Engineering ecosystems and synthetic ecologies†
Michael T.
Mee
ab and
Harris H.
Wang
*cd
aDepartment of Biomedical Engineering, Boston University, Massachusetts, USA
bDepartment of Genetics, Harvard Medical School, Boston, Massachusetts, USA
cWyss Institute for Biologically Inspired Engineering, Harvard University, Boston, Massachusetts, USA. E-mail: harris.wang@wyss.harvard.edu
dDepartment of Systems Biology, Harvard Medical School, Boston, Massachusetts, USA
First published on 22nd May 2012
Abstract
Microbial ecosystems play an important role in nature. Engineering these systems for industrial, medical, or biotechnological purposes are important pursuits for synthetic biologists and biological engineers moving forward. Here we provide a review of recent progress in engineering natural and synthetic microbial ecosystems. We highlight important forward engineering design principles, theoretical and quantitative models, new experimental and manipulation tools, and possible applications of microbial ecosystem engineering. We argue that simply engineering individual microbes will lead to fragile homogenous populations that are difficult to sustain, especially in highly heterogeneous and unpredictable environments. Instead, engineered microbial ecosystems are likely to be more robust and able to achieve complex tasks at the spatial and temporal resolution needed for truly programmable biology.
Michael T. Mee obtained his undergraduate degree in Bioresource Engineering from McGill University in 2009. He is pursuing his doctoral studies in the Department of Biomedical Engineering at Boston University. Under the supervision of Prof. George Church in the Department of Genetics at Harvard Medical School, he is currently working on developing technologies to improve the engineering of microbial ecosystems. |
 Harris H. Wang | Harris H. Wang is a Fellow at the Wyss Institute for Biologically Inspired Engineering and an Instructor in the Department of Systems Biology at Harvard Medical School. Harris holds B.S. degrees in Physics and Mathematics from MIT and a PhD in Biophysics from Harvard University. He also received a joint degree from Harvard-MIT Health Sciences and Technology in Medical Engineering and Medical Physics. Harris has been developing foundational technologies in automated genome engineering to rapidly program cells with improved functions and new traits. Harris is one of ten young investigators to receive the first NIH Director's Early Independence Award (DP5) and is listed in Forbes 30 under 30 in Science in 2012. Harris is engineering microbial communities for applications in medical therapeutics, bioremediation, and bioenergy. |
Introduction
Microbes constitute the most abundant and diverse set of organisms on Earth.1,2 By generating and turning over organic material, they play a dominant role in performing key biochemical reactions essential to sustaining the biosphere.3 As such, these micron-sized cells have evolved an impressive array of strategies that have allowed them to grow in almost any environment on the planet.4 Microbes, however, do not live alone. Rather, they live in crowded environments in association with other microbes, competing for resources, sharing metabolism, and forming a complex, dynamic and evolving microbial ecosystem.5,6In nature, stable microbial consortia are generally composed of members that have specialized physiologies and are tasked with different roles. These intertwined roles transform individuals that would otherwise compete into a group that lives in concert.7 Many such microbial ecosystems have evolved to be highly refractory to perturbations in the environment and are able to repopulate themselves when depleted in numbers. We are now beginning to appreciate the myriad of sophisticated processes and behaviors that manifest in microbial consortia, some of which mirror many essential features found in higher-level metazoans and multicellular organisms.8 Understanding how individual microbes form communities will bring new and important insight to the evolution of multicellularity.9 A grand challenge in applied biology is to develop the knowledge and technology necessary to build these self-adaptive systems that can perform complex tasks at the micron-scale. Therefore, engineering microbial communities is an important endeavor, ripe for pursuit by synthetic biologists.
Over the past decade, the field of synthetic biology has aimed to make biology easier to engineer.10,11 Under the paradigm of traditional engineering, new conceptual frameworks were devised to describe the organization of genetic regulation and cellular machinery to build new metabolisms.12,13 New tools for the synthesis, assembly, and engineering of genes have been scaled to whole genomes to enable faster prototyping of biological designs.14 Standardized inventories of useful genes and other biological components are growing rapidly.15 All of these efforts help us develop a better understanding of the cell and the underlying design principles for engineering it. Scaling these efforts to communities of cells will require the development of new frameworks, methods and technologies.
In this review, we discuss recent advances in biological engineering at the level of cell populations and microbial consortia. We detail specific parameters that are crucial for building and engineering communities of cells that can exhibit sophisticated and robust behaviors and how these parameters can be synthesized into theoretical and predictive models for forward-design and engineering. We highlight new population measurement approaches and genome-perturbation techniques that facilitate the functional dissection of complex interactions occurring in microbial consortia. Finally, we discuss applications of synthetic and engineered microbial ecosystems in areas of biotechnology, bioenergy, and medicine (Fig. 1).
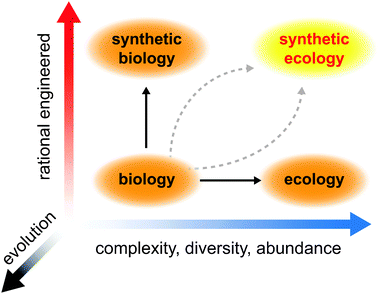 | ||
| Fig. 1 Development of synthetic ecology requires insights gained through manipulating simple biological systems and analyzing complex ecological systems. Evolution must be factored into these pursuits, not only as a destabilizing force but also as a means to optimize our engineered designs. | ||
Engineering parameters
What goes on in microbial communities can be quite complicated to understand, appearing almost irreducibly complex. Therefore, engineering such a system is a daunting task. Even when grossly approximating a cell as a linear input–output unit, we are confronted with the observation that interactions between cells generate behaviors that are non-linear, asynchronous, and heterogeneous. Toward building a framework for engineering synthetic microbial ecosystems, we outline a set of essential parameters that we believe are core features of a microbial community. These parameters should be the subject of analysis, perturbation, and optimization when building synthetic ecosystems de novo. Based on recent literature about natural and engineered ecologies, we highlight these parameters with regard to their significance, relationship with one another, and tunability from a synthetic perspective. These parameters help to build a framework for microbial communities where the individual members interact with one another through exchange of material, energy, and information (Fig. 2).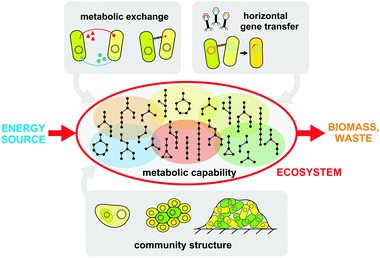 | ||
| Fig. 2 A summary of the crucial parameters that impact a microbial ecosystem. These parameters determine the ecosystem's ability to convert an energy source into biomass and waste, and are prime targets for engineering and optimization. Metabolic capabilities are distributed across different members as defined by metabolotypes (shaded and colored ovals). Metabolic exchange can occur via metabolite transport across cellular membranes or through intercellular bridges. Community structure can be tuned by adjusting the degree of aggregation and formation of extracellular structures such as biofilms. Horizontal gene transfer enables genomic innovation and the rise of new capabilities within the population. | ||
Metabolic capabilities and metabolotypes
Metabolism is the core essence of life at all scales, from individual enzymatic reactions in each cell all the way to the ecosystem as a whole. In nature, the goal of metabolism is to extract energy from substrates, use them to synthesize biomass, and leave behind waste byproducts. For any given environment, we can argue that the residing consortium of cells performs a set of input–output operations to generate biomass and waste from an initial source of energy (e.g. sunlight, sugar, other biomass, etc.). The black-box operation that the consortium performs may in fact be very complicated depending on the metabolic capability and efficiency of the members, as well as their abundance and diversity. In fact, many different arrangements can be functionally equivalent because microbes house a staggering array of metabolic capabilities in a near infinite number of combinations. Over the past decades, we have cataloged a significant portion of all possible chemical and enzymatic reactions that biology can perform in databases such as KEGG16 or MetaCyc.17 With computers and in silico models, we can now recreate cellular metabolism for well-studied organisms.18–22 Therefore, a deeper understanding of how metabolism scales to communities of cells can now be achieved.The total metabolic capability of a microbial community arises from the summation of capabilities of each individual member. Identification of a cell’s metabolism is not a trivial task, however. Traditional taxonomic classification of microbial species by 16S rRNA23 profiling is a poor reflector of metabolic functionality. For example, communities that are only 15% similar as profiled by 16 S may be 70% similar in terms of metabolic capability as determined by metagenomic sequencing.24 Furthermore, we have a poor understanding of how metabolic capabilities that are distributed across different individuals can impact the community as a whole. We do know that with sufficient functional redundancy in the population, system-level behavior can be stably maintained even though individuals may vary in abundance.25,26 Therefore, to have a clear picture of community-level metabolism, it is essential to identify the total list of metabolic genes, how they are allocated among individual members, and the level of redundancy in the system. Additionally, to improve the design of controllable and robust systems, greater understanding of the thermodynamics of these interacting metabolisms is needed. Individual metabolisms can impact the physiological environment (e.g. pH or oxygen level), as well as generate compounds that affect metabolism. These effects combine to shift the thermodynamic environment of cells with interacting metabolisms, altering the rate of growth and product yields.27 We believe that the metabolotype, or the range of metabolic capabilities of any individual cell, may be a more relevant identifier of consortium members than the standard 16 S phylogenetic signature. Metabolotype can be derived from the genotype via comparative genomic analyses28 or from the phenotype via experimental characterizations.29 Engineering metabolotypes may provide important avenues to tune the metabolic capacity, dynamics, and diversity of the ecosystem.
Intercellular exchange of metabolites and signals
In order to understand intercellular metabolic interactions (i.e. those occurring between cells), we need to understand the trafficking of metabolites across the cell membrane. The cell membrane provides an essential function: trapping enzymes and metabolites within the cytosol to increase their effective local concentration, thereby increasing their rate of catalysis. Any metabolic interaction between cells must require metabolites and intermediates to cross the membrane barrier. For most valuable metabolites, passive diffusion across the membrane barrier is very limited and active transport systems are needed. These molecular transport pumps vary in terms of specificity (general vs. specific pumps), directionality (symport, antiport), and energy requirement (ATP-dependency).30–32 Controlling these transport processes is an important thrust in microbial ecosystem engineering.While most cells have a myriad of transporters that import metabolites, far fewer transporters that export metabolites out of the cell have been identified. It is thought that most exporters (or efflux pumps) mainly serve to remove toxic or antagonistic compounds such as antibiotics from the cell.33 More recent studies have suggested that these exporters are important in the maintenance of cellular homeostasis by regulating intracellular metabolite concentrations.34 For example, a number of exporters exist to prevent excessive accumulation of different amino acids such as R, Y, W, F, L, M, K, I.35–40 From the microbial community perspective, these transport systems are critical in enabling selective, and potentially programmable, metabolite sharing between cells with different metabolotypes. In addition to extracellular exchange, other strategies for metabolite sharing exist. Nano-tubules or pilus-based structures enable direct cell-to-cell exchange by establishment of cytosolic bridges.41,42 These systems allow larger macromolecules such as polypeptides, proteins and DNA/RNA to be exchanged, thus providing additional means to metabolically connect individual cells within a community.
Microbes interact not only through interdependent metabolisms, but also by coordinated behaviors. Group behavior differentiates microbial communities that are merely collections of individuals from those that truly work in a concerted fashion. Coordinating behavior at the population level requires chemical signals and intercellular communication systems such as quorum sensing.43 Quorum sensing is the ability of cells to detect population density by measuring the concentration of a membrane-permeable chemical signal. As our knowledge of the diversity and mechanisms of how these small-molecule sensing systems grow, we are beginning to appreciate their important role in the formation and maintenance of microbial communities. For example, indole, a metabolic intermediate in tryptophan biosynthesis, serves also to promote resistance to antibiotics by generating persisters within a microbial community through intercellular signaling.44,45 Communication molecules triggering genetic programs across a population may elicit additional synchronized behaviors, such as cell division, differentiation, and aggregation.46–48 From an engineering perspective, we can co-opt these chemical communication systems for synthetic ecosystems. Using synthetic quorum sensing circuits, Basu et al. generated cell communities that exhibited different spatially-defined phenotypes in response to chemical gradients.49 These circuits have been further developed for edge detection systems that allow cells to sense the state of adjacent neighbors and respond accordingly,50 as well as for macro-scale synchronization of behavior across physical distances 1000 times greater than the length of a cell.51 These examples of engineered synthetic communities illustrate that controllable cell–cell signaling can enable the design of even more complex systems.
Aggregation and physical structures
Metabolic exchange and intercellular interactions require cells to be in close proximity. Cellular aggregation, by cell–cell contact or generation of extracellular matrices (known as biofilms), is a common strategy that natural microbial communities use to increase their local cell density.52 Often, cell aggregates directly lead to the formation of biofilms.53 Biofilm structures are particularly common as they anchor communities to a surface, allowing them to thrive more stably than in an otherwise mixed environment. By strengthening the local interactions in a community, these extracellular structures further enrich for ecosystems that behave cooperatively and in concert.54 Biofilms also decrease permeability of toxins and antimicrobial compounds thereby protecting the entire community.55 These structures provide tantalizing opportunities for synthetic engineering. For example, Brenner and Arnold developed an engineered biofilm community with increased cooperative growth and resilience to fluctuating environments.56 These systems should be further engineered for directed reciprocity – the ability for individuals to recognize and foster cooperative partners. Directed reciprocity is often found in naturally structured communities such as plant-mycorrhizal ecosystems57 and other symbiotic systems.58An extreme case of cell–cell association is endosymbiosis.59 The engulfment of one cell by another and the sustainment of such association can lead to the development of complementary physiologies. It is thought that eukaryotic organelles such as the chloroplast and the mitochondria were the result of endosymbiosis.60 Metabolic interdependency of endosymbionts often rely on exchange of essential metabolites (e.g. amino acids) as is the case for insect endosymbionts such as Tremblaya & Moranella in mealybugs,61 Buchnera in aphids62 and Sulcia in cicadas.63 While these systems clearly present fascinating examples of extreme interdependency, we have yet to fully understand the evolutionary processes that lead to endosymbiosis.64 Therefore, forward engineering of such systems remains a significant challenge.
Mutation and gene flow
The genetic makeup of the cell is not static but subject to constant change. In a microbial consortium, an individual's metabolic capabilities can change over time due to evolution and horizontal gene transfer (HGT).65,66 Small changes to the genome arise from mutations generated during replication or from DNA-damaging agents. Larger changes may arise from mobile genetic elements that move around the same genome and between different genomes.66 Small-scale mutations (e.g. point mutations, indels) generally affect the activity, specificity, or expression of proteins, so they are more likely to impact the cell’s physiology incrementally.67 Truly novel traits rarely evolve independently and are more likely to be acquired horizontally from another cell.66,68 HGT enables the cell to adopt new traits that require large leaps in sequence space, such as new biosynthesis capabilities. These processes can occur via conjugation, natural transformation, recombination, or transduction.67 So what influences the rate of genetic exchange in communities? Using comparative genomics, Smillie et al. argued that shared ecology is the most important factor that facilitates genetic exchange.69 The rate of HGT can also be accelerated in structured environments when neighboring cells are in close proximity and are more related phylogenetically.69 The level at which Darwinian selection occurs will affect the distribution and abundance of metabolotypes in the population. In order to effectively engineer ecosystems that behave predictably and stably over time, we must be able to either insulate the system from genetic mutations or harness natural selection to help maintain the engineered and desired state.An ecosystem engineering example
Here, we provide an example of microbial community engineering based on the parameters discussed above. We focus on biosynthesis because it is an important component of metabolism. On the population level, just as for individual cells, biosynthesis is optimized relative to cost and utility.70 Redundant or unnecessary biosynthetic pathways may reduce the metabolic efficiency of the population and are likely removed through Darwinian evolution.59 Using comparative genomics, we can computationally predict the biosynthetic capabilities of organisms that have fully sequenced genomes. Presence or absence of genes needed for biosynthesis of essential metabolites can be tabulated. Using the Integrated Microbial Genomes (IMG) database of sequenced organisms71 (3062 Bacteria, 121 Archaea, 124 Eukarya) and an algorithm for biosynthesis prediction, we discovered huge variation in biosynthetic capabilities for essential metabolites such as amino acids. The algorithm annotates an organism's biosynthetic capabilities based on sequence homology of its genome to genes in established databases.72–75 When plotting a histogram of organisms that are capable of biosynthesizing zero to all 20 standard amino acids, we find a wide distribution (Fig. 3a). The Bacteria domain tends to have organisms that on average can completely biosynthesize 7.9 out of 20 amino acids de novo. The average is 8.3 amino acids for Archaea and 4.1 for Eukarya. The histogram for Bacteria seems to be bimodal (Fig. 3a), suggesting that further classification is needed. Organisms in the Archaea domain on average have a slightly higher biosynthetic range for amino acids. This perhaps is due to their more ancient origin as a domain. Unsurprisingly, organisms in the Eukarya domain appear to make fewer amino acids since they derive most essential amino acids from nutrient-rich diets. As a reference, humans can only make 10 out of the 20 amino acids. It is important to note that these computational estimates of prototrophy are likely to be at the low end. More accurate comparative genomic analysis using better populated and more annotated databases will likely identify more biosynthetic genes. Nonetheless, the observation that most organisms cannot make all of their essential metabolites importantly highlights the interrelatedness of ecosystems. For each amino acid, we can further analyze whether the full biosynthetic pathway is intact across different organisms (Fig. 3b). We find that glutamic acid (E), glycine (G), and asparagine (N) tend be synthesized in most organisms while tyrosine (Y), phenylalanine (F), lysine (K), and histidine (H) tend to be made in few organisms. These trends appear to hold across Bacteria, Archaea, and Eukarya suggesting more universal processes at play. It is interesting to note that the more infrequently synthesized amino acids are also more costly to produce than those that are synthesized by most organisms, suggesting a level of cost-to-utility optimization.76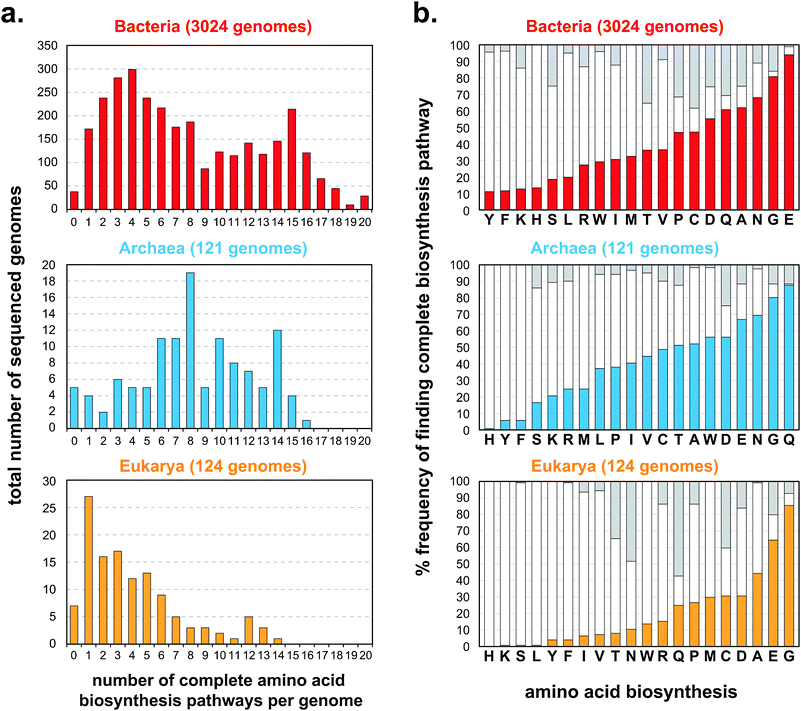 | ||
| Fig. 3 Diversity of amino acid biosynthetic capabilities across all sequenced organisms from the Integrated Microbial Genomes (IMG) database,71 separated based on the three domains (Bacteria, red, top panel; Archaea, blue, middle panel; Eukarya, orange, bottom panel). (a) Predicted frequencies at which species have the ability to synthesize zero to all 20 standard amino acids. (b) For each amino acid, frequencies at which complete biosynthetic pathways are found across each domain are shown in solid colored bars (Bacteria, red, top panel; Archaea, blue, middle panel; Eukarya, orange, bottom panel). White bars indicate fractions in each domain where one or more biosynthetic gene is missing. Gray bars indicate unknown annotations. | ||
Amino acid exchange has in fact been used to build synthetic microbial ecosystems. Hosoda et al. built a syntrophic cross-feeding community composed of E. coli strains that were either auxotrophic for isoleucine or leucine.77 Wild-type E. coli (e.g. K12 lineage) can normally make all 20 amino acids, but when genetically manipulated can be made auxotrophic (i.e. ΔilvE, or ΔleuB). Neither cell-type is able to grow independently, but when placed together in sufficient abundance will grow synergistically. Such a system has also been built to exchange arginine and tyrosine between engineered yeast strains.78 Wintermute and Silver more systematically quantified this syntrophic exchange using 46 auxotrophic E. coli strains to generate 1035 cross-feeding interactions.79 Metabolites that exchanged across different biosynthetic pathways led to more growth than those that exchanged along the same pathway. By examining the energetic cost and benefits of metabolite exchange, it was determined that syntrophic pairs achieved higher growth when the exchanged metabolite is less expensive to produce and is required in low amounts.79 These and other design principles enable us to model the effects of metabolite exchange and supply on biomass generation for forward engineering. Therefore, amino acids are a versatile set of metabolites whose exchange can enrich for consortium-level associations. Interdependencies can be engineered by exploiting biosynthetic configurations of these essential metabolites, which can further be tuned with transporters systems. These engineered communities present a framework for programming structures and dynamics into microbial ecosystems and serve to improve our ability to engineer metabolism at the population-level.
Theoretical and quantitative models
Theoretical and quantitative models are valuable analysis tools for studying natural and synthetic microbial ecosystems.80,81 While numerous important contributions have been made in this area, they have been for the most part limited by analytical, computational or algorithmic complexity. Since natural ecosystems are highly heterogeneous and nonlinear, molecular-resolution simulations of population-level interactions remain infeasible with current computational resources. Nonetheless, significant progress has been made for in silico reconstruction of cell physiology.82 Scaling these models from single cells to ecosystem, however, often demands a compromise in generality. Certain models may highlight individual population-level behavior better than others, but are doing so by sacrificing consideration of another important parameter. Here, we describe four classes of quantitative models that have been developed for understanding microbial ecosystems (Fig. 4) and highlight the importance of each.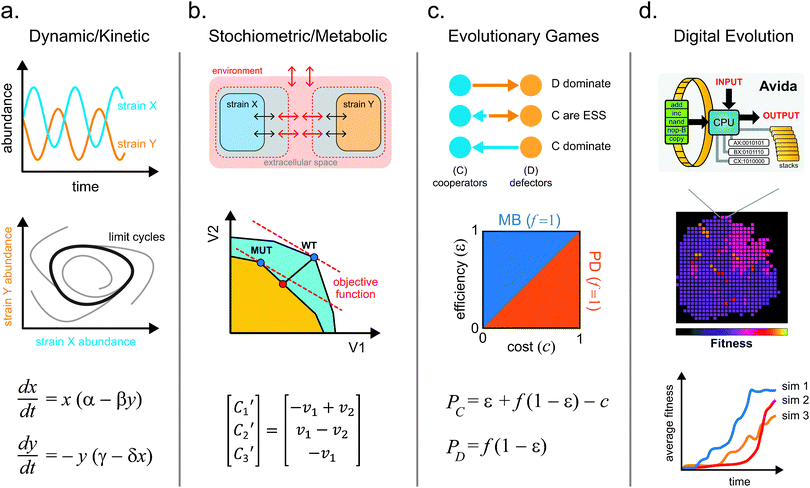 | ||
| Fig. 4 The four main classes of quantitative models that are used to study microbial ecosystems. (a) Kinetic models describe changes in system variables (e.g. abundance) with simple differential equations that can exhibit interesting dynamics such as oscillations and limit cycles. (b) Stoichiometric models can be applied to study optimal metabolic flux using objective functions to guide the design of intercellular metabolite exchange. (c) Evolutionary games can be used to analyze phenotypic strategies within a microbial community using payoff calculations. These models aid in elucidating key variables that influence the domination or coexistence of microbial strategies. (d) Digital evolution systems help to simulate microbial evolution, traversal of fitness landscapes, development of complex traits, and contributions of epistatic and pleiotropic effects to fitness. | ||
Dynamic models
Dynamic models are used to predict changes in a system as a function of time. They can be used at various scales from individual metabolites, to proteins, all the way to groups of cells.83 In general, concentration or abundance of each component in the system is tracked over time as they interact with one another. In dynamic models, every process in the system is described by a differential equation. Variables in the equations represent the time-varying parameters being modeled. Coefficients in the equations define the type (e.g. positive or negative) and strength of each interaction. The classical example of such a model is the Lotka-Volterra predator-prey system.84 In this system, two subpopulations exist, the predator and the prey. The predator consumes the prey, which leads to depletion of the prey population. A significant depletion of the prey population leads to starvation and decline of the predator population. When the predator population is low, the prey population is then able to thrive, thereby bringing the ecosystem through cycles of boom and bust. The dynamic model is able to capture the expected phasic oscillation in abundance of predator and prey subpopulations and determine parameters in which such associations may exist (Fig. 4a).85 This model can be scaled to whole populations as long as proper assumptions are made (e.g. linear vs. nonlinear parameter relationships). For example, dynamic models have been successfully applied to study macro-scale systems such as freshwater lake ecosystems.86 These models also enable perturbation studies where starting conditions (such as population size) can be varied, and solutions are obtained. The largest limitation to these models is that analytical solutions for most nonlinear differential equations with more than two variables are not readily available. Numerical solutions require additional mathematical and computational tools that need to be further developed. Nonetheless, these models are helpful for us to develop first order intuition about the dynamics of the system.Stoichiometric metabolic models
Stoichiometric models have been developed to study metabolism at the cellular level.87 These models describe metabolism of individual cells using matrices containing stoichiometric coefficients of all metabolic reactions and sets of optimization constraints. Stoichiometric representation of metabolism can be analyzed by various approaches88,89 such as Flux Balance Analysis (FBA).90 In contrast to dynamic models, FBA assumes that the system is at steady state such that all metabolite concentrations are time-invariant. This assumption is likely valid for cells grown in exponential phase.91 The solution to the system is described by a series of steady state fluxes for each reaction. By combining all possible fluxes, we can generate a multidimensional flux space that describes the entire metabolic capacity of the cell (Fig. 4b). An objective statement is used to define a given flux or criterion, such as flux to biomass (approximating growth rate), for which the multidimensional flux space can be optimized. Through linear optimization, the model predicts metabolic fluxes that maximize the objective function (e.g. biomass). This model has been extensively applied to in silico metabolic reconstruction of a variety of organisms.18–22 Stolyar et al. used a FBA model to describe a methanogenic community of M. maripaludis and D. vulgaris that exchanged metabolites hydrogen and formate.92 The metabolisms of the two strains are divided into two separate compartments that exchange metabolites via a third common compartment. This model successfully predicted the ratio of M. maripaludis to D. vulgaris during growth and suggested that hydrogen was essential for syntrophy while formate could be removed from the co-culture interaction.92Two developments have greatly improved stoichiometric models of microbial communities: the application of multi-level objective statements,87,93 and inclusion of dynamics.94 Multi-level objective statements can be formulated to describe different and potentially competing flux conditions. This approach has been used to model synthetic ecosystems of three or more members, where objective statements are defined separately for both the strain and the community.93 By simultaneously optimizing these objective functions, the model captures the selective forces that act on individuals and the community. For example, growth of individual species can be sacrificed to promote maximal community growth.93 Thus, models with multi-level objectives more accurately describe metabolite exchange. To account for dynamics in the system, population abundance and metabolite concentrations can be separated into different FBA models and solved independently at every time step in an approach called dynamic multi-species metabolic modeling (DMMM).95 As substrate concentrations change over time, DMMM is able to adjust the substrate utilization mode of each strain to the present conditions by switching to the appropriate stoichiometric matrix. This method is able to capture scenarios of resource competition and identify metabolites whose limited exchange affect population dynamics.95 These and other stoichiometric models, such as elementary mode analysis (EMA),96 enable full-scale quantitative models of ecosystems that are predictive and important for forward engineering.
Evolutionary game models
In contrast to dynamic and metabolic models, evolutionary game models focus on describing strategic decision-making of interacting agents and successfulness of their strategies (Fig. 4c).97 Rules of the evolutionary game define the payout that each player receives for every possible combination of strategies (e.g. cooperate, cheat). Each player’s payout represents the individual's fitness, and the highest value “wins” the game. For example, microbial phenotypes can often be described as altruistic (A) or selfish (S); evolutionary games can model how such behaviors arise.97 While we would assume that selfish exploitation of the environment may be a winning strategy, the natural world is paradoxically filled with organisms that exhibit cooperative behavior.98 For microbial communities, the fitness of every individual in a population is determined by the net payout from all pairwise games with all other individuals. The initial proportion of individuals adopting a given strategy is an input for this model. These games are then iterated over time with a given strategy changing in abundance based on the fitness of individuals who hold the strategy compared to the average population fitness. As the marginal cost of cooperating and benefit of cheating lead to changing payouts, the two strategies will dynamically vary and affect the outcome of the game.99 From these models, we find that populations that are dominated by altruists will often have a higher fitness than those dominated by selfish exploiters.100For microbial ecosystems, evolutionary game theory models allow us to investigate how system parameters impact microbial interactions and dynamics of competing strategies. These models have been used to predict the evolutionary steady state of engineered yeast populations that exhibit altruistic or selfish strategies through the snowdrift game.101 In such a game, the altruists secrete an invertase enzyme that hydrolyses a polysaccharide to generate diffusible glucose products that are available to the entire population. The selfish individuals forgo the cost of secreting the enzyme, but rely on the glucose generated by the altruistic strains. Modulating the cost of cooperation resulted in shifts in the final population structure. Altruists dominated when cost of cooperation was very low. Altruists and cheaters coexisted at median costs of cooperation, while cheaters dominated at high costs.101 To further take into account spatial structures, agent-based game models are used to restrict interactions to individuals in close physical proximity.102 Clusters of cells that exhibit cooperative strategies will derive more benefit due to spatial confinement, and thus will be further enriched in the population. These and other evolutionary game models100 will be important quantitative tools to guide ecosystem engineering.
Digital evolution
Long-term bacterial evolution experiments have been used to track how phenotypes and genotypes change in a constantly selective environment.103 Similarly, in silico simulations of evolution have been developed (Fig. 4d).104 Earlier forms of these simulations derive from cellular automata approaches, such as the Game of Life.105 Cells in the cellular automata live in a two-dimensional environment. Reproductive success or cell death is governed by the density and configuration of the local population. Discrete time steps are iterated over the population to simulate the process of life. A more sophisticated implementation of digital evolution, called Avida, has been described.106 Avida is inspired by an earlier system Tierra, in which digital organisms contain computer programs that compete for Central Processing Units (CPUs) and access to memory in order to reproduce.107 In Avida, digital organisms have their own memory space and virtual CPUs to perform tasks.108 Each digital organism has a circular “genome” composed of a collection of 26 possible discrete basic programs (Nand, IO, swap etc.) that are executed in series. When certain combinations of these basic programs are executed in the correct order, one of several logic operations is performed. Strains able to execute higher complexity operations are rewarded with more energy and therefore replicate faster. As cells replicate, mutations are introduced, which result in programs being added, removed, or moved. This leads to new operational capabilities. Because the history of each organism’s genotype and phenotype are chronicled, digital evolution models enable better understanding of how individuals traverse a fitness landscape as complex traits evolve. These artificial life models also enable the reversion of individual and combinations of mutations to study epistasis. Key conclusions106 reinforced by these models include: (1) deleterious mutations may be needed to develop complex traits; (2) even though complex traits are fragile to mutations, they fix in the population because they provide significant fitness benefit, and (3) development of complexity requires selection of traits with intermediate complexity to allow gradual transition through the fitness landscape. Since complex phenotypes are a hallmark feature of microbes, this framework will likely provide useful insights to improve engineering of ecosystems through digital simulations. These approaches are now being extended to simulation population-level behavior.109,110Experimental tools
Over the last decade, the field of microbial ecology has been swept by a wave of new technologies, significantly reshaping the traditional investigative approach. These advances have centered on key developments in microfabrication, high-throughput sequencing, genome engineering, and synthetic circuit design. These new methods allow for better in vitro and in vivo models, culture-independent identification and quantification of individual species across populations, and generation of targeted genotypes for functional studies (Fig. 5). Forward engineering of synthetic microbial ecosystems will rely heavily on these techniques.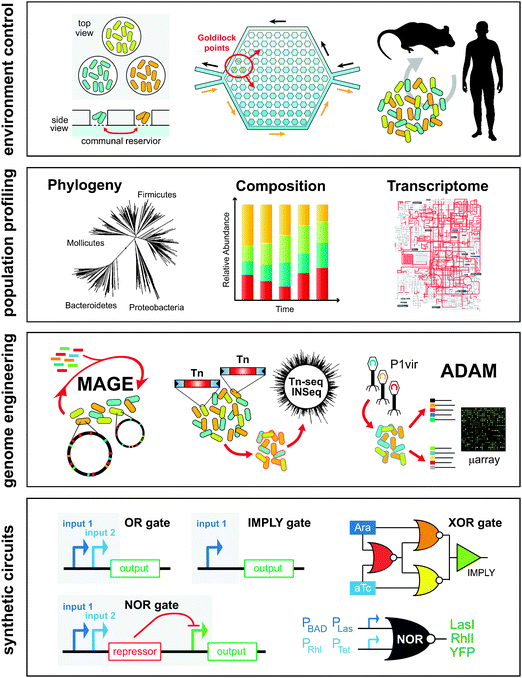 | ||
| Fig. 5 Experimental tools enable engineering of microbial ecosystems from the population level down to the DNA level. In vitro tools such microfluidics and microchambers or in vivo mice models enable precise control of the environment. High-throughput sequencing and transcriptomics enable parallel interrogation of phylogeny, composition, and gene expression of cell populations. Techniques such as multiplexed genome engineering and transposon mutagenesis enable forward engineering and accelerated evolution of cell populations at the genetic level. New genetic circuitry and synthetic biology frameworks enable the development of multi-component genetic programs that are executed across populations of cells. | ||
In vitro models
Going beyond traditional cultivation techniques using petri dishes and culture flasks, advances in microfabrication and microfluidics have produced a variety of cheap lab-chip devices that can be used to cultivate and analyze microbes grown in massively parallel micron-sized chambers and channels.111,112 These devices are particularly useful for generating physicochemical conditions found in heterogeneous ecological niches to study behaviors such as quorum sensing or antibiotic susceptibility. For example, Zhang et al. developed a microfluidic chip that contained 1200 interconnected wells to probe the development of ciprofloxacin antibiotic resistance.113 Local antibiotic gradients generated “Goldilocks points” in the microchamber where motile strains gathered and developed notable ciprofloxacin resistance (10 mg ml−1) – 200 times the minimum inhibitory concentration. This phenomenon was not observed in the absence of such antibiotic gradients when grown in standard flasks as no resistance strains developed. This work highlights the importance of local heterogeneity in the evolution of microbial populations and development of antibiotic resistance.Microfluidic chambers can also be used to study chemical signaling and nutritional cross-feeding between different microbes. Kim et al. developed a fluidic chip that contained arrays of spatially separated micro-wells with selectively permeable bottoms placed over a common liquid reservoir.114 Through size exclusion, metabolites could diffuse to neighboring wells while the bacteria producing them remained in each well. Using this system, the authors built a synthetic consortium of three bacteria, Azotobacter vinelandii, Bacillus licheniformis, and Paenibacillus curdlanolyticus, which normally do not grow together in nature. In a defined environment that is nitrogen and carbon depleted, and in the presence of antibiotics, the consortium exhibited reciprocal syntrophy because each species performed a specialized function that benefited the entire group. A. vinelandii fixed gaseous nitrogen into amino acids. B. licheniformis degraded the antibiotic penicillin. P. curdlanolyticus generated carbon sources needed by the consortium by degrading carboxymethyl-cellulose. In this co-culture, spatial structures and local interactions amongst the members defined the viability of the ecosystem. These interactions can be further elucidated at the single-cell level by using agarose tracks in channels that are the width of one cell.115,116 Through optical microscopy, growth of individual cells by linear extension along the channel can be tracked over 40 generations. Syntrophic exchange between strains of E. coli auxotrophic for different amino acids enabled growth in separate parallel channels.115 Highlighting the importance of locality in syntrophic exchange, the co-culture growth rate was shown to decrease sharply when the distance between complementary strains in neighboring channels increased by more than a few cell lengths.
In addition to microchambers and microchannels, microdroplet technology is also useful in probing interspecies interactions.117 Groups of cells can be encapsulated in monodispersed aqueous-phase droplets using a T-junction microfluidic channel with an oil-phase. Through syntrophic cross-feeding, auxotrophic E. coli strains can grow in these microdroplets and be analyzed by microscopy.117 These approaches will improve cultivation of new microbes by recapitulating microenvironments in which otherwise unculturable microbes can grow in the presence of metabolically compatible partners.
In vivo models
Experimental models that recapitulate natural environments lend crucial insights into structure and function of microbial communities in their native habitats. Tractable live animal models, such as gnotobiotic germ-free (GF) mice, have been used extensively to investigate the relationship between the mammalian gut and the resident microbial community.118 Gnotobiotic mice can be inoculated with defined and sequenced microbes that are trackable to investigate processes of gut colonization, food metabolism, and community stability. In one such recent study, Faith et al. introduced 10 representative strains of the human microbiota into GF mice that are fed with defined diets of macronutrients.119 Four classes of foods were given to mice: proteins, fats, polysaccharides, and sugars. The 10-member microbial consortium was tracked by analysis of faecal samples after transition to different diets. The researchers found that a simple linear model could predict over 60% of the variation in species abundance due to diet perturbations. The use of synthetic microbial communities in live animal models provides a feasible way to untangle the web of complex interactions that may go on in the population. Furthermore, in vivo mice models are amenable to genetic modifications to produce important disease phenotypes such as ob/ob120 or Tlr2(-/-),121 which can be used to tease out host-microbe interactions.Simple evolutionary models of antibiotic antagonism, such as the classic non-transitive rock–paper–scissors (RPS) game, have also been demonstrated by studying engineered E. coli strains in GF-mice. Kirkup and Riley122 used three types of strains: one that produces bactericidal colcins (P) that preferentially kill off sensitive strains (S) versus resistant strains (R). Sensitive strains can outcompete resistant strains, which in turn can outcompete colcin-producing strains. GF-mice associated with the microbial consortium showed cycling between the three phenotypes, which illustrated the RPS model and the in vivo role of colcin as an antibiotic. More interestingly this synthetic consortium model suggests that antibiotic-mediated antagonism can serve to promote microbial diversity in the mammalian gut.
Population quantification techniques
Precipitous reduction in cost and exponential growth in throughput of next-generation DNA sequencing technologies have revolutionized molecular biology.123 Sequencing has been used extensively for cataloging the composition, abundance, and metabolic potential of microbes from a variety of natural environments such as soil,124 ocean,125 acid mines,126 and the human body.127 Molecular barcoding allows large numbers of samples to be multiplexed and can be combined with time-series measurements to capture temporal changes across the entire population.128,129 Furthermore, transcriptome sequencing methods such as RNA-seq allow us to measure detailed transcriptional profiles of consortium members under different environmental conditions.130 Resequencing genomes from long-term evolution studies have also increased in popularity.103 These investigations help to identify genetic mutations that arise due to adaptation to new environments131,132 and help to reveal genetic heterogeneity within the population.133 Goodarzi et al. developed the genetic footprinting technique, array-based discovery of adaptive mutations (ADAM), which enabled selective identification of mutations that provide a competitive advantage within a cell population.134 Combining sequencing and functional measurements, this method reconstructs beneficial phenotypes to increase the scope of adaptive lab evolution studies and enhance understanding of genetic interactions in complex populations.Genome engineering
Construction and engineering of sophisticated synthetic ecosystems require facile modification of microbial genomes. Transposable elements have long been used as an efficient way to produce mutants of various phenotypes by random insertion into the genome.135 Libraries of such transposon-mutated strains diverge in genotype and phenotype, but when pooled together can begin to resemble a microbial consortium. Using high-throughput DNA sequencing, large libraries of transposon mutants can be interrogated efficient. Goodman et al. combined the use of transposon mutagenesis, high-throughput sequencing and gnotobiotic mice in a technique called Insertion Sequencing (IN-Seq) to probe the function of Bacteroides thetaiotaomicron in the mouse gut.136 Populations of B. thetaiotaomicron cells that were mutated by Himar1 mariner transposons were assessed by Illumina sequencing. The modified Himar1 inverted repeat sites contained MmeI-compatible sequences. Upon MmeI digestion of genomic DNA from the mutant population, high-throughput sequencing can be used to determine two 18-bp pairwise genomic fragments that correspond to the transposon insertion. Abundance levels of each mutant can be tracked and distinguished from one another, as well as from defined microbes in other phylum such as Firmicutes or Actinobacteria. Other similar techniques for high-throughput transposon sequencing include Tn-seq,137 high-throughput insertion tracking by deep sequencing (HITS)138 and transposon-directed insertion-site sequencing (TraDIS)139 have also been developed.Often, engineering members of a synthetic consortium requires precise genetic manipulation of the genome instead of random mutagenesis. Recent advances in oligo-mediated genomic engineering such as Multiplex Automated Genome Engineering (MAGE) has enabled efficient, parallel, and site-specific modification of genomes across many target sites.140–142 By using pools of oligos, MAGE can generate genetic diversity in the population at a rate of 4.3 × 109 modified bases per day, which enables combinatorial generation of divergent and complementary phenotypes within population clades.140 MAGE relies on the transformation of small chemically synthesized oligonucleotides (∼50–90 bp) into the genome that then proceed to integrate into the chromosome during replication in an Okazaki-like fashion. Single-stranded DNA binding proteins and recombinases greatly facilitate this process and are often found as a part of viral integration machinery.143 Rapid generation of cells that exhibit a variety of physiologies is not only feasible but can be automated. Therefore, these approaches are crucial to the construction of viable and stable synthetic communities. Oligo-mediated genomic engineering has shown promise in a variety of organisms including Escherichia coli,144Pseudomonas syringae,145Pantoea ananatis,146 and other gram-negative bacteria,147 as well as Mycobacterium tuberculosis,148 lactic acid bacteria,149 and yeast.150
Synthetic computing circuits
Construction of genetic circuits that perform computational operations has been a long-standing goal in synthetic biology.151 Recent advances in genetic circuit design have now been extended to libraries of cells, which can be modularly combined to perform basic logic functions. Earlier work demonstrated that population-level behavior can be programmed using feedback genetic circuits and quorum sensing molecules152,153 but needed precise population-synchronization for robust behavior.154 More recently, two groups developed multicellular computing systems.155,156 Regot et al. constructed a library of engineered yeast cell-types that could sense different extracellular input signals such as NaCl, doxycycline, galactose, oestradiol and produce chemical ‘wiring molecules’ such as pheromones to communicate with one another.155 These cell-types were made into AND and inverted IMPLIES logic functions to implement Boolean operations. For example, Cell 1 when presented with an input such as NaCl, will produce the wire molecule, pheromone, which is received by Cell 2. Cell 2 will produce a detectable fluorescence output only when it senses the pheromone and a second input such as oestradiol. The NaCl AND oestradiol operation is achieved with this two-cell implementation. By combining different cell-types, the authors generated a variety of logic gates (AND, NOR, OR, NAND, XNOR, XOR). More impressively, complex circuits including a multiplexer and a 1-bit adder with carry were built using additional chemical wires and cell-types. Based on a similar design scheme, Tamsir et al. constructed libraries of E. coli cells with simple NOR logic gates and connected them using quorum sensing molecules.156 The NOR gate was built using two tandem promoters that served as orthogonal inputs to drive the transcription of a repressor element. This simple implementation was used to build more complex circuits, which the authors demonstrated by performing logic operations on solid plates with different spatially defined colony types.156 These results support the notion that cellular consortia may be used to perform complex tasks more efficiently than single-cell implementations, further advocating the development of synthetic consortia as a platform technology.Applications of synthetic consortia
Microbial consortia can potentially be programmed to perform useful tasks in both natural and artificial environments at spatial and temporal scales well beyond the capabilities of any individual member. Numerous applications may warrant such systems, ranging both in sophistication and in scale. Engineered microbes have long been used for industrial production of chemicals and pharmaceuticals.157 These reactions tend to occur in fermentation chambers using genetically identical strains. All multi-step reactions need to be carried out intracellularly or would require separate fermentation pipelines. For complex feedstocks such as cellulosic biomass, single-strain fermentation reactions are unlikely to suffice. On the other hand multi-species communities can degrade these complex substrates efficiently.158 Thus, future microbial fermentation systems are likely to shift to more heterogeneous population of engineered strains with diversified metabolic capabilities.159Engineered consortia can be designed to degrade complex feedstock while simultaneously producing valued products. Using a symbiotic co-culture of engineered yeasts and Actinotalea fermentans, a cellulolytic bacterium, Bayer et al. were able to convert unprocessed switchgrass, corn stover, sugar cane bagasse, and poplar into methyl halide, a biofuel precursor.160A. fermentans fermented cellulose to acetate and ethanol, but its growth was inhibited by these toxic waste products. However, engineered yeast was used to reduce acetate level by utilizing it for energy to produce methyl halide through heterologous expression of a methyl halide transferase. Thus, interdependence was established between the two strains to alleviate growth inhibition toward production of a biofuel. This type of division of labor is a powerful approach for processing complex substrates – a strategy commonly adopted in natural microbial consortia.161
Applications in coordinated toxin detection and bioremediation may also benefit from synthetic consortia. By engineering auto-synchronization in populations of oscillating cells, Prindle et al. developed a liquid crystal display (LCD)-like macroscopic clock that could sense arsenic concentrations and respond by changing the oscillatory period.51 The researchers nested two modes of cell signaling to expand the scale at which coordinated events manifest across the population. Slower local synchronization proceeded via a well-established quorum sensing genetic circuit to form colonies called “biopixels.” Arrays of these small colonies were synchronized across a large scale with a weaker but faster redox signaling system using hydrogen peroxide. Using an extra positive-feedback element that was linked to an arsenic-responsive promoter, the oscillatory system became a macroscopic arsenic biosensor that fluoresced at different periods depending on the arsenic concentration. By combining the two modes of cellular communication across thousands of microwell channels, the authors developed a proof-of-principle biochip that may potentially be used as a handheld arsenic detector.
For applications in medical therapeutics, engineered microbial gut consortia will likely be an important area of development. Recent studies have highlighted the important role of human-associated microbial communities in maintaining health and causing diseases,162–164 especially in the gastrointestinal (GI) tract where food and drugs are metabolized. The gut environment is home to the highest density of microbes in the body (up to 1011 cells/gram) and irregularities in the microbial composition are linked to diseases including Crohn’s,165,166 inflammatory bowel disease,167 obesity,26 diabetes,168 infections,169 and maldigestion.170 Traditional therapeutic strategies using probiotics have failed to generate consistent results largely due to a lack of understanding for the design principles needed to maintain engineered microbes in vivo. New approaches in synthetic consortia engineering will likely succeed where previous attempts have failed. Few successes in this area are already encouraging. Steidler et al. engineered an orally administered Lactococcus lactis strain that excreted human interleukin-10 in the GI tract.171 This engineered probiotic strain reduced the degree of induced colitis in mice models, paving the way for human clinical trials for IBD.172 Saeidi et al. showed that engineered E. coli could detect the human pathogen Pseudomonas aeruginosa via a quorum sensing pathway.173P. aeruginosa often colonize the respiratory and GI tracts, leading to chronic and fatal diseases. Upon pathogen detection, the programmed E. coli self-lyse and release pyocin, a narrow-spectrum bacteriocin that kills P. aeruginosa. In another study,174 the administration of non-pathogenic engineered E. coli that communicate to coordinate the secretion of cholera quorum sensing molecules (i.e. autoinducer-1) resulted in increased survival of murine models to Vibrio cholera infection from 0% to >92%. Future applications of human-microbiome engineering may include enhancing catabolism of troublesome but common metabolites (e.g. lactose and gluten), precise microbial modulation of the immune system, and removal of multi-drug resistant pathogens by selective toxin release.
Concluding remarks
The prospect is bright for synthetic biologists to build ecosystem that reproducibly exhibit complex behavior. Yet there remain many challenges ahead that reflect our incomplete understanding of the many governing principles that underlie microbial physiology, ecology, and evolution. A better working knowledge of the different parameters that drive social interaction in cell populations will be needed. As most intercellular interactions exhibit non-linear relationships based on spatial, temporal, thermodynamic, and energetic constraints, we expect that new theoretical frameworks need to be developed to describe these complex, dynamic, and heterogeneous ecosystems. New techniques that facilitate massively parallel synthesis, engineering, and analysis of microbial consortia at single-cell resolution will be critical for predictive programming of synthetic communities. As we progress toward engineering biological systems of ever-increasing sophistication, social and ethical concerns surrounding the creation of non-natural life forms and ecosystems will require open dialogue between researchers and the public on the risks and rewards of these activities in the post-Darwinian era of biology.Acknowledgements
H.H.W. acknowledges funding from the Wyss Institute Technology Development Fellowship and the National Institutes of Health Director's Early Independence Award (grant 1DP5OD009172-01).References
- M. Achtman and M. Wagner, Nat. Rev. Microbiol., 2008, 6, 431–440 CAS.
- P. D. Schloss and J. Handelsman, Microbiol. Mol. Biol. Rev., 2004, 68, 686–691 CrossRef.
- P. G. Falkowski, T. Fenchel and E. F. Delong, Science, 2008, 320, 1034–1039 CrossRef CAS.
- C. Fraser, E. J. Alm, M. F. Polz, B. G. Spratt and W. P. Hanage, Science, 2009, 323, 741–746 CrossRef CAS.
- N. Klitgord and D. Segre, Curr. Opin. Biotechnol., 2011, 22, 541–546 CrossRef CAS.
- M. E. Hibbing, C. Fuqua, M. R. Parsek and S. B. Peterson, Nat. Rev. Microbiol., 2010, 8, 15–25 CrossRef CAS.
- M. J. Pocock, D. M. Evans and J. Memmott, Science, 2012, 335, 973–977 CrossRef CAS.
- K. R. Foster, Nat. Rev. Genet., 2011, 12, 193–203 CrossRef CAS.
- I. Ispolatov, M. Ackermann and M. Doebeli, Proc. R. Soc. London, Ser. B, 2012, 279, 1768–1776 CrossRef.
- D. Endy, Nature, 2005, 438, 449–453 CrossRef CAS.
- A. S. Khalil and J. J. Collins, Nat. Rev. Genet., 2010, 11, 367–379 CrossRef CAS.
- B. Canton, A. Labno and D. Endy, Nat. Biotechnol., 2008, 26, 787–793 CrossRef CAS.
- M. H. Medema, R. van Raaphorst, E. Takano and R. Breitling, Nat. Rev. Microbiol., 2012, 10, 191–202 CrossRef CAS.
- P. A. Carr and G. M. Church, Nat. Biotechnol., 2009, 27, 1151–1162 CrossRef CAS.
- K. M. Muller and K. M. Arndt, Methods Mol. Biol., 2012, 813, 23–43 Search PubMed.
- M. Kanehisa, S. Goto, Y. Sato, M. Furumichi and M. Tanabe, Nucleic Acids Res., 2012, 40, D109–D114 CrossRef CAS.
- R. Caspi, T. Altman, K. Dreher, C. A. Fulcher, P. Subhraveti, I. M. Keseler, A. Kothari, M. Krummenacker, M. Latendresse, L. A. Mueller, Q. Ong, S. Paley, A. Pujar, A. G. Shearer, M. Travers, D. Weerasinghe, P. Zhang and P. D. Karp, Nucleic Acids Res., 2012, 40, D742–D753 CrossRef CAS.
- Y. K. Oh, B. O. Palsson, S. M. Park, C. H. Schilling and R. Mahadevan, J. Biol. Chem., 2007, 282, 28791–28799 CrossRef CAS.
- J. L. Reed, T. D. Vo, C. H. Schilling and B. O. Palsson, GenomeBiology, 2003, 4, R54 CrossRef.
- C. H. Schilling, M. W. Covert, I. Famili, G. M. Church, J. S. Edwards and B. O. Palsson, J. Bacteriol., 2002, 184, 4582–4593 CrossRef CAS.
- N. C. Duarte, M. J. Herrgard and B. O. Palsson, Genome Res., 2004, 14, 1298–1309 CrossRef CAS.
- N. C. Duarte, S. A. Becker, N. Jamshidi, I. Thiele, M. L. Mo, T. D. Vo, R. Srivas and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1777–1782 CrossRef CAS.
- C. R. Woese and G. E. Fox, Proc. Natl. Acad. Sci. U. S. A., 1977, 74, 5088–5090 CrossRef CAS.
- C. Burke, P. Steinberg, D. Rusch, S. Kjelleberg and T. Thomas, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 14288–14293 CrossRef CAS.
- A. Fernandez, S. Huang, S. Seston, J. Xing, R. Hickey, C. Criddle and J. Tiedje, Appl. Environ. Microbiol., 1999, 65, 3697–3704 CAS.
- P. J. Turnbaugh, M. Hamady, T. Yatsunenko, B. L. Cantarel, A. Duncan, R. E. Ley, M. L. Sogin, W. J. Jones, B. A. Roe, J. P. Affourtit, M. Egholm, B. Henrissat, A. C. Heath, R. Knight and J. I. Gordon, Nature, 2009, 457, 480–484 CrossRef CAS.
- T. R. Zuroff and W. R. Curtis, Appl. Microbiol. Biotechnol., 2012, 93, 1423–1435 CrossRef CAS.
- C. S. Goh, T. A. Gianoulis, Y. Liu, J. Li, A. Paccanaro, Y. A. Lussier and M. Gerstein, BMC Genomics, 2006, 7, 257 CrossRef.
- B. R. Bochner, P. Gadzinski and E. Panomitros, Genome Res., 2001, 11, 1246–1255 CrossRef CAS.
- M. H. Saier, Jr., Microbiol. Mol. Biol. Rev., 2000, 64, 354–411 CrossRef CAS.
- E. L. Borths, B. Poolman, R. N. Hvorup, K. P. Locher and D. C. Rees, Biochemistry, 2005, 44, 16301–16309 CrossRef CAS.
- J. S. Patzlaff, T. van der Heide and B. Poolman, J. Biol. Chem., 2003, 278, 29546–29551 CrossRef CAS.
- X. Z. Li and H. Nikaido, Drugs, 2009, 69, 1555–1623 CrossRef CAS.
- A. Burkovski and R. Kramer, Appl. Microbiol. Biotechnol., 2002, 58, 265–274 CrossRef CAS.
- I. Franke, A. Resch, T. Dassler, T. Maier and A. Bock, J. Bacteriol., 2003, 185, 1161–1166 CrossRef CAS.
- H. Cruz-Ramos, G. M. Cook, G. Wu, M. W. Cleeter and R. K. Poole, Microbiology, 2004, 150, 3415–3427 CrossRef CAS.
- V. Doroshenko, L. Airich, M. Vitushkina, A. Kolokolova, V. Livshits and S. Mashko, FEMS Microbiol. Lett., 2007, 275, 312–318 CrossRef CAS.
- E. Peeters, P. Nguyen Le Minh, M. Foulquie-Moreno and D. Charlier, Mol. Microbiol., 2009, 74, 1513–1526 CrossRef CAS.
- L. Eggeling and H. Sahm, Arch. Microbiol., 2003, 180, 155–160 CrossRef CAS.
- E. A. Kutukova, V. A. Livshits, I. P. Altman, L. R. Ptitsyn, M. H. Zyiatdinov, I. L. Tokmakova and N. P. Zakataeva, FEBS Lett., 2005, 579, 4629–4634 CrossRef CAS.
- G. P. Dubey and S. Ben-Yehuda, Cell, 2011, 144, 590–600 CrossRef CAS.
- C. S. Hayes, S. K. Aoki and D. A. Low, Annu. Rev. Genet., 2010, 44, 71–90 CrossRef CAS.
- B. L. Bassler and R. Losick, Cell, 2006, 125, 237–246 CrossRef CAS.
- H. H. Lee, M. N. Molla, C. R. Cantor and J. J. Collins, Nature, 2010, 467, 82–85 CrossRef CAS.
- N. M. Vega, K. R. Allison, A. S. Khalil and J. J. Collins, Nat. Chem. Biol., 2012, 8, 431–433 CrossRef CAS.
- C. M. Rath and P. C. Dorrestein, Curr. Opin. Microbiol., 2011 Search PubMed.
- P. D. Straight and R. Kolter, Annu. Rev. Microbiol., 2009, 63, 99–118 CrossRef CAS.
- E. A. Shank and R. Kolter, Curr. Opin. Microbiol., 2011, 14, 741–747 CrossRef CAS.
- S. Basu, Y. Gerchman, C. H. Collins, F. H. Arnold and R. Weiss, Nature, 2005, 434, 1130–1134 CrossRef CAS.
- J. J. Tabor, H. M. Salis, Z. B. Simpson, A. A. Chevalier, A. Levskaya, E. M. Marcotte, C. A. Voigt and A. D. Ellington, Cell, 2009, 137, 1272–1281 CrossRef.
- A. Prindle, P. Samayoa, I. Razinkov, T. Danino, L. S. Tsimring and J. Hasty, Nature, 2012, 481, 39–44 CrossRef CAS.
- L. Hall-Stoodley, J. W. Costerton and P. Stoodley, Nat. Rev. Microbiol., 2004, 2, 95–108 CrossRef CAS.
- L. Hall-Stoodley and P. Stoodley, Curr. Opin. Biotechnol., 2002, 13, 228–233 CrossRef CAS.
- W. Harcombe, Evolution, 2010, 64, 2166–2172 Search PubMed.
- C. A. Fux, J. W. Costerton, P. S. Stewart and P. Stoodley, Trends Microbiol., 2005, 13, 34–40 CrossRef CAS.
- K. Brenner and F. H. Arnold, PLoS One, 2011, 6, e16791 CAS.
- E. T. Kiers, M. Duhamel, Y. Beesetty, J. A. Mensah, O. Franken, E. Verbruggen, C. R. Fellbaum, G. A. Kowalchuk, M. M. Hart, A. Bago, T. M. Palmer, S. A. West, P. Vandenkoornhuyse, J. Jansa and H. Bucking, Science, 2011, 333, 880–882 CrossRef CAS.
- E. G. Ruby, Nat. Rev. Microbiol., 2008, 6, 752–762 CrossRef CAS.
- J. P. McCutcheon and N. A. Moran, Nat. Rev. Microbiol., 2012, 10, 13–26 CAS.
- L. Margulis, Am. Sci., 1971, 59, 230–235 CAS.
- J. P. McCutcheon and C. D. von Dohlen, Curr. Biol., 2011, 21, 1366–1372 CrossRef CAS.
- A. K. Hansen and N. A. Moran, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 2849–2854 CrossRef CAS.
- J. P. McCutcheon, B. R. McDonald and N. A. Moran, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 15394–15399 CrossRef CAS.
- S. D. Dyall, M. T. Brown and P. J. Johnson, Science, 2004, 304, 253–257 CrossRef CAS.
- V. Burrus and M. K. Waldor, Res. Microbiol., 2004, 155, 376–386 CrossRef CAS.
- L. S. Frost, R. Leplae, A. O. Summers and A. Toussaint, Nat. Rev. Microbiol., 2005, 3, 722–732 CrossRef CAS.
- J. P. Gogarten and J. P. Townsend, Nat. Rev. Microbiol., 2005, 3, 679–687 CrossRef CAS.
- M. W. van Passel, P. R. Marri and H. Ochman, PLoS Comput. Biol., 2008, 4, e1000059 Search PubMed.
- C. S. Smillie, M. B. Smith, J. Friedman, O. X. Cordero, L. A. David and E. J. Alm, Nature, 2011, 480, 241–244 CrossRef CAS.
- D. R. Smith and M. R. Chapman, MBio, 2010, 1 Search PubMed.
- V. M. Markowitz, I. M. Chen, K. Palaniappan, K. Chu, E. Szeto, Y. Grechkin, A. Ratner, B. Jacob, J. Huang, P. Williams, M. Huntemann, I. Anderson, K. Mavromatis, N. N. Ivanova and N. C. Kyrpides, Nucleic Acids Res., 2012, 40, D115–D122 CrossRef CAS.
- R. L. Tatusov, E. V. Koonin and D. J. Lipman, Science, 1997, 278, 631–637 CrossRef CAS.
- T. N. Petersen, S. Brunak, G. von Heijne and H. Nielsen, Nat. Methods, 2011, 8, 785–786 CrossRef CAS.
- M. Punta, P. C. Coggill, R. Y. Eberhardt, J. Mistry, J. Tate, C. Boursnell, N. Pang, K. Forslund, G. Ceric, J. Clements, A. Heger, L. Holm, E. L. Sonnhammer, S. R. Eddy, A. Bateman and R. D. Finn, Nucleic Acids Res., 2012, 40, D290–D301 CrossRef CAS.
- S. Hunter, P. Jones, A. Mitchell, R. Apweiler, T. K. Attwood, A. Bateman, T. Bernard, D. Binns, P. Bork, S. Burge, E. de Castro, P. Coggill, M. Corbett, U. Das, L. Daugherty, L. Duquenne, R. D. Finn, M. Fraser, J. Gough, D. Haft, N. Hulo, D. Kahn, E. Kelly, I. Letunic, D. Lonsdale, R. Lopez, M. Madera, J. Maslen, C. McAnulla, J. McDowall, C. McMenamin, H. Mi, P. Mutowo-Muellenet, N. Mulder, D. Natale, C. Orengo, S. Pesseat, M. Punta, A. F. Quinn, C. Rivoire, A. Sangrador-Vegas, J. D. Selengut, C. J. Sigrist, M. Scheremetjew, J. Tate, M. Thimmajanarthanan, P. D. Thomas, C. H. Wu, C. Yeats and S. Y. Yong, Nucleic Acids Res., 2011, 40, D306–D312 CrossRef.
- H. Akashi and T. Gojobori, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 3695–3700 CrossRef CAS.
- K. Hosoda, S. Suzuki, Y. Yamauchi, Y. Shiroguchi, A. Kashiwagi, N. Ono, K. Mori and T. Yomo, PLoS One, 2011, 6, e17105 CAS.
- W. Shou, S. Ram and J. M. Vilar, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 1877–1882 CrossRef CAS.
- E. H. Wintermute and P. A. Silver, Mol. Syst. Biol., 2010, 6, 407 CrossRef.
- T. Koide, W. L. Pang and N. S. Baliga, Nat. Rev. Microbiol., 2009, 7, 297–305 CAS.
- J. Raes and P. Bork, Nat. Rev. Microbiol., 2008, 6, 693–699 CrossRef CAS.
- A. M. Feist, M. J. Herrgard, I. Thiele, J. L. Reed and B. O. Palsson, Nat. Rev. Microbiol., 2009, 7, 129–143 CAS.
- W. W. Chen, M. Niepel and P. K. Sorger, Genes Dev., 2010, 24, 1861–1875 CrossRef CAS.
- P. van den Ende, Science, 1973, 181, 562–564 CAS.
- F. K. Balagadde, H. Song, J. Ozaki, C. H. Collins, M. Barnet, F. H. Arnold, S. R. Quake and L. You, Mol. Syst. Biol., 2008, 4, 187 CrossRef.
- S. Sahasrabudhe and A. E. Motter, Nat. Commun., 2011, 2, 170 CrossRef.
- N. E. Lewis, H. Nagarajan and B. O. Palsson, Nat. Rev. Microbiol., 2012, 10, 291–305 CAS.
- N. D. Price, J. L. Reed, J. A. Papin, S. J. Wiback and B. O. Palsson, J. Theor. Biol., 2003, 225, 185–194 CrossRef CAS.
- J. A. Papin, J. Stelling, N. D. Price, S. Klamt, S. Schuster and B. O. Palsson, Trends Biotechnol., 2004, 22, 400–405 CrossRef CAS.
- C. H. Schilling and B. O. Palsson, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 4193–4198 CrossRef CAS.
- A. Varma and B. O. Palsson, Appl. Environ. Microbiol., 1994, 60, 3724–3731 CAS.
- S. Stolyar, S. Van Dien, K. L. Hillesland, N. Pinel, T. J. Lie, J. A. Leigh and D. A. Stahl, Mol. Syst. Biol., 2007, 3, 92 CrossRef.
- A. R. Zomorrodi and C. D. Maranas, PLoS Comput. Biol., 2012, 8, e1002363 CAS.
- R. Mahadevan, J. S. Edwards and F. J. Doyle,3rd, Biophys. J., 2002, 83, 1331–1340 CrossRef CAS.
- K. Zhuang, M. Izallalen, P. Mouser, H. Richter, C. Risso, R. Mahadevan and D. R. Lovley, ISME J., 2011, 5, 305–316 CrossRef.
- R. Taffs, J. E. Aston, K. Brileya, Z. Jay, C. G. Klatt, S. McGlynn, N. Mallette, S. Montross, R. Gerlach, W. P. Inskeep, D. M. Ward and R. P. Carlson, BMC Syst. Biol., 2009, 3, 114 CrossRef.
- S. A. West, A. S. Griffin, A. Gardner and S. P. Diggle, Nat. Rev. Microbiol., 2006, 4, 597–607 CrossRef CAS.
- J. L. Sachs, U. G. Mueller, T. P. Wilcox and J. J. Bull, Q. Rev. Biol., 2004, 79, 135–160 CrossRef.
- C. Hauert, F. Michor, M. A. Nowak and M. Doebeli, J. Theor. Biol., 2006, 239, 195–202 CrossRef.
- M. A. Nowak, Science, 2006, 314, 1560–1563 CrossRef CAS.
- J. Gore, H. Youk and A. van Oudenaarden, Nature, 2009, 459, 253–256 CrossRef CAS.
- C. D. Nadell, K. R. Foster and J. B. Xavier, PLoS Comput. Biol., 2010, 6, e1000716 Search PubMed.
- T. M. Conrad, N. E. Lewis and B. O. Palsson, Mol. Syst. Biol., 2011, 7, 509 CrossRef.
- C. O. Wilke, J. L. Wang, C. Ofria, R. E. Lenski and C. Adami, Nature, 2001, 412, 331–333 CrossRef CAS.
- M. Gardner, Sci. Am., 1970, 120–123 CrossRef.
- R. E. Lenski, C. Ofria, R. T. Pennock and C. Adami, Nature, 2003, 423, 139–144 CrossRef CAS.
- T. S. Ray, in Artificial Life II, 1992, pp. 371–408 Search PubMed.
- C. Ofria, C. T. Brown and C. Adami, Introduction to Artificial Life, 1998, 297–350 Search PubMed.
- S. S. Chow, C. O. Wilke, C. Ofria, R. E. Lenski and C. Adami, Science, 2004, 305, 84–86 CrossRef CAS.
- G. Yedid, C. A. Ofria and R. E. Lenski, J. Evol. Biol., 2008, 21, 1335–1357 CrossRef CAS.
- A. J. Link, K. J. Jeong and G. Georgiou, Nat. Rev. Microbiol., 2007, 5, 680–688 CrossRef CAS.
- F. K. Balagadde, L. You, C. L. Hansen, F. H. Arnold and S. R. Quake, Science, 2005, 309, 137–140 CrossRef CAS.
- Q. Zhang, G. Lambert, D. Liao, H. Kim, K. Robin, C. K. Tung, N. Pourmand and R. H. Austin, Science, 2011, 333, 1764–1767 CrossRef CAS.
- H. J. Kim, J. Q. Boedicker, J. W. Choi and R. F. Ismagilov, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 18188–18193 CrossRef CAS.
- J. R. Moffitt, J. B. Lee and P. Cluzel, Lab Chip, 2012, 12, 1487–1494 RSC.
- N. Q. Balaban, J. Merrin, R. Chait, L. Kowalik and S. Leibler, Science, 2004, 305, 1622–1625 CrossRef CAS.
- J. Park, A. Kerner, M. A. Burns and X. N. Lin, PLoS One, 2011, 6, e17019 CAS.
- J. J. Faith, F. E. Rey, D. O'Donnell, M. Karlsson, N. P. McNulty, G. Kallstrom, A. L. Goodman and J. I. Gordon, ISME J., 2010, 4, 1094–1098 CrossRef.
- J. J. Faith, N. P. McNulty, F. E. Rey and J. I. Gordon, Science, 2011, 333, 101–104 CrossRef CAS.
- R. E. Ley, F. Backhed, P. Turnbaugh, C. A. Lozupone, R. D. Knight and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11070–11075 CrossRef CAS.
- R. Kellermayer, S. E. Dowd, R. A. Harris, A. Balasa, T. D. Schaible, R. D. Wolcott, N. Tatevian, R. Szigeti, Z. Li, J. Versalovic and C. W. Smith, FASEB J., 2011, 25, 1449–1460 CrossRef CAS.
- B. C. Kirkup and M. A. Riley, Nature, 2004, 428, 412–414 CrossRef CAS.
- D. Medini, D. Serruto, J. Parkhill, D. A. Relman, C. Donati, R. Moxon, S. Falkow and R. Rappuoli, Nat. Rev. Microbiol., 2008, 6, 419–430 CAS.
- R. Mackelprang, M. P. Waldrop, K. M. DeAngelis, M. M. David, K. L. Chavarria, S. J. Blazewicz, E. M. Rubin and J. K. Jansson, Nature, 2011, 480, 368–371 CrossRef CAS.
- E. J. Biers, S. Sun and E. C. Howard, Appl. Environ. Microbiol., 2009, 75, 2221–2229 CrossRef CAS.
- S. L. Simmons, G. Dibartolo, V. J. Denef, D. S. Goltsman, M. P. Thelen and J. F. Banfield, PLoS Biol., 2008, 6, e177 Search PubMed.
- J. Peterson, S. Garges, M. Giovanni, P. McInnes, L. Wang, J. A. Schloss, V. Bonazzi, J. E. McEwen, K. A. Wetterstrand, C. Deal, C. C. Baker, V. Di Francesco, T. K. Howcroft, R. W. Karp, R. D. Lunsford, C. R. Wellington, T. Belachew, M. Wright, C. Giblin, H. David, M. Mills, R. Salomon, C. Mullins, B. Akolkar, L. Begg, C. Davis, L. Grandison, M. Humble, J. Khalsa, A. R. Little, H. Peavy, C. Pontzer, M. Portnoy, M. H. Sayre, P. Starke-Reed, S. Zakhari, J. Read, B. Watson and M. Guyer, Genome Res., 2009, 19, 2317–2323 CrossRef.
- D. MacLean, J. D. Jones and D. J. Studholme, Nat. Rev. Microbiol., 2009, 7, 287–296 Search PubMed.
- M. Hamady and R. Knight, Genome Res., 2009, 19, 1141–1152 CrossRef CAS.
- P. J. Turnbaugh, C. Quince, J. J. Faith, A. C. McHardy, T. Yatsunenko, F. Niazi, J. Affourtit, M. Egholm, B. Henrissat, R. Knight and J. I. Gordon, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7503–7508 CrossRef CAS.
- H. H. Chou, H. C. Chiu, N. F. Delaney, D. Segre and C. J. Marx, Science, 2011, 332, 1190–1192 CrossRef CAS.
- A. I. Khan, D. M. Dinh, D. Schneider, R. E. Lenski and T. F. Cooper, Science, 2011, 332, 1193–1196 CrossRef CAS.
- O. Tenaillon, A. Rodriguez-Verdugo, R. L. Gaut, P. McDonald, A. F. Bennett, A. D. Long and B. S. Gaut, Science, 2012, 335, 457–461 CrossRef CAS.
- H. Goodarzi, A. K. Hottes and S. Tavazoie, Nat. Methods, 2009, 6, 581–583 CrossRef CAS.
- F. Hayes, Annu. Rev. Genet., 2003, 37, 3–29 CrossRef CAS.
- A. L. Goodman, N. P. McNulty, Y. Zhao, D. Leip, R. D. Mitra, C. A. Lozupone, R. Knight and J. I. Gordon, Cell Host Microbe, 2009, 6, 279–289 CAS.
- T. van Opijnen, K. L. Bodi and A. Camilli, Nat. Methods, 2009, 6, 767–772 CrossRef CAS.
- J. D. Gawronski, S. M. Wong, G. Giannoukos, D. V. Ward and B. J. Akerley, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 16422–16427 CrossRef CAS.
- G. C. Langridge, M. D. Phan, D. J. Turner, T. T. Perkins, L. Parts, J. Haase, I. Charles, D. J. Maskell, S. E. Peters, G. Dougan, J. Wain, J. Parkhill and A. K. Turner, Genome Res., 2009, 19, 2308–2316 CrossRef CAS.
- H. H. Wang, F. J. Isaacs, P. A. Carr, Z. Z. Sun, G. Xu, C. R. Forest and G. M. Church, Nature, 2009, 460, 894–898 CrossRef CAS.
- H. H. Wang and G. M. Church, Methods Enzymol., 2011, 498, 409–426 CAS.
- H. Wang, H. B. Kim, L. Cong, D. Bang and G. M. Church, Nat. Methods, 2012, 9, 591–593 CrossRef CAS.
- S. Datta, N. Costantino, X. Zhou and D. L. Court, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1626–1631 CrossRef CAS.
- H. M. Ellis, D. Yu, T. DiTizio and D. L. Court, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 6742–6746 CrossRef CAS.
- B. Swingle, Z. Bao, E. Markel, A. Chambers and S. Cartinhour, Appl. Environ. Microbiol., 2010, 76, 4960–4968 CrossRef CAS.
- J. I. Katashkina, Y. Hara, L. I. Golubeva, I. G. Andreeva, T. M. Kuvaeva and S. V. Mashko, BMC Mol. Biol., 2009, 10, 34 CrossRef.
- B. Swingle, E. Markel, N. Costantino, M. G. Bubunenko, S. Cartinhour and D. L. Court, Mol. Microbiol., 2010, 75, 138–148 CrossRef CAS.
- J. C. van Kessel and G. F. Hatfull, Nat. Methods, 2007, 4, 147–152 CrossRef CAS.
- J. P. van Pijkeren and R. A. Britton, Nucleic Acids. Res., 2012 Search PubMed.
- Y. W. Kow, G. Bao, J. W. Reeves, S. Jinks-Robertson and G. F. Crouse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 11352–11357 CrossRef CAS.
- T. K. Lu, A. S. Khalil and J. J. Collins, Nat. Biotechnol., 2009, 27, 1139–1150 CrossRef CAS.
- L. You, R. S. Cox, 3rd, R. Weiss and F. H. Arnold, Nature, 2004, 428, 868–871 CrossRef CAS.
- K. Brenner, D. K. Karig, R. Weiss and F. H. Arnold, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 17300–17304 CrossRef CAS.
- T. Danino, O. Mondragon-Palomino, L. Tsimring and J. Hasty, Nature, 2010, 463, 326–330 CrossRef CAS.
- S. Regot, J. Macia, N. Conde, K. Furukawa, J. Kjellen, T. Peeters, S. Hohmann, E. de Nadal, F. Posas and R. Sole, Nature, 2011, 469, 207–211 CrossRef CAS.
- A. Tamsir, J. J. Tabor and C. A. Voigt, Nature, 2011, 469, 212–215 CrossRef CAS.
- H. Alper and G. Stephanopoulos, Nat. Rev. Microbiol., 2009, 7, 715–723 CrossRef CAS.
- S. Kato, S. Haruta, Z. J. Cui, M. Ishii and Y. Igarashi, Appl. Environ. Microbiol., 2005, 71, 7099–7106 CrossRef CAS.
- J. Shong, M. R. Jimenez Diaz and C. H. Collins, Curr. Opin. Biotechnol., 2012 Search PubMed.
- T. S. Bayer, D. M. Widmaier, K. Temme, E. A. Mirsky, D. V. Santi and C. A. Voigt, J. Am. Chem. Soc., 2009, 131, 6508–6515 CrossRef CAS.
- F. Warnecke, P. Luginbuhl, N. Ivanova, M. Ghassemian, T. H. Richardson, J. T. Stege, M. Cayouette, A. C. McHardy, G. Djordjevic, N. Aboushadi, R. Sorek, S. G. Tringe, M. Podar, H. G. Martin, V. Kunin, D. Dalevi, J. Madejska, E. Kirton, D. Platt, E. Szeto, A. Salamov, K. Barry, N. Mikhailova, N. C. Kyrpides, E. G. Matson, E. A. Ottesen, X. Zhang, M. Hernandez, C. Murillo, L. G. Acosta, I. Rigoutsos, G. Tamayo, B. D. Green, C. Chang, E. M. Rubin, E. J. Mathur, D. E. Robertson, P. Hugenholtz and J. R. Leadbetter, Nature, 2007, 450, 560–565 CrossRef CAS.
- P. J. Turnbaugh, R. E. Ley, M. Hamady, C. M. Fraser-Liggett, R. Knight and J. I. Gordon, Nature, 2007, 449, 804–810 CrossRef CAS.
- L. Dethlefsen, M. McFall-Ngai and D. A. Relman, Nature, 2007, 449, 811–818 CrossRef CAS.
- J. K. Nicholson, E. Holmes and I. D. Wilson, Nat. Rev. Microbiol., 2005, 3, 431–438 CrossRef CAS.
- H. Sokol, P. Seksik, J. P. Furet, O. Firmesse, I. Nion-Larmurier, L. Beaugerie, J. Cosnes, G. Corthier, P. Marteau and J. Dore, Inflammatory Bowel Dis., 2009, 15, 1183–1189 CrossRef CAS.
- C. Manichanh, L. Rigottier-Gois, E. Bonnaud, K. Gloux, E. Pelletier, L. Frangeul, R. Nalin, C. Jarrin, P. Chardon, P. Marteau, J. Roca and J. Dore, Gut, 2006, 55, 205–211 CrossRef CAS.
- S. Nell, S. Suerbaum and C. Josenhans, Nat. Rev. Microbiol., 2010, 8, 564–577 CrossRef CAS.
- A. Giongo, K. A. Gano, D. B. Crabb, N. Mukherjee, L. L. Novelo, G. Casella, J. C. Drew, J. Ilonen, M. Knip, H. Hyoty, R. Veijola, T. Simell, O. Simell, J. Neu, C. H. Wasserfall, D. Schatz, M. A. Atkinson and E. W. Triplett, ISME J., 2011, 5, 82–91 CrossRef CAS.
- S. T. Walk and V. B. Young, Interdiscip. Perspect. Infect. Dis., 2008, 2008, 125081 Search PubMed.
- T. He, K. Venema, M. G. Priebe, G. W. Welling, R. J. Brummer and R. J. Vonk, Eur. J. Clin. Invest., 2008, 38, 541–547 CrossRef CAS.
- L. Steidler, W. Hans, L. Schotte, S. Neirynck, F. Obermeier, W. Falk, W. Fiers and E. Remaut, Science, 2000, 289, 1352–1355 CrossRef CAS.
- L. Steidler, P. Rottiers and B. Coulie, Ann. N. Y. Acad. Sci., 2009, 1182, 135–145 CrossRef CAS.
- N. Saeidi, C. K. Wong, T. M. Lo, H. X. Nguyen, H. Ling, S. S. Leong, C. L. Poh and M. W. Chang, Mol. Syst. Biol., 2011, 7, 521 CrossRef.
- F. Duan and J. C. March, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11260–11264 CrossRef CAS.
Footnote |
| † Published as part of a themed issue dedicated to Emerging Investigators. |
| This journal is © The Royal Society of Chemistry 2012 |