Dynamic optimization of signal transductionvia intrinsic disorder†‡
L. Michel
Espinoza-Fonseca
*
Department of Biochemistry, Molecular Biology and Biophysics, University of Minnesota, Minneapolis, MN 55455, USA. E-mail: mef@ddt.biochem.umn.edu
First published on 14th November 2011
Abstract
It is widely accepted that the inherent flexibility of intrinsically disordered proteins (IDPs) correlates with essential functions in the cell such as signaling. However, the mechanisms by which disorder dynamically facilitates and optimizes signal transduction remain unclear. In this study, we have used a computational protocol to evaluate the interplay between the intrinsic disorder of p27kip1 and the collective motions of its binding partners, cyclin dependent kinase 2 (CDK2) and cyclin A (CA). We found that the synergy between intrinsic disorder of p27kip1 and the essential collective motions of the CDK2–CA complex introduces a set of sequential steps to dynamically optimize signal transduction. Our observations indicate that optimized p27kip1-mediated signaling originates from a combination of adaptive folding, and the cooperativity between its residual disorder and the functional collective motions of the CDK2–CA complex.
It has long been established that proteins populate a limited set of discrete conformations in their native state. This dynamic nature has often been correlated with various roles of proteins in the cell, such as molecular recognition,1–3 enzymatic catalysis,4–7 and signaling.8 However, in recent years a group of very flexible proteins, referred to as intrinsically disordered proteins (IDPs), have been described in the literature as key players in the cellular machinery. Indeed, these proteins, which are characterized by the lack of a well-defined structure in solution, play a number of important roles in the cell, such as signaling and regulation.9–12 In particular, a large number of IDPs exert their biological functions by binding to biomolecular targets such as proteins9 and nucleic acids.13 Hence, the intrinsic flexibility confers IDPs with favorable mechanistic characteristics such as high specificity when binding to multiple targets14 and fast binding kinetics,15,16 which are indispensable for precise and rapid signaling in the cell.
It has been suggested that a very important feature of IDPs is their ability to act as an efficient and specific “fly-caster” which accelerates IDP binding.15 Once the encounter complex is formed, IDPs are reeled in to the surface of the binding partner to ultimately undergo a disorder-to-order transition. This model has been challenged by recent observations demonstrating that IDPs can form dynamic complexes that sample an ensemble of rapidly interconverting conformations devoid of structure in the bound state.17 Such a dynamic nature of the bound complex has been proposed to play a major role in fine tune binding of IDPs.18 Despite these studies, there is limited understanding of how IDPs dynamically connect functional motions and signal transduction in the cell. Therefore, important issues need to be resolved: do IDPs influence the collective motions of their binding partner? Does intrinsic disorder optimize protein function for dynamic integration of signaling in the cell? To address these issues, we used a computational protocol to evaluate the effect of a disorder protein, p27kip1, on the collective motions of its binding partners, cyclin dependent kinase 2 (CDK2) and cyclin A (CA). p27kip1 has a very important biological role as it binds with high affinity to the CDK2–CA complex via a coupled binding and folding mechanism, resulting in the occlusion of the ATP-binding site of CDK219,20 and subsequent blocking of the progression from G1 to S phase of the cell division cycle. We found that the synergy between intrinsic disorder of p27kip1 and the functional collective motions of the CDK2–CA complex dynamically optimizes signal transduction. The implications of this finding are discussed in this communication.
We generated a conformational ensemble of the CDK2–CA complex bound to p27kip1 using the program CONCOORD. CONCOORD generates conformational ensembles based on geometric restrictions around a known structure.21 The coordinates of the p27kip1–CDK2–CA complex (Fig. 1B) were obtained from the protein data bank (accession code: 1JSU). In addition, we also generated a conformational ensemble of CDK2–CA using the X-ray structure that includes ATP bound to CDK2 (PDB: 1JST; Fig. 1A). We used this ensemble to compare the motions of the active CDK2–CA complex [i.e. bound to ATP (ATP–CDK2–CA)] with those of CDK2–CA bound to p27kip1. In both cases, CDK2 is phosphorylated at position Thr160, which is necessary to activate CDK2 by providing structural stability of the ATP binding site. We further used principal component analysis (PCA)22 to extract the collective motions of p27kip1–CDK2–CA and ATP–CDK2–CA and to evaluate the changes in the essential collective motions of CDK2–CA induced by p27kip1. Rendering and visualization of the protein complexes were performed using VMD 1.9.23 A full description of the methods can be found in the ESI.‡
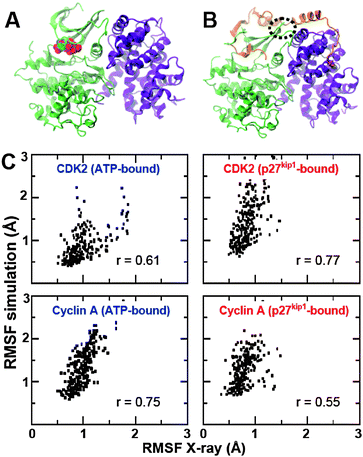 | ||
| Fig. 1 (A) Crystal structure of the CDK2–CA complex bound to ATP (spheres); (B) 3-D structure of the p27kip1–CDK2–CA complex. CDK2 (green), CA (violet) and p27kip1 (orange) are shown as ribbons. (C) Comparison of Cα RMSF values calculated for the ensembles generated by CONCOORD and the ones derived from crystallographic B-factors. For clarity, separate scatter plots were constructed for CDK2 and CA. | ||
We generated two ensembles for p27kip1–CDK2–CA and ATP–CDK2–CA complexes (500 structures each) and validated them against available experimental data. Validation was performed by quantitatively comparing the root mean square fluctuation (RMSF) values of the Cα atoms between the ensembles generated by CONCOORD and those calculated from crystallographic B-factors. We found that CONCOORD-generated ensembles display relatively larger RMSF values compared to experimental ones, which suggests that the fluctuations of Cα atoms in the generated ensembles might be overestimated (Fig. 1C). It is also possible that the dynamic behavior of CDK2 and CA might be restricted by crystal packing forces, and therefore the ensembles generated by CONCOORD could represent the actual conformational space of the complexes. Nevertheless, we found a good correlation between calculated and experimental RMSF values of CDK2 and CA (Fig. 1C). These observations indicate that CONCOORD gives a good approximation of the conformational space of ATP–CDK2–CA and p27kip1–CDK2–CA.
Next, we performed PCA and evaluated the contribution of the first principal components (or degrees of freedom) to the variance in the conformational space of ATP–CDK2–CA and p27kip1–CDK2–CA complexes (Fig. 2A). We found that the first 20 principal components account for more than 95% of the motions of ATP–CDK2–CA and p27kip1–CDK2–CA complexes (Fig. 2A). Interestingly, we found remarkable similarity in the cumulative contribution of the principal components between ATP–CDK2–CA and p27kip1–CDK2–CA complexes. This suggests that the conformational space of CDK2–CA is probably similar in the ATP- and p27kip1-bound states. To quantitatively test this idea, we calculated the inner products of the principal components of ATP–CDK2–CA and p27kip1–CDK2–CA. We used inner products as they can be used to quantify the mutual collinearity of the principal components. The inner product matrix of the first 10 principal components (which account for 90% of the total motions of the complex) is shown in Fig. 2B. The inner product matrix indicates that 50% of the first 10 principal components of CDK2–CA are relatively different between the ATP- and p27kip1-bound complexes. Interestingly, we found that the first four principal components of ATP-bound CDK2–CA are collinear to the first four components of the p27kip1-bound complex (Fig. 2B). Generally, only the first few principal components (that is those with the largest eigenvalues) are of practical interest, since they usually have the largest contribution to the motions of a protein. Because ∼80% of the total motions are represented by the first four principal components in the complexes, the strong collinearity of these components indicates that p27kip1 binding does not affect the most relevant motions of CDK2–CA.
 | ||
| Fig. 2 Comparison of the first principal components of ATP–CDK2–CA and p27kip1–CDK2–CA ensembles. (A) Cumulative contribution of the first 20 principal components to the variance in the conformational space of ATP–CDK2–CA and p27kip1–CDK2–CA. (B) Inner product matrix of the first 10 principal components of CDK2–CA bound to ATP or p27kip1. A value close or equal to 1 indicates that the principal components exist in the same conformational sub-space (or dimension) whereas values close or equal to 0 indicate that a pair of principal components are poorly correlated or uncorrelated. | ||
To better illustrate this observation, we created a representation, in the form of porcupine plots, of the collective motions extracted from the first two principal components (Fig. 3). We selected the first two principal components because they belong to the essential phase space, according to the quantitative classification described in a recent study (i.e. PCs with a R2 coefficient ≤ 0.9 when fitted to a Gaussian distribution).24 Analysis of the porcupine plots showed differences in the magnitude of Cα atom displacements between the ATP- and p27kip1-bound complexes. For instance, the displacement of the Cα atoms of CDK2 extracted from the first principal component is substantially smaller in the p27kip1-bound complex (Fig. 3). Despite these differences, we found that the directionality of the collective motions described by the first two principal components is very similar between ATP- and p27kip1-bound CDK2–CA complexes. It is interesting to note that the directionality of the motions described by the third and fourth principal components is also very similar between the complexes (data not shown), indicating that p27kip1 binding does not alter the intrinsic collective motions of CDK2–CA. In both complexes, the motion described by the first principal component corresponds to a global twisting of CDK2 and CA in opposite directions (Fig. 3A and B). This motion is strongly coupled to the opening/closing of the ATP binding site (N-lobe of CDK2). Likewise, the second principal component corresponds to a hinge motion of the N- and C-lobes of CDK2 (Fig. 3C and D). Because it was recently shown that slow motions can influence the chemical step of enzyme catalysis,7 we suggest that the hinge motion of CDK2 is likely to be coupled to the phosphorylation of peptide substrates.
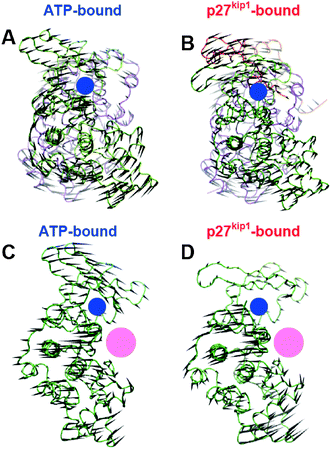 | ||
| Fig. 3 Representation of the essential collective motions described by the first two principal components. (A) and (B) correspond to the first principal component of ATP–CDK2–CA and p27kip1–CDK2–CA complexes, respectively. (C) and (D) illustrate the motions described by the second principal component of ATP–CDK2–CA and p27kip1–CDK2–CA, respectively. CDK2 (green), CA (violet) and p27kip1 (orange) are rendered as tubes, and the black cones represent the direction and amplitude of the motion for each Cα in the complexes. The location of the ATP- and peptide substrate-binding sites is shown by blue and pink circles, respectively. | ||
An important question arises from these observations: how does active CDK2–CA conserve its essential collective motions in complex with p27kip1? Upon binding, a large surface area of p27kip1 becomes buried (∼5750 Å2) and the interaction energy is very favorable (∼−12 kcal mol−1).19,20 Yet, strong protein–protein interactions with significantly smaller buried surface area, such as the homodimerization of triosephosphate isomerase, were found to profoundly affect the intrinsic dynamics of the bound complex.25 Crystallographic B-factors of p27kip1 bound to CDK2–CA indicate that while most of the p27kip1 becomes relatively ordered upon association, there is a short segment of p27kip1 (residues Arg50-Glu54) that remains highly disordered in the bound state (Fig. S1, ESI‡). Interestingly, this disordered segment is located exactly at the interface of the CDK2–CA complex (Fig. 1B, dashed oval). This adaptive mechanism of folding and binding allows p27kip1 to behave as a single-segment-multi-domain protein, which is necessary to adapt to the essential collective motions of CDK2–CA.
How does this adaptive binding translate into function optimization of p27kip1 for the dynamic signaling optimization? Based on our observations and a previously suggested model,26,27 we propose a mechanism that integrates intrinsic disorder of p27kip1 and essential collective motions of CDK2–CA to optimize signal transduction (Fig. 4). This mechanism consists of several steps that allow signaling to proceed efficiently and fluidly. In the first step of this mechanism, p27kip1 adaptively binds CDK2–CA and occludes the ATP binding site. However, the residual flexibility of the segment Arg50-Glu54 of p27kip1 allows the complex to retain the essential collective motions characteristic of active CDK2–CA (Fig. 4A and B). This adaptive binding is crucial for subsequent steps of the signaling mechanism. In the bound state, the conservation of the opening/closing motion of the ATP-binding site facilitates the partial unbinding events of the occluding segment of the p27kip1CDK2–CA complex (Fig. 4C). This motion plays an important role in partially restoring CDK2–CA activity as it facilitates phosphorylation of Tyr88 of p27kip1 by a number of tyrosine kinases, thus preventing its occluding segment from binding.28 In the absence of this motion it is unlikely that Tyr88 can be phosphorylated because only a small fraction of its side chain is solvent accessible in the complex (<35%). Given the inability of phosphorylated Tyr88 to occlude the ATP-binding site, ATP binds to CDK2. As a result, CDK2–CA re-gains partial catalytic activity, as it has been shown experimentally.28 Subsequently, the intrinsically disordered C-terminus of p27kip1 binds to the peptide substrate binding site (Fig. 4D). Finally, the hinge motion of the N- and C-lobes of CDK2 (Fig. 3D) facilitates phosphorylation of p27kip1 at position Thr187, which initiates p27kip1degradation through the ubiquitin–proteasome system and full reactivation of CDK2–CA (Fig. 4A and D). Because the essential collective motions of CDK2–CA appear to be tightly coupled to steps along the catalytic pathway of CDK2, we suggest that the dynamic synergy between p27kip1 and CDK2–CA introduces a set of sequential steps that guarantee a fluid and rapid signal transduction necessary for the activation of cell division.
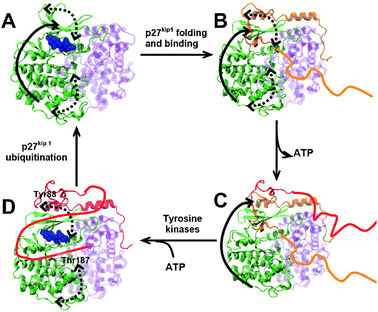 | ||
| Fig. 4 Proposed mechanism of dynamic optimization of signal transduction of p27kip1. (A) Active CDK2–CA natively undergoes functional motions, which are coupled to the opening/closing of the ATP-binding site (solid arrow) and the hinge motion of the N and C lobes of CDK2 (dashed arrows). ATP (blue spheres) is bound to CDK2 (green). (B) Upon coupled folding and binding of p27kip1, the complex retains the functional motions observed in the active form of CDK2–CA. (C) Because the directionality of opening/closing motion of the ATP-binding site (solid arrow) is not affected by p27kip1 binding, this motion facilitates the spontaneous conformational transitions of p27kip1 that occlude (orange ribbons) and release (red ribbons) the ATP-binding site. In response to a number of cell proliferation signals, p27kip1 is phosphorylated by tyrosine kinases at Tyr88. (D) ATP binds to CDK2 facilitated by the motion coupled to the opening/closing of the ATP binding site; finally, the hinge motions of CDK2 (dashed arrows) facilitate the binding and phosphorylation of the intrinsically disordered C-terminus of p27kip1, which initiates p27kip1 ubiquitination and reactivation of CDK2–CA. | ||
In conclusion, we propose that optimized signaling-mediated functions of p27kip1 originate from a combination of adaptive folding, and the cooperativity between its residual disorder and the functional collective motions of the CDK2–CA complex. Similar approaches could also be combined with network models29 and game theory of proteins30 to create a more detailed description of IDP-mediated mechanisms of signal transduction. What is absolutely clear is that complementary experimental and computational studies will be needed to further understand the relationship between functional dynamics of IDPs interaction networks and the optimization of signal transduction in the cell.
Acknowledgements
The author was supported by the American Heart Association and the Minnesota Supercomputing Institute at the University of Minnesota.Notes and references
- O. F. Lange, et al. , Science, 2008, 320, 1471–1475 CrossRef CAS.
- A. Bakan and I. Bahar, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14349–14354 Search PubMed.
- D. D. Boehr, R. Nussinov and P. E. Wright, Nat. Chem. Biol., 2009, 5, 789–796 CrossRef CAS.
- K. A. Henzler-Wildman, et al. , Nature, 2007, 450, 913–916 CrossRef CAS.
- K. A. Henzler-Wildman, et al. , Nature, 2007, 450, 838–844 CrossRef CAS.
- D. D. Boehr, D. McElheny, H. J. Dyson and P. E. Wright, Science, 2006, 313, 1638–1642 CrossRef CAS.
- G. Bhabha, et al. , Science, 2011, 332, 234–238 Search PubMed.
- R. G. Smock and L. M. Gierasch, Science, 2009, 324, 198–203 CrossRef CAS.
- V. N. Uversky and A. K. Dunker, Biochim. Biophys. Acta, 2010, 1804, 1231–1264 CAS.
- V. N. Uversky, Protein Sci., 2002, 11, 739–756 CrossRef CAS.
- P. Tompa, Trends Biochem. Sci., 2002, 27, 527–533 CrossRef CAS.
- P. E. Wright and H. J. Dyson, J. Mol. Biol., 1999, 293, 321–331 CrossRef CAS.
- H. J. Dyson, Mol. Biosyst., 2011 10.1039/C1MB05258F.
- J. Liu, J. R. Faeder and C. J. Camacho, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 19819–19823 CAS.
- B. A. Shoemaker, J. J. Portman and P. G. Wolynes, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 8868–8873 CrossRef CAS.
- Y. Huang and Z. Liu, J. Mol. Biol., 2009, 393, 1143–1159 CrossRef CAS.
- V. N. Uversky, Chem. Soc. Rev., 2011, 40, 1623–1634 RSC.
- P. Tompa and M. Fuxreiter, Trends Biochem. Sci., 2008, 33, 2–8 CrossRef CAS.
- E. R. Lacy, et al. , Nat. Struct. Mol. Biol., 2004, 11, 358–364 CrossRef CAS.
- A. A. Russo, P. D. Jeffrey, A. K. Patten, J. Massague and N. P. Pavletich, Nature, 1996, 382, 325–331 CrossRef CAS.
- B. L. de Groot, et al. , Proteins: Struct., Funct., Genet., 1997, 29, 240–251 CrossRef CAS.
- A. Amadei, A. B. Linssen and H. J. Berendsen, Proteins: Struct., Funct., Genet., 1993, 17, 412–425 CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef , 27–28.
- C. K. Materese, C. C. Goldmon and G. A. Papoian, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 10659–10664 Search PubMed.
- S. Cansu and P. Doruker, Biochemistry, 2008, 47, 1358–1368 CrossRef CAS.
- C. A. Galea, Y. Wang, S. G. Sivakolundu and R. W. Kriwacki, Biochemistry, 2008, 47, 7598–7609 CrossRef CAS.
- C. A. Galea, et al. , J. Mol. Biol., 2008, 376, 827–838 CrossRef CAS.
- M. Grimmler, et al. , Cell (Cambridge, Mass.), 2007, 128, 269–280 CrossRef CAS.
- P. Csermely, Trends Biochem. Sci., 2004, 29, 331–334 CrossRef CAS.
- I. A. Kovacs, M. S. Szalay and P. Csermely, FEBS Lett., 2005, 579, 2254–2260 CrossRef CAS.
Footnotes |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: Guest Editor M. Madan Babu. |
| ‡ Electronic supplementary information (ESI) available: Full description of methods and Fig. S1. See DOI: 10.1039/c1mb05412k |
| This journal is © The Royal Society of Chemistry 2012 |