Meta-structure correlation in protein space unveils different selection rules for folded and intrinsically disordered proteins†
Yandi
Naranjo
a,
Miquel
Pons
*ab and
Robert
Konrat
*c
aLaboratory of Biomolecular NMR, Institute for Research in Biomedicine (IRB Barcelona), Parc Científic de Barcelona, Baldiri Reixac, 10, 08028, Barcelona, Spain
bDepartament de Química Orgànica, Universitat de Barcelona, Martí i Franquès, 1-11, 08028, Barcelona, Spain. E-mail: mpons@ub.edu
cDepartment of Structural and Computational Biology, Max F. Perutz Laboratories, University of Vienna, Vienna Biocenter Campus 5, A-1030 Vienna, Austria. E-mail: robert.konrat@univie.ac.at
First published on 23rd November 2011
Abstract
The number of existing protein sequences spans a very small fraction of sequence space. Natural proteins have overcome a strong negative selective pressure to avoid the formation of insoluble aggregates. Stably folded globular proteins and intrinsically disordered proteins (IDPs) use alternative solutions to the aggregation problem. While in globular proteins folding minimizes the access to aggregation prone regions, IDPs on average display large exposed contact areas. Here, we introduce the concept of average meta-structure correlation maps to analyze sequence space. Using this novel conceptual view we show that representative ensembles of folded and ID proteins show distinct characteristics and respond differently to sequence randomization. By studying the way evolutionary constraints act on IDPs to disable a negative function (aggregation) we might gain insight into the mechanisms by which function-enabling information is encoded in IDPs.
Intrinsically Disordered Proteins (IDPs) are now recognized as a major class of natural proteins following a long invisibility period arising mainly from their failure to adopt a single, well defined conformation capable of forming three-dimensional crystal lattices. IDPs are especially abundant in eukaryotes and predominantly associated to regulatory processes.1 These two facts are unlikely to be unrelated and the exploitation of IDPs may be one of the molecular features that enabled evolution beyond simple unicellular prokaryotes. The important functional roles of IDPs are supported, among others, by evidences about their tight regulations at all levels.1,2
Recognition of the widespread occurrence of IDPs was made possible by the access to the exhaustive list of primary sequences of complete organisms through full genome sequencing and the development of bioinformatic tools that could correctly predict the probability of disorder from the primary sequence.3–14 Hallmarks of protein disorder are the low frequency of aromatic and other hydrophobic residues combined with a high frequency of charged, polar and structure breaking residues.
The growing evidence that a unique folding may not be required to perform a function in the case of IDP suggests that the structure–activity paradigm has to be expanded into a more general one involving more sophisticated concepts about ensemble averaging and sampling of accessible conformational space. While the rules by which information is encoded in flexible proteins are still elusive, successful evolution of protein sequences (survival of the fittest) in general points to the relevance of a common property: avoiding the formation of amyloids. In other words, amyloid formation (or its prevention) can be considered a common functional requirement to which all proteins have adapted. Although there exist different ways to achieve it, looking for common principles that apply to whole classes of proteins may provide some insights into the information-encoding capabilities of IDPs.
In this article we explore two aspects of IDPs that can be derived from the analysis of primary sequences. First we compare the occurrence of amyloidogenic regions, as predicted by the Waltz algorithm,15 in folded and intrinsically disordered proteins. Second, we introduce the average meta-structure correlation maps (AMCM) and compare natural and randomized sequences to unveil conserved features that are different in IDPs and folded proteins beyond their different amino acid compositions. We suggest that the observed differences may reflect different strategies to avoid amyloid formation by folded and disordered proteins.
Methods
The propensity for forming insoluble amyloids was assessed by the Waltz algorithm.15 The Waltz program allows for the analysis of amyloidogenesis exclusively from the primary sequence and gives the location of sequences predicted to have high propensities to form amyloids.The meta-structure concept was recently introduced by one of us to extract implicit structural information encoded in the sequence and derived from topological pairwise propensities extracted from the analysis of proteins of known structure.16 It was shown that although meta-structures can be derived directly from sequences, they are much more conserved in evolutionarily related proteins.16 In the meta-structure concept, a 3D protein structure is perceived as a network of residue interactions, in which nodes refer to residues and edges indicate the existence of (through space) neighbourhood relationships. The mutual topological relationship between two residues (A, B) is quantified by the shortest path length across the network and characteristically depends on the amino acid types (A, B) and their primary sequence distance, lAB. The frequency of different topological relationships was evaluated in a subset of structures taken from the PDB database and stored as pairwise statistical distribution functions. The meta-structure analysis employs this statistically derived topological information to extract higher order information implicitly contained in the primary sequence, the secondary structure parameter and the local compaction: local 2nd structure information is quantified by a parameter that takes positive values for helical structures and negative values for predicted beta sheets. Compactness refers to the tendency of local regions of the sequence to be buried from solvent access. We refer to the sequence of pairs of local secondary structure and compaction values as the protein meta-structure. Although only structured protein parts were used in the derivation of the pairwise distribution function, the methodology is not limited to structured proteins but rather provides quantitative information about the most probable network topology of a given protein, folded or unfolded.
For a given protein, the average value of each of the meta-structure parameters can be calculated to provide a global pair of parameters. Note that since the residue-specific meta-structure parameters reflect the sequence context, the global average values are sensitive to the actual protein sequence, and do not reflect simply the protein composition. The pairs of average meta-structure parameters for a given protein can be represented as a point in a two dimensional plot, which we refer to as an average meta-structure correlation map (AMCM).
The potential of the meta-structure approach for high-throughput IDP identification was already demonstrated.16 Average residue compactness values of proteins were introduced as measures for protein foldedness. While stably folded proteins display average compactness values of about 300, significantly smaller values (<200) are found for structurally flexible proteins (intrinsically disordered/unstructured). Applications of this approach to proteins from different kingdoms (archea, prokaryotes and eukaryotes) corroborated the widely accepted notion that lower organisms (archea and prokaryotes) have only a few unstructured proteins (1.7%–3.5%), whereas for eukaryotes a significant fraction of the proteome falls into this category (from 13.9% to 21.5%).16 This criterion (average compactness value <200) was used to define a set of human IDP proteins (see below).
In this study, folded proteins are represented by a non-redundant representation of 27![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 780 proteins from the Protein Data Bank (which we will refer to as PDB). A set of highly disordered proteins was selected by calculating the meta-structure parameters for all human proteins and selecting those with an average compactness value of <200. This set (referred to as IDP200) contains 1012 proteins. Of these, 50.4% have more than 90% of their residues disordered and 97% are predicted to be disordered in more than 50% of their sequence, according to the VL2 predictor10 accessed through the DisProt server.17 A third set was formed by 164 proteins (which we call DIS50) selected from the DISPROTdatabase12 and annotated to be more than 50% disordered. On a per residue basis, 40.8% of the DIS50 proteins are predicted by VL2 to have more than 90% of their residues in disordered regions and 85.9% of the proteins are more than 50% disordered. 23 out of the 164 sequences of DIS50 were also present in IDP200.
780 proteins from the Protein Data Bank (which we will refer to as PDB). A set of highly disordered proteins was selected by calculating the meta-structure parameters for all human proteins and selecting those with an average compactness value of <200. This set (referred to as IDP200) contains 1012 proteins. Of these, 50.4% have more than 90% of their residues disordered and 97% are predicted to be disordered in more than 50% of their sequence, according to the VL2 predictor10 accessed through the DisProt server.17 A third set was formed by 164 proteins (which we call DIS50) selected from the DISPROTdatabase12 and annotated to be more than 50% disordered. On a per residue basis, 40.8% of the DIS50 proteins are predicted by VL2 to have more than 90% of their residues in disordered regions and 85.9% of the proteins are more than 50% disordered. 23 out of the 164 sequences of DIS50 were also present in IDP200.
A comparison between the predicted disorder and compactness at a residue level for DIS50 and IDP200 is shown in Fig. 1. For the DIS50 residues, the frequency of compactness values for residues with a disorder score larger than 50% shows a Gaussian shape centered at 200, while the distribution of the residues predicted to be ordered is centered at 300, the same average value found for PDB proteins. For the IDP200 set, which is biased to average compactness values below 200, the distribution of predicted disordered values is centered at around 150 while the small fraction of residues predicted to be ordered has compactness values centered at around 250. Thus we conclude that meta-structure derived compactness values can be used as reliable parameters for identifying disordered segments in proteins.
 | ||
| Fig. 1 Frequency distribution of compactness values of individual residues obtained from meta-structure analysis of the proteins in the DIS50 (left) and IDP200 (right) databases. The compactness distribution of residues predicted to be disordered or ordered is represented in black or red, respectively. | ||
A fourth set (AFR) was generated by collecting the regions of the proteins of the first three sets predicted to form amyloids by Waltz. Reference sets containing randomized sequences for each protein were prepared and are referred to as RPDB, RIDP200, RDIS50, and RAFR. An additional set of 592 sequences (β-protein interactors or BPI) contains natural proteins experimentally identified to be efficiently captured in vivo by amyloid forming peptides as described by Olzscha et al.18 and was used to validate the amyloidogenesis analysis.
Results and discussion
Firstly, we computed from the Waltz server output the percentage of sequences in the PDB, DIS50 and IDP200 sets that contain at least one amyloidogenic region (Fig. 2). As a comparison we also included the BPI set of proteins captured by three different amyloid forming artificial proteins.18 A large proportion of the sequences of proteins in the PDB are predicted to have at least one amyloidogenic region confirming that folding is indeed preventing amyloid formation. Consistently, the fraction of amyloidogenic sequences computed by Waltz in the β-protein interactors set is larger than the average of the PDB, confirming the capacity of the Waltz algorithm to predict amyloidogenic regions.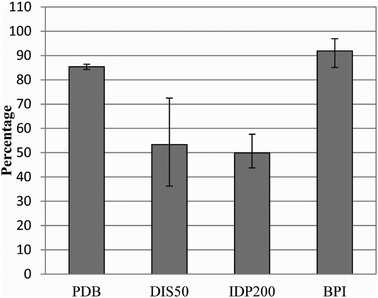 | ||
| Fig. 2 Percentage of sequences with amyloidogenic regions in different datasets. Amyloidogenic regions were predicted using the Waltz algorithm. The β-protein interactors correspond to the sequences of the proteins captured by amyloid forming artificial peptides described in ref. 18. Sequences were extracted using the Human Protein Reference Database identifier and include different isoforms (www.hprd.org). Error bars were generated by comparing random subsets of 10% of the databases. | ||
The propensity to form amyloids by unfolded proteins in the IDP sets is much lower than that of the proteins present in the PDB, in agreement with the notion that alternative strategies to avoid amyloid formation are required for unfolded proteins. Next we computed the total length of the amyloidogenic sequences and the fraction they represent of the total sequence for natural proteins and for a matching set made of randomized sequences. Fig. 3 shows histograms of the frequencies of predictions of different proportions of amyloidogenic segments with respect to the total protein length. The relative importance of amyloidogenic regions with respect to the total sequence is low for a large fraction of the natural sequences present in the PDB and IDP sets have very low tendencies to form amyloids. PDB sequences show a bimodal distribution with a sharp peak of non-aggregation prone sequences, followed by a broad distribution with a maximum at 4% and a slow decay with a substantial number of sequences comprising more than 20% of amyloidogenic regions. The IDP sets can be described by a much narrower distribution with most sequences having less than 10–12% of amyloidogenic regions. The bimodal distribution in the PDB set probably reflects an additional negative selection by the structural biology researchers being able to solve preferentially those proteins that have favorable solution properties.
 | ||
| Fig. 3 Number of sequences containing different percentage of residues in amyloidogenic regions. The total number of residues present in the different amyloidogenic regions predicted by Waltz using the natural sequences and one randomized version of each protein were compared. Bin width is 2%. Only sequences for which the Waltz server provided a valid output for both the natural and randomized versions were used and the total number of valid points is given in each graph. | ||
The sets of randomized sequences show a general tendency to increased amyloidogenesis. This is more pronounced in the PDB. The tendency of RIDP200 to form amyloids is similar to the matching natural sequences. This observation is in agreement with the idea that naturally disordered proteins have an intrinsic lower tendency to aggregate than denatured globular proteins, which primarily comes from the residue composition of disordered proteins, including the avoidance of hydrophobic residues. However, general sequence constraints or preferences for IDPs have been observed,19 and are additionally shown in the meta-structure correlation results given below.
Secondly, the proteins from the different data sets were subjected to a meta-structure analysis. The meta-structure information from each protein in the different sets was represented as points in an Average Meta-Structure Correlation Map (AMCM) in which the average value of the meta-structure derived residue compactness is plotted against the average secondary structure as predicted also from meta-structures. Fig. 4 shows AMCM of naturally occurring proteins. The average compactness of the PDB set is, not surprisingly, larger than the DIS50 set. The IDP200 is, by construction, limited to compaction values lower than 200. For comparison we also calculated the AMCM plot for individual segments predicted to be prone to form amyloids (AFR set). The larger scatter is presumably due to the fact that here smaller peptide fragments are analyzed instead of entire protein averages. The compactness values of the aggregation-prone fragments of the AFR set are shifted to higher values than those of the sets of complete proteins from which they were derived.
 | ||
| Fig. 4 The average meta-structure correlation map (AMCM) is a meaningful representation of protein space. The AMCM shows the correlation between sequence-derived compactness values and 2nd structure parameters. The two meta-structure parameters are calculated based on the primary sequence of a given protein and given as a protein average. Large compactness values are found for compact 3D structures with dense side-chain interaction networks, whereas small compactness values are indicative of flexible polypeptide chains devoid of significant stabilizing interactions. 2nd structure parameters are defined as follows: positive: α-helix, negative: β-strand. Naturally occurring and random sequences are given in black and red, respectively (for details see the text). | ||
A clear additional distinction is observed between the sets of globular and disordered complete proteins. The PDB set shows a negative correlation between the secondary structure and average compactness indicating that the most compact structures are enriched in β sheets (Pearson' correlation coefficient: −0.36; slope of the best linear fit: −0.19 for proteins with average compactness values between 200 and 400). In contrast, both IDP enriched sets show a clear positive correlation (Pearson' correlation coefficients (slopes): DIS50: 0.62 (0.56); IDP200: 0.46 (0.66), calculated for proteins displaying average compactness values above 100). Thus, more compact structures are associated to increasing helical contents, presumably reflecting the building principle of IDPs comprising locally defined structural elements. Only a few IDPs have negative secondary structure values. Amyloid forming peptides of the AFR set show a large scatter of secondary structure parameters and a low correlation between the two meta-structure parameters.
The observed correlations in full proteins suggest that intricate side-chain interactions leading to large compactness values are preferentially associated to β-strand formation in folded proteins. In contrast, IDP compaction is preferentially achieved by forming less aggregation-prone α-helices.
The location of protein sequences in the average meta-structure correlation maps shows a better correlation with their folding characteristics than the individual parameters. Fig. 5 shows a smoothed representation obtained by clustering of neighbor points in the AMCMs of the different protein sets. It can be seen that folded proteins, IDPs and aggregation prone peptides are located in distinct and only partially overlapping sub-spaces. We thus believe that the AMCM analysis provides a meaningful representation of “protein space”. In addition to the analysis given here we anticipate further applications of the AMCM approach to global, large-scale analysis of the architecture and organization of the accessible protein space.
 | ||
| Fig. 5 The structural heterogeneity of protein space is indicated by distinct cluster formation in the average meta-structure correlation map. Amyloidogenic fragments (AFR) are indicated in red, folded proteins (PDB) in blue and IDPs in green (IDP200) or violet (DIS50). | ||
A comparison of the AMCM of natural and randomized sequences shows significant differences between globular and disordered protein sequences. In the data set of randomized IDP sequences the observed correlation between average compactness and 2nd structure changes from positive to negative (Pearson' coefficients: DIS50 0.62; RDIS50: −0.25; IDP200: 0.46; RIDP200: −0.20). In contrast, random protein sequences of folded proteins maintain a negative correlation similar to natural protein sequences (Pearson' coefficients: PDB −0.36; RPDB: −0.50). These observations point to a different origin of the correlations in AMCMs of IDPs and globular proteins.
The physical background rules for IDPs involve both a bias in the amino acid composition, minimizing the occurrence of non-polar residues, and restrictions at the primary structure level that links increased compaction with the minimization of the formation of β-sheets. We suggest that the origin of the correlation between meta-structure parameters in IDPs is predominantly avoiding aggregation when local structures are formed, although other contributions to the low propensity to adopt β-structures by IDP cannot be ruled out, including the modulation of conformational entropy which is easier to achieve through helical conformations than with β-sheets.20
IDPs and globular proteins have been selected during evolution using two distinct strategies to avoid the development of deleterious intermolecular associations. Interestingly, while folded proteins partially overlap with amyloidogenic peptides, IDP sequences are distinctly different and clearly set apart from the reservoir of sequences prone to aggregation. This statement is in contrast to the widespread belief that disorder increases the tendency to aggregation and disease. However, while a number of disordered proteins are associated with amyloidosis leading to devastating diseases, it is not obvious that the fraction of naturally disordered proteins that can form amyloids is larger than the fraction of naturally occurring stably folded proteins that aggregate upon misfolding. Most globular proteins show a tendency to aggregate when solution conditions or point mutations disturb their natural folding while this is less obvious for naturally disordered proteins. Thus, while folding of globular proteins offers protection against aggregation, disordered proteins have naturally evolved alternative strategies to avoid amyloidogenesis.
An analogy could be drawn between the sequence universe and the surface of the Earth. Most of the Earth is covered with water. Likewise, most sequences would naturally form amyloids. Natural globular/folded proteins and IDPs can be associated to different “emerged continents”. Both have in common that they are not “covered with water”/forming amyloids yet they are clearly different. Shores are regions that can be easily flooded by small changes in the environment and would represent “dangerous” sequences prone to form amyloids. They are present in both folded proteins and IDPs. Therefore, the observation of amyloids associated to some IDPs should not hide the fact that the vast majority of IDPs are not aggregating and this is the physical background on which function for IDPs can be built.
The structure–function relationship is one of the guiding paradigms of structural biology. IDPs challenge this principle as their function is associated with the availability of multiple conformations and the sampling of a wide conformational space within characteristic time scales thereby allowing for concerted recognition events by diverse receptors or chemical reactions to proceed between closely placed complementary reacting centres.
Naturally occurring IDPs in part encode functionality in the form of restrictions of the “random-coil” conformational space actually sampled by a particular IDP sequence. In order to eventually decipher how IDPs information is encoded and stored in the primary sequence a more thorough analysis of network topologies will be valuable. We suggest that meta-structure analysis, in addition to providing a tool to separate IDPs from folded protein structures from the amino acid sequences, may be used in the future to differentiate between functionally related subsets of IDPen route to the challenging goal of uncovering how other functions are encoded in IDPs.
Acknowledgements
This work was supported in part by the FWF (SFB-17; P22125-B12, P20549-N19), FEDER-MICINN (BIO2010-15683, and acción integrada AT2009-0013), Generalitat de Catalunya (2009SGR1352) and the 7FP Bio-NMR project (contract 261863). YN has benefitted from predoctoral fellowships from the Generalitat de Catalunya and the Spanish FPU program.References
- (a) H. J. Dyson and P. E. Wright, Nat. Rev. Mol. Cell Biol., 2005, 6, 197–208 CrossRef CAS; (b) H. Xie, S. Vucetic, L. M. Iakoucheva, C. J. Oldfield, A. K. Dunker, V. N. Uversky and Z. Obradovic, J. Proteome Res., 2007, 6, 1882–1898 CrossRef CAS; (c) A. K. Dunker and Z. Obradovic, Nat. Biotechnol., 2001, 19, 805–806 CrossRef CAS; (d) P. Tompa, FEBS Lett., 2005, 579, 3346–3354 CrossRef CAS; (e) J. J. Ward, J. S. Sodhi, L. J. McGuffin, B. F. Buxton and D. T. Jones, J. Mol. Biol., 2004, 337, 65–645 Search PubMed.
- J. Gsponer, M. E. Futschik, S. A. Teichmann and M. M. Babu, Science, 2008, 322, 1365–1368 CrossRef CAS.
- P. Lieutaud, B. Canard and S. Longhi, BMC Genomics, 2008, 9(Suppl 2), S25 CrossRef.
- Z. Dosztanyi, V. Csizmok, P. Tompa and I. Simon, Bioinformatics, 2005, 21, 3433–3434 CrossRef CAS.
- K. Coeytaux and A. Poupon, Bioinformatics, 2005, 21, 1891–1900 CrossRef CAS.
- Z. R. Yang, R. Thomson, P. McNeil and R. M. Esnouf, Bioinformatics, 2005, 21, 3369–3376 CrossRef CAS.
- R. Linking, L. J. Jensen, F. Diella, P. Bork, T. J. Gibson and R. B. Russell, Structure, 2003, 11, 1453–1459 CrossRef CAS.
- J. Prilusky, C. E. Felder, T. Zeev-Ben-Mordehai, E. H. Rydberg, O. Man, J. S. Beckmann, I. Silman and J. L. Sussman, Bioinformatics, 2005, 21, 3435–3438 CrossRef CAS.
- R. Linding, R. B. Russell, V. Neduva and T. J. Gibson, Nucleic Acids Res., 2003, 31, 3701–3708 CrossRef CAS.
- S. Vucetic, C. J. Brown, A. K. Dunker and Z. Obradovic, Proteins, 2003, 52, 573–584 CrossRef CAS.
- Z. Obradovic, K. Peng, S. Vucetic, P. Radivojac and A. K. Dunker, Proteins: Struct., Funct., Bioinf., 2005, 61(Suppl 7), 176–182 Search PubMed.
- M. Sickmeier, J. A. Hamilton, T. LeGall, V. Vacic, M. S. Cortese, A. Tantos, B. Szabo, P. Tompa, J. Chen, V. N. Uversky, Z. Obradovic and A. K. Dunker, Nucleic Acids Res., 2007, 35, D786–D793 CrossRef CAS.
- T. Ishida and K. Kinoshita, Bioinformatics, 2008, 24, 1344–1348 CrossRef CAS.
- A. Campen, R. M. Williams, C. J. Brown, J. Meng, V. N. Uversky and A. K. Dunker, Protein Pept. Lett., 2008, 15, 956–963 CrossRef CAS.
- S. Maurer-Stroh, M. Debulpaep, N. Kuemmerer, M. Lopez de la Paz, I. C. Martins, J. Reumers, K. L. Morris, A. Copland, L. Serpell, L. Serrano, J. W. Schymkowitz and F. Rousseau, Nat. Methods, 2010, 7, 237–242 CrossRef CAS.
- R. Konrat, Cell. Mol. Life Sci., 2009, 66, 3625–3639 Search PubMed.
- http:// http:// www.dabi.temple.edu/disprot/predictor.php (last accessed 1.11.2011).
- H. Olzscha, S. M. Schermann, A. C. Woerner, S. Pinkert, M. H. Hecht, G. G. Tartaglia, M. Vendruscolo, M. Hayer-Hartl, F. U. Hartl and R. M. Vabulas, Cell, 2011, 144, 67–78 Search PubMed.
- S. Lise and D. T. Jones, Proteins: Struct., Funct., Bioinf., 2005, 58, 144–150 Search PubMed.
- P. Tompa, Structure and Function of Intrinsically Disordered Proteins, Chapman and Hall/CRC, 2010 Search PubMed.
Footnote |
| † Published as part of a Molecular BioSystems themed issue on Intrinsically Disordered Proteins: guest editor M. Madan Babu. |
| This journal is © The Royal Society of Chemistry 2012 |