Structure, photophysics, and photooxidation of crowded diethynyltetracenes†
Jingjing
Zhang
a,
Syena
Sarrafpour
a,
Terry E.
Haas
a,
Peter
Müller
b and
Samuel W.
Thomas
III
*a
aDepartment of Chemistry, Tufts University, 62 Talbot Avenue, Medford, MA 02155, USA. E-mail: sam.thomas@tufts.edu; Fax: +1-617-627-3443; Tel: +1-617-627-3771
bDepartment of Chemistry, Massachusetts Institute of Technology, 77 Massachusetts Ave, Cambridge, MA 02139, USA
First published on 17th February 2012
Abstract
This paper describes a previously unreported class of sterically crowded tetracene derivatives that have both phenyl and ethynyl substituents. The steric crowding above and below the tetracene core prevents overlap between the extended π-systems of the acenes. Substituent effects cause these tetra-substituted tetracenes to have absorbance and fluorescence spectra red shifted from either disubstituted derivatives or rubrenes, such that they have spectra similar to diarylpentacenes, but with higher quantum yields of fluorescence and greater photostability. These new molecules also undergo cycloaddition reactions with 1O2, giving regioisomeric mixtures of endoperoxides, and in contrast to longer acenes, the ethynyl substituents show only a modest stabilizing effect to photooxidation. Ethynylated tetracenes also exhibited photochromism, with their endoperoxides undergoing cycloreversion to yield the acene starting material at room temperature in the dark.
Introduction
This paper describes the synthesis, optical properties, and cycloaddition reactivity of a previously unreported class of substituted tetracenes: 5,12-diethynyl-6,11-diphenyltetracenes. Linear acenes of three or more rings have important roles in a number of applications. Acenes are among the materials that have the best performance in organic electronics:1 thin films of pentacene and single crystals of rubrene show efficient charge transport properties. Rubrene and other acenes are common luminescent materials in light-emitting devices2 and in chemiluminescent glowsticks. In addition to their useful properties in devices, depending on their substitution, acenes can undergo facile cycloaddition reactions with dieneophiles.3–6 Cycloaddition reactions with singlet oxygen (1O2)7,8 have been identified as a mechanism of acene degradation,9 and also play a key role in schemes for sensing 1O2 both from our group10 and others.11,12 Acene-based cycloaddition reactions with other dieneophiles are also useful for constructing structural scaffolds such as iptycenes.13Primary pathways of acene decomposition include endoperoxide formation with singlet oxygen and photodimerization. There have been a variety of substitution strategies to increase the stability of acene derivatives, including substitution with halogens,9,14 thioethers,15–17 sterically demanding arenes,9 and heteroaromatics;18 ethynyl substitution, however, such as in 6,13-bis(triisopropylsilylethynyl)pentacene (TIPS pentacene) is among the most popular.19–23 Silylethynyl groups have been critical in stabilizing large acenes, such as hexacene,24,25 heptacene,23,24 and nonacene26 that are otherwise too reactive to isolate and characterize. Work on silylethynyl hexacenes has shown that for these longer acenes, dimerization, and not photooxidation, appears to be the primary mode of decomposition.25 In addition, heptacenes that bear both silylethynyl and phenyl substituents are reported to be the most stable such derivatives.23,27
Ethynyl substitution red-shifts absorbance and luminescence spectra of acenes because of increased π-conjugation, and also improves solubility, which enables solution processing to replace vacuum deposition for constructing devices. Our group is interested in strategies for tuning both emission color and cycloaddition reactivity of acenes for applications in 1O2 detection or photo-tunable solid-state emitters. The goal of this study was to develop soluble, red-emitting, 1O2-reactive acenes that are significantly more stable than 6,13-diarylpentacenes, which are too reactive for reliable handling of thin films under typical laboratory conditions. Tetracenes are an interesting class of molecules for this purpose because they have reactivities and optical properties that are intermediate relative to similarly substituted anthracene and pentacene derivatives. A number of classes of substituted tetracenes have been reported, such as 5,12-diethynyltetracenes,28,29 tetraalkyltetracenes,30–32 dialkoxytetracenes,33 and substituted rubrene derivatives.34,35 Herein we report the preparation, photophysical properties, and reactivity of what we believe to be a new class of sterically crowded substituted tetracenes.
Experimental section
General considerations
The following chemicals were purchased from commercial sources and used as received: 1,3-diphenylisobenzofuran (Acros), 1,4-naphthoquinone (Aldrich), 5,12-naphthacenequinone (Acros), 6,13-pentacenedione (TCI), phenylacetylene (Aldrich), 4-methoxyphenylacetylene (Alfa Aesar), Sn(II) chloride dihydrate (Alfa Aesar) and poly(9,9-di-n-dodecylfluorenyl-2,7-diyl) (Aldrich). Dry tetrahydrofuran (THF) was obtained using a column purification system.All synthetic manipulations were performed under standard air-free conditions under an atmosphere of argon gas with magnetic stirring unless otherwise mentioned. Flash chromatography was performed using silica gel (230–400 mesh) as the stationary phase. NMR spectra were acquired on a Bruker Avance III 500 or Bruker DPX-300 spectrometer. Chemical shifts are reported relative to residual protonated solvent (7.27 ppm for CDCl3). High-resolution mass spectra (HRMS) were obtained at the MIT Department of Chemistry Instrumentation Facility using a peak-matching protocol to determine the mass and error range of the molecular ion.
Synthesis
1T (0.26 g, 31%) 1H NMR (500 MHz, CDCl3): δ 8.58–8.57 (m, 2H), 7.56–7.54 (d, 6H), 7.46–7.43 (t, 6H), 7.30–7.27 (m, 10H), 7.23 (s, 5H). 13C NMR (125 MHz, CDCl3): δ 141.3, 137.9, 134.3, 132.8, 131.7, 131.2, 128.6, 128.3, 128.1, 128.1, 128.0, 127.7, 127.2, 127.1, 125.8, 124.3, 119.1, 108.9, 89.7. HRMS calcd for C46H28 (M + H)+, 581.2264; found, 581.2280.
2T (0.33 g, 35%) 1H NMR (500 MHz, CDCl3): δ 8.57–8.56 (m, 2H), 7.55–7.54 (d, 6H), 7.50–7.43 (m, 6H), 7.35–7.31 (m, 6H), 7.22–7.20 (m, 6H), 6.82–6.80 (d, 4H), 3.82 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 159.6, 141.5, 137.9, 134.3, 133.2, 132.8, 131.2, 128.6, 128.2, 127.9, 127.7, 127.2, 126.9, 125.6, 119.2, 116.6, 113.7, 109.0, 88.7, 55.5. HRMS calcd for C48H32O2 (M + H)+, 641.2475; found, 641.2463.
3T (0.16 g, 16%) 1H NMR (500 MHz, CDCl3): δ 8.56–8.54 (m, 2H), 7.60–7.57 (m, 10H), 7.56–7.47 (m, 6H), 7.40–7.39 (d, 4H), 7.33–7.31 (m, 4H). 13C NMR (125 MHz, CDCl3): δ 141.2, 137.9, 134.3, 132.8, 131.8, 131.4, 128.5, 128.4, 127.5, 127.2, 126.1, 125.1, 118.9, 107.3, 91.8. HRMS calcd for C48H26F6 (M + H)+, 717.2011; found, 717.2028.
4T (0.18 g, 17%) 1H NMR (500 MHz, CDCl3): δ 8.66–8.65 (m, 2H), 7.64–7.62 (m, 2H), 7.51–7.48 (m, 10H), 7.45–7.43 (m, 2H), 7.27–7.24 (m, 2H), 1.07 (s, 42H). 13C NMR (125 MHz, CDCl3): δ 141.0, 137.5, 135.0, 133.2, 130.9, 128.6, 128.1, 128.0, 127.0, 126.7, 125.6, 119.4, 112.1, 105.3, 19.1, 12.2. HRMS calcd for C52H60Si2 (M + H)+, 741.4306; found, 741.4301.
1D (0.32 g, 48%) 1H NMR (500 MHz, CDCl3): δ 9.29 (s, 2H), 8.69–8.67 (m, 2H), 8.12–8.09 (m, 2H), 7.85–7.82 (m, 4H), 7.58–7.55 (m, 2H), 7.49–7.46 (m, 8H).
2D (0.41 g, 54%) 1H NMR (500 MHz, CDCl3): δ 9.28 (s, 2H), 8.70–8.69 (m, 2H), 8.10–8.08 (m, 2H), 7.80–7.78 (m, 4H), 7.57–7.55 (m, 2H), 7.49–7.44 (m, 2H), 7.04–7.02 (m, 4H), 3.92 (s, 6H). 13C NMR (125 MHz, CDCl3): δ 160.2, 133.4, 132.3, 132.2, 130.1, 128.8, 127.6, 126.6, 126.2, 126.0, 118.5, 116.0, 114.5, 103.5, 86.2, 55.6. HRMS calcd for C36H24O2 (M + H)+, 489.1849; found, 489.1853.
3D (0.45 g, 52%) 1H NMR (500 MHz, CDCl3): δ 9.25 (s, 2H), 8.66–8.64 (m, 2H), 8.13–8.11 (m, 2H), 7.95–7.94 (m, 4H), 7.77–7.75 (m, 4H), 7.62–7.60 (m, 2H), 7.53–7.51 (m, 2H). 13C NMR (125 MHz, CDCl3): δ 132.7, 132.5, 132.1, 130.1, 128.7, 127.4, 127.2, 126.6, 126.2, 125.8, 125.2, 123.1, 122.8, 118.4, 102.1, 89.5. HRMS calcd for C36H18F6 (M + H)+, 565.1385; found, 565.1396.
4D 28 (0.39 g, 43%) 1H NMR (500 MHz, CDCl3): δ 9.31 (s, 2H), 8.64–8.62 (m, 2H), 8.03–8.01 (m, 2H), 7.56–7.54 (m, 2H), 7.48–7.46 (m, 2H), 1.33 (s, 42H).
1P 36 (0.33 g, 53%) 1H NMR (500 MHz, CDCl3): d 9.31 (s, 4H), 8.07–8.05 (m, 4H), 7.93–7.91 (m, 4H), 7.55–7.52 (m, 4H), 7.50–7.48 (m, 2H), 7.44–7.42 (m, 4H).
2P (0.32 g, 43%) 1H NMR (500 MHz, CDCl3): d 9.27 (s, 4H), 8.05–8.02 (m, 4H), 7.85–7.82 (m, 4H), 7.41–7.38 (m, 4H), 7.06–7.03 (m, 4H), 3.91 (s, 6H). 13C NMR (125 MHz, CDCl3) d 160.3, 133.4, 132.3, 130.37, 128.8, 126.3, 126.0, 118.2, 116.0, 114.5, 104.7, 87.0, 55.6. HRMS calcd for C40H26O2 M+, 538.1927; found, 538.1924.
3P 37 (0.35 g, 44%) 1H NMR (500 MHz, CDCl3) d 9.29 (s, 4H), 8.10–8.08 (m, 4H), 8.03–8.02 (m, 4H), 7.81–7.80 (m, 4H), 7.48–7.46 (m, 4H).
4P 38 (0.33 g, 40%) 1H NMR (500 MHz, CDCl3): d 9.31 (s, 4H), 7.99–7.97 (m, 4H), 7.43–7.41 (m, 4H), 1.38 (s, 42H).
Optical experiments
Electronic absorbance spectra were acquired with a Varian Cary-100 instrument in double-beam mode using a solvent-containing cuvette for background subtraction spectra. Irradiation of the methylene blue photosensitizer to generate 1O2 was performed with a 200W Hg/Xe lamp (Newport-Oriel) equipped with a condensing lens, recirculating water filter, manual shutter, a 665 nm high-pass filter and a focusing plano-convex lens (focal length 15 cm) in the light path.Kinetics
A stock solution of methylene blue was prepared in CHCl3 to give an absorbance of 1.0 at its peak. Ethynyl derivatives (1T–4T, 1D–4D, 4P), 5,12-bis(4-methoxyphenyl) tetracene 5, or 6,13-bis(4-methoxyphenyl) pentacene 6 was dissolved in CHCl3 at a concentration of 0.005 M. 5,12-Bis(4-methoxyphenyl)tetracene 5 was used as reference for 1T–4T, 1D–4D, and 6,13-bis(4-methoxyphenyl) pentacene 6 was used as the reference for 4P. When the 0.005 M solution was diluted 100-fold with the methylene blue stock solution, the final concentration of the acene in the sample was 0.05 mM. With the sample placed 15 cm from the light source, the solution was irradiated using a 665 nm high-pass filter and convex lens in increments of 15 s for 120 s (1T–4T) or increments of 45 s for 360 s (1D–4D, 4P); the absorbance spectrum was measured after each interval of irradiation.Synthetic photooxidation
The acene was dissolved in CDCl3 with methylene blue and bubbled with O2. The solution was irradiated with the Hg/Xe lamp using a high-pass filter and plano-convex lens. When all starting material has reacted, methylene blue was removed by quick filtration with silica gel in a filter funnel.4T endoperoxides: HRMS calcd for C52H60O2Si2 (M + H)+, 773.4205; found, 773.4190. 3D endoperoxides: HRMS calcd for C36H18F6O2 (M + H)+, 597.1284; found, 597.1298.
Crystal structure determination
Low-temperature diffraction data (φ-and ω-scans) were collected on a Bruker-AXS X8 Kappa Duo diffractometer coupled to a Smart Apex2 CCD detector with Mo-Kα radiation (λ = 0.71073 Å) from an IμS microsource. All structures were solved by direct methods using SHELXS39 and refined against F2 on all data by full-matrix least squares with SHELXL-9740 using established refinement techniques.41 For details of data quality and structure refinement, see ESI.Results and discussion
Synthesis of acenes
Fig. 1 shows the four derivatives of the diethynyldiphenyltetracene core (tetra-substituted tetracenes, compounds with suffix “T”) prepared, each with different terminal groups bound to the alkyne substituents. Cycloaddition between 1,3-diphenylisobenzofuran and napthoquinone yielded the common diphenyl-tetracenequinone intermediate.34,35 Addition of appropriately substituted acetylide anions, prepared by deprotonation of the terminal acetylene with n-BuLi, followed by reduction of the resulting diols with SnCl2 gave the target acenes 1–4T. Similar strategies, using commercially available tetracenequinone or pentacenequinone, gave analogously substituted 5,12-diethynyltetracenes (1D–4D) and 6,13-diethynylpentacenes (1P–4P). Yields of the tetrasubstituted compounds were 15–35%, whereas those of the disubstituted compounds were somewhat higher. We ascribe the lower yield of the T-series compounds to steric hindrance presented by the terphenyl moiety on the quinone. | ||
| Fig. 1 Structures of acenes studied in this work. | ||
X-ray crystal structures
Single crystal X-ray structures of compounds 2T and 4T highlight the influence of the substituents on the structures of this new class of substituted acenes (Fig. 2 and 3). Both of these sterically crowded molecules relieve intramolecular congestion through structural distortions. In addition to all the phenyl substituents on the terphenyl moieties being rotated away from a conformation coplanar with the tetracene backbone, the substituents are bent away from each other, which has been observed in other tetra-substituted tetracenes.28,35 The arylethynyl groups of 2T are rotated out of plane by 73° and 66° in opposite directions, in contrast to less congested 5,12-diarylethynyltetracenes, in which the aromatic groups bound to the sp-hybridized carbons are coplanar with the acene backbone.29 The bulkier triisopropylsilyl groups of 4T force even greater distortions, with the tetracene backbone of 4T having a significant twist of 31° (Fig. 3). This type of twisting is known to occur in single crystals of sterically congested acenes.42 We suspect that in solution the acene backbone of 2T may also be twisted, but that crystal packing forces planarize the tetracene in the single crystal, as is known for rubrene.434T crystallizes in the chiral space group P212121 with only one of the two possible twisted enantiomers present in the crystal we analyzed. | ||
| Fig. 2 X-ray crystal structure of 2T with atomic labeling scheme. Hydrogen atoms omitted for clarity. Thermal ellipsoids shown at 50% probability. Packing diagram visualized in a projection along tetracene cores. | ||
 | ||
| Fig. 3 X-ray crystal structure of 4T. Methyl groups omitted from lower structure for clarity. Ellipsoids shown at 50% probability. | ||
Although 2T shows interactions between the anisyl rings in neighboring columns along the crystallographic a-axis, neither 2T nor 4T exhibits significant π–π interactions between the acene backbones in their crystal structures. Steric hindrance presented above and below the acene plane by the twisted arene rings prevents such interactions. The closest to such an interaction is a distance of 4.63 Angstroms between parallel mean planes defined by the tetracene ring systems of 2T. The pitch between these molecules is so large, however, that the distance between centroids of the tetracenes is 7.02 Å (Fig. 2).
Photophysical properties
Table 1 summarizes absorbance and fluorescence parameters for all tetracenes investigated; the trends discussed below comparing 2T and 2D are consistent with the other pairs of similarly substituted acenes. Fig. 4 contrasts the absorbance and fluorescence spectra of tetrasubstituted tetracene 2T and with two related substituted acenes—5,12-dianisylethynyl tetracene (2D) and 6,13-dianisylpentacene (6)—in dilute dichloromethane solution. Relative to the disubstituted tetracene 2D that does not have pendant phenyl rings, compound 2T shows a 30 nm red shift (587 nm vs. 557 nm, 0.12 eV lower energy) in the peak of the lowest energy vibronic band of absorbance, and a ∼50 nm (0.16 eV) shift in the onset of absorbance. This red shift, resulting from the two additional phenyl rings substituted on the tetracene backbone, is analogous to that between 5,12-diphenyltetracene (494 nm) and rubrene (528 nm). Recent theoretical calculations on tetracene and rubrene have attributed this bathochromic shift upon substitution with phenyl rings to a destabilization of the HOMO by inductive effects from the phenyl rings and twisting of the tetracene backbone.44 In addition, similar to rubrene, the absorbance spectrum of 2T shows less vibronic resolution than 2D, and a larger Stokes Shift, consistent with additional vibrational modes involving rotations of the phenyl substituents.44 The optical HOMO–LUMO gap of 2T is similar to 6 (1.9 eV), a highly reactive pentacene derivative, derivatives of which have been used as red emitters in light-emitting devices.45| λmax (ab)b | λmax (em) | ΦFc | kreld | |
|---|---|---|---|---|
| a All photophysical data collected in CH2Cl2; kinetics data collected in CHCl3. b Wavelength of maximum absorbance of the 0,0 band. c Measured relative to cresyl violet in methanol (ΦF = 0.54) or Coumarin 6 in ethanol (ΦF = 0.78). d Rate of reaction, relative to 5, with 1O2 prepared by irradiation of methylene blue in the presence of O2. | ||||
| 1T | 578 nm | 616 nm | 0.73 | 0.39 |
| 2T | 586 nm | 626 nm | 0.63 | 0.30 |
| 3T | 577 nm | 625 nm | 0.71 | 0.26 |
| 4T | 575 nm | 602 nm | 0.75 | 0.40 |
| 1D | 552 nm | 565 nm | 0.76 | 0.31 |
| 2D | 559 nm | 575 nm | 0.85 | 0.40 |
| 3D | 556 nm | 571 nm | 0.87 | 0.18 |
| 4D | 534 nm | 542 nm | 0.67 | 0.23 |
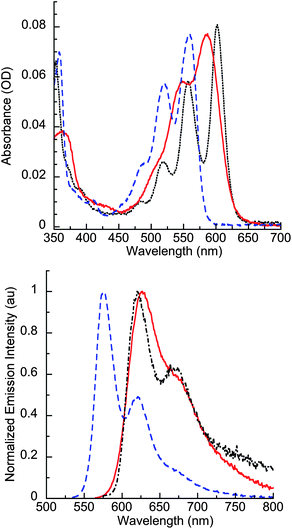 | ||
| Fig. 4 Absorbance (top) and photoluminescence (bottom) spectra of 2T (red solid lines), 2D (blue dashed lines), and 6 (black dotted lines) in CH2Cl2. Phenyl substitution on 2T red-shifts the absorption and emission spectra such that they resemble that of pentacene 6. | ||
The fluorescence spectrum of 2T and related acenes in CH2Cl2 follows a similar pattern to that found in absorbance (Fig. 4): it is strongly red-shifted from most other previously reported tetracene derivatives, including 2D, and has a maximum wavelength of emission (626 nm) similar to 6 (620 nm). We attribute these effects on the optical properties of 2T to a combination of the extended π-conjugation from the phenylethynyl substituents and the inductive effects of the phenyl rings, which because of steric interactions are nearly orthogonal to the tetracene core. The emission spectra of 4T and 4D, which do not have aromatic rings bound to the alkyne substituents, are blue shifted from similar structures that have arylethynyl substituents, consistent with reduced delocalization in molecules with fewer conjugated π-bonds. Increased conjugation from ethynyl groups also accounts for the red shifted absorbance and fluorescence that all of the T-series compounds described here have relative to rubrene.43 All four of the tetrasubstituted tetracenes described here are highly fluorescent, with quantum yields of fluorescence (ΦF) > 0.6, while ΦF of 6 is 0.24. When doped into a host thin film of poly(9,9-didodecylfluorene) at 20% loading (w/w), 2T accepted energy from the photoexcited host to give a ratio of emission intensities of 6![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (guest:host, see ESI). The spectrum of the emissive acene in this film was nearly identical to that observed in solution.
:1 (guest:host, see ESI). The spectrum of the emissive acene in this film was nearly identical to that observed in solution.
1O2-mediated oxidation of tetracenes
Ethynyl substituents have been the most popular method for stabilizing pentacenes and higher-order acenes that are otherwise susceptible to photooxidation. To quantitatively evaluate the effect of alkynes on the photooxidation of these tetracenes, we determined the relative rates of reaction of these acenes with 1O2. Each acene, at a concentration of 5.0 × 10−5 M in air-equilibrated CHCl3, was exposed to 1O2 by irradiation of the sensitizer methylene blue (MB) with a peak absorbance of 1.0 using a 200 W Hg/Xe lamp and a 665 nm high-pass filter. These conditions insured that MB was the only chromophore in solution absorbing any incident light. Control experiments in the absence of MB showed no reaction of acenes under otherwise identical irradiation conditions.Table 1 summarizes the results of these experiments. Each reaction with photogenerated 1O2 followed pseudo first-order kinetics; the slopes of best-fit lines to plots of ln(absorbance) vs. time (Fig. 5) gave relative rate constants krel. The previously reported 5,12-dianisyltetracene (5) was the standard (krel = 1) for these kinetics experiments.10 All of the ethynylated tetracenes (T and D) derivatives react with 1O2 with similar relative rate constants that are 2.5–5 times slower than diaryltetracene 5. Trifluoromethyl-substituted derivatives 3T and 3D were the least reactive of each of their respective classes (T or D) of tetracenes.27 This structure-property relationship is consistent with the findings of both Chi and Ong, who used 4-trifluoromethylphenyl substituents to improve the stability of a heptacene and a pentacene, respectively, and attributed these observations to a lowering of the energy of the HOMO.27,37
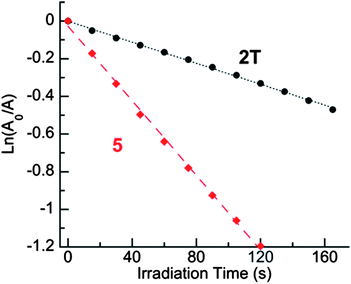 | ||
| Fig. 5 Semilogarithmic kinetic plots showing relative rates of reaction of 5 (red diamonds) and 2T (black circles) as a function of irradiation time of MB in air-equilibrated CHCl3. | ||
Although there was some reduction in reactivity between these ethynylated tetracenes compared to non-ethynylated tetracene 5, consistent with the electron withdrawing alkyne groups decreasing the reactivity of acenes towards 1O2, the magnitude of this difference in stability towards photogenerated 1O2 is one order of magnitude less than that reported for the difference in photolytic stability between 6,13-diphenylpentacene and TIPS-pentacene (4P).9 Therefore, the ethynyl groups on TIPS-pentacene have a greater stabilizing effect than the ethynyl groups on these tetracenes. In addition, there was no statistically significant stabilization of any of the ethynyl-substituted tetracenes described here compared to 5 upon direct irradiation at λ > 400 nm in the absence of MB.
These observations are consistent with the conclusions of Maliakal, who found that the principal reason for stabilization of ethynylated pentacenes is the selective stabilization of the pentacene LUMO. These lower energy LUMOs result in low triplet energies, making them slow to donate energy to O2, and make photoinduced electron transfer from the excited acene to O2 less energetically favorable.46,47 Tetracene derivatives have a larger band gap and higher energy LUMO than larger acenes. We therefore propose that unlike similarly substituted pentacene derivatives, the triplet energies of these ethynylated tetracenes are not of low enough energies to either prevent 1O2 sensitization or accept energy from 1O2, resulting in only a modest stabilization towards photooxidation relative to non-ethynylated tetracene 5. The reported observation of the endoperoxide of silylethyne-substituted anthradithiophenes (ADTs),48 together with an only slight increase in stability to photooxidation that these substituents provide to the ADT backbone,36 is consistent with the proposed model.
Regioselective reversibility of 1O2 cycloaddition
Analytical data of products of MB-mediated photooxidation of the tetracenes were consistent with mixtures of endoperoxides. For D-series tetracenes, 1H NMR spectra showed clean mixtures of two endoperoxides, with characteristic singlets from bridgehead (δ 6.6 ppm) and aromatic protons (δ 8.2 ppm) corresponding to 1O2 cycloaddition across the unsubstituted and ethynylated positions, respectively (Fig. 6).49 Therefore, although there was a modest 2:1 regiochemical preference for oxidation of the unsubstituted position, silyl acetylenes did not protect ethynylated positions from photooxidation.23T-series compounds did not have these clearly identifiable characteristic NMR signals because of the high degree of substitution on the acenes; nevertheless, high resolution mass spectra (HRMS) of their products of oxidation by photosensitization with methylene blue were consistent with endoperoxides.
 | ||
| Fig. 6 NMR spectrum of 4D (top), its products of photooxidation by methylene blue-mediated 1O2 photosensitization (middle) in CDCl3, and the same sample three weeks later, showing cycloreversion of the endoperoxide substituted across the ethynylated positions; vertical arrows signify NMR signals assigned to 4D formed via cycloreversion. | ||
Cycloreversion of endoperoxides of tetracene derivatives typically requires heating or irradiation with short wavelengths of light.50,51 In contrast, isolated mixtures of ethynylated tetracene endoperoxides developed the red color of the acene starting material upon storage in the dark, neat, under otherwise ambient conditions. In addition, for the one acene that we investigated in detail (4D, selected for the ease of NMR peak assignments), there was a large difference in the rate of the cycloreversion for the two endoperoxide regioisomers: the endoperoxoide on the ethynylated positions underwent cycloreversion, while it appeared that the other regioisomer was unreactive. The 1H NMR spectrum of the endoperoxides of 4D, which initially showed a 2:1 molar ratio of the two endoperoxides (as shown in Fig. 6), showed approximately 10% conversion to the acene and a 3:1 ratio of endoperoxides after four days in the dark. After three weeks, the ratio of endoperoxides was 20:1, with the endoperoxide on the unsubstituted positions the major regioisomer present, and the unoxidized acene representing about 25% of the molecules in the sample. No peaks other than those readily assigned to the endoperoxides or the acene were observed. This observation is consistent with a recent report by Fudickar and Linker, who found that endoperoxides of 9,10-diethynylanthracenes gave 1O2 through cycloreversion on a short time scale.52 We suspect that a transition state with propargyl radical character is responsible for the cycloreversion of the reactive regioisomer.50,52,53
Conclusions
We have prepared new sterically congested tetracene derivatives with absorbance and fluorescence similar to diarylpentacenes, but that are more efficient emitters and more resistant to photooxidation while still retaining sensitivity to 1O2. We anticipate that the steric congestion around the tetracene core of the T series compounds, as reflected in the lack of acene-acene interactions in their crystal structures, should prevent “butterfly” dimerization,25,54 which would otherwise be a source of false positives in 1O2-responsive materials. Research into applications of these acenes in 1O2-responsive materials, including reversibly responsive materials enabled by their photochromism, is currently under way in our laboratory.Acknowledgements
The authors acknowledge DARPA for a Young Faculty Award (N66001-09-1-2116) and Tufts University for support. X-ray diffraction instrumentation was purchased with the help of funding from the National Science Foundation (CHE-0946721).Notes and references
- J. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS.
- C. H. Chuen and Y. T. Tao, Appl. Phys. Lett., 2002, 81, 4499–4501 CrossRef CAS.
- Z. Y. Jia, S. Li, K. Nakajima, K. Kanno and T. Takahashi, J. Org. Chem., 2011, 76, 293–296 CrossRef CAS.
- G. P. Miller and J. Briggs, Org. Lett., 2003, 5, 4203–4206 CrossRef CAS.
- G. P. Miller, J. Mack and J. Briggs, Org. Lett., 2000, 2, 3983–3986 CrossRef CAS.
- D. H. Zhao and T. M. Swager, Org. Lett., 2005, 7, 4357–4360 CrossRef CAS.
- J. M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS.
- B. Northrop, K. Houk and A. Maliakal, Photochem. Photobiol. Sci., 2008, 7, 1463 CAS.
- I. Kaur, W. L. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS.
- J. Zhang, S. Sarrafpour, R. H. Pawle and S. W. Thomas, Chem. Commun., 2011, 47, 3445–3447 RSC.
- K. Tanaka, T. Miura, N. Umezawa, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, J. Am. Chem. Soc., 2001, 123, 2530–2536 CrossRef CAS.
- N. Umezawa, K. Tanaka, Y. Urano, K. Kikuchi, T. Higuchi and T. Nagano, Angew. Chem., Int. Ed., 1999, 38, 2899–2901 CrossRef CAS.
- J. H. Chong and M. J. MacLachlan, Chem. Soc. Rev., 2009, 38, 3301–3315 RSC.
- Z. Chen, P. Müller and T. Swager, Org. Lett., 2006, 8, 273–276 CrossRef CAS.
- I. Kaur, N. Stein, R. Kopreski and G. Miller, J. Am. Chem. Soc., 2009, 131, 3424–3425 CrossRef CAS.
- I. Kaur, W. Jia, R. P. Kopreski, S. Selvarasah, M. R. Dokmeci, C. Pramanik, N. E. McGruer and G. P. Miller, J. Am. Chem. Soc., 2008, 130, 16274–16286 CrossRef CAS.
- I. Kaur, M. Jazdzyk, N. N. Stein, P. Prusevich and G. P. Miller, J. Am. Chem. Soc., 2010, 132, 1261–1263 CrossRef CAS.
- A. A. Gorodetsky, M. Cox, N. J. Tremblay, I. Kymissis and C. Nuckolls, Chem. Mater., 2009, 21, 4090–4092 CrossRef CAS.
- O. Griffith, J. Anthony, A. Jones and D. Lichtenberger, J. Am. Chem. Soc., 2009, 132, 580–586 CrossRef.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS.
- M. Payne, J. Delcamp, S. Parkin and J. Anthony, Org. Lett., 2004, 6, 1609–1612 CrossRef CAS.
- Y. Li, Y. Wu, P. Liu, Z. Prostran, S. Gardner and B. Ong, Chem. Mater., 2007, 19, 418–423 CrossRef CAS.
- D. Chun, Y. Cheng and F. Wudl, Angew. Chem., Int. Ed., 2008, 47, 8380–8385 CrossRef CAS.
- M. Payne, S. Parkin and J. Anthony, J. Am. Chem. Soc., 2005, 127, 8028–8029 CrossRef CAS.
- B. Purushothaman, S. Parkin and J. Anthony, Org. Lett., 2010, 12, 2060–2063 CrossRef CAS.
- B. Purushothaman, M. Bruzek, S. R. Parkin, A.-F. Miller and J. E. Anthony, Angew. Chem., Int. Ed., 2011, 50, 7013–7017 CrossRef CAS.
- H. Qu and C. Chi, Org. Lett., 2010, 12, 3360–3363 CrossRef CAS.
- S. Odom, S. Parkin and J. Anthony, Org. Lett., 2003, 5, 4245–4248 CrossRef CAS.
- R. Schmidt, S. Gottling, D. Leusser, D. Stalke, A. Krause and F. Wurthner, J. Mater. Chem., 2006, 16, 3708–3714 RSC.
- C. Kitamura, Y. Abe, T. Ohara, A. Yoneda, T. Kawase, T. Kobayashi, H. Naito and T. Komatsu, Chem. Eur. J., 2009, 890–898 Search PubMed.
- C. Kitamura, T. Ohara, A. Yoneda, T. Kawase, T. Kobayashi and H. Naito, Chem. Lett., 2011, 40, 58–59 CrossRef CAS.
- C. Kitamura, H. Tsukuda, A. Yoneda, T. Kawase, T. Kobayashi and H. Naito, Eur. J. Org. Chem., 2010, 2010, 3033–3040 CrossRef.
- J. Reichwagen, H. Hopf, A. Del Guerzo, C. Belin, H. Bouas-Laurent and J. Desvergne, Org. Lett., 2005, 7, 971–974 CrossRef CAS.
- J. Dodge, J. Bain and A. Chamberlin, J. Org. Chem., 1990, 55, 4190–4198 CrossRef CAS.
- A. S. Paraskar, A. R. Reddy, A. Patra, Y. H. Wjjsboom, O. Gidron, L. J. W. Shimon, G. Leitus and M. Bendikov, Chem.–Eur. J., 2008, 14, 10639–10647 CrossRef CAS.
- C. Kim, P. Y. Huang, J. W. Jhuang, M. C. Chen, J. C. Ho, T. S. Hu, J. Y. Yan, L. H. Chen, G. H. Lee, A. Facchetti and T. J. Marks, Org. Electron., 2010, 11, 1363–1375 CrossRef CAS.
- Y. Li, Y. Wu, P. Liu, Z. Prostran, S. Gardner and B. Ong, Chem. Mater., 2007, 19, 418–423 CrossRef CAS.
- J. Anthony, J. Brooks, D. Eaton and S. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, A46, 467–473 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, A64, 112–122 CrossRef CAS.
- P. Müller, Crystallogr. Rev., 2009, 15, 57–83 CrossRef.
- R. A. Pascal, Chem. Rev., 2006, 106, 4809–4819 CrossRef CAS.
- A. S. Paraskar, A. R. Reddy, A. Patra, Y. H. Wjjsboom, O. Gidron, L. J. W. Shimon, G. Leitus and M. Bendikov, Chem.–Eur. J., 2008, 14, 10639–10647 CrossRef CAS.
- T. Petrenko, O. Krylova, F. Neese and M. Sokolowski, New J. Phys., 2009, 11, 015001 CrossRef.
- L. Picciolo, H. Murata and Z. Kafafi, Appl. Phys. Lett., 2001, 78, 2378 CrossRef CAS.
- A. Maliakal, K. Raghavachari, H. Katz, E. Chandross and T. Siegrist, Chem. Mater., 2004, 16, 4980–4986 CrossRef CAS.
- B. Northrop, K. Houk and A. Maliakal, Photochem. Photobiol. Sci., 2008, 7, 1463 CAS.
- Y. S. Chung, N. Shin, J. Kang, Y. Jo, V. M. Prabhu, S. K. Satija, R. J. Kline, D. M. DeLongchamp, M. F. Toney, M. A. Loth, B. Purushothaman, J. E. Anthony and D. Y. Yoon, J. Am. Chem. Soc., 2010, 133, 412–415 CrossRef.
- Z. Liang, W. Zhao, S. Wang, Q. Tang, S. Lam and Q. Miao, Org. Lett., 2008, 10, 2007–2010 CrossRef CAS.
- W. Fudickar and T. Linker, Chem. Commun., 2008, 1771–1773 RSC.
- J. M. Aubry, C. Pierlot, J. Rigaudy and R. Schmidt, Acc. Chem. Res., 2003, 36, 668–675 CrossRef CAS.
- W. Fudickar and T. Linker, Chem.–Eur. J., 2011 DOI:10.1002/chem.201102230.
- N. J. Turro, M. F. Chow and J. Rigaudy, J. Am. Chem. Soc., 1981, 103, 7218–7224 CrossRef CAS.
- P. Coppo and S. Yeates, Adv. Mater., 2005, 17, 3001–3005 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: kinetic data, crystallographic information, and solid-state emission. CCDC reference numbers 853371 and 853372. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2jm16173g |
| This journal is © The Royal Society of Chemistry 2012 |