DOI:
10.1039/C2JM33620K
(Paper)
J. Mater. Chem., 2012,
22, 18204-18213
A green-yellow emitting oxyfluoride solid solution phosphor Sr2Ba(AlO4F)1−x(SiO5)x:Ce3+ for thermally stable, high color rendition solid state white lighting
Received
6th June 2012
, Accepted 16th July 2012
First published on 26th July 2012
Abstract
A near-UV excited, oxyfluoride phosphor solid solution Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x has been developed for solid state white lighting applications. An examination of the host lattice, and the local structure around the Ce3+ activator ions through a combination of density functional theory, synchrotron X-ray and neutron powder diffraction and total scattering, and electron paramagnetic resonance, points to how chemical substitutions play a crucial role in tuning the optical properties of the phosphor. The maximum emission wavelength can be tuned from green (λem = 523 nm) to yellow (λem = 552 nm) by tuning the composition, x. Photoluminescent quantum yield is determined to be 70 ± 5% for some of the examples in the series. Excellent thermal properties were found for the x = 0.5 sample, with the photoluminescence intensity at 160 °C only decreased to 82% of its room temperature value. Phosphor-converted LED devices fabricated using an InGaN LED (λmax = 400 nm) exhibit high color rendering white light with Ra = 70 and a correlated color temperature near 7000 K. The value of Ra could be raised to 90 by the addition of a red component, and the correlated color temperature lowered to near 4000 K.
1 Introduction
Advances in light emitting diode (LED) technologies,1,2 namely the development of high efficiency blue and near-UV emitting InGaN,3 has led to devices with greatly diminished energy demands. The use of these solid state devices for white lighting in place of conventional incandescent or fluorescent light sources has become quite appealing for a wide range of applications.4 Solid state white lighting offers many advantages over traditional lighting including longer lifetimes, reduced energy consumption, and an environmentally friendly design without the need for mercury.5
The phosphor material used in solid state white lighting devices greatly affects the overall device, including the luminous efficacy, color rendition, thermal and chemical stability, and the color quality of the white light.6,7 Highly efficient phosphors are needed to convert the LED emission with little or no energy loss, apart from what is inevitably lost to the Stokes shift. Specific luminescence properties are needed to produce an optimal white light with a high color rendering index (Ra). These properties include strong absorption in the region of the LED emission and a broad emission profile across all regions of the visible spectrum. The thermal stability of the phosphor is also important, since thermal quenching of phosphor luminescence can lead to decreased efficiency and color instability at the operating temperatures of modern devices.
The most commonly used phosphor for solid state white lighting, is cerium-doped yttrium aluminum garnet Y3Al5O12 (YAG:Ce3+), developed by Blasse and Bril in 1967.8,9 Its widespread use stems from its strong absorption of blue light, ideal for use with an InGaN LED, and its very efficient yellow emission (QY ≈ 85% or greater) in addition to excellent chemical stability. Yet YAG:Ce3+ is not ideal as it suffers from low color rendition due to weak luminescence in the red spectral region, and from thermal quenching10 at elevated temperatures. A variety of other phosphors have been developed including oxides,11–13 oxyfluorides,14,15 oxynitrides16–18 and nitrides.19–22 Some promising phosphor candidates include LaSr2AlO5:Ce3+ (LSA:Ce3+)11 and the isostructural Sr2BaAlO4F:Ce3+ (SBAF:Ce3+)14 recently developed by our group. SBAF:Ce3+ is a highly efficient phosphor with a quantum yield approaching 100% and good thermal properties. However, the emission from SBAF:Ce3+ is too blue to create a suitable white light. Furthermore, it degrades upon contact with moisture. It was shown by Im et al.15 that the oxyfluoride Sr3AlO4F:Ce3+ compound (isostructural with SBAF:Ce3+) exhibited increased moisture stability, and a red shift in emission when introduced into a solid solution with the nearly isostructural oxide compound, Sr3SiO5:Ce3+.
In this contribution, we report the green-to-yellow emitting phosphor solid solution, Sr2Ba(AlO4F)1−x(SiO5)x:Ce3+ comprising Sr2BaAlO4F as the oxyfluoride end-member and the nearly isostructural Sr3SiO5 as the oxide end member. The structural, optical, and thermal properties have been examined, along with the quality of white LEDs fabricated using the solid solution phosphor in conjunction with a near-UV InGaN LED chip. The solid solution strategy suggested here greatly improves certain key properties of these promising oxyfluoride phosphors.
2 Experimental and computational methods
Samples of Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x (x = 0.1, 0.2, …0.9) were prepared by mixing stoichiometric amounts of SrCO3 (99.5% purity, Materion), CeO2 (99.9% purity, Cerac), BaCO3 (99.9% purity, Cerac), BaF2 (99% purity, Cerac), Al2O3 (high purity, sub-micron SM8, Baikowski) and SiO2 (amorphous fumed, Degussa Aerosil TT600). Powders were intimately mixed by grinding in an agate mortar with acetone for approximately 30 min, then pressed into pellets. Pellets were placed on sacrificial powder in alumina crucibles and heated at 1350 °C for 4 h in a reducing atmosphere of 5% H2/95% N2 with heating and cooling ramps of 10 h.
Density functional theory (DFT) calculations were performed using the Vienna ab initio Simulation Package (VASP)23–26 in order to predict site preferences for the Ce3+ ion in the host oxyfluoride compound. Projector augmented waves (PAW)27,28 were employed to describe the atomic cores and the valence electrons were described using plane-waves. The exchange–correlation energy was calculated using the generalized gradient approximation (GGA) functional within the Perdew–Burke–Ernzerhof (PBE) formulation.29,30 Calculations for bulk Sr2BaAlO4F and for the substitutions were performed on the conventional unit cell, using a 4 × 4 × 4 Monkhorst–Pack k-point sampling. A cutoff energy of 550 eV was used in all calculations. Relaxations were deemed to have converged when the forces on all the atoms were less than 0.01 eV Å−1.
Synchrotron X-ray diffraction data were collected at the 11-BM instrument at the Advanced Photon Source (APS), Argonne National Laboratory using a wavelength of λ = 0.424629 Å. Rietveld analysis was carried out using the XND Rietveld code.31 Time-of-flight neutron powder diffraction data were collected on the Nanoscale Ordered Materials Diffractometer (NOMAD) at the Spallation Neutron Source (SNS), Oak Ridge National Laboratory. Least-squares refinement was performed on the pair-distribution function (PDF) to obtain average structure information. Electron paramagnetic resonance (EPR) spectra were collected on powder samples using a Bruker X-Band EPR spectrometer (ν = 9.486 GHz), equipped with a helium flow cryostat. Samples were placed in quartz tubes and data was acquired at 10 K.
Photoluminescence (PL) spectra were obtained on a Perkin Elmer LS55 spectrophotometer equipped with a xenon lamp. Spectra were measured by mixing the sample with KBr (≥99% purity, FT-IR grade, Sigma-Aldrich) in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10 ratio of sample to KBr and pressing into a pellet. Photoluminescence quantum yield (PLQY) was measured using phosphor powders encapsulated in silicone resin (GE Silicones, RTV-615) and deposited on transparent quartz substrates (Chemglass). The samples were placed inside a Spectralon™-coated integrating sphere (6 inch diameter, Labsphere) and excited using 405 nm light generated by a solid-state laser (Crystalaser DL-405-40-S) operated at a power between 1 mW and 2 mW. The light was collected by a quartz lens and directed onto a calibrated Si photodiode (Newport 818-UV) using filters (Omega Filters) to collect respective wavelengths. Data collection and processing procedures were conducted similar to those described by Greenham et al.32 Temperature dependence of the photoluminescence was measured in the temperature range from 77 K to 493 K using a home-built fluorimeter and incorporating a cryostat for low temperature measurements and a heating stage for high temperature data collection.
:10 ratio of sample to KBr and pressing into a pellet. Photoluminescence quantum yield (PLQY) was measured using phosphor powders encapsulated in silicone resin (GE Silicones, RTV-615) and deposited on transparent quartz substrates (Chemglass). The samples were placed inside a Spectralon™-coated integrating sphere (6 inch diameter, Labsphere) and excited using 405 nm light generated by a solid-state laser (Crystalaser DL-405-40-S) operated at a power between 1 mW and 2 mW. The light was collected by a quartz lens and directed onto a calibrated Si photodiode (Newport 818-UV) using filters (Omega Filters) to collect respective wavelengths. Data collection and processing procedures were conducted similar to those described by Greenham et al.32 Temperature dependence of the photoluminescence was measured in the temperature range from 77 K to 493 K using a home-built fluorimeter and incorporating a cryostat for low temperature measurements and a heating stage for high temperature data collection.
Electroluminescence spectra, (CIE) chromaticity coordinates, and color rendering index (Ra) were collected by incorporating the samples into a device. A “capping” strategy was used, described previously by our group,15,33 in which the phosphor powder (5 wt% to 6 wt%) was mixed into silicon resin (GE Silicones, RTV-615) and molded into a cap that was then placed over a silicone encapsulated InGaN LED chip (λmax = 400 nm, 8 mm, FrozenCPU). A commercially available red phosphor (1 wt%) from Mitsubishi Chemical Corporation was also added for select measurements. The measurements were conducted in an integrating sphere using a forward bias current of 20 mA.
3 Results and discussion
Sr2BaAlO4F (SBAF) crystallizes in the tetragonal space group I4/mcm (no. 140) while the nearly isostructural Sr3SiO5 (SSO) crystallizes in the tetragonal space group P4/ncc (no. 130). In the SBAF structure, Sr2+ ions reside in the 8h site, Ba2+ ions the 4a site, Al3+ ions the 4b site, O2− ions the 16l site and F− ions the 4c site.34 In the SSO structure, Sr2+ ions reside in the 8f and 4c sites, Si4+ ions the 4b site and O2− ions the 16g and 4c sites.35Fig. 1 portrays the structural differences between the two end members, highlighted by the tilted SiO4 tetrahedra in the SSO structure which lends this compound to the lower symmetry space group. In both structures, there are two sites that Ce3+ can be substituted into based on ionic size and coordination number,36,37 summarized in Table 1. The first is the 8h site in the SBAF structure and the 8f site in the SSO structure, which is coordinated with six oxygen and two fluorine atoms, in which the slightly smaller Ce3+ ion can reside. The second is the 4a site in the SBAF structure and the 4c site in the SSO structure, which is coordinated with eight oxygen and two fluorine atoms, in which the smaller Ce3+ ion can also reside.
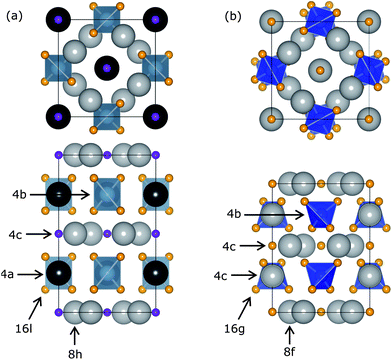 |
| | Fig. 1 Crystal structures of (a) Sr2BaAlO4F and (b) Sr3SiO5 viewed along the c-axis (top) and the a-axis (bottom) with labeled Wyckoff positions. Sr2+/Ba2+ (grey, black) and Al3+/Si4+ (blue) ions occupy similar sites in both end members, while the O2− (orange) and F− (purple) ions have slightly different positions leading to a lower symmetry structure for Sr3SiO5. | |
Table 1 Comparison of effective ionic radii shows that Ce3+ can potentially be substituted into two different crystallographic sites in both the SBAF and SSO structures, replacing Sr2+ and/or Ba2+ ions
| Structure |
Site |
CN |
Effective ionic radii (Å) |
| Sr2+ |
Ba2+ |
Ce3+ |
| SBAF |
8h |
8 |
1.26 |
1.42 |
1.143 |
| SSO |
8f |
|
|
|
|
| SBAF |
4a |
10 |
1.36 |
1.52 |
1.25 |
| SSO |
4c |
|
|
|
|
DFT was used to calculate site preference for substituting La3+ into the SBAF lattice. La3+ substitution was chosen to model Ce3+ substitution, since they are similar in ionic size and charge, (CN = 8, reff,Ce3+ = 1.143 Å, reff,La3+ = 1.160 Å; CN = 10, reff,Ce3+ = 1.25 Å, reff,La3+ = 1.27 Å) and La3+ does not require calculations of f-electrons, as Ce3+ would. Two types of substitutions were examined: La3+ substituting into the 8h site in place of Sr2+ and La3+ substituting into the 4a site in place of Ba2+. Charge compensation for replacing a +2 species with La3+ was taken into consideration and balanced, through F−/O2− ratios, as we would expect to occur. Total ground state energy calculations were performed for bulk SBAF, and the two substitution cases. The conventional unit cell was used, corresponding to 4 formula units and a total of 36 atoms. The first substitution renders the formula Sr1.75BaLa0.25AlO4.25F0.75 and the second Sr2Ba0.75La0.25AlO4.25F0.75. To determine the lowest energy ground state for the unit cells with substitutions, ionic positions, unit cell volume and shape were relaxed.
Total energies for bulk SBAF and substitutions are presented in Table 2. Both types of substitutions result in a larger negative total energy, indicating the substitution is favorable and will occur. Fig. 2 shows the ionic displacements of the coordinating ions that take place when La substitutions are made. The substitution of La3+ in the 8h site in place of Sr2+ is slightly more energetically favorable than La3+ in the 4a site in place of Ba2+, but only by 0.14 eV. From this, we can expect Ce3+ substitution in SBAF to occur on both sites, with a slightly higher preference for the Sr2+ 8h site. This agrees well with experimental data obtained by Im et al.14 of the emission spectrum Sr1.975BaCe0.025AlO4F at 77 K deconvoluted into four Gaussian curves, corresponding to Ce3+ occupying two distinct sites and a slightly higher preference for CeSr substitution considering the Ce3+ ion is closer in size to the Sr2+ ion than the Ba2+ ion.
Table 2 Ground state energies per conventional unit cell for the SBAF structure without substitution, and with one La3+ ion per unit cell substituted into various crystallographic sites, calculated using density functional theory
| Wyckoff position |
Substitution |
Total energy per unit cell (eV) |
| Bulk SBAF |
|
−233.37 |
| 8h |
LaSr |
−239.17 |
| 4a |
LaBa |
−239.03 |
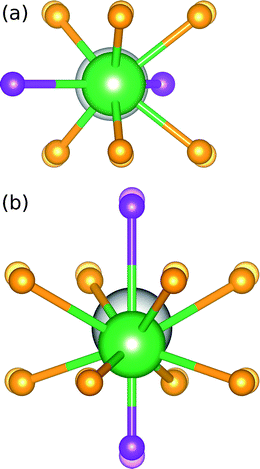 |
| | Fig. 2 Ionic displacements of the coordinating ions when La3+ substitutions are made in place of (a) Sr2+ in the 8h site and (b) Ba2+ in the 4a site, where the substituted La3+ ions are represented by green spheres and the translucent ions represent the ions in the bulk structure prior to substitutions. | |
Synchrotron X-ray diffraction patterns of prepared samples show all are nearly single phase compounds similar to both the SBAF and SSO compounds. The small amount of impurities are likely (Sr,Ba)F2 and (Sr,Ba)2SiO4 and do not appear to contribute to the luminescence. Ce3+-substituted (Sr,Ba)2SiO4 results in weak blue emission, which we have not observed.38 For all compositions, the amount of substituted Ce3+ was held constant corresponding to the optimal amount found for SBAF:Ce3+,14 rendering the formula Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x. Across the solid solution series, increasing values of x correspond to Si4+ ions (CN = 4, reff,Si4+ = 0.26 Å) replacing Al3+ ions (CN = 4, reff,Al3+ = 0.39 Å) and simultaneously O2− ions (CN = 6, reff,O2− = 1.40 Å) replacing F− ions (CN = 6, reff,F− = 1.33 Å). The diffraction patterns in Fig. 3 illuminate the structural evolution as x increases. Rietveld refinements were conducted using both the SBAF structure and the SSO structure as models. Disorder between Sr2+ and Ba2+ in the 8h and 4a sites in the SBAF model and the 8h and 4c sites in the SSO model was considered due to similar ionic size and was included in refinements.39 In further discussions, the Sr/Ba site with a multiplicity of 8 will be referred to as the Sr1/Ba1 site and the Sr/Ba site with a multiplicity of 4 the Sr2/Ba2 site in both structural models. Site disordering between O2− and F− was not considered since it has been found previously from NMR studies15 that F− resides in a single crystallographic site. Refinements yielded successful convergence with similar figures of merit for both the SBAF and the SSO structural models. Fig. 4 shows the similarity in fit for the x = 0.9 sample using each model. This inability to distinguish the space group is most likely due to the fact that the differences arise from displaced O2− and F− ions, both are which are not sensitive to X-ray scattering techniques. Neutron scattering data was also collected for samples x = 0.1, x = 0.5, and x = 0.9, in an effort to enhance the structural model. Pair-distribution functions (PDF) were refined using a least-squares fitting method. Fits to the PDFs are shown in Fig. 5, where the changing peak intensities are indications of a change in the average structure as x increases. The PDFs fit well to both the SBAF and SSO structures, again arising from the similar neutron scattering lengths of oxygen (5.803 fm) and fluorine (5.654 fm). The structural parameters obtained from the PDF refinement agree well with the results from Rietveld refinement of the synchrotron X-ray diffraction data.
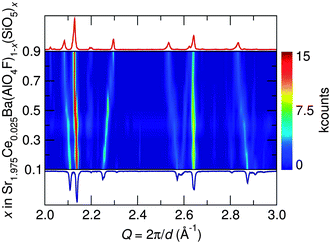 |
| | Fig. 3 Portion of the synchrotron X-ray diffraction patterns, collected at room temperature, for the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x series as a function of x clearly illustrates the structural evolution across the solid solution as demonstrated by the shifts in diffraction peak positions. | |
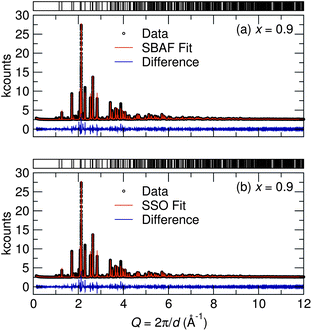 |
| | Fig. 4 Rietveld refinements of synchrotron X-ray diffraction data collected at room temperature for the x = 0.9 sample show good fits to both (a) the SBAF structure and (b) the SSO structure. This inability to distinguish the space group is likely due to the fact that the differences arise from displaced O2− and F− ions, neither of which influence X-ray scattering greatly. | |
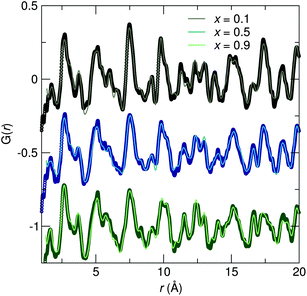 |
| | Fig. 5 Refinements of neutron scattering PDF data collected at room temperature for the x = 0.1, x = 0.5, and x = 0.9 samples show changes in peak intensities across the solid-solution series, corresponding to changes in the average structure, as also seen in synchrotron X-ray diffraction data. | |
The shift in diffraction peaks in Fig. 3 indicates changing lattice parameters, namely a linear increase in the a and b cell parameters and a decrease in the c cell parameter, shown in Fig. 6. Lattice parameters for the x = 0.1 sample were determined to be a = 6.9120(2) Å and c = 11.1634(4) Å with V = 533.31(2) Å3 and for the x = 0.9 sample a = 7.0127(1) Å and c = 10.9486(3) Å with V = 538.47(3) Å3, and intermediate compounds varying linearly obeying the Végard law. Refined structural parameters for x = 0.5 fit to the SBAF structure and to the SSO structure are displayed in Table 3 and select interatomic distances in Table 4. The simultaneous expansion and contraction of the unit cell in different directions causes increased tilting of the (Al,Si)O4 tetrahedra and greater distortion of the (Sr,Ba)(O,F)8 and (Sr,Ba)(O,F)10 polyhedra. A polyhedral distortion index,40D, can be defined as
| | 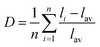 | (1) |
where
li is the distance from the central atom to the
ith coordinating atom and
lav is the average bond length.
Fig. 7 shows the changing polyhedral volume and distortion as
x increases and
Fig. 8 depicts the distorted polyhedra for the
x = 0.1 and
x = 0.9 samples, obtained from fits to the SBAF structure model. Polyhedral volumes were calculated through the method outlined by Swanson and Peterson.
41 This distortion of the active sites where Ce
3+ resides greatly affects the optical properties of the solid-solution phosphor, as discussed later in this paper.
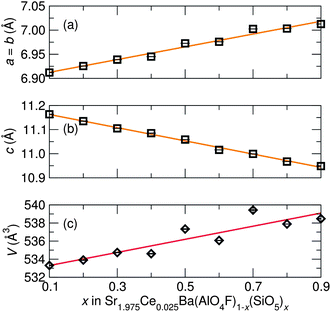 |
| | Fig. 6 Unit cell parameters of Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x from Rietveld refinement of room temperature synchrotron X-ray diffraction data show (a) an expansion in the a lattice parameter and (b) a contraction in the c lattice parameter with (c) an overall increase in cell volume as x increases, reflecting the effects of the Al3+/Si4+ and O2−/F− substitutions. | |
Table 3 Refined structural parameters for Sr2.975Ce0.025BaAl0.5Si0.5O4.5F0.5 (x = 0.5) determined by Rietveld refinement of powder synchrotron X-ray diffraction data collected at room temperature and using a fit to (a) the SBAF model and (b) the SSO model. The values in parentheses represent the average standard deviation
| Atom |
Site |
x
|
y
|
z
|
Occupancy |
|
Space group: I4/mcm (no. 140), Z = 4, a = 6.9726(2) Å, c = 11.0586(4) Å, V = 537.64(3) Å3.
Space group: P4/ncc (no. 130), Z = 4, a = 6.9698(2) Å, c = 11.0611(4) Å, V = 537.33(3) Å3.
|
| Sr2.975Ce0.025BaAl0.5Si0.5O4.5F0.5a |
| Sr1 |
8h |
0.1758(1) |
x + 1/2 |
0 |
0.90(1) |
| Ba1 |
8h |
0.1758(1) |
x + 1/2 |
0 |
0.10(1) |
| Sr2 |
4a |
0 |
0 |
1/4 |
0.21(1) |
| Ba2 |
4a |
0 |
0 |
1/4 |
0.79(1) |
| Al |
4b |
0 |
1/2 |
1/4 |
0.5 |
| Si |
4b |
0 |
1/2 |
1/4 |
0.5 |
| O |
16l |
0.6324(4) |
x + 1/2 |
0.1494(4) |
1 |
| F |
4c |
0 |
0 |
0 |
1 |
| Sr2.975Ce0.025BaAl0.5Si0.5O4.5F0.5b |
| Sr1 |
8f |
0.1750(1) |
x
|
1/4 |
0.88(1) |
| Ba1 |
8f |
0.1750(1) |
x
|
1/4 |
0.12(1) |
| Sr2 |
4c |
0 |
1/2 |
0.4992(7) |
0.24(1) |
| Ba2 |
4c |
0 |
1/2 |
0.4992(7) |
0.76(1) |
| Al |
4b |
0 |
0 |
0 |
0.5 |
| Si |
4b |
0 |
0 |
0 |
0.5 |
| O |
16g |
0.855(1) |
0.121(1) |
0.0985(4) |
1 |
| F |
4c |
0 |
1/2 |
0.250(5) |
1 |
Table 4 Selected interatomic distances corresponding to active site polyhedra found in Sr2.975Ce0.025BaAl0.5Si0.5O4.5F0.5 (x = 0.5) using a fit to (a) the SBAF model and (b) the SSO model from Rietveld refinements of room temperature synchrotron X-ray diffraction data. The values in parentheses represent the average standard deviation
| Atoms |
Distance (Å) |
| Sr2.975Ce0.025BaAl0.5Si0.5O4.5F0.5 |
| Sr1/Ba1–O |
(×4) 2.728(3) |
| (×2) 2.512(3) |
| Sr1/Ba1–F |
(×2) 2.571(1) |
| Sr2/Ba2–O |
(×8) 2.943(3) |
| Sr2/Ba2–F |
(×2) 2.765(0) |
| Sr2.975Ce0.025BaAl0.5Si0.5O4.5F0.5 |
| Sr1/Ba1–O |
(×2) 2.815(6) |
| (×2) 2.666(6) |
| (×2) 2.531(6) |
| Sr1/Ba1–F |
(×2) 2.573(1) |
| Sr2/Ba2–O |
(×4) 2.033(7) |
| (×4) 2.829(7) |
| Sr2/Ba2–F |
(×1) 2.78(6) |
| (×1) 2.75(6) |
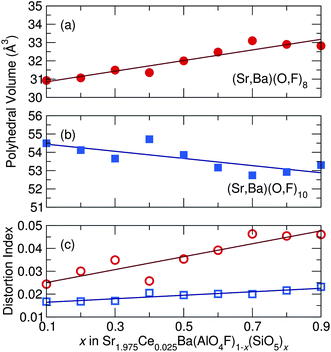 |
| | Fig. 7 As x increases across the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x solid solution series, the (a) (Sr,Ba)(O,F)8 average polyhedral volume increases, the (b) (Sr,Ba)(O,F)10 average polyhedral volume decreases, while the (c) polyhedral distortion of both sites increases as x increases, accounting for the experimentally seen red-shift in emission wavelength. | |
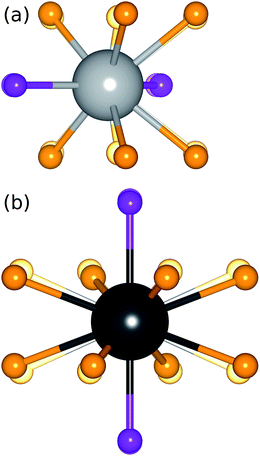 |
| | Fig. 8 Increased distortion of the active site polyhedra, the origin of the red-shifted emission, as x increases across the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x solid solution series can be seen by comparing (a) the Sr1/Ba1 site, viewed along the c-axis, and (b) the Sr2/Ba2 site, viewed along the a-axis, for compositions x = 0.1 (translucent spheres) and x = 0.9 (solid spheres). | |
Bond valence sums (BVS),42 displayed in Fig. 9, were calculated from refinements of synchrotron X-ray diffraction data using the SBAF model to again help determine the most favorable site for Ce3+. The shorter bonds of the Sr1/Ba1 site result in a BVS of around 2 for Sr and Ce on the Sr1/Ba1 site, but a BVS of around 3 for Ba. This shows the valence state of +2 for Sr is satisfied on the Sr1/Ba1 site, but Ba is very over-bonded. This agrees with the refined occupancies for site mixing between Sr and Ba, with more Sr residing in the Sr1/Ba1 site (75–80%) than in the Sr2/Ba2 site. Conversely, the BVS of Ba on the Sr2/Ba2 site is around 2, while that of Sr and Ce is closer to 1.25. This further supports Ba residing on the Sr2/Ba2 site and Sr on the Sr1/Ba1 site, since Sr would be very under-bonded on the Sr2/Ba2 site. Using the average structure Sr/Ba–O/F bond lengths from Rietveld refinement, Ce appears under-bonded in both sites. On the Sr1/Ba1 site, Ce has a BVS of 2, slightly less than the BVS of Ce of 1.25 on the Sr2/Ba2 site. The under-bonding can be reduced by contraction of the Ce–O/F bond lengths, making the local environment of Ce more amenable to the optically active charge state of +3. A contraction of the Sr1/Ba1 bond lengths of around 5% from the average structure facilitates an increase in the BVS of Ce on the Sr1/Ba1 site from around 2 to around 3, which is small compared to what is required on the Sr2/Ba2 site. An average bond length contraction of 11.5% on the Sr2/Ba2 site brings the Ce BVS near 3. The smaller contraction of Ce–O/F bonds of the Sr1/Ba1 site compared with Ce–O/F bonds of the Sr2/Ba2 site required to make the BVS of Ce = 3 indicates Ce should be more easily incorporated on the Sr1/Ba1 site.
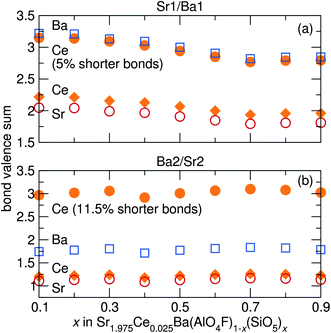 |
| | Fig. 9 Bond valence sums of Ba, Sr, and Ce on (a) the Sr1/Ba1 site and (b) the Sr2/Ba2 site in Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x, calculated using refined bond distances from synchrotron X-ray diffraction data, show Sr preferentially resides in the Sr1/Ba1 site, while Ba prefers the Sr2/Ba2 site. The BVS of Ce on both sites with slightly shorter bonds are shown, indicating Ce3+ is more easily incorporated into the Sr1/Ba1 site due to the smaller contraction in bond length required. | |
The structural information gleaned from synchrotron X-ray diffraction is useful for analysis of the average structure of the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x phosphors, but the small amount of Ce3+ substituted into the lattice prohibits using conventional scattering techniques to probe the structure of the lattice around Ce3+. Electron paramagnetic resonance (EPR) spectroscopy provides a way to selectively probe unpaired spins in the system, such as Ce3+, enabling a detailed examination of the local structure of Ce3+ in the lattice. In the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x structure, the Sr1/Ba1 site has point-group symmetry of 2, while the Sr2/Ba2 site has point-group symmetry of ![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) . Both sites have axial symmetry, resulting in only two components of the g-tensor, g∥ and g⊥.43,44 The powder EPR spectra for the compositions x = 0.1, 0.3, 0.5, 0.7, and 0.9, shown in Fig. 10, display very wide lineshapes, indicative of a large distribution of local Ce3+ environments. This may be from the site mixing of Sr and Ba in the lattice as observed from the synchrotron X-ray analysis, or may be from the charge compensation mechanisms that are expected to be present due to the substitution of Ce3+ for Sr2+/Ba2+. The peak positions do not appear to move substantially with composition, likely due to the shielded nature of the 4f orbitals, which are not strongly affected by the local bonding environment. The small features near 1700 G in the x = 0.9 spectra and near 3300 G in the x = 0.3, x = 0.5, and x = 0.7 spectra are from Ce3+ in impurity phases. Table 5 lists the components of the g-tensors of both sites for all compositions.
. Both sites have axial symmetry, resulting in only two components of the g-tensor, g∥ and g⊥.43,44 The powder EPR spectra for the compositions x = 0.1, 0.3, 0.5, 0.7, and 0.9, shown in Fig. 10, display very wide lineshapes, indicative of a large distribution of local Ce3+ environments. This may be from the site mixing of Sr and Ba in the lattice as observed from the synchrotron X-ray analysis, or may be from the charge compensation mechanisms that are expected to be present due to the substitution of Ce3+ for Sr2+/Ba2+. The peak positions do not appear to move substantially with composition, likely due to the shielded nature of the 4f orbitals, which are not strongly affected by the local bonding environment. The small features near 1700 G in the x = 0.9 spectra and near 3300 G in the x = 0.3, x = 0.5, and x = 0.7 spectra are from Ce3+ in impurity phases. Table 5 lists the components of the g-tensors of both sites for all compositions.
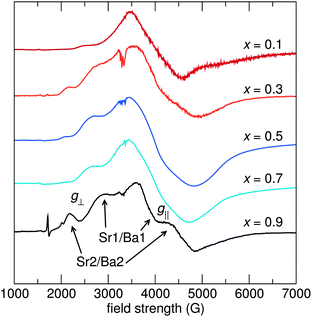 |
| | Fig. 10 Powder EPR spectra of Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x taken at 10 K on an X-band EPR spectrometer (ν = 9.4 GHz) indicate a large distribution of local Ce3+ environments, with a disproportionate amount of Ce3+ favoring the Sr1/Ba1 site over the Sr2/Ba2 site. | |
Table 5 Components of the g-tensor from 10 K powder EPR measurements of Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x shown in Fig. 10
| Sr1/Ba1 site (8h) |
Sr2/Ba2 site (4a) |
| Sample |
g
∥
|
g
⊥
|
Sample |
g
∥
|
g
⊥
|
|
x = 0.1 |
2.71 |
1.71 |
x = 0.1 |
2.69 |
1.66 |
|
x = 0.3 |
2.53 |
1.67 |
x = 0.3 |
3.12 |
1.56 |
|
x = 0.5 |
2.54 |
1.69 |
x = 0.5 |
3.22 |
1.71 |
|
x = 0.7 |
2.55 |
1.70 |
x = 0.7 |
3.24 |
1.70 |
|
x = 0.9 |
2.31 |
1.67 |
x = 0.9 |
3.09 |
1.58 |
The larger peaks have been attributed to the Sr1/Ba1 site since this site is twice as abundant in the crystal structure compared to the Sr2/Ba2 site and analyses of bond valence sums and DFT calculations indicate that Ce3+ is more easily incorporated into the Sr1/Ba1 site. The other peaks, attributed to the Sr2/Ba2 site, are much smaller than the Sr1/Ba1 peaks, indicating much more Ce3+ resides on the Sr1/Ba1 site. The ratio of Ce3+ on the Sr1/Ba1:Sr2/Ba2 sites changes non-systematically with x, as exemplified by the changing intensity in the peak near 2200 G from the Sr2/Ba2 site compared with the intensity of the peaks from the Sr1/Ba1 site. This peak at 2200 G is the least prevalent in the x = 0.7 material, which also has the lowest quantum efficiency of all the compositions studied here. The intensity of this peak, and the inferred amount of Ce3+ substituted on the Sr2/Ba2 site, correlates well with the quantum efficiency reported later in this paper – high quantum efficiency correlates with a large Sr2/Ba2 substitution level. This seems counter-intuitive, since multiple substitution sites should increase excitation/emission overlap, thereby decreasing quantum efficiency. The correlation between high quantum efficiency and large relative Ce3+ substitution on the Sr2/Ba2 site may be due to a higher quantum efficiency of Ce3+ on this site compared to the Sr1/Ba1 site, or Ce3+ on the Sr2/Ba2 may sensitize Ce3+ emission on the Sr1/Ba1 site.
Excitation and emission spectra for all samples were collected using the relative maxima in emission/excitation intensity found for each sample. The resulting excitation and emission spectra are displayed in Fig. 11, along with that of YAG:Ce3+ for comparison, and photoluminescence data in Table 6. The maximum excitation wavelength exhibits a slight red-shift from 398 nm to 409 nm as x increases, while the maximum emission wavelength and the Commission Internationale de L'Eclairage (CIE) chromaticity coordinates, shown in Fig. 12, exhibit a large red shift as x increases, from λem = 523 nm for x = 0.1 to λem = 552 nm for x = 0.9. This same trend in photoluminescent properties as x increases was also seen in the isostructural oxyfluoride solid solution developed by Im et al.15 Yet the introduction of Ba2+ into the host crystal results in a greater red shift in emission compared to the previous solid solution phosphor. The local environment around the Ce3+ activator ions is altered by the substitution of different cations and anions into the host lattice. In this compound, emission arises from electronic transitions in Ce3+ from the 5d (2D3/2, 2D5/2) excited state to the 4f (2F5/2, 2F7/2) ground state.45 The change in bond lengths from Ce3+ ions to their O2− and F− ligands, the degree of distortion of the Ce(O,F)8 and Ce(O,F)10 polyhedra, and the change in lattice covalency dictated by the relative change in charged species all result in changes in the crystal field splitting of the Ce3+ 5d levels. The crystal field splitting46 can be modeled by
| | 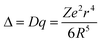 | (2) |
where Δ or
Dq is the crystal field for octahedral symmetry,
R is the distance between the central ion and its
ligands,
Z is the charge or valence of the anion,
e is the charge of the electron, and
r is the radius of the
d wavefunction.
Fig. 7, discussed above, illustrates the changing polyhedral environments around the Ce
3+ activator ions. The (Sr,Ba)(O,F)
8 average polyhedral volume increases by about 2.5 Å
3, which would indicate a blue-shift based on conventional crystal field splitting, while the (Sr,Ba)(O,F)
10 average polyhedral volume decreases, which would lead to a red-shift. Yet with twice as many (Sr,Ba)(O,F)
8 polyhedra than (Sr,Ba)(O,F)
10 in the unit cell and a greater change in volume, we would expect and overall blue-shift in emission wavelength. Yet, the experimentally seen red-shift contradicts this expected blue-shift.
46,47 One explanation for this unconventional red-shift in emission, which has been seen in garnet systems,
48–51 is the increased distortion of the active site polyhedra causing an increased crystal field splitting and red-shift. Although the (Sr,Ba)(O,F)
8 average polyhedral volume does increase, the coordination around the Sr1/Ba1 and Sr2/Ba2 sites becomes increasingly anisotropic as
x increases due to alloying of Al
3+/Si
4+.
52–54Fig. 8 depicts this increased polyhedral distortion around the Sr1/Ba1 site and the Sr2/Ba2 site for the
x = 0.9 composition compared to the
x = 0.1 composition. Additionally, increased lattice covalency (nephalauxetic effect),
55–59 caused by replacing the more electronegative F
− with the less electronegative O
2−, contributes to a red-shift in both the excitation and emission. The slight change in excitation wavelength in the phosphor series and the drastic change in emission wavelength represent the changing crystal field around Ce
3+ sites and the changing covalency of the lattice having different effects on the Ce
3+ excited state and ground state energy levels.
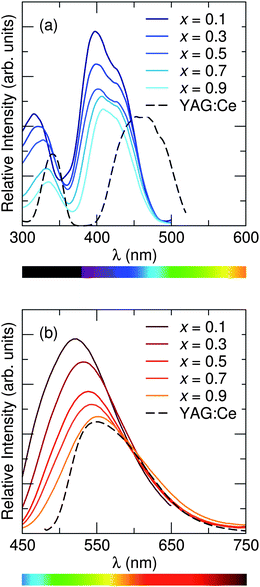 |
| | Fig. 11 Photoluminescence (a) excitation spectra and (b) emission spectra of Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x, collected at room temperature, show that the maximum excitation wavelength exhibits a slight red shift across the solid solution series as x increases, while the maximum emission wavelength shows an even greater shift to longer wavelengths. Excitation/emission spectra were recorded using the maximum emission/excitation wavelength. | |
Table 6 Optical properties of the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x solid solution phosphor at room temperature depict an easily tunable system that can vary emission color from green to yellow with a broad emission spectrum and high quantum yield
| Sample |
λ
ex (nm) |
λ
em (nm) |
FWHM (nm) |
CIE x, y |
PLQY (%) |
|
x = 0.1 |
398 |
523 |
117 |
0.31, 0.49 |
70 |
|
x = 0.3 |
399 |
532 |
121 |
0.35, 0.51 |
70 |
|
x = 0.5 |
402 |
539 |
125 |
0.36, 0.52 |
63 |
|
x = 0.7 |
407 |
542 |
128 |
0.38, 0.52 |
54 |
|
x = 0.9 |
409 |
552 |
133 |
0.41, 0.52 |
67 |
| YAG:Ce3+ |
465 |
550 |
111 |
0.43, 0.55 |
87 |
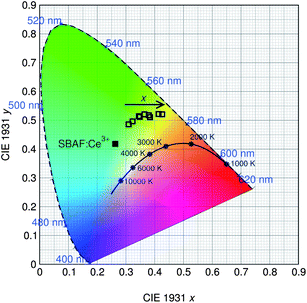 |
| | Fig. 12 CIE color coordinates of the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x phosphor series, along with the SBAF:Ce3+ end member, show the true color of the phosphor taking into account the broad emission spectra. Emission of the phosphor is tuned from green to yellow as x increases due to the increased distortion of the active site polyhedra. | |
The shift in emission wavelength with composition allows for the color properties of a solid state device to be easily tuned for different applications. The emission spectra of all compositions of the phosphor series are greatly red shifted from the green emission of the SBAF:Ce3+ end member, making these compositions promising candidates for white lighting. Additionally, the full-width at half-maximum (FWHM) of the emission profile is relatively broad for all samples, ranging from 117 nm to 133 nm as x increases, covering a wide range of the visible spectrum which will lead to high color rendering white LEDs. The x = 0.9 composition in particular can be used to create high quality white light without the need for an additional red component. The moisture stability of the compounds was also increased from the SBAF end member. The compositions x = 0.5 and greater are stable under ambient laboratory storage conditions.
Photoluminescent quantum yield (PLQY) was measured to be 70% at room temperature for the most efficient samples, x = 0.1 and x = 0.3, with the lowest being 54% for x = 0.7. Thermal performance of the photoluminescence for x = 0.5 and x = 0.9 samples are shown in Fig. 13. High temperature emission spectra were collected from room temperature to 220 °C to examine the thermal quenching of luminescence due to increased non-radiative transition probability in the configurational coordinate diagram45 with increasing temperature. The maximum PL intensity at 160 °C decreased to 82% and 52% of their room temperature values for samples x = 0.5 and x = 0.9, respectively, while YAG:Ce3+ decreases to 64% of its room temperature value at 160 °C. The activation energy for thermal quenching can be calculated using the Arrhenius equation60,61 which obeys
| | 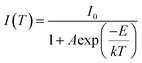 | (3) |
where
I0 is the initial intensity,
I(
T) is the intensity at a given temperature
T,
A is a constant,
E is the activation energy for thermal quenching and
k is the Boltzmann constant. The
activation energy for thermal quenching was determined to be 0.31 eV and 0.14 eV for
x = 0.5 and
x = 0.9, respectively. The
x = 0.5 composition therefore exhibits impressive thermal stability, retaining more of its PL intensity at elevated temperatures than commercial YAG:Ce
3+. It has been suggested previously that in the fluoride compounds,
14 the high efficiency and low thermal quenching arise from the softer phonon modes associated with fluorine atoms in the host lattice.
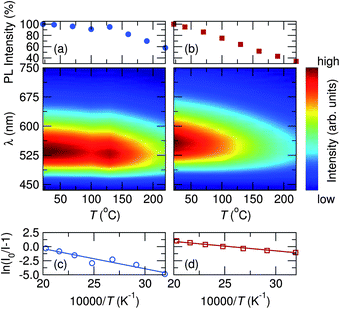 |
| | Fig. 13 Temperature dependent PL data for x = 0.5 (circles) and x = 0.9 (squares) compositions of the Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x phosphor series show (a and b) weak thermal quenching from room temperature to 220 °C and (c and d) a large activation energy for thermal quenching. | |
The quality of the resulting white light produced by using this solid solution phosphor was examined by incorporating the phosphor into a device with an InGaN LED (λmax = 400 nm). The “capping” strategy used here is depicted in Fig. 14. This phosphor deployment strategy allows for optimal light extraction in addition to reproducibility in measurements since the same LED can be used for all subsequent measurements. This strategy also allows us to calculate an energy efficiency of the phosphor when used with an LED. The energy efficiency33 of the phosphor (η) can be calculated using
| | 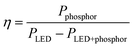 | (4) |
where
Pphosphor is the integrated radiometric power of the phosphor emission, and
PLED+phosphor and
PLED are the integrated radiometric powers of the near-UV
photon emission spectra with and without the phosphor cap, respectively. This energy efficiency includes the conversion and scattering efficiency of the phosphor as well as the extraction efficiency of the phosphor cap.
 |
| | Fig. 14 Photograph of the phosphor silicone ‘cap’ on a silicone-encapsulated LED chip when (a) the device is off and (b) illuminated, demonstrated using the x = 0.9 phosphor sample. | |
Fig. 15 shows electroluminescence spectra and CIE chromaticity coordinates for devices made using samples x = 0.5 and x = 0.9. For x = 0.5, devices were also made by adding a commercially available red phosphor from Mitsubishi Chemical Corporation to obtain a white light closer to the Planckian locus of black body radiation. Table 7 lists the calculated values for phosphor energy efficiency, luminous efficacy, correlated color temperature (CCT) and color rendering (Ra). At room temperature, values for η ranged from 37% to 43% and values for the luminous efficacy ranged from 12 lm W−1 to 20 lm W−1. The relatively low luminous efficacy numbers, compared to the blue-LED + YAG:Ce combination, is a consequence of the near-UV LEDs being not as bright, combined with the larger Stokes shift. High color rendering indices were obtained for all samples due to the broad nature of the emission profile, namely x = 0.9 with Ra = 70 and a CCT = 6815 K, with additional improvements through adding a red component to x = 0.5 for an Ra = 90 with CCT = 4113 K. The results indicate that this solid solution phosphor has strong potential applications in developing high-efficiency, thermally stable, warm-white LEDs.
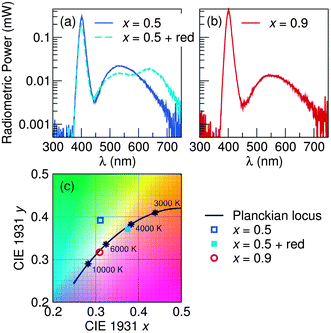 |
| | Fig. 15 Electroluminescence data collected at room temperature for devices assembled using the phosphor compositions (a) x = 0.5 and (b) x = 0.9, in conjunction with a near-UV InGaN LED (λmax = 400 nm) operating under a forward bias current of 20 mA. The (c) CIE chromaticity coordinates are plotted for each device, showing the color quality of the white light produced. | |
Table 7 Properties calculated from electroluminescence data collected at room temperature for the phosphor-converted white light using a InGaN LED chip (λmax = 400 nm) operating under a forward bias current of 20 mA. Data for YAG:Ce3+ was collected using a blue InGaN LED (λmax = 450 nm) also operating under a forward bias current of 20 mA
| Sample |
η (%) |
Efficacy (lm W−1) |
CIE |
CCT (K) |
R
a
|
|
x = 0.5 |
40 |
19 |
0.31, 0.39 |
6264 |
68 |
|
x = 0.9 |
37 |
12 |
0.31, 0.32 |
6815 |
70 |
|
x = 0.5 + red |
43 |
16 |
0.37, 0.37 |
4113 |
90 |
| YAG:Ce3+ |
51 |
122 |
0.37, 0.42 |
4555 |
65 |
4 Conclusions
In conclusion, a Ce3+-doped oxyfluoride solid solution phosphor has been prepared from the Sr2BaAlO4F and Sr3SiO5 end members. Sr1.975Ce0.025Ba(AlO4F)1−x(SiO5)x phosphors have easy color tunability, by changing the composition x, with near-UV excitation and green/yellow emission wavelengths optimal for solid state white lighting applications. With a highly efficient photoluminescent quantum yield of 70% at room temperature and photoluminescence intensity decreasing to only 82% of its room temperature value at 160 °C, this solid solution phosphor exhibits impressive thermal stability. Devices fabricated using InGaN LEDs with λmax = 400 nm showed excellent white light characteristics, with a color rendering of Ra = 70 and CCT near 6815 K, eliminating the need for an additional red component. As such, this newly developed solid solution phosphor shows great potential for use in high efficiency, thermally stable white LEDs.
Acknowledgements
We thank Alexander Birkel for helpful discussions. Fellowship support to KAD and NCG from the ConvEne IGERT Program (NSF-DGE 0801627) is gratefully acknowledged. The research reported here made use of MRL Central Facilities, supported by the MRSEC Program of the NSF under Award no. DMR 1121053. We also acknowledge support from the UCSB Center for Scientific Computing (NSF DMR-1121053 and NSF CNS-0960316). Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract no. DE-AC02-06CH11357. The research at Oak Ridge National Laboratory's Spallation Neutron Source was sponsored by the Scientific User Facilities Division, Office of Basic Energy Sciences, U. S. Department of Energy.
References
- S. Pimputkar, J. S. Speck, S. P. DenBaars and S. Nakamura, Nat. Photonics, 2009, 3, 180–182 CrossRef CAS.
- S. Nakamura, MRS Bull., 2009, 34, 101–107 CrossRef CAS.
- T. Hashimoto, F. Wu, J. S. Speck and S. Nakamura, Nat. Mater., 2007, 6, 568–571 CrossRef CAS.
- E. F. Schubert and J. K. Kim, Science, 2005, 308, 1274–1278 CrossRef CAS.
- T. Jüstel, H. Nikol and C. Ronda, Angew. Chem., Int. Ed., 1998, 37, 3084–3103 CrossRef.
- S. Ye, F. Xiao, Y. Pan, Y. Ma and Q. Zhang, Mater. Sci. Eng., R, 2010, 71, 1–34 CrossRef.
- C. Feldmann, T. Jüstel, C. Ronda and P. Schmidt, Adv. Funct. Mater., 2003, 13, 511–516 CrossRef CAS.
- G. Blasse and A. Bril, Appl. Phys. Lett., 1967, 11, 53–55 CrossRef CAS.
- G. Blasse and A. Bril, J. Chem. Phys., 1967, 47, 5139 CrossRef CAS.
- V. Bachmann, C. Ronda and A. Meijerink, Chem. Mater., 2009, 21, 2077–2084 CrossRef CAS.
- W. B. Im, Y.-I. Kim, N. N. Fellows, H. Masui, G. A. Hirata, S. P. DenBaars and R. Seshadri, Appl. Phys. Lett., 2008, 93, 091905 CrossRef.
- W. B. Im, N. N. Fellows, S. P. DenBaars and R. Seshadri, J. Mater. Chem., 2009, 19, 1325–1330 RSC.
- W. B. Im, Y. Fourré, S. Brinkley, J. Sonoda, S. Nakamura, S. P. DenBaars and R. Seshadri, Opt. Express, 2009, 17, 22673–22679 CrossRef CAS.
- W. B. Im, S. Brinkley, J. Hu, A. Mikhailovsky, S. P. DenBaars and R. Seshadri, Chem. Mater., 2010, 22, 2842–2849 CrossRef CAS.
- W. B. Im, N. George, J. Kurzman, S. Brinkley, A. Mikhailovsky, J. Hu, B. F. Chmelka, S. P. DenBaars and R. Seshadri, Adv. Mater., 2011, 23, 2300–2305 CrossRef CAS.
- K. Uheda, Key Eng. Mater., 2009, 403, 15–18 CrossRef CAS.
- Y. Q. Li, A. C. A. Delsing, G. D. With and H. T. Hintzen, Powder Diffr., 2005, 3242–3248 CAS.
- R.-J. Xie and N. Hirosaki, Sci. Technol. Adv. Mater., 2007, 8, 588–600 CrossRef CAS.
- A. A. Setlur, W. J. Heward, M. E. Hannah and U. Happek, Chem. Mater., 2008, 20, 6277–6283 CrossRef CAS.
- J. A. Kechele, C. Hecht, O. Oeckler, P. J. Schmidt, W. Schnick and D. Mu, Chem. Mater., 2009, 21, 1288–1295 CrossRef CAS.
- C. Hecht, F. Stadler, P. J. Schmidt, V. Baumann and W. Schnick, Chem. Mater., 2009, 21, 1595–1601 CrossRef CAS.
- H. Höppe, H. Lutz, P. Morys, W. Schnick and A. Seilmeier, J. Phys. Chem. Solids, 2000, 61, 2001–2006 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1993, 47, 558 CrossRef CAS.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter, 1994, 49, 14251 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter, 1996, 54, 11169 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B: Condens. Matter, 1994, 50, 17953 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1997, 78, 1396 CrossRef CAS.
- G. Baldinozzi, J.-F. Bérar and G. Calvarin, J. Phys.: Condens. Matter, 1997, 9, 9731–9744 CrossRef CAS.
- N. Greenham, I. Samuel, G. Hayes, R. Phillips, Y. Kessener, S. Moratti, A. Holmes and R. Friend, Chem. Phys. Lett., 1995, 241, 89–96 CrossRef CAS.
- S. E. Brinkley, N. Pfaff, K. A. Denault, Z. Zhang, H. T. B. Hintzen, R. Seshadri, S. Nakamura and S. P. DenBaars, Appl. Phys. Lett., 2011, 99, 241106 CrossRef.
- T. Vogt, P. Woodward, B. Hunter, A. Prodjosantoso and B. Kennedy, J. Solid State Chem., 1999, 144, 228–231 CrossRef CAS.
- J. K. Park, C. H. Kim, S. H. Park, H. D. Park and S. Y. Choi, Appl. Phys. Lett., 2004, 84, 1647–1649 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- Y. Q. Jia, J. Solid State Chem., 1991, 187, 184–187 CrossRef.
- P. V. Kelsey Jr and J. J. Brown Jr, J. Electrochem. Soc., 1976, 123, 1384 CrossRef.
- A. Prodjosantoso, B. Kennedy, T. Vogt and P. Woodward, J. Solid State Chem., 2003, 172, 89–94 CrossRef CAS.
- W. H. Baur, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1974, 30, 1195–1215 CrossRef CAS.
- D. K. Swanson and R. C. Peterson, Can. Mineral., 1980, 18, 153–156 CAS.
- N. E. Brese and M. O'Keeffe, Acta Crystallogr., Sect. B: Struct. Sci., 1991, 47, 192–197 CrossRef.
- L. Pidol, O. Guillot-Noël, A. Kahn-Harari, B. Viana, D. Pelenc and D. Gourier, J. Phys. Chem. Solids, 2006, 67, 643–650 CrossRef CAS.
-
Electron Paramagnetic Resonance: A Practitioner's Toolkit, ed. M. Brustolon and E. Giamello, Wiley, 2008 Search PubMed.
-
G. Blasse and B. C. Grabmaier, Luminescent Materials, Springer-Verlag, Berlin, 1994 Search PubMed.
- P. D. Rack and P. H. Holloway, Mater. Sci. Eng., R, 1998, 21, 171–219 CrossRef.
-
Phosphor Handbook, ed. S. Shionoya and W. M. Yen, CRC Press, New York, 1998 Search PubMed.
- J. L. Wu, G. Gundiah and A. Cheetham, Chem. Phys. Lett., 2007, 441, 250–254 CrossRef CAS.
- W. W. Holloway, Jr and M. Kestigian, Phys. Lett. A, 1967, 25, 614–615 CrossRef.
- W. W. Holloway, Jr and M. Kestigian, J. Opt. Soc. Am., 1969, 59, 60 CrossRef.
- J. M. Robertson, M. W. van Tol, W. H. Smits and J. P. H. Heynen, Philips J. Res., 1981, 36, 15–30 CAS.
-
T. Egami and S. J. L. Billinge, Underneath the Bragg Peaks: Structural Analysis of Complex Materials, Pergamon, Amsterdam, 2003 Search PubMed.
- T. Egami, Annu. Rev. Mater. Res., 2007, 37, 297–315 CrossRef CAS.
- I. K. Jeong, F. Mohiuddin-Jacobs, V. Petkov, S. J. L. Billinge and S. Kycia, Phys. Rev. B: Condens. Matter, 2001, 63, 205202 CrossRef.
-
C. K. Jörgensen, Modern Aspects of Ligand Field Theory, North-Holland, Amsterdam, 1971 Search PubMed.
- P. Dorenbos, Phys. Rev. B: Condens. Matter, 2000, 62, 15640–15649 CrossRef CAS.
- P. Dorenbos, Phys. Rev. B: Condens. Matter, 2001, 64, 125117 CrossRef.
- P. Dorenbos, Phys. Rev. B: Condens. Matter, 2002, 65, 235110 CrossRef.
- P. Dorenbos, J. Andriessen and C. van Eijk, J. Solid State Chem., 2003, 171, 133–136 CrossRef CAS.
- S. Bhushan and M. V. Chukichev, J. Mater. Sci. Lett., 1988, 7, 319–321 CrossRef CAS.
- Y. Chen, B. Liu, C. Shi, G. Ren and G. Zimmerer, Nucl. Instrum. Methods Phys. Res., Sect. A, 2005, 537, 31–35 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.