–0.75 μM range for LnCaP and 0.35–1.00 μM range for MDA MB231. This confirms the results from the Berners-Price groups. Unfortunately, no selectivity for cancer cells was proven as complexes were very cytotoxic towards the normal human urothelial cell line SV-HUC-1. All these results indicate that cationic Au complexes are good anticancer candidates.
Heteroleptic Au(NHC)Cl complex 52 was studied by Panda and Ghosh et al.19 and was virtually inactive against HeLa cell proliferation (1.7% at 10 μM). As its silver analogue, its biological interest was rather in the antibacterial field. Similarly, in the recent paper by Nolan et al.,54 the 51 NHC bioconjugates—with 1-thio-β-d-glucose tetraacetate (45f),53 amino acids, β-estradiol, saccharin—unfortunately, all the complexes but 45f were inactive on the tested cell lines (LNCaP: prostate carcinoma; MDA MB231). 45f proved on the contrary to be active (LNCaP: IC50 = 1.25 μM; MDA MB231: IC50 = 0.82 μM;)
In the course of our studies15 interesting cytotoxicities for complex 53 were found on five cancerous cell lines. While cytotoxicity was lesser than for its silver analogue 40, complex 53 was still displaying IC50s in the same range than those of DDP (Table 9). Complex 53 was also relatively toxic for cisPt resistant cell lines: only 3- and 17-fold losses were observed for MCF-7R and HL60R (cell lines overexpressing P glycoprotein, MDR1 efflux protein) respectively, compared to the corresponding sensitive lines.
| KB | HL60 | HL60R | MCF-7 | MCF-7R | |
|---|---|---|---|---|---|
| 53 | 4.3 | 0.29 | 5.1 | 1.29 | 4.5 |
| Cisplatin | 2.2 | 6.78 | ND | 10.4 | 4.6 |
Ott and co-workers have reported recently a series of benzimidazole-derived heteroleptic Au(NHC)Cl complexes 54.56 The benzimidazol-2-ylidene core was designed by analogy with the structurally related phosphole complex GoPI, for which a crystal structure of a covalent thiolate adduct with glutathione reductase (GR)—highly structurally analogous to TrxR—was published. After having checked that reaction of the gold complexes with glutathione was not or barely occurring in a buffered medium at 37 °C, the complexes were proven to be good inhibitors of TrxR as well as moderate inhibitors of GR. TrxR-GR selectivity was in the 20- to 75-fold range as for the 46 complexes. In cellulo experiments outlined the activity of the gold complexes which were in the low micromolar range (Table 10).
| MCF-7 | HT-29 | HCT-116 | HEP-G2 | |
|---|---|---|---|---|
| 54a | 7.5 | 13.3 | 6.7 | 4.9 |
| 54b | 4.6 | 6.4 | 8.4 | 11.2 |
| 54c | 10.2 | 11.8 | 10.0 | 14.9 |
| 54d | 10.3 | 12.3 | 24.6 | 60.7 |
At the biological mechanistic level, the mechanism of action implied ROS generation, disruption of mitochondrial integrity followed by DNA fragmentation and ultimately apoptosis and necrosis. Effects were induced in rather short timeframes (few hours) and were generally irreversible.
Alternatively, in the context of identifying new potential targets for cancer treatment, Barrios et al.57 considered the inhibition of Protein Tyrosine Phosphatase (PTP) by Au(I)–NHC 55 and Auranofin. The active site of this protein contains a Cys residue, and the mechanism of action relies on the gold–sulfur affinity. PTPs are implied in several pathophysiological pathways. They have been shown to be involved in cancer, among other diseases. These studies revealed that 55 is a better inhibitor of PTP than Auranofin: IC50s are comprised between 10 and 40 μM on four PTP types. From the mechanistic point of view, 55 was found to behave as a competitive and reversible inhibitor of PTPs.
The cytotoxicity of 56, an amino-linked gold–NHC, was reported (Table 11). This complex displays up to 8 fold increased activity vs. cisplatin on breast cancer (MCF-7, MDA-MB-231) and glioblastoma (U-87 MG).20
| MCF-7 | MDA-MB-231 | U-87 MG | |
|---|---|---|---|
| 56 | 16.3 | 14.2 | 1.26 |
The similar activity in cells possessing estrogen receptors (ER) or not, respectively, MCF-7 and MDA-MB-231, suggests an ER-independent pathway. A S phase cycle arrest was also evidenced as well as a blocked DNA replication. Western blot analysis highlighted the activation of PARP, caspase-3 and -9 and the reduction of the level of Bcl-2. Contrary to compounds 54, the complex 56 does not target TrxR but rather interferes with DNA in a binding mode significantly different from that of cisplatin. The DNA interaction produces a down-regulation of cyclins A, B1 and cdk2 and P21, an enhancement of p53, phospho-p53(ser15) and bak. It is believed that the down regulation of p21 by 56 would render the cells more sensitive to p53-mediated apoptosis.
Che and co-workers reported the use of gold(III) complexes including NHC ligands as anticancer agents (Scheme 17).58 The strategy followed by this research group includes the use of metallic elements to control the structure of molecules able to interact with biological targets. This strategy is inspired by “classical” medicinal chemistry approaches and benefits from geometrical properties enforced by the metal that are impossible to obtain with purely organic compounds: i.e. for gold(III), square planar geometry. It is also reminiscent of nature using metallic elements in enzymes either with catalytic or structural functions. Recently, mononuclear 57 and dinuclear 58 complexes containing 2,6-diphenylpyridine and one NHC ligand were reported. The phosphine analogues of these complexes were previously described59 and the phosphine-to-NHC replacement was expected to stabilize the gold(III) state. The mode of action differs from the one established for other gold complexes. The coordinatively saturated gold(III) complexes where shown to bind calf thymus DNA by UV-vis and fluorescence spectroscopies. Biological assays showed that the mode of binding was similar to that of organic intercalating agents such as ethidium bromide. Further experiments showed that Che's gold complexes were able to poison Topoisomerase I (TopoI) as do organic drugs such as camptothecin (CPT) or topotecan. In particular, the gold species formed ternary gold complex–TopoI–DNA adducts, and DNA strand breakage occurs at that point. The activities were correlated with the molecular structure by in silico docking. The best gold complex was the simplest mononuclear complex 57a, which exhibited an activity similar to that of CPT on the tested cell lines (Table 12). Its activity was only slightly decreased in the CPT-resistant cell-line KB-CPT-100 and selectivity between a healthy lung cell line CCD-19Lu and tumour cells was outlined. In vivo studies for this complex were performed and it was shown that tumour growth was significantly suppressed in nude mice bearing PLC tumour (hepatocellular carcinoma): injection of 57a at 10 mg kg−1 week−1 for 4 weeks resulted in the impressive suppression of 47% of tumor growth.
 | ||
| Scheme 17 Au(III)–NHC complex displaying anti-cancer activity. | ||
| HepG2 | KB | KB-CPT-100 | SUNE1 | NCI-H460 | CCD-19Lu | |
|---|---|---|---|---|---|---|
| 57a | 0.37 | 0.56 | 1.2 | 0.25 | 0.17 | 25 |
| Cisplatin | 10.5 | 9.8 | 10.1 | 4.9 | 3.5 | 51 |
| Camptothecin | 0.57 | 0.52 | 33 | 0.37 | 0.09 | 99 |
In summary, in cellulo activity of Au(I)–NHC complexes is now well studied. Au(III)–NHCs is emerging as well, at the moment with a structural role. Overall, the numerous works published on the subject in the last few years demonstrate the dynamism of this area. The gold–NHC family with its bioactivity inherently different from that of platinum or palladium compounds might furnish in a near future one or several useful drugs. We are still at the dawn of the in vivo evaluation of selected gold–NHC compounds.
Other metals: ruthenium, rhodium
After their report in 200860 focusing on peptide–metal–NHC conjugates (metal: Ru, Rh), Lemke and Metzler-Nolte reported very recently thiazolylalanine-containing tripeptides as metallocarbene precursors (59–61, Scheme 18).61 The thiazole moieties were converted to thiazolium salts after reaction with ethyl bromide. Subsequently, they were reacted with [(p-cymene)RuCl2]2 or [Cp*RhCl2]2 in the presence of silver oxide to yield ruthenium(II) or rhodium(I) carbene complexes. Although no biological activity for the complexes has been yet reported, the compounds seem quite promising since antiproliferative activity of Ru(II) complexes is well-known.62 | ||
| Scheme 18 Examples of peptide–metal–NHC chimeras (metal: Ru, Rh) by Lemke and Metzler-Nolte. | ||
The toxicity of classical and abnormal Ru–NHCs was recently investigated on zebrafish embryos on which results can be accepted as predictive for a human drug concern (Scheme 19).63 The results clearly reveal that the ruthenium complexes 62–65 are safe, with LD50 > 100 μg mL−1 and thus can be considered as nontoxic.64 To the best of our knowledge, this is the only report focussing on abnormal NHCs.
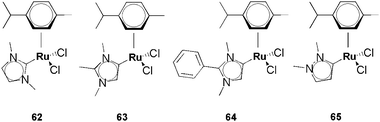 | ||
| Scheme 19 Ru–NHCs tested on Zebrafish embryos. | ||
Conclusions
All the reports concerning the anti-cancer properties of metal–NHC complexes—including important and novel reports in the last two years—have confirmed their value as metal-based drugs. For each metal of interest, some metal–NHC complexes exhibit cytotoxic potencies similar to cisplatin, and frequently higher. In general metal–NHCs are efficient as apoptosis-triggering species. Most importantly the first examples—in the silver, platinum and gold series—highlight their in vivo efficiency on xenograft models. Whereas for silver complexes the mode of action has not yet been unraveled, Pt(II)–, Pd(II)–, Au(I)– and Cu(I)–NHCs follow most frequently a metal-dependent pathway to induce cancerous cell-death. Undoubtedly, this promising field of investigation focusing on anticancer metal–NHC complexes will continue to blossom as ADC and abnormal carbenes are emerging. The first results on anticancer metal–NHC complexes concerned complexes selected from or closely related to the pool of NHC-based catalysts. We believe that the next stage in the rise of anticancer NHC complexes is the targeting of cancerous cells vs. healthy ones and, ultimately, specific organelles within the cells. To this end, complexes containing tailor-made ligands may be synthesized using straightforward methods: bioconjugates such as metal–NHC–peptide or metal–NHC–hormone species.60,61,65 This important direction may provide selectivity which is highly desirable for the discovery of clinically useful metal–NHC drugs.Notes and references
- B. Rosenberg, C. L. Van and T. Krigas, Nature, 1965, 205, 698–699 CrossRef CAS.
- Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, ed. B. Lippert, Verlag Helvetica Chimica Acta, Zürich, 1999 Search PubMed.
- N. J. Wheate, S. Walker, G. E. Craig and R. Oun, Dalton Trans., 2010, 39, 8113–8127 RSC.
- S. J. Berners-Price, Angew. Chem., Int. Ed., 2011, 50, 804–805 CrossRef CAS.
- (a) Bioinorganic Medicinal Chemistry, ed. E. Alessio, Wiley-VCH, Weinheim, 2011 Search PubMed; (b) G. Gasser, I. Ott and N. Metzler-Nolte, J. Med. Chem., 2011, 54, 3–25 CrossRef CAS; (c) M. J. Hannon, Pure Appl. Chem., 2007, 79, 2243–2261 CrossRef CAS.
- J. M. Rademaker-Lakhai, D. B. D. Van, D. Pluim, J. H. Beijnen and J. H. M. Schellens, Clin. Cancer Res., 2004, 10, 3717–3727 CrossRef CAS.
- C. G. Hartinger, M. A. Jakupec, S. Zorbas-Seifried, M. Groessl, A. Egger, W. Berger, H. Zorbas, P. J. Dyson and B. K. Keppler, Chem. Biodiversity, 2008, 5, 2140–2155 CAS.
- A. Vessieres, S. Top, W. Beck, E. Hillard and G. Jaouen, Dalton Trans., 2006, 529–541 RSC.
- P. M. Abeysinghe and M. M. Harding, Dalton Trans., 2007, 3474–3482 RSC.
- (a) A. J. Arduengo, III, R. L. Harlow and M. Kline, J. Am. Chem. Soc., 1991, 113, 361–363 CrossRef; (b) K. Öfele, J. Organomet. Chem., 1968, 12, 42–43 CrossRef; (c) H. W. Wanzlick and H. J. Schoenherr, Angew. Chem., Int. Ed. Engl., 1968, 7, 141–142 CrossRef CAS.
- To the best of our knowledge, the first metal carbene complex was synthesised at the early nineteen century by Chugaev: (a) L. Chugaev and M. Skanavy-Grigorieva, J. Russ. Chem. Soc., 1915, 47, 776 Search PubMed; (b) L. Chugaev, M. Skanavy-Grigorieva and A. Posniak, Z. Anorg. Allg. Chem., 1925, 148, 37 CrossRef CAS. These complexes were fully characterised later: (c) A. Burke, A. L. Balch and J. H. Enemark, J. Am. Chem. Soc., 1970, 92, 2555 CrossRef CAS. For a general review on M–NHCs see: (d) P. de Frémont, N. Marion and S. P. Nolan, Coord. Chem. Rev., 2008, 253, 862–892 CrossRef. For modern applications of metal–NHCs, see: (e) N-Heterocyclic Carbenes in Synthesis, ed. S. P. Nolan, WILEY-VCH, Weinheim, 2006 Search PubMed; (f) W. A. Herrmann, Angew. Chem., Int. Ed., 2002, 41, 1290–1309 CrossRef CAS; (g) N. Marion, S. Díez-González and S. P. Nolan, Angew. Chem., Int. Ed., 2007, 46, 2988–3000 CrossRef CAS; (h) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606–5655 CrossRef CAS.
- First examples can be found in: B. Cetinkaya, E. Cetinkaya, H. Kucukbay and R. Durmaz, Arzneim. Forsch., 1996, 46, 821–823 CAS.
- (a) C. M. Che, R. W.-Y. Sun, L. F. Chow and J. Yan, USPTO Patent Application 20110098264, 2011 Search PubMed; (b) W. J. Youngs, K. Hindi and D. A. Medvetz, WO/2008/150830, 2008 Search PubMed; (c) W. J. Youngs, K. M. Hindi, D. A. Medvetz, M. Panzner and C. Tessier, WO/2009/015112, 2009 Search PubMed; (d) W. J. Youngs, C. A. Tessier, J. Garrison, C. Quezada, A. Meilaye, M. Pazner and S. Durmus, WO/2005/023760, 2005 Search PubMed; (e) W. J. Youngs, C. A. Tessier, W. G. Kofron, R. S. Simons and J. C. Garrison, US/2004/0097723, 2004 Search PubMed.
- P. Mailliet, A. Marinetti and M. Skander, WO/2009/118475, 2009 Search PubMed.
- M.-L. Teyssot, A.-S. Jarrousse, M. Manin, A. Chevry, S. Roche, F. Norre, C. Beaudoin, L. Morel, D. Boyer, R. Mahiou and A. Gautier, Dalton Trans., 2009, 6894–6902 RSC.
- (a) M. C. Deblock, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, in N-Heterocyclic Carbenes, The Royal Society of Chemistry, 2011, pp. 119–133 Search PubMed; (b) K. M. Hindi, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Chem. Rev., 2009, 109, 3859–3884 CrossRef CAS.
- G. Sava, A. Bergamo and P. J. Dyson, Dalton Trans., 2011, 40, 9069–9075 RSC.
- S. Ray, J. Asthana, J. M. Tanski, M. M. Shaikh, D. Panda and P. Ghosh, J. Organomet. Chem., 2009, 694, 2328–2335 CrossRef CAS.
- S. Ray, R. Mohan, J. K. Singh, M. K. Samantaray, M. M. Shaikh, D. Panda and P. Ghosh, J. Am. Chem. Soc., 2007, 129, 15042–15053 CrossRef CAS.
- C.-H. Wang, W.-C. Shih, H. C. Chang, Y.-Y. Kuo, W.-C. Hung, T.-G. Ong and W.-S. Li, J. Med. Chem., 2011, 54, 5245–5249 CrossRef CAS.
- M. Skander, P. Retailleau, B. Bourrié, L. Schio, P. Mailliet and A. Marinetti, J. Med. Chem., 2010, 53, 2146–2154 CrossRef CAS.
- G. Berthon-Gelloz, O. Buisine, J.-F. Brière, G. Michaud, S. Stérin, G. Mignani, B. Tinant, J.-P. Declercq, D. Chapon and I. E. Markó, J. Organomet. Chem., 2005, 690, 6156–6168 CrossRef CAS.
- R. Wai-Yin Sun, A. Lok-Fung Chow, X.-H. Li, J. J. Yan, S. Sin-Yin Chui and C.-M. Che, Chem. Sci., 2011, 2, 728–736 RSC.
- E. Chardon, G. L. Puleo, G. Dahm, G. Guichard and S. Bellemin-Laponnaz, Chem. Commun., 2011, 47, 5864–5866 RSC.
- H. Struthers, T. L. Mindt and R. Schibli, Dalton Trans., 2010, 39, 675–696 RSC.
- A. I. Moncada, M. A. Khan and L. M. Slaughter, Tetrahedron Lett., 2005, 46, 1399–1403 CrossRef CAS.
- G. Alves, L. Morel, M. El-Ghozzi, D. Avignant, B. Legeret, L. Nauton, F. Cisnetti and A. Gautier, Chem. Commun., 2011, 47, 7830–7832 RSC.
- C. Marzano, M. Pellei, F. Tisato and C. Santini, Anti-Cancer Agents Med. Chem., 2009, 9, 185–211 CAS.
- M. Barceló-Oliver, Á. García-Raso, Á. Terrón, E. Molins, M. J. Prieto, V. Moreno, J. Martínez, V. Lladó, I. López, A. Gutiérrez and P. V. Escribá, J. Inorg. Biochem., 2007, 101, 649–659 CrossRef.
- M. Pitié, B. Donnadieu and B. Meunier, Inorg. Chem., 1998, 37, 3486–3489 CrossRef.
- D. S. Sigman, A. Mazumder and D. M. Perrin, Chem. Rev., 1993, 93, 2295–2316 CrossRef CAS.
- (a) C. Marzano, V. Gandin, M. Pellei, D. Colavito, G. Papini, G. G. Lobbia, G. E. Del, M. Porchia, F. Tisato and C. Santini, J. Med. Chem., 2008, 51, 798–808 CrossRef CAS; (b) D. Suresh, M. S. Balakrishna, K. Rathinasamy, D. Panda and J. T. Mague, Dalton Trans., 2008, 2285–2292 RSC.
- S. Díez-González, A. Correa, L. Cavallo and S. P. Nolan, Chem.–Eur. J., 2006, 12, 7558–7564 CrossRef.
- G. M. Ehrenfeld, J. B. Shipley, D. C. Heimbrook, H. Sugiyama, E. C. Long, B. J. H. Van, d. M. G. A. Van, N. J. Oppenheimer and S. M. Hecht, Biochemistry, 1987, 26, 931–942 CrossRef CAS.
- M.-L. Teyssot, A.-S. Jarrousse, A. Chevry, H. A. De, C. Beaudoin, M. Manin, S. P. Nolan, S. Diez-Gonzalez, L. Morel and A. Gautier, Chem.–Eur. J., 2009, 15, 314–318 CrossRef CAS.
- M. Pitié, A. Croisy, D. Carrez, C. Boldron and B. Meunier, ChemBioChem, 2005, 6, 686–691 CrossRef.
- (a) T. J. Siciliano, M. C. Deblock, K. M. Hindi, S. Durmus, M. J. Panzner, C. A. Tessier and W. J. Youngs, J. Organomet. Chem., 2011, 696, 1066–1071 CrossRef CAS; (b) D. A. Medvetz, K. M. Hindi, M. J. Panzner, A. J. Ditto, Y. H. Yun and W. J. Youngs, Metal-Based Drugs, 2008, 2008, 384010 CrossRef.
- K. M. Hindi, T. J. Siciliano, S. Durmus, M. J. Panzner, D. A. Medvetz, D. V. Reddy, L. A. Hogue, C. E. Hovis, J. K. Hilliard, R. J. Mallet, C. A. Tessier, C. L. Cannon and W. J. Youngs, J. Med. Chem., 2008, 51, 1577–1583 CrossRef CAS.
- (a) S. Patil, J. Claffey, A. Deally, M. Hogan, B. Gleeson, L. M. Menéndez Méndez, H. Müller-Bunz, F. Paradisi and M. Tacke, Eur. J. Inorg. Chem., 2010, 1020–1031 CrossRef CAS; (b) S. Patil, A. Deally, B. Gleeson, F. Hackenberg, H. Müller-Bunz, F. Paradisi and M. Tacke, Z. Anorg. Allg. Chem., 2011, 637, 386–396 CrossRef CAS; (c) S. Patil, A. Deally, B. Gleeson, H. Müller-Bunz, F. Paradisi and M. Tacke, Metallomics, 2011, 3, 74–88 RSC; (d) S. Patil, A. Deally, B. Gleeson, H. Müller-Bunz, F. Paradisi and M. Tacke, Appl. Organomet. Chem., 2010, 24, 781–793 CrossRef CAS; (e) S. Patil, K. Dietrich, A. Deally, B. Gleeson, H. Müller-Bunz, F. Paradisi and M. Tacke, Helv. Chim. Acta, 2010, 93, 2347–2364 CrossRef CAS.
- (a) S. Roland, C. Jolivalt, T. Cresteil, L. Eloy, P. Bouhours, A. Hequet, V. Mansuy, C. Vanucci and J. M. Paris, Chem.–Eur. J., 2011, 17, 1442–1446 CrossRef CAS; (b) M.-L. Teyssot, A.-S. Jarrousse, A. Chevry, A. De Haze, C. Beaudoin, M. Manin, S. P. Nolan, S. Díez-González, L. Morel and A. Gautier, unpublished results.
- (a) S. J. Berners-Price and A. Filipovska, Metallomics, 2011, 3, 863–873 RSC; (b) C.-M. Che and R. W.-Y. Sun, Chem. Commun., 2011, 47, 9554–9560 RSC; (c) I. Ott, Coord. Chem. Rev., 2009, 253, 1670–1681 CrossRef CAS.
- (a) C. K. Mirabelli, R. K. Johnson, C. M. Sung, L. Faucette, K. Muirhead and S. T. Crooke, Cancer Res., 1985, 45, 32–39 CAS; (b) T. M. Simon, D. H. Kunishima, G. J. Vibert and A. Lorber, Cancer Res., 1981, 41, 94–97 CAS; (c) B. M. Sutton, E. McGusty, D. T. Waltz and M. J. DiMartino, J. Med. Chem., 1972, 15, 1095–1098 CrossRef CAS.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS.
- M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, J. Organomet. Chem., 2005, 690, 5625–5635 CrossRef CAS.
- P. J. Barnard and S. J. Berners-Price, Coord. Chem. Rev., 2007, 251, 1889–1902 CrossRef CAS.
- C. Gabbiani, G. Mastrobuoni, F. Sorrentino, B. Dani, M. P. Rigobello, A. Bindoli, M. A. Cinellu, G. Pieraccini, L. Messori and A. Casini, Med. Chem. Commun., 2011, 2, 50–54 RSC.
- S. Urig, K. Fritz-Wolf, R. Réau, C. Herold-Mende, K. Tóth, E. Davioud-Charvet and K. Becker, Angew. Chem., Int. Ed., 2006, 45, 1881–1886 CrossRef CAS.
- (a) P. J. Barnard, M. V. Baker, S. J. Berners-Price and D. A. Day, J. Inorg. Biochem., 2004, 98, 1642–1647 CrossRef CAS; (b) J. L. Hickey, R. A. Ruhayel, P. J. Barnard, M. V. Baker, S. J. Berners-Price and A. Filipovska, J. Am. Chem. Soc., 2008, 130, 12570–12571 CrossRef CAS; (c) M. V. Baker, P. J. Barnard, S. J. Berners-Price, S. K. Brayshaw, J. L. Hickey, B. W. Skelton and A. H. White, Dalton Trans., 2006, 3708–3715 RSC.
- P. J. Barnard, L. E. Wedlock, M. V. Baker, S. J. Berners-Price, D. A. Joyce, B. W. Skelton and J. H. Steer, Angew. Chem., Int. Ed., 2006, 45, 5966–5970 CrossRef CAS.
- P. J. Barnard, M. V. Baker, S. J. Berners-Price, B. W. Skelton and A. H. White, Dalton Trans., 2004, 1038–1047 RSC.
- U. E. I. Horvath, G. Bentivoglio, M. Hummel, H. Schottenberger, K. Wurst, M. J. Nell, C. E. J. van Rensburg, S. Cronje and H. G. Raubenheimer, New J. Chem., 2008, 32, 533–539 RSC.
- P. C. Kunz, C. Wetzel, S. Kogel, M. U. Kassack and B. Spingler, Dalton Trans., 2011, 40, 35–37 RSC.
- P. de Frémont, E. D. Stevens, M. D. Eelman, D. E. Fogg and S. P. Nolan, Organometallics, 2006, 25, 5824–5828 CrossRef.
- J. Weaver, S. Gaillard, C. Toye, S. Macpherson, S. P. Nolan and A. Riches, Chem.–Eur. J., 2011, 17, 6620–6624 CrossRef CAS.
- S. Gaillard, A. M. Z. Slawin and S. P. Nolan, Chem. Commun., 2010, 46, 2742–2744 RSC.
- R. Rubbiani, I. Kitanovic, H. Alborzinia, S. Can, A. Kitanovic, L. A. Onambele, M. Stefanopoulou, Y. Geldmacher, W. S. Sheldrick, G. Wolber, A. Prokop, S. Wölfl and I. Ott, J. Med. Chem., 2010, 53, 8608–8618 CrossRef CAS.
- D. Krishnamurthy, M. R. Karver, E. Fiorillo, V. Orru, S. M. Stanford, N. Bottini and A. M. Barrios, J. Med. Chem., 2008, 51, 4790–4795 CrossRef CAS.
- J. J. Yan, A. L.-F. Chow, C.-H. Leung, R. W.-Y. Sun, D.-L. Ma and C.-M. Che, Chem. Commun., 2010, 46, 3893–3895 RSC.
- C. K.-L. Li, R. W.-Y. Sun, S. C.-F. Kui, N. Zhu and C.-M. Che, Chem.–Eur. J., 2006, 12, 5253–5266 CrossRef CAS.
- J. Lemke and N. Metzler-Nolte, Eur. J. Inorg. Chem., 2008, 3359–3366 CrossRef CAS.
- J. Lemke and N. Metzler-Nolte, J. Organomet. Chem., 2011, 696, 1018–1022 CrossRef CAS.
- I. Bratsos, T. Gianferrara, E. Alessio, C. G. Hartinger, M. A. Jakupec and K. B. Keppler, in Bioinorganic Medicinal Chemistry ed. E. Alessio, Wiley-VCH, Weinheim, 2011, pp. 151–174 Search PubMed.
- P. M. Eimon and A. L. Rubinstein, Expert Opin. Drug Metab. Toxicol., 2009, 5, 393–401 CrossRef CAS.
- J. M. Alfaro, A. Prades, M. del Carmen Ramos, E. Peris, J. Ripoll-Gomez and M. Poyatos, Zebrafish, 2010, 7, 13–21 CrossRef CAS.
- (a) J. F. Jensen, K. Worm-Leonhard and M. Meldal, Eur. J. Org. Chem., 2008, 3785–3797 CAS; (b) G. Xu and S. R. Gilbertson, Org. Lett., 2005, 7, 4605–4608 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |