 Open Access Article
Open Access ArticleAn enantio-selective chromatographic stationary phase for S-ibuprofen prepared by stoichiometric molecular imprinting
Panagiotis
Manesiotis
*,
Qendresa
Osmani
and
Peter
McLoughlin
Pharmaceutical and Molecular Biotechnology Research Centre, Waterford Institute of Technology, Waterford, Ireland. E-mail: pmanesiotis@wit.ie; Fax: +353 (0)51 302679; Tel: +353 (0)51 306167
First published on 1st May 2012
Abstract
Molecularly Imprinted Polymers (MIPs) against S-ibuprofen were synthesised using a tailor made functional monomer, 2-acrylamido-4-methylpyridine, following extensive pre-polymerisation studies of template–monomer complexation. An apparent association constant of 340 ± 22 M−1 was calculated that was subsequently corrected to account for dimerisation of ibuprofen (Kdim = 320 ± 95 M−1) resulting in an intrinsic association constant of 715 ± 16 M−1, consistent with previously reported values. Using the synthesised imprinted polymer as a stationary phase, complete resolution of a racemic mixture of ibuprofen was achieved in predominantly aqueous mobile phases. An imprinting factor of 10 was observed, and was found to be in agreement with the difference in the average number of binding sites between MIP and blank polymers, calculated by staircase frontal chromatography. The imprinted polymers exhibited enhanced selectivity for the templated drug over structurally related NSAIDs. When applied as sorbents in solid-phase extraction of ibuprofen from commercial tablets, urine and blood serum samples, recoveries up to 92.2% were achieved.
1. Introduction
Molecular imprinting is a technique that enables the generation of specific binding sites within the matrix of a synthetic organic polymer, analogous to the binding sites found in nature's own specific receptors, enzymes and antibodies.1–6 Commonly, the generation of such imprinted sites within the polymer matrix is achieved by utilising non-covalent interactions between the template of interest and a complementary functional monomer at the pre-polymerisation stage. Hence, upon initiation of the polymerisation reaction, it is theorised that the template is held in close proximity to the growing polymer chains and a cast is generated around it that upon its extraction, reveals the binding site that is then capable of reversibly binding the templated analyte or closely related compounds. The strength of template–monomer interactions is therefore critical for the fidelity of the binding sites formed. Commercially available monomers such as methacrylic acid (MAA) and vinyl-4-pyridine (4-VPy) have been used extensively in MIP synthesis with varying degrees of success. With few exceptions,7 interactions between these functional monomers and templates are weak, single point hydrogen bonds, and an excess of functional monomer is commonly employed in order to drive the equilibrium towards complex formation. This excess of functional monomer results in random distribution of functionality within the polymer and consequently non-specific binding of the analytes to the polymer matrix at the application stage. Additionally, the limited strength of template–monomer and subsequently template–imprinted polymer interactions has limited their applications to predominantly organic media, as addition of an aqueous modifier disrupts the weak hydrogen bonding and leads to excessive non-specific binding.These shortfalls have been overcome by designing functional monomers capable of forming strong interactions with a particular template, thus eliminating the need for an excess of functional monomer and therefore reducing the degree of non-specific binding.8–11 Stoichiometric ratios of the functional monomer and the template are used at the pre-polymerisation stage, forming strong pre-polymerisation complexes, which subsequently generate imprinted polymers with high affinity binding sites.
Ibuprofen is a member of the non-steroidal anti-inflammatory drugs (NSAIDs) and is commonly used for relief from arthritis, fever and as a generic pain reliever. Being one of the most popular drugs of its category, it has attracted significant research interest over the past years, and a number of reports of its targeting by molecular imprinting are found in literature. An early study by Kempe and Mosbach investigated the enantio-selective properties of S-naproxen imprinted polymers prepared using 4-VPy as the functional monomer.12 Enantio-separation of a racemic mixture of naproxen was achieved using an organic mobile phase, with a separation factor of 1.65. Caro et al. prepared ibuprofen imprinted polymers using a similar monomer composition and used it as an SPE sorbent for selective extraction of ibuprofen, naproxen, benzoic acid, fenoprofen and diclofenac from spiked river water samples.13 The polymer was capable of selective extraction of the tested analytes and performed similarly to a commercial SPE sorbent. However, chromatographic evaluation of the imprinted polymer revealed rather low imprinting factors for ibuprofen and naproxen. In another study, Haginaka et al. prepared S-ibuprofen imprinted beads, generated by multi-step swelling and a thermal polymerisation method.14 The imprinted beads were employed as a HPLC chromatographic stationary phase using a mixture of organic–aqueous mobile phases. An imprinting factor of 2.08 was calculated for S-ibuprofen, while it was lower for structurally related compounds. Furthermore, only partial enantio-separation of ibuprofen enantiomers was achieved. In a more recent study Spegel et al. prepared imprinted polymers by in situ polymerisation for enantiomeric separation of ibuprofen using capillary electrochromatography.15 Although a range of polymers with varying compositions of the starting materials were prepared using 4-VPy as the functional monomer, only partial separation of enantiomers was achieved.
In contrast to previous reports detailed above, where commercially available 4-VPy was used as the functional monomer, the work presented here details the design and preparation of S-ibuprofen-specific stoichiometric imprinted polymers using a tailor-made functional monomer, 2-acrylamido-4-methylpyridine.16 Initially, an in-depth investigation of pre-polymerisation complexes was performed using NMR spectroscopy, whereby it was possible to deconvolute the apparent association constant from the dimerisation of ibuprofen itself, reported here for the first time. The subsequently synthesised imprinted polymer was employed as a chromatographic stationary phase. Using a 30 mm HPLC column and a predominantly aqueous mobile phase, the imprinted polymer was capable of complete enantiomeric separation of racemic ibuprofen in less than 30 minutes. Furthermore, selectivity of the imprinted polymer was demonstrated by the superior specific binding of S-ibuprofen compared to its structurally related compounds (Scheme 1) with calculated imprinting factors of up to 10. The imprinted polymer was also used as a selective SPE sorbent for the extraction of ibuprofen from commercial tablets, blood serum and urine samples with recoveries of up to 92.2%.
 | ||
| Scheme 1 Proposed mode of interaction between the functional monomer (AMP) and S-ibuprofen plus the chemical structures of related NSAIDs. | ||
2. Experimental
2.1. Materials and methods
Ethyleneglycol dimethacrylate (EDMA), 1,1′-azobis(cyclohexanecarbonitrile) (ACCN), 2-amino-4-picoline, triethylamine, deuterated chloroform (CDCl3), HPLC grade solvents, acetonitrile, methanol, dichloromethane, toluene, and anhydrous tetrahydrofuran were purchased from Sigma-Aldrich (Wicklow, Ireland). Acryloyl chloride was purchased from VWR International (Dublin, Ireland). Water was purified by a SG Ultra Clear TWF UV Plus TM water purification system (Whitewater, Blackrock, Ireland). EDMA was purified by washing with 10% NaOH, washing with brine, drying over anhydrous magnesium sulfate, and subsequent distillation under reduced pressure. The functional monomer, 2-acrylamido-4-methylpyridine (AMP), was synthesised as reported previously by reacting equimolar amounts of amino-4-picoline and acryloyl chloride in the presence of triethylamine.16,17NMR spectra were obtained using a Jeol ECX 400 spectrometer (Tokyo, Japan). HPLC measurements were performed using an Agilent 1200 system equipped with a diode array detector. Surface area analysis was performed using a Micromeritics Gemini VI Nitrogen sorption analyser (Particular Sciences, Dublin, Ireland).
2.2. Pre-polymerisation studies
Monomer–template, monomer–monomer and template–template complexation were studied prior to polymer synthesis to establish the type and strength of interactions present in the pre-polymerisation solution, by means of a 1H NMR titration and dilution studies in CDCl3. In the first instance, to a solution of AMP (0.001 mol L−1) were added increasing amounts of the guest S-ibuprofen. The complexation-induced shift (CIS) of the host amide proton was followed, and titration curves were then constructed. The raw titration data was fitted to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 binding isotherm, and the association constant was obtained by nonlinear regression of the isotherms using WinEQNMR2 software.18 Secondly, a standard solution of AMP or S-ibuprofen (0.1 mol L−1) was sequentially diluted to 0.001 mol L−1 and the self-association induced shift of the amide proton (AMP) or the proton on the methylene adjacent to the carboxyl group (S-ibuprofen) was followed. The stoichiometry of the AMP–S-ibuprofen complex was confirmed using Job's method of continuous variation.19 Hence, equimolar solutions (0.01 mol L−1) of AMP and S-ibuprofen were mixed in different ratios and plots of Δδ against the molar fraction of AMP multiplied by the CIS (XAMP × Δδ) were constructed.
:1 binding isotherm, and the association constant was obtained by nonlinear regression of the isotherms using WinEQNMR2 software.18 Secondly, a standard solution of AMP or S-ibuprofen (0.1 mol L−1) was sequentially diluted to 0.001 mol L−1 and the self-association induced shift of the amide proton (AMP) or the proton on the methylene adjacent to the carboxyl group (S-ibuprofen) was followed. The stoichiometry of the AMP–S-ibuprofen complex was confirmed using Job's method of continuous variation.19 Hence, equimolar solutions (0.01 mol L−1) of AMP and S-ibuprofen were mixed in different ratios and plots of Δδ against the molar fraction of AMP multiplied by the CIS (XAMP × Δδ) were constructed.
2.3. Preparation of imprinted polymers
A stoichiometric 1:1 ratio of functional monomer (AMP) and template plus 20 equivalents of the cross-linker were employed in the synthesis of the imprinted polymers. 0.206 g (1 mmol) of S-ibuprofen and 0.163 g (1 mmol) of AMP were dissolved in 5.6 mL of toluene followed by addition of 4 g (20 mmol) of EDMA in a 20 mL screw-cap vial. The solution was degassed using N2 while cooling in an ice-bath for 5 minutes. Finally, 0.04 g of the free radical initiator ACCN were added and the vial was sealed and placed in a UV reactor at 4 °C for 24 hours. A non-imprinted polymer (NIP) was synthesised in the same manner with omission of the template. The resulting monoliths were smashed to coarse particles and washed with methanol for 24 hours using a Soxhlet apparatus. Finally, the polymers were manually ground and sieved collecting the 25–38 μm fraction for packing into HPLC columns and SPE cartridges.
2.4. Chromatographic evaluation
Imprinted or non-imprinted particles of 25–38 μm were slurry packed in 30 mm × 4.6 mm i.d. HPLC columns using a 4:1 methanol:water mixture. The columns were then connected to a HPLC instrument and equilibrated with acetonitrile until a stable baseline was obtained. Analyses were performed at 0.5 mL min−1 by injecting 5 μL of a 0.1 mmol L−1 solution of each analyte and recording its elution profile at 220 or 240 nm. All injections were repeated at least three times, alternating between the different analytes to avoid column overloading. Imprinting factors (kMIP/kNIP) for each analyte as well as separation factors (kS/kR) for the template were calculated.
Staircase frontal chromatography was performed using solutions of S-ibuprofen in acetonitrile as the mobile phase, ranging from 0.001 to 0.1 mol L−1, and mixing them in a step-wise fashion of 10% increments with pure acetonitrile to produce a staircase frontal chromatogram with a total of 30 steps. From each step the corresponding amount of bound analyte was calculated and the collected results were plotted against the corresponding concentration of S-ibuprofen to produce binding isotherms that were fitted using the Freundlich binding model.20,21
2.5. Solid phase extractions
25 mg of MIP and NIP particles were dry packed in 3 mL SPE cartridges using 20 μm porous PTFE frits. Equilibration of the columns, loading and washing were performed using 2 mL aliquots of the corresponding solutions and elution of the retained analytes with 1 mL of CH3CN–1% CH3COOH. Full vacuum was applied between each step for 3 minutes in order to dry the stationary phases. The collected fractions were analysed by HPLC using a 150 mm × 4.6 mm 5 μm C18 column and isocratic elution using a mixture of methanol:water:acetic acid 90:5:5 with the flow-rate set at 1 mL min−1. Under these conditions ibuprofen eluted at 1.9 minutes. Commercial ibuprofen tablets were ground to a fine powder and dissolved in ultrapure water prior to analysis. Blood serum samples were prepared by allowing the collected blood to clot at room temperature for approximately 45 minutes, followed by centrifugation at 3000 rpm for 15 minutes, and careful collection of the clear top layer. Serum samples were stored at −80 °C. SPE analysis of serum samples was performed in spiked samples (200 μg mL−1) after dilution with ultra-pure water. Urine samples were filtered, spiked and analysed following dilution with ultra-pure water. The protocol for biological sample analysis was approved by the Institutional Ethics Committee and written consent was obtained.
3. Results and discussion
3.1. Pre-polymerisation studies
In a previous report on the use of acrylamido pyridines for the recognition of carboxylic acids, Hall et al. performed an extensive study on the influence of substitution of the pyridine ring on binding strength.16 2-Acrylamido-4-methylpyridine was found to be the strongest receptor and hence it was selected as the functional monomer for the present study. A 1H NMR titration revealed that its apparent association constant with S-ibuprofen (Kapp) was 340 ± 22 M−1 in CDCl3 at 20 °C (Fig. 1). This is significantly lower that the constant calculated for the binding of propanoic acid (777 ± 84 M−1), used as model in the previous study. This observation cannot be explained by either the difference in the pKa of S-ibuprofen (pKa = 4.9) and propanoic acid (pKa = 4.7), or the larger molecular size and flexibility of ibuprofen. A further set of NMR based experiments was therefore performed, this time investigating the dimerisation of both AMP and ibuprofen, facilitated by the self-complementary H-bond donor–acceptor motif present in both molecules. The first dilution study revealed very weak, yet detectable, self-association of AMP calculated at approximately 10 ± 0.8 M−1. This was deemed too weak to have any significant bearing on the overall equilibrium and was considered negligible. However, the dilution study of ibuprofen (Fig. 1, insert) revealed a significant dimerisation equilibrium with Kdim = 320 ± 95 M−1, very similar to the apparent association constant calculated for the AMP–ibuprofen complex. When this second equilibrium was taken into account, we were able to re-plot the original 1:1 binding isotherm and using non-linear regression an intrinsic Ka = 715 ± 16 M−1 was calculated for the AMP–ibuprofen complex, in agreement with the previously reported value. To our knowledge, this is the first time that the dimerisation of ibuprofen has been studied, certainly in the context of molecular imprinting, although the dimerisation of carboxylic acids is a well-studied phenomenon.22
 | ||
| Fig. 1 1H NMR titration binding isotherm of S-ibuprofen with AMP: experimental data (triangles), corresponding fitted curve (dashed line), data corrected for self-association of ibuprofen (squares) and corresponding fitted curve (solid line). Insert: S-ibuprofen self-association isotherm obtained by 1H NMR dilution study and corresponding fitted curve. | ||
The stoichiometry of the AMP–ibuprofen complex was verified by a Job plot experiment. This showed a maximum at XAMP = 0.5, verifying the 1:1 stoichiometry as expected by the two point interaction postulated in Scheme 1 and supporting our use of a stoichiometric imprinting protocol.
3.2. Preparation and characterisation of imprinted polymers
Imprinted and non-imprinted polymers were synthesised using a 1:1:20 ratio of S-ibuprofen:AMP:EDMA by photochemical initiation at 4 °C using UV irradiation. Toluene was chosen as the porogen, in order to facilitate and stabilise template–monomer interactions due to its non-polar nature, but also to yield polymers with relatively high surface area. The similar mesoporous nature of the polymers was confirmed by N2 porosimetry with a surface area of 398 m2 g−1 and an average pore diameter of 71 Å for the MIP and 431 m2 g−1 with average pore diameter of 69.5 Å for the NIP. Both MIP and NIP monoliths appeared semi-transparent and a slight colour difference was observed upon visual examination under visible and UV light, possibly due to a complexation induced change in the absorption characteristics of AMP.
Following Soxhlet extraction of the polymers, 13C solid state NMR analysis of the materials was performed with nearly identical spectra obtained, as seen in Fig. 2. Spin-lattice (T1) relaxation times were also measured by inversion recovery experiments and were found to be 0.6 seconds for the MIP and 0.55 seconds for the NIP, demonstrating the comparable physical character of the two polymers.
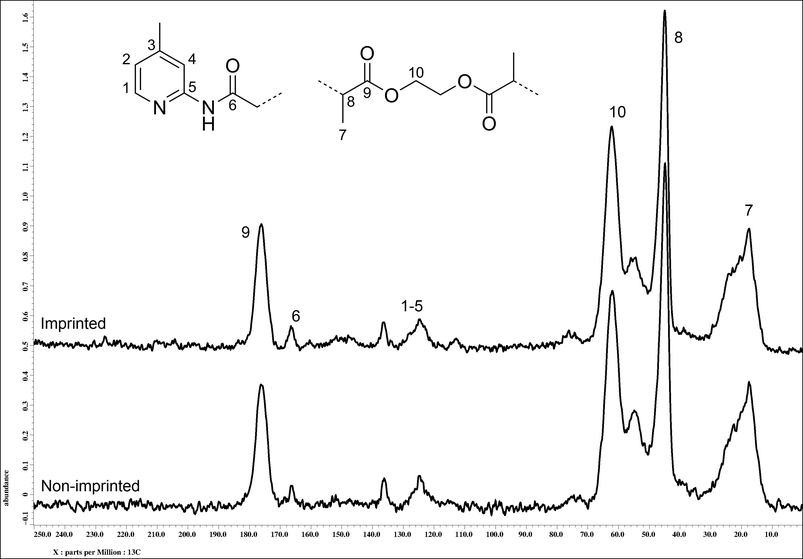 | ||
| Fig. 2 13C solid state NMR spectra (100 MHz) of imprinted (top) and non-imprinted polymers (bottom) with assignment of major peaks. | ||
3.3. Chromatographic evaluation of imprinted polymers
HPLC evaluation of the synthesised materials was initially performed in pure acetonitrile. S-Ibuprofen was the longest retained analyte with a corresponding imprinting factor (kMIP/kNIP) of 5. The R-enantiomer was significantly less retained and a kMIP/kNIP = 3.2 was measured. When racemic ibuprofen was injected a split peak was observed corresponding to the R and S enantiomers, with a separation factor α = kS/kR of 1.5. This finding suggested that complete enantio-separation could be possible, if the elution conditions were further optimised.The retention profiles of the other injected analytes also revealed some interesting trends. The much smaller analyte 2-phenylpropanoic acid, as well as larger indoprofen, were less retained, similarly to the geometrically different naproxen and ketoprofen (Table 1). It is however noteworthy that of all analytes tested, S-flurbiprofen and S-naproxen, both comparable in size and shape to the templated S-ibuprofen, exhibited slightly higher retention than the other analytes tested, highlighting a size-exclusion effect taking place within the polymer's binding sites. Furthermore, the slightly more acidic character of these analytes, as calculated using Marvin software package,23 might also be responsible for their marginally longer retention. It should be noted here that in all cases no enantio-separation was observed when racemic mixtures were injected.
:60)
| Analyte | CH3CN | CH3CN–H2O (40:60) |
||
|---|---|---|---|---|
| Imprinted | Non-imprinted | Imprinted | Non-imprinted | |
| S-Ibuprofen | 6.5 | 2.2 | 17.0 | 2.5 |
| R-Ibuprofen | 4.5 | 2.2 | 10.2 | 2.5 |
| S-Flurbiprofen | 7.3 | 4.6 | 16.3 | 2.4 |
| R-Flurbiprofen | 7.3 | 4.6 | 12.0 | 2.4 |
| S-Naproxen | 7.5 | 3.6 | 9.2 | 2.0 |
| S-Ketoprofen | 3.9 | 5.6 | 5.8 | 1.7 |
| R-Ketoprofen | 3.9 | 5.6 | 5.8 | 1.7 |
| S-2-PPA | 4.5 | 3.6 | 3.5 | 1.4 |
| R-2-PPA | 4.5 | 3.6 | 3.5 | 1.4 |
| S-Indoprofen | 7.5 | 6.0 | 4.3 | 1.5 |
| R-Indoprofen | 7.5 | 6.0 | 4.3 | 1.5 |
In order to optimise the mobile phase composition, the percentage of water in acetonitrile was incremented in steps of 10%. When 60% water content was used, complete resolution of ibuprofen enantiomers was achieved in approximately 30 minutes and an α = 1.8 was calculated, while the imprinting factor for S-ibuprofen was equal to 10, a 5-fold increase compared to previously reported literature values. The composition of each of the two peaks was verified by injection of pure S-ibuprofen and it was verified that this was the longer retained enantiomer, while the non-templated R-enantiomer eluted first. At water contents higher than 70% chromatographic run times exceeded 60 minutes and the quality of the peaks diminished significantly, hence 60% water content was used for the further evaluation of the polymers. The obtained imprinting factors are presented in Fig. 3.
 | ||
| Fig. 3 Imprinting factors (kMIP/kNIP) for ibuprofen and structurally related compounds in CH3CN and CH3CN–H2O (40:60) (2-PPA:2-phenylpropanoic acid). | ||
As expected, the relative hydrophobic character of NSAIDs resulted in significantly longer retention of all injected analytes, compared to the analyses in pure organic medium, with S-flurbiprofen and S-naproxen being retained the longest compared to the other structurally related analytes. Moreover the imprinted polymer exhibited enantio-selectivity for flurbiprofen as its racemic mixture was nearly completely resolved (α = 1.4). This result was attributed to the comparable size and shape of this analyte to the templated structure.
Further optimisation of the chromatographic conditions was attempted by means of temperature control. Thus, a series of injections of rac-ibuprofen was performed at 20, 40 and 60 °C and the elution profiles were recorded. It was observed that although peak symmetry improved and run times were reduced from 30 to 10 minutes as the temperature increased, the resolution was dramatically affected and separation factors decreased from 1.8 at 20 °C to 1.5 at 40 °C and 1.35 at 60 °C. It was thus decided that further experiments should be run at 20 °C (Fig. 4).
 | ||
| Fig. 4 Elution profiles of 0.1 mmol L−1rac-ibuprofen injected on the S-ibuprofen imprinted column. From bottom to top: column temperature 20 °C, 40 °C and 60 °C. Dashed line: 0.1 mmol L−1S-ibuprofen injected on S-ibuprofen imprinted column at 20 °C. Traces are shown in scale but have been off-set on the y-axis for clarity. Insert: 1 mmol L−1rac-ibuprofen injection at 20 °C. Mobile phase: CH3CN–H2O (40:60), flow-rate: 0.5 mL min−1. | ||
Once the optimum chromatographic conditions were established, direct analysis of a commercial ibuprofen tablet was attempted. The tablet was ground to a fine powder and 20 mg were dissolved in 10 mL of CH3CN–H2O 40:60. This solution was injected directly on the MIP LC column and while all excipients eluted near the solvent front, we were able to resolve the two enantiomers of ibuprofen found in the formulation and achieve accurate determination of the drug concentration.
Finally, a staircase frontal chromatography experiment was performed, using the optimised chromatographic conditions described above. A wide range of S-ibuprofen concentrations was tested from 10−6 mol L−1 to 10−3 mol L−1 and a total of 30 points were collected for each column (MIP and NIP). The amounts of bound analyte during each step were then plotted against the corresponding analyte concentration and two binding isotherms were constructed (Fig. 5). The imprinted polymer clearly exhibits significantly higher affinity for S-ibuprofen throughout the range of concentrations tested, compared to the non-imprinted polymer. The plotted curves were fitted to the Freundlich isotherm q = aCm, where q is the amount of adsorbed analyte, a is a coefficient, C the concentration of the analyte in the mobile phase and m the heterogeneity index. Parameters a and m where calculated by non-linear regression of the experimental data and using the equations detailed in the article by Umpleby et al.,21 the average affinity constant K and the average number of binding sites N were extracted. Interestingly, the Freundlich model calculated that both MIP and NIP polymers have the same average affinity constant of 1.47 ± 0.02 × 104 M−1. However, the difference in the performance of the two polymers regarding the retention of S-ibuprofen as established by chromatographic evaluation, can be attributed to the starkly different average number of binding sites, with the MIP containing 2.19 ± 0.01 μmol g−1 and the NIP 0.23 ± 0.01 μmol g−1. The similarity of affinity constants could be ascribed to the fact that both polymers contain the same ingredients, and more importantly the same functional monomer, while the introduction of S-ibuprofen at the pre-polymerisation mixture is responsible for the generation of specific recognition points, hence the nearly 10-fold difference in average number of binding sites, clearly demonstrated by the corresponding affinity distribution plot (Fig. 5, insert). This difference is in remarkable agreement with the calculated imprinting factor for S-ibuprofen discussed previously.
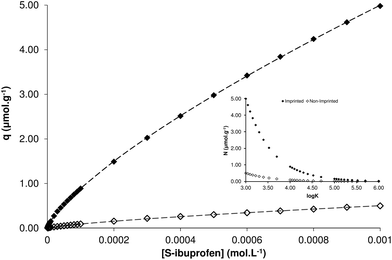 | ||
| Fig. 5 Binding isotherms of S-ibuprofen on MIP (◆) and NIP (◇) columns and corresponding curves fitted to the Freundlich isotherm model (dashed lines). Insert: affinity distribution plots. | ||
3.4. Solid phase extractions
The synthesised polymers were subsequently tested in MI-SPE in order to establish whether they could be used as selective sorbents for the extraction of ibuprofen from complex samples. Initially, extractions of aqueous standard solutions (200 μg mL−1) were attempted and the percentage of acetonitrile as the organic modifier in the wash step was varied between 0 and 40%, while elution was performed with CH3CN containing 1% acetic acid. It was found that ibuprofen recoveries decreased as the percentage of CH3CN in the wash step increased and when pure water or up to 20% CH3CN was used recoveries averaged 92%, decreased to 85% at 30% CH3CN and 63% at 40% CH3CN. Interestingly, the selectivity of the MIP increased in the same order, as the recoveries on the MIP and NIP were equal at 0 and 20% CH3CN, a ratio of 1.4 was reached at 40% (Fig. 6). This result exemplifies the role of the organic modifier in the wash step of MI-SPE, commonly referred to as the “molecular recognition” step: in purely aqueous mobile phases non-specific hydrophobic binding dominates while the addition of an organic solvent plays an important role in making the selective binding sites accessible to the template molecules. | ||
| Fig. 6 Ibuprofen recovery (%) dependence on CH3CN content in the wash step (bars) and corresponding imprinting factors (dashed line). | ||
Based on these results and the common assumption that imprinted polymers perform best when applied in the solvent which was used as a porogen during their synthesis, we decided to use toluene as the organic solvent at the molecular recognition step. Hence, after loading the sample, the polymer phase was washed with 2 mL of toluene, followed by complete drying of the bed and finally elution of the retained analyte. Indeed, this protocol resulted in recoveries averaging 86.3% on the MIP column and 29.1% on the NIP, a ratio of 3, verifying the above hypothesis.
We then modified this protocol adding an aqueous wash after loading the sample to remove any water soluble compounds from the column, and applied it in the extraction of commercial ibuprofen tablets, as well as spiked (200 μg mL−1) blood serum and urine samples. The results are summarised in Table 2. Recoveries from ibuprofen tablets were comparable to the ones obtained from aqueous standards although the differences between MIP and NIP were smaller. Furthermore, significantly reduced recoveries were initially achieved from urine and blood serum samples averaging just 24.1% at physiological pH. This was ascribed to the acidic character of ibuprofen that is found in the carboxylate anion form at pH values near 7. In this form, the donor–acceptor array is no longer present, hence the analyte is not complementary to the functional monomer. Consequently, when the pH was reduced to 4 by addition of a few drops of 1 M aqueous HCl, recoveries of up to 92.2% were achieved, confirming the proposed two-point hydrogen bond complexation.
| Sample | Imprinted | Non-imprinted |
|---|---|---|
| H2O | 86.3 ± 2 | 29.1 ± 2 |
| Tablet | 84.7 ± 3 | 50.0 ± 4 |
| Blood serum | 92.2 ± 4 | 56.8 ± 2 |
| Urine | 87.3 ± 3 | 47.1 ± 4 |
It should be noted here that throughout this series of SPE experiments, totalling at least 100 extraction cycles, the same 25 mg MIP cartridge was used and no loss in performance or reproducibility was evident, verifying the robustness and reusability of imprinted materials compared to biological affinity media.
4. Conclusions
A complete pre-polymerisation study of the 2-acrylamido-4-methylpyridine–ibuprofen system was performed by 1H NMR titration and dilution studies. The apparent association constant for the 1:1 complex was found to be lower than the expected value and this was attributed to significant dimerisation of ibuprofen in the studied solution. Stoichiometrically imprinted polymers for S-ibuprofen were synthesised using a 1:1 ratio of the functional monomer and the drug. The materials exhibited strong affinity and selectivity for the templated structure versus other related NSAIDs, both in organic and aqueous media, containing up to 60% water, and imprinting factors of 5 and 10 respectively were obtained. Complete resolution of ibuprofen enantiomers was achieved in predominantly aqueous mobile phases, using 30 mm columns, minute solvent consumption and in relatively short run times. Chromatographic separations performed at elevated temperatures resulted in much faster runs at the expense of resolution, albeit elution profiles obtained at 40 °C could perhaps be further fine-tuned to achieve complete enantiomer resolution in less than 15 minutes. MI-SPE experiments using just 25 mg of packing material resulted in excellent recoveries of the drug from aqueous standard solutions, commercial tablets, urine and blood serum samples, while the only sample pre-treatment necessary was a simple dilution and adjustment of the pH to ensure the drug was present in the desirable neutral carboxylic acid form.
The results presented here, when directly compared to previous reports found in literature, clearly demonstrate that imprinted materials with superior performance are readily accessible using simple, custom-made building blocks, optimised for each targeted analyte.
Acknowledgements
Financial support from Enterprise Ireland's Applied Research Enhancement Programme (Contract number: RE-2008-11) is gratefully acknowledged.References
- B. Sellergren, Molecularly Imprinted Polymers. Man Made Mimics of Antibodies and their Applications in Analytical Chemistry, Elsevier Science B.V., Amsterdam, 2001 Search PubMed.
- J. Steinke, D. Sherrington and I. Dunkin, Adv. Polym. Sci., 1995, 123, 80 CrossRef.
- P. Manesiotis and G. Theodoridis, in Encyclopedia of Chromatography, ed. J. Cazes, Taylor & Francis, 3rd edn, 2009, pp. 24–30 Search PubMed.
- C. Alexander, H. S. Andersson, L. I. Andersson, R. J. Ansell, N. Kirsch, I. A. Nicholls, J. O'Mahony and M. J. Whitcombe, J. Mol. Recognit., 2006, 19, 106–180 CrossRef CAS.
- G. Wulff, Angew. Chem., Int. Ed. Engl., 1995, 34, 1812–1832 CrossRef CAS.
- M. J. Whitcombe and E. N. Vulfson, Adv. Mater., 2001, 13, 467–478 CrossRef.
- R. J. Ansell, D. Y. Wang and J. K. L. Kuah, Analyst, 2008, 133, 1673–1683 RSC.
- A. Hall, P. Manesiotis, M. Emgenbroich, M. Quaglia, E. De Lorenzi and B. Sellergren, J. Org. Chem., 2005, 70, 1732–1736 CrossRef CAS.
- P. Manesiotis, C. Borrelli, C. S. A. Aureliano, C. Svensson and B. Sellergren, J. Mater. Chem., 2009, 19, 6185–6193 RSC.
- P. Manesiotis, A. Hall, J. Courtois, K. Irgum and B. Sellergren, Angew. Chem., Int. Ed., 2005, 44, 3902–3906 CrossRef CAS.
- G. Wulff and K. Knorr, Bioseparation, 2001, 10, 257–276 CrossRef CAS.
- M. Kempe and K. Mosbach, J. Chromatogr., A, 1994, 664, 276–279 CrossRef CAS.
- E. Caro, R. M. Marce, P. A. G. Cormack, D. C. Sherrington and F. Borrull, J. Sep. Sci., 2005, 28, 2080–2085 CrossRef CAS.
- J. Haginaka, H. Sanbe and H. Takehira, J. Chromatogr., A, 1999, 857, 117–125 CrossRef CAS.
- P. Spegel, L. Schweitz, L. I. Andersson and S. Nilsson, Chromatographia, 2009, 69, 277–285 CAS.
- A. J. Hall, M. Quaglia, P. Manesiotis, E. De Lorenzi and B. Sellergren, Anal. Chem., 2006, 78, 8362–8367 CrossRef CAS.
- J. Y. Ju, C. S. Shin, M. J. Whitcombe and E. N. Vulfson, Biotechnol. Bioeng., 1999, 64, 232–239 CrossRef CAS.
- M. J. Hynes, J. Chem. Soc., Dalton Trans., 1993, 311–312 RSC.
- K. A. Connors, Binding Constants; The Measurement of Molecular Complex Stability, John Wiley & Sons, New York, 1987 Search PubMed.
- Y. B. Chen, M. Kele, P. Sajonz, B. Sellergren and G. Guiochon, Anal. Chem., 1999, 71, 928–938 CrossRef CAS.
- R. J. Umpleby II, S. C. Baxter, M. Bode, J. K. Berch, R. N. Shah and K. D. Shimizu, Anal. Chim. Acta, 2001, 435, 35–42 CrossRef.
- G. Allen and E. F. Caldin, Q. Rev. Chem. Soc., 1953, 7, 255–278 RSC.
- ChemAxon, Marvin 5.6.0.1, http://www.chemaxon.com/, 2011 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |