 Open Access Article
Open Access ArticleElucidating the morphological and structural evolution of iron oxide nanoparticles formed by sodium carbonate in aqueous medium†
Cristina
Blanco-Andujar
ab,
Daniel
Ortega
ab,
Quentin A.
Pankhurst
ab and
Nguyen Thi Kim
Thanh
*ab
aDepartment of Physics and Astronomy, University College London, Gower Street, London, UK WC1E 6BT. E-mail: ntk.thanh@ucl.ac.uk; Fax: +44 (0)207-670-2920; Tel: +44 (0)207-491-6509
bDavy-Faraday Research Laboratory, The Royal Institution of Great Britain, 21 Albemarle Street, London, UK W1S 4BS
First published on 18th April 2012
Abstract
Ferrimagnetic iron oxides are the common choice for many current technologies, especially those with application in biology and medicine. Despite the comprehensive knowledge accumulated about their chemistry in the bulk state, the sequence of changes taking place during the precipitation of iron oxide nanoparticles in aqueous media is much less extensive. We show that using sodium carbonate as a co-precipitating agent for the synthesis of uncoated iron oxide nanoparticles, the reaction proceeds sufficiently slowly to enable a detailed study of both the reaction pathway and products. The effect of pH, temperature and reaction time on particle size, morphology, crystalline phase and its magnetic properties was investigated. The obtained nanoparticles showed an increase in average particle size of about 10 nm per pH unit for the magnetite phase leading to 6.9 ± 0.4 nm, 18 ± 3 nm and 28 ± 5 nm for pH 8, 9 and 10 respectively. Goethite was initially formed by an olation mechanism at room temperature, followed by a slow transformation into magnetite over a 24 h period, as tracked by X-ray diffraction. In another set of experiments where the reaction temperatures were varied, magnetite was obtained directly by the oxolation mechanism at temperatures above 45 °C. The optimization of the experimental parameters led to superparamagnetic nanoparticles with a high saturation magnetization of 82 A m2 kg−1 at 300 K when synthesized at pH 9.
1 Introduction
The first reported synthetic magnetite nanoparticles were made in 1852 by Lefort via a co-precipitation method.1 Since then, the synthesis of iron oxide nanoparticles has seen a vast development, giving rise to a wide variety of chemical methods such as inverse microemulsion,2,3 sol–gel synthesis,4 flow injection,5 electrospray synthesis,6 the sonochemical method,7 hydrothermal synthesis8 or thermal decomposition.9 The shape, size and composition of nanoparticles (NPs) can be tuned depending on reaction conditions, such as pH, temperature, atmosphere, use of surfactants, ionic strength of the medium or relative ratio of the reagents. Nonetheless, most of the above mentioned methods usually incorporate complex post-synthesis purification and present low reproducibility, which limits their scalability.As such, among the available methods for the synthesis of iron oxide nanoparticles, co-precipitation is the most commonly used method10,11 due to its simplicity and the possibility of obtaining large quantities of nanoparticles in a single batch. However, control over particle size, morphology and composition is limited as particle growth is kinetically controlled. Additionally, factors such as the nature of the precursor salts used (chlorides, perchlorates, sulfates, nitrates, etc.), Fe(II)/Fe(III) ratio, pH and ionic strength of the medium are known to affect particle growth.12–16 The use of bases such as sodium hydroxide or ammonia constitutes a typical choice in many protocols; this is due to their benefit as effective precipitating agents directly leading to iron oxide with an inverse spinel structure. In fact, in recent years research directed towards bioapplications of magnetic nanoparticles has been based on Massart's method,17 which is one of the most cited co-precipitation procedures in aqueous media. Within these bioapplications, magnetic hyperthermia has been a particular focus of interest since it was revealed that magnetic resonance contrast agents presented good heating performance under the action of an AC field. Nonetheless, commercially available nanoparticles prepared using the precipitation method were found to present significant batch-to-batch performance differences. This was pointed out by studies focused on their use in magnetic hyperthermia-based treatments.18
This limited control on the production characteristics of magnetic nanoparticles has been a drawback for some applications to date. This is particularly so for biomedical applications, where stringent quality control and adherence to GMP (good manufacturing practice) standards are a determinant of whether or not a material may be granted regulatory approval for use. At the same time, there are many opportunities for the use of magnetic nanoparticles in biomedicine and healthcare. The main interest within the medical field has focused on superparamagnetic nanoparticles, which are single-domain particles that experience a thermal randomization of their magnetic moments at room temperature in the absence of an externally applied magnetic field. In biomedicine they have been used as contrast agents in magnetic resonance imaging (MRI),19 for drug delivery20,21 or biosensing22 or, most recently, magnetic particle imaging (MPI),23,24 among other applications.25,26 The ferrimagnetic iron oxides – i.e. maghemite (γ-Fe2O3) and magnetite (Fe3O4) – have become the preferred materials due to their relative ease of synthesis, their high magnetic moment and their biocompatibility, being already approved by FDA (US Food and Drug Administration). In this regard, it is worth noting that some of the most successful and acclaimed methods for synthesizing iron oxide nanoparticles27,28 mostly rely on the use of certain precursors that severely reduce their chances to be employed in preparing biocompatible systems. To solve this, thorough post-processing techniques have been commonly used for purification or phase-transfer, but these could certainly result in a partial distortion of the final properties and/or an increase in the production costs, therefore hindering the scalability of the process. Bearing in mind all these considerations, a much simpler and water-based synthesis would be more desirable for producing biocompatible nanoparticles.
Finally, despite the considerable number of publications on the synthesis of iron oxide nanoparticles, there is still a lack of reports dealing with both structure and morphology evolution in nanoparticles at different points of the synthesis from the same initial precursor solution. This could be due to the fact that the formation of iron oxide nanoparticles with inverse spinel structure is a very fast process, which hampers the possibility of tracking this event.13,14 To that aim, a weak base such as sodium carbonate, previously used for studying the early hydrolysis of ferric nitrate solutions,29 would be particularly convenient to slow down the precipitation process.
This work shows the suitability of sodium carbonate for studying the sequence of morphological and structural changes taking place during the precipitation of iron oxide nanoparticles in aqueous media. Particular attention has been paid to the influence of pH, reaction time and temperature over the structural and magnetic properties of the resulting products. A detailed tracking of the subsequent changes has been performed by X-ray diffractometry, transmission electron microscopy and SQUID magnetometry. The set of results provides valuable information that can be used either to improve existing preparation methodologies or elaborate new ones, more appropriate for mass production.
2 Experimental
Ferric chloride hexahydrate (FeCl3·6H2O, >99%), ferrous chloride tetrahydrate (FeCl2·4H2O, 99%) and sodium carbonate (Na2CO3, 99%) were obtained from Sigma Aldrich, UK. Double distilled (dd) degassed water was used for all the experiments. All reagents were used as purchased without any further modification. Iron oxide nanoparticles were synthesized by co-precipitation of ferric and ferrous chloride salts with sodium carbonate under inert atmosphere in a Schlenk line. Briefly, FeCl2·4H2O (0.1988 g, 0.001 mol, 0.02 M) and FeCl3·6H2O (0.5406 g, 0.002 mol, 0.04 M) were dissolved in degassed ddH2O under a nitrogen atmosphere. Sodium carbonate aqueous solution (1 M) was added dropwise until the target pH was reached. The addition of sodium carbonate was carried out at different temperatures between room temperature and 70 °C. The initial samples were taken after the addition of the base and labelled as t = 0 h. All the remaining nanoparticle suspensions were kept under nitrogen for 24 h before any further work was carried out. The obtained nanoparticles were washed by centrifugation (7500 rpm, 10 min, 3 times, 20 ml ddH2O) and redispersed in ddH2O leading to neutral pH. A fraction of the as-obtained product was freeze-dried before XRD characterization.Iron oxide nanoparticles were synthesized at pH 9 as previously described and the obtained reaction mixture split into aliquots. Samples were washed by centrifugation (7500 rpm, 10 min, 3 times, 10 ml ddH2O), redispersed in ddH2O and the obtained product freeze-dried before XRD analysis. The initial sample was assigned as time zero and the remaining samples were processed every hour for a period of 24 h.
Infrared spectra of freeze-dried samples were collected using a Spectrum 100 instrument with Ge/Ge universal ATR from Perkin Elmer. Spectra were acquired in the 650–4000 cm−1 region with a resolution of 2 cm−1, accumulating 16 scans. The morphology, particle size and size distribution of iron oxide and iron oxyhydroxide nanoparticles were examined with a JEOL JEM 1200-EX transmission electron microscope operated at an acceleration voltage of 120 kV. Samples were prepared by dropping the aqueous dispersion onto a carbon-coated copper grid and allowed to air-dry. XRD was conducted on an X-ray diffractometer PanAlytical, using CoKα radiation λ = 1.789010 Å. Samples were prepared by pressing dried powders on a zero background silicon wafer and diffraction patterns were collected within 20–100 2θ (degrees) range. Magnetization curves of iron oxide nanoparticles were carried out in a Quantum Design hybrid Superconducting Quantum Interference Device-Vibrating Sample Magnetometer (SQUID-VSM) at 300 K, with applied fields up to 7 T.
3 Results
3.1 Effect of pH on particle size and morphology
Co-precipitation of a stoichiometric mixture of ferrous and ferric chloride salts was carried out at pH values between 6 and 10 at room temperature to study the effect of pH on the properties of the obtained materials. The experiments were carried out following the procedures detailed in the Experimental section. | ||
| Fig. 1 TEM image of goethite nanoparticles synthesized at RT and pH 6 observed under (a) low magnification and (b) higher magnification. | ||
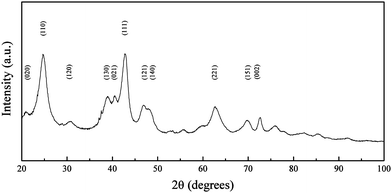 | ||
| Fig. 2 XRD pattern of goethite nanoparticles synthesized at RT and pH 6. Peaks are indexed according to the reference patterns for goethite (pdf ref. 00-029-0713). | ||
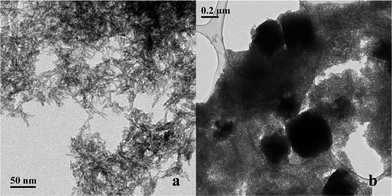 | ||
| Fig. 3 TEM image of multiphase nanoparticles synthesized at RT and pH 7 with (a) goethite acicular nanoparticles and (b) siderite square crystals over goethite nanoparticles. | ||
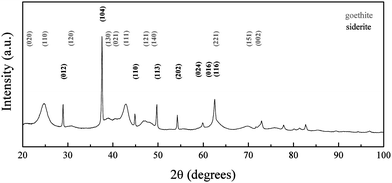 | ||
| Fig. 4 XRD pattern of iron oxide nanoparticles synthesized at RT and pH 7. Peaks are indexed according to the reference patterns for goethite (pdf ref. 00-029-0713) and siderite (pdf ref. 00-029-0696). | ||
 | ||
| Fig. 5 TEM image of nanoparticles synthesized at RT and pH 8 with (a) nearly square shaped forming aggregates and (b) remaining acicular goethite nanoparticles present as seen in the inset. | ||
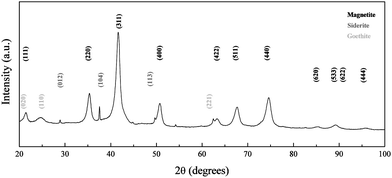 | ||
| Fig. 6 XRD pattern of iron oxide nanoparticles synthesized at RT and pH 8. Peaks are indexed according to the reference patterns for goethite (pdf ref. 00-029-0713), magnetite (pdf ref. 01-088-0315) and siderite (pdf ref. 00-029-0713). | ||
 | ||
| Fig. 7 TEM image of nanoparticles synthesized at RT and pH 9: (a) at low magnification and (b) higher magnification. | ||
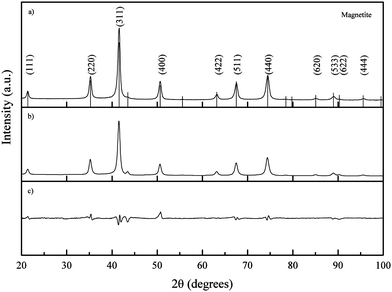 | ||
| Fig. 8 (a) Experimental XRD pattern of iron oxide nanoparticles synthesized at RT and pH 9; peaks are indexed according to the reference pattern for magnetite (pdf ref. 01-088-0315); (b) calculated diffraction pattern and (c) difference between calculated and experimental patterns. | ||
 | ||
| Fig. 9 TEM images of iron oxide NPs synthesized at RT and pH 10 showing (a) the predominant square shape and (b) remaining acicular goethite nanoparticles present (magnified view in the inset). | ||
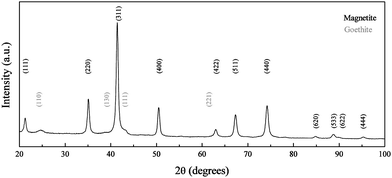 | ||
| Fig. 10 XRD pattern of iron oxide nanoparticles synthesized at RT and pH 10. Peaks have been indexed according to the reference patterns for goethite (pdf ref. 00-029-0713) and magnetite (pdf ref. 01-088-0315). | ||
Considering the overall process, when a mixture of ferrous and ferric ions is present in solution the condensation process is initially oriented towards a specific iron phase, mainly green rusts with a hydrotalcite structural type.30 Nonetheless, green rusts are highly reactive intermediates and the system will tend to evolve to a spinel-type structure. This preferential orientation is dependent on the Fe(II)/Fe(III) ratio.31–33 On the one hand, Tronc et al.34 reported the production of goethite when Fe(II)/Fe(III) < 0.1 due to dissolution of non-stable complexes that recrystallized into goethite; however, when the ratio was >![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 0.1 pure magnetite was obtained. On the other hand, Jolivet et al.35 found that when using a ratio >0.35 only the oxide phase was present in solution.
0.1 pure magnetite was obtained. On the other hand, Jolivet et al.35 found that when using a ratio >0.35 only the oxide phase was present in solution.
In the present work, the precipitation of iron oxide nanoparticles was carried out with a Fe(II)/Fe(III) ratio of 0.5, which should orientate the condensation of the iron complexes towards the formation of a unique oxide phase, either magnetite or maghemite. The use of carbonate had a profound effect on the condensation process of the aquo-complexes as the precipitation process was performed gradually, which prevented drastic changes of pH, minimizing multiple bursts of nucleation as well. Nevertheless, this strategy allowed for subsequent metastable equilibria of the different complexes in solution and at the same time permitted the interaction of the available species with the carbonate present leading to competing reactions. The investigation of the reaction equilibria at pH values between 6 and 10 allowed for the study of the synthesis evolution when carried out to completion to yield iron oxide.
Condensation of ferrous ions does not occur below pH 6, remaining as aquo-complexes of the form [Fe(OH)h(H2O)6−h](2−h)+ with h = 0 up to that point, whereas ferric ions would react under strongly acidic pH (pH ≥ 1) due to the speciation of the aquo-complex ([Fe(OH)h(H2O)6−h](3−h)+).33 The study was therefore started at pH 6, as for a mixture of Fe(II) and Fe(III) both species would be able to react at this point. When the reaction was carried out at pH 6, the Fe(II) present in solution was not available for reaction with the formed aquo-complexes of Fe(III), which led to the initial precipitation of Fe(III) into goethite acicular nanoparticles via an olation mechanism involving the condensation of the hydroxo ligands associated with the metal centre acting as a nucleophile in the presence of an aquo ligand (Fig. 1). As the pH of the reaction was increased to pH 7, the equilibrium of Fe(II) with the CO32− (eqn (1)) present in solution was favoured towards the precipitation of siderite (as shown in Fig. 3).36
| Fe2+ + CO32− ⇌ Fe(CO3)(s) | (1) |
| Fe2+ + CO32− + OH− ⇌ [Fe(CO3)(OH)]− | (2) |
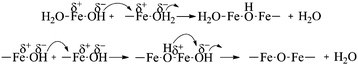 | (3) |
At pH 8, the equilibrium of Fe(II) towards the precipitation of siderite (eqn (1)) will compete with the formation of ferrous aquo-complexes (mainly [Fe(OH)(H2O)5]+), which would be available to react with the Fe(III) aquo-complexes present in the medium (eqn (3)). Simultaneously, as the pH of the solution is increased, the equilibrium of Fe(II) with carbonate towards the formation of siderite is hindered by the presence of hydroxyl groups leading to the formation of a water soluble complex (eqn (2)). Stoichiometric mixtures of Fe(II) and Fe(III) drive the reaction towards the formation of iron oxide with an inverse spinel structure and, as the competing precipitation of siderite is minimized, the condensation of the aquo-complexes will be favoured. Consequently, when the reaction was carried out at pH 8, no crystals of siderite were observed (Fig. 5) and the main iron phase found by XRD was magnetite (Fig. 6). The obtained iron oxide nanoparticles were found as short rods or square particles forming aggregates with a clear resemblance to the initial goethite, which could act as a seed for the formation of the iron oxide phase via a dissolution–precipitation mechanism.37
At pH 9, only magnetite nanoparticles were observed forming aggregates of about 34 nm (Fig. 7) as seen at pH 8. The constituent particles were on average 10 nm bigger than the ones obtained at pH 8, which could indicate that within this stage particle growth is the prevailing process. The decrease in the aggregate size with the subsequent increase in particle size would indicate that particle growth takes place by Ostwald ripening. For increasing pH values the concentration of hydroxyl groups in solution will subsequently increase and so will the ratio [Fe(OH)(H2O)5]+/[Fe(H2O)6]2+. The kinetics of the formation of iron oxide nanoparticles will be favoured due to the increase in the concentration of Fe(II) available as aquo-complex, leading the reaction to completion after 24 h with no remaining goethite (Fig. 8). Simultaneously, the rise in the concentration of hydroxyl groups in solution will favour the reaction of Fe(II) with the carbonate and the hydroxyl groups (eqn (2)) to the detriment of the precipitation of siderite. As a result, the siderite phase was not found at pH 9 as observed on the TEM (Fig. 7) and XRD (Fig. 8).
At pH 10, the obtained material was expected to continue the trend previously described for pH 9; however, goethite particles were confirmed by TEM (Fig. 9) and XRD (Fig. 10). The use of carbonate, a weak base, meant doubling the volume of added base solution in order to achieve pH 10. The increase of the total volume of the reaction, and therefore the decrease in the concentration of the species present in solution, led to a decrease in the rate of the kinetically controlled reaction and therefore the transformation from the oxyhydroxide phase to the oxide phase was slower. This difference was confirmed by TEM, as the studied sample from the reaction at pH 9 at time 24 h (Fig. 7) was free from goethite nanoparticles, whereas at pH 10 and t = 24 h acicular goethite nanoparticles were found (Fig. 9b). Pure magnetite nanoparticles could be isolated by magnetic separation from the reaction products formed at pH 8, 9 and 10. An overview of the morphology, phase and particle size changes with pH can be seen in Table 1.
| pH | Particle size (nm) | Morphology | Phase |
|---|---|---|---|
| 6 | 30 ± 4 (length)/3.2 ± 0.4 (width) | Acicular | Goethite |
| 7 | 31 ± 1 (length)/4 ± 1 (width) | Acicular | Goethite and siderite |
| 440 ± 65 | Nearly square | ||
| 8 | 6.9 ± 0.4 (nanoparticle)/50–70 (aggregates) | Square forming aggregates/acicular | Magnetite, goethite and siderite |
| 9 | 18 ± 3 (nanoparticle)/34 (aggregates) | Spheroidal | Magnetite |
| 10 | 28 ± 5 | Square and acicular | Magnetite and goethite |
3.2 Phase change study during reaction time
As previously mentioned, the formation of iron oxide with an inverse spinel crystal structure from a stoichiometric mixture of ferrous and ferric ions, as used in the present work, had been previously established as too rapid to allow the study of the reaction mechanism.32 However, the use of a weak base allowed for the slow precipitation of the nanoparticles and allowed for the observation of the crystalline phase changes of the formed material over time.The diffraction pattern obtained from the reaction mixture aliquoted after the addition of the base (t = 0 h) matched the reference diffraction pattern of goethite when reaction was carried out at pH 9 (Fig. 11). Changes on crystal structure over time were followed. No significant differences were found for the first two hours after the addition of the base. On the third hour, the appearance of reflections that did not correspond to the initial crystal phase was observed; in fact, (220), (311) and (400) reflections of magnetite started to appear. As the reaction continued, goethite reflections slowly disappeared as the magnetite ones became predominant. On the fourth hour, goethite (221) reflection started to broaden due to the shift towards the family of planes (422) of magnetite. A similar situation was observed from the third hour for the goethite (022) reflections, shifting towards the magnetite (111) family of planes represented by the 21.38° peak. The diffraction pattern showed no trace of a secondary phase 24 h after the reaction was carried out and the reflections matched those of magnetite. The experimental data therefore accounted for the transformation of goethite nanoparticles towards magnetite.
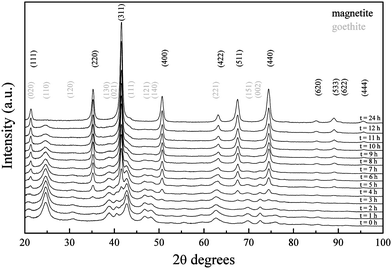 | ||
| Fig. 11 XRD pattern shift over 24 h for iron oxide nanoparticles synthesized at RT and pH 9. Diffraction patterns have been offset along the y axis for a better comparison. | ||
This was also confirmed by the ATR-FTIR spectra. The as-synthesized sample presented the characteristic goethite νOH stretch, δOH in plane bend and γOH out of plane bend bands, at 3140, 892 and 795 cm−1;38 which were not present 24 h after the synthesis had been carried out (Fig. S1†) due to the evolution of the crystal structure over time. As the band observed around 3500 cm−1 for magnetite is also associated to coordinated OH groups from the physisorbed water on the nanoparticle surface,39,40 it cannot be used for identification purposes.
In order to check the evolution of the magnetic properties of the freeze-dried products within the studied 24 h period (Fig. 12), hysteresis loops corresponding to both ends of the time range were measured at 300 and 5 K. On the one hand, the 300 K magnetization curve of the freeze-dried material at t = 0 h showed the typical linear dependence with the applied field shown by paramagnets. Although an antiferromagnetic response is expected from goethite, which is the main phase in the sample as revealed by XRD patterns (Fig. 11), paramagnetic or superparamagnetic behaviour can be observed when forming nanoparticles due to uncompensated spins in the corresponding magnetic sublattices at the surface of the nanoparticles, similar to the case of hematite.41 On the other hand, a ferromagnetic-like curve with σs = 77 A m2 kg−1 was obtained for the sample after 24 h, which roughly corresponds to the magnetic saturation of bulk maghemite (78 A m2 kg−1 at 300 K). This result clearly supports the spontaneous oxidation process undergone by the magnetite NPs seen after the preparation in the form of a black to brown transition, which perforce takes place if no specific measures, such as coating with suitable molecules, are adopted to prevent it. The 5 K loop for the same sample exhibits a much higher coercivity compared to the 300 K one (Fig. 12, inset), as a consequence of the exchange between randomly frozen spins during the lower temperature end of the blocking process of nanoparticles.
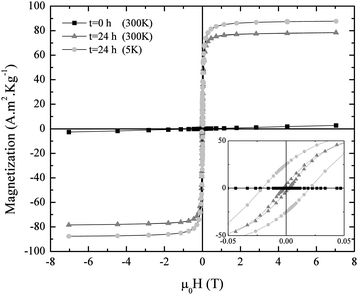 | ||
| Fig. 12 Magnetization curves of iron oxide nanoparticles synthesized at RT and pH 9 aliquoted at t = 0 h and t = 24 h, and measured at 300 K and 5 K. The inset shows a zoom into the low magnetic field region. | ||
Changes in the crystal structure of the obtained materials were also studied at pH 10 (Fig. S2†). The phase transformation observed at pH 9 was also found at pH 10; however, it was slower. As previously described in this work, the increase in the total volume of the reaction solution and subsequent decrease in the concentration of the species present in solution led to a small decrease in the rate of the kinetically controlled reaction showing traces of goethite at t = 24 h.
3.3 Phase change study with reaction temperature
XRD patterns obtained from the as-synthesized iron oxide nanoparticles (t = 0 h) produced at pH 9 and temperatures between RT and 35 °C agreed with the reference diffraction pattern of goethite (Fig. 13). When the reaction was carried out at 40 °C, a decrease in the intensity of the (111) peak was observed. The synthesis of iron oxide nanoparticles at 45 °C led to a black suspension that spontaneously turned light brown after the standard washing procedure due to the oxidation towards maghemite. The instability of the formed material resulted in a deterioration of crystallinity with a remarkable decrease in the intensity of the (130), (021) and (111) peaks compared to the products obtained at lower temperatures between RT and 40 °C. When the reaction was carried out between 50 and 70 °C, the obtained nanoparticles exhibited a diffraction pattern that matched magnetite at t = 0 h. The diffraction pattern of the samples after 24 h did not show any differences and therefore is independent of the temperature of the reaction.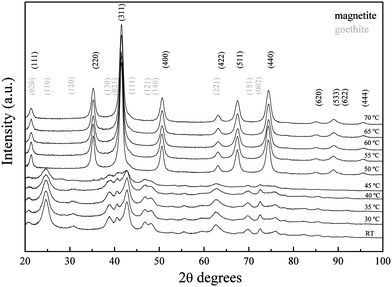 | ||
| Fig. 13 XRD collected from samples synthesized at temperatures between RT and 70 °C at t = 0 h. Diffraction patterns have been offset along the y axis. | ||
The mechanism of goethite formation is known to take place through olation or condensation of hydroxo and aquohydroxo-complexes in solution.32 Conversely, spinel structures are formed through oxolation, a two-step reaction involving an initial condensation of hydroxo complexes followed by β-elimination of a proton that leads to the formation of the oxo-bridge. This second step is favoured at high temperatures, which would increase the tendency of the system to proceed via an oxolation mechanism. The obtained diffraction patterns from the iron oxide nanoparticles at different temperatures between RT and 70 °C showed that the product formed after the addition of sodium carbonate was initially goethite, but when the reaction was carried out below and above 45 °C the obtained product was magnetite. This would indicate that there is a remarkable difference in the reaction mechanism and the oxolation is clearly favoured above 45 °C.
The magnetic properties of the samples were found to be affected also by the reaction temperature. At 300 K (Fig. 14a) the sample synthesized at RT exhibited a lower magnetization saturation (σs) compared to the one synthesized at 60 °C, but on the contrary shows both higher coercivity (Hc) and magnetization remanence (Mr). This difference in the magnetic parameters (Table 2) can be explained in terms of both the different particle size and oxidation degree towards maghemite between both samples. Particles observed under TEM synthesised at RT were 18.1 ± 2.4 nm in size, whereas particles prepared at 60 °C were 12.3 ± 1.8 nm in size (Fig. 15). On the one hand, the σS > 78 A m2 kg−1 indicates that there is still some non-oxidized magnetite in the particles, most likely in their core. On the other hand, the smaller particle size of the sample synthesized at 60 °C makes it closer to a pure superparamagnetic behaviour than the RT one, and hence the lower coercivity and remanence values. In addition to purely size effects, the magnetite–maghemite ratio also plays a role in the magnetic properties of these samples. By virtue of the different magnetocrystalline anisotropy and exchange stiffness constant typical from each phase, maghemite is expected to form single domain particles at lower critical sizes, but these particles become superparamagnetic at bigger sizes compared to magnetite.42 At 5 K (Fig. 14b) both samples show similar values of Hc and Mr, but the increase is much higher in the case of the sample synthesized at 60 °C. This indicates that for the latter, a much larger fraction of nanoparticles became blocked after decreasing the temperature, reflecting a size distribution centred at lower values than those of the RT sample.
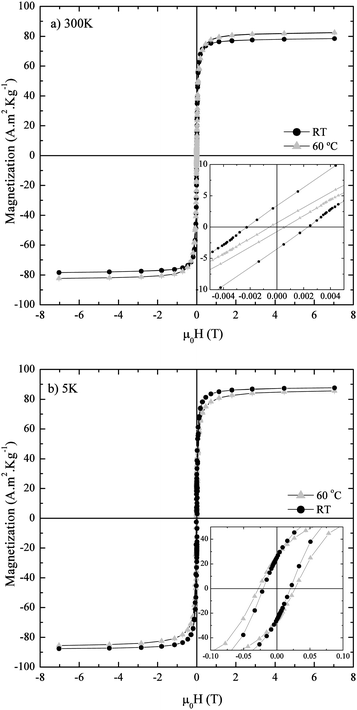 | ||
| Fig. 14 Magnetization curves of iron oxide nanoparticles synthesized at pH 9 and T = RT and T = 60 °C measured at (a) 300 K and (b) 5 K. | ||
| Sample | Measurement temperature (K) | σ s (A m2 kg−1) | H c (T) | M r (A m2 kg−1) |
|---|---|---|---|---|
| Synthesized at RT | 300 | 74.3 | 0.002 | 3.4 |
| 5 | 86.5 | 0.02 | 25.3 | |
| Synthesized at 60 °C | 300 | 80.6 | 5.4 × 10−4 | 0.7 |
| 5 | 83.7 | 0.03 | 25.9 |
 | ||
| Fig. 15 TEM images of iron oxide NPs synthesized at pH 9 and (a) RT and (b) 60 °C. | ||
4 Conclusions
Sodium carbonate has been found to be a suitable co-precipitating agent for the selective synthesis of stable iron oxide and oxyhydroxide nanoparticles from a stoichiometric mixture of Fe(II) and Fe(III) at different pH values. The moderate carbonate concentration contributed to a more gradual precipitation that allowed for tracking the reaction changes reducing the abrupt pH variations within the reaction medium leading to a narrow particle size distribution. The mild reaction conditions and the slow process permitted the study of the nanoparticle formation, revealing the initial occurrence of goethite nanoparticles that evolved to the spinel-type structure after 24 h when the reaction was carried out below 45 °C. Conversely, above that temperature, the obtained product was iron oxide with an inverse spinel structure, which evidenced a change in the preferential reaction mechanism from olation to oxolation.At the same time, the reaction temperature was found to affect the final magnetic properties of the NPs mainly via a decrease in the average particle size, which also resulted in smaller coercivity values, closer to pure superparamagnetic behaviour. The reaction pH has been found to be a decisive factor, leading to variations both in particle size and morphology. Pure acicular goethite particles were obtained at pH 6, whereas pH 9 was found to be optimum for producing magnetite. A further increase of pH leads to well-defined square shaped particles, but owing to the larger reaction volume there was a very small local reversion to goethite. The size of the obtained magnetite nanoparticles could be controlled with the reaction pH. An increase of ca. 10 nm per pH unit was found leading to 6.9 ± 0.4 nm, 18 ± 3 nm and 28 ± 5 nm for pH 8, 9 and 10 respectively.
These results establish that a sodium carbonate-based co-precipitation method allows for the preparation of nanoparticles in aqueous media with tuneable size, morphology and crystal phase. Not only does this synthetic route permit the formation of magnetite nanoparticles, but more importantly it offers a benchmark for the study of the physicochemical changes that a solution of Fe(II)/Fe(III) experiences throughout a reaction. Furthermore, changes throughout the reaction could be studied in detail, allowing for an improved understanding of the reaction variability that has limited reproducibility in the production of commercial particles. The information gathered could be used in achieving more controllable and reproducible processing, which may allow progress towards more diverse GMP manufacturing of magnetic nanoparticles. If this were to be achieved it would represent a major advance in the field, allowing for the biomedical applications of magnetic nanoparticles to be extended and broadened beyond the currently limited range of FDA-approved materials.
Acknowledgements
Nguyen T. K. Thanh thanks the Royal Society for her University Research Fellowship. Cristina Blanco-Andujar thanks UCL and RI for her PhD studentship. Finally the authors thank Dr Paul Southern for his help on the magnetic measurements and discussion.References
- M. J. Lefort, Comptes rendus hebdomadaires des séances de l'Académie des Sciences, 1852, 34, 488–491 Search PubMed.
- A. B. Chin and I. I. Yaacob, J. Mater. Process. Technol., 2007, 191, 235–237 CrossRef CAS.
- P. A. Dresco, V. S. Zaitsev, R. J. Gambino and B. Chu, Langmuir, 1999, 15, 1945–1951 CrossRef CAS.
- N. J. Francois, S. Allo, S. E. Jacobo and M. E. Daraio, J. Appl. Polym. Sci., 2007, 105, 647–655 CrossRef CAS.
- G. Salazar-Alvarez, M. Muhammed and A. A. Zagorodni, Chem. Eng. Sci., 2006, 61, 4625–4633 CrossRef CAS.
- S. Basak, D. R. Chen and P. Biswas, Chem. Eng. Sci., 2007, 62, 1263–1268 CrossRef CAS.
-
S. S. Papell, US Pat., 3215572, 1965 Search PubMed.
- T. J. Daou, G. Pourroy, S. Begin-Colin, J. M. Greneche, C. Ulhaq-Bouillet, P. Legare, P. Bernhardt, C. Leuvrey and G. Rogez, Chem. Mater., 2006, 18, 4399–4404 CrossRef CAS.
- D. Maity, S. N. Kale, R. Kaul-Ghanekar, J. M. Xue and J. Ding, J. Magn. Magn. Mater., 2009, 321, 3093–3098 CrossRef CAS.
- J. M. D. Coey and D. Khalafal, Phys. Status Solidi A, 1972, 11, 229 CrossRef CAS.
- C. Ross, Annu. Rev. Mater. Res., 2001, 31, 203–235 CrossRef CAS.
- S. Laurent, D. Forge, M. Port, A. Roch, C. Robic, L. V. Elst and R. N. Muller, Chem. Rev., 2008, 108, 2064–2110 CrossRef CAS.
- R. M. Cornell and U. Schwertmann, The iron oxides, Wiley-VCH Publishers, Weinheim, Germany, 2003 Search PubMed.
- R. M. Cornell and U. Schwertmann, The Iron Oxides in the Laboratory: Preparation and Characterization, Wiley-VCH Publishers, Weinheim, Germany, 2000 Search PubMed.
- J.-P. Jolivet, C. Froidefond, A. Pottier, C. Chaneac, S. Cassaignon, E. Tronc and P. Euzen, J. Mater. Chem., 2004, 14, 3281–3288 RSC.
- L. Vayssières, C. Chanéac, E. Tronc and J. P. Jolivet, J. Colloid Interface Sci., 1998, 205, 205–212 CrossRef.
- R. Massart, IEEE Trans. Magn., 1981, 17, 1247–1248 CrossRef.
- M. Kallumadil, M. Tada, T. Nakagawa, M. Abe, P. Southern and Q. A. Pankhurst, J. Magn. Magn. Mater., 2009, 321, 3650–3651 CrossRef CAS.
- H. B. Na, I. C. Song and T. Hyeon, Adv. Mater., 2009, 21, 2133–2148 CrossRef CAS.
- B. Chertok, B. A. Moffat, A. E. David, F. Q. Yu, C. Bergemann, B. D. Ross and V. C. Yang, Biomaterials, 2008, 29, 487–496 CrossRef CAS.
- M. Arruebo, R. Fernandez-Pacheco, M. R. Ibarra and J. Santamaria, Nano Today, 2007, 2, 22–32 CrossRef.
- E. Y. Sun, L. Josephson, K. A. Kelly and R. Weissleder, Bioconjugate Chem., 2006, 17, 109–113 CrossRef CAS.
- J. Weizenecker, B. Gleich, J. Rahmer, H. Dahnke and J. Borgert, Phys. Med. Biol., 2009, 54, L1–L10 CrossRef CAS.
- D. E. Markov, H. Boeve, B. Gleich, J. Borgert, A. Antonelli, C. Sfara and M. Magnani, Phys. Med. Biol., 2010, 55, 6461–6473 CrossRef CAS.
- Q. A. Pankhurst, N. K. T. Thanh, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2009, 42, 224001 CrossRef.
- N. T. K. Thanh, Magnetic Nanoparticles: From Fabrication to Clinical Applications, Taylor and Francis, 2011 Search PubMed.
- S. H. Sun and H. Zeng, J. Am. Chem. Soc., 2002, 124, 8204–8205 CrossRef CAS.
- T. Hyeon, S. S. Lee, J. Park, Y. Chung and H. Bin Na, J. Am. Chem. Soc., 2001, 123, 12798–12801 CrossRef CAS.
- T. G. Spiro, S. E. Allerton, J. Renner, A. Terzis, R. Bils and P. Saltman, J. Am. Chem. Soc., 1966, 88, 2721 CrossRef CAS.
- J.-M. R. Génin, G. Bourrié, F. Trolard, M. Abdelmoula, A. Jaffrezic, P. Refait, V. Maitre, B. Humbert and A. Herbillon, Environ. Sci. Technol., 1998, 32, 1058–1068 CrossRef.
- J. P. Jolivet, E. Tronc and C. Chaneac, C. R. Geosci., 2006, 338, 488–497 CrossRef CAS.
- J. P. Jolivet, M. Henry, J. Livage and E. Bescher, Metal Oxide Chemistry and Synthesis: From Solution to Solid State, Wiley Publishers, Chichester, England, 2000 Search PubMed.
- J. P. Jolivet, C. Chaneac and E. Tronc, Chem. Commun., 2004, 481–487 RSC.
- E. Tronc, P. Belleville, J. P. Jolivet and J. Livage, Langmuir, 1992, 8, 313–319 CrossRef CAS.
- J. P. Jolivet, P. Belleville, E. Tronc and J. Livage, Clays Clay Miner., 1992, 40, 531–539 CAS.
- D. W. King, Environ. Sci. Technol., 1998, 32, 2997–3003 CrossRef CAS.
- Y. Tamaura, K. Ito and T. Katsura, J. Chem. Soc., Dalton Trans., 1983, 189–194 RSC.
- P. Cambier, Clay Miner., 1986, 21, 191–200 CAS.
- J. T. Keiser, C. W. Brown and R. H. Heidersbach, J. Electrochem. Soc., 1982, 129, 2686–2689 CrossRef CAS.
- G. Busca, V. Lorenzelli, G. Ramis and R. J. Willey, Langmuir, 1993, 9, 1492–1499 CrossRef CAS.
- A. H. Morrish, Canted Antiferromagnetism: Hematite, World Scientific Pub Co Inc, 1994 Search PubMed.
- J. Coey, Magnetism and Magnetic Materials, Cambridge University Press Cambridge, UK, 2010 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ATR-FTIR spectra of iron oxide nanoparticles prepared at RT at t = 0 and t = 24 h; XRD patterns within 24 hours for samples synthesized at pH = 10 and RT. See DOI: 10.1039/c2jm31295f |
| This journal is © The Royal Society of Chemistry 2012 |