Preparation and properties of two-legged ladder polymers based on polydiacetylenes†
Hideyuki
Tabata
,
Hiroaki
Tokoyama
,
Hideo
Yamakado
and
Tsunehisa
Okuno
*
Department of Material Science and Chemistry, Wakayama University, 930 Sakaedani, Wakayama, 640-8510, JAPAN. E-mail: okuno@center.wakayama-u.ac.jp
First published on 31st October 2011
Abstract
A novel diacetylene derivative, N,N′-bis[5-(3-tolylaminocarbonyloxy)-1,3-pentadiynyl]-N,N′-diphenyl-1,4-phenylenediamine (1), was prepared, where two diacetylene groups were connected by a 1,4-phenylenediamine moiety. The molecules stacked one-dimensionally and showed solid-state-polymerization reactivity above 80 °C. The decay of the monomers at 100 °C proceeded gradually without a marked induction period and was fully completed after 400 h. The obtained polymer was a crystalline solid judged by powder X-ray diffraction (PXRD) patterns and SEM imaging. The conjugated π-system of the obtained polymer was classified as a two-legged conjugated ladder. The polymer showed a broad absorption from the visible to the near IR region, indicating a decrease in the optical band gap of ca. 1.0 eV, because of the expansion of the π-conjugated system from a one-dimensional system to a ladder. The ladder polymer showed high conductivity after I2 doping from σ293K = 5 × 10−12 S cm−1 to 1.2 × 10−5 S cm−1. The conductivity depended heavily on the pressure of iodine and reached σ373K = 2.3 × 10−1 S cm−1. The activation energy of the ladder polymer was also estimated as 360 meV.
Introduction
Organic conducting materials containing polymeric materials, have attracted interest from the viewpoint of application in electric devices such as OFET1,2 or EL.3,4 Polydiacetylenes (PDAs), which are classified as conjugated polymers,5 have attracted attention not only for applications in chemosensors,6 but also for their physical properties originating from their one-dimensional π-conjugated systems, such as non-linear optics7 or conductivity.8,9PDAs are prepared by solid-state-polymerization of diacetylene monomers.5,10,11 The polymerization has certain reaction characteristics. First, the polymerization proceeds by heat or by irradiation of high energy photons without catalyst. Second, the relative orientation of the monomer is quite important because molecular motion is restricted in the crystal. Even when the orientation is satisfied, the high activation energy for polymerization originating from large molecular motion sometimes prevents the molecules from polymerizing. Third, the polymerization has an induction period. This is also because the polymerization is a lattice-controlled reaction. Crystals store structural strain during an induction period, and the transformation from a mother to a daughter lattice accelerates the reaction.
A novel methodology developed by Okawa and Aono, where the control of polymerization was performed by STM tips, made it possible to produce PDA nanowires at will.12–14 Therefore PDAs were focused as one of the best candidates for making nanowires. The potential conductivity of PDAs along the nanowire was also estimated to be 3–5 × 10−6 S cm−1 by the double-tip-STM method,15,16 which is comparable to that of Si or some organic conducting polymers, although anisotropy of the conductivity in PDAs is significantly large. Namely, conductivity parallel to the backbone is significantly larger than that perpendicular to the backbone. It was found that the resistivity across the boundaries of PDAs was also large.
The conductivities of doped PDAs were not so large compared with other conductive polymers.17 Two significant reasons are thought to exist in the PDA system: One is the little degree of doping and the other is the large band gap of the π-conjugated system of simple PDAs. The degree of doping could be overcome by making the polymer in the form of small powders18 or as a film.19
In order to decrease their band gaps, Matsuda et al. expanded the π-system of PDAs by introducing aromatic19 or heteroaromatic rings20,21 directly as side groups. The conductivity of the bulk PDAs reached to 1.1 × 10−4 S cm−1.19 The expansion of π-system of PDAs was found to be effective for conductivity. However, it is mentioned that the direct introduction of aromatic rings to the backbone tends to decrease the flexibility for structural change and deactivate reactivity.22,23
As another methodology for expansion of the π-system, making ladder polymers was thought to be effective. By expanding of their dimensions, the planarity of the polymers increases and the band gap of the polymer decreases. In addition, making a ladder system enables prevention of the termination of π-conjugation, which is inevitable for simple PDAs because of the lattice mismatch between mother and daughter crystals.
Nakanishi et al. examined the polymerization of simple tetraacetylene derivatives.23–26 However, polymerization did not give a regular PDA. In further studies of hexaacetylene derivatives, polymerization proceeded successfully at the 1,4- and 9,12-positions to give a ladder polymer where central two acetylene units act as a linker.23,27 However, the conductivity of the ladder polymer has not been reported yet.
In order to increase the conductivity, it is important to decrease the oxidation potential of the polymer by introducing a suitable linker. We report herein the preparation and the properties of conjugated two-legged ladder PDAs, where two PDA backbones were connected by an electron donating unit as a linker. (Fig. 1)
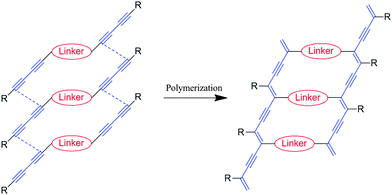 | ||
| Fig. 1 A schematic presentation for construction of conjugated ladder polymers. | ||
Results and discussion
Our molecular and crystal designs are summarized below. Basically, the configuration of the 1,4-carbons of diacetylenes changes from sp to sp2 after solid-state-polymerization. The structural strain caused by configuration changes has to be reduced by some mechanism in the solid state. In reported PDAs, methylene groups adjacent to the diacetylenic moiety play a significant role in reduction of crystal strain.11 However aromatic hydrocarbons are thought to be rigid, it was thought to be unsuitable as a linker.22,28 We have investigated ynamine moieties, where nitrogen atoms are bonded directly to acetylene groups.29 It is well-known that nitrogen atoms in arylamines can have both sp2 and sp3 configuration because the energy difference between the two configurations is small. Therefore nitrogen atoms are able to act as a cushion to reduce structural strain like methylene groups. Further, nitrogen atoms have a lone pair which can conjugate the π-system between a linker and PDAs backbone. Arylamines are known to be electron donors. Therefore inclusion of arylamines as a linker satisfies both requirements.According to an aforementioned strategy, we designed the novel diacetylene monomer 1 as shown in Fig. 2(a), where N,N′-phenylenediamine unit was introduced as a conjugated linker. Since phenylenediamines (PDs) are well-known to be electron donors,30 the linker should contribute to the decrease in the ionization potential. Arylurethane units were also introduced in the both ends in order to arrange molecular stacking by utilizing intermolecular hydrogen bonds to repeating intervals of ca. 5 Å.31
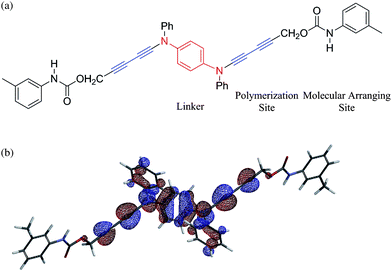 | ||
| Fig. 2 (a) Molecular design of the diacetylene monomer as a building block of a conjugated ladder polymer (b) HOMO of the monomer 1 obtained by DFT calculation, where its energy level of HOMO is estimated to be −5.2 eV. | ||
The properties of the monomer 1 were obtained by the DFT method. The HOMO of 1 was shown in Fig. 2(b). The HOMO of 1 was found to localize at the central PD unit and diacetylene moieties. The energy level of HOMO was estimated to ca. −5.2 eV, supporting that the linker can act as an electron donor.
The preparation of the monomer 1 is shown in Scheme 1.32–34 Treatment of N,N′-diacetyl-N,N′-diphenyl-1,4-phenylenediamine with phosphorus pentachloride in toluene gave N,N′-bis(trichloroethenyl)-N,N′-diphenyl-1,4-phenylenediamine (2) in 55% yield. Treatment of 2 with n-butyl lithium in tetrahydrofuran gave N,N′-diethynyl-N,N′-diphenyl-1,4-phenylenediamine (3) as a pale yellow solid in 50% yield. Hay coupling between 3 and excess of 2-propyn-1-ol afforded N,N′-bis(5-hydroxy-1,3-pentadiynyl)-N,N′-diphenyl-1,4-phenylenediamine (4) in 50% yield. Condensation of 4 with m-tolylisocyanate gave N,N′-bis[5-(3-tolylaminocarbonyloxy)-1,3-pentadiynyl]-N,N′-diphenyl-1,4-phenylenediamine (1) as a white powder in 61% isolated yield. Recrystallization from a mixed solution of dichloromethane–cyclohexane (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 v/v) gave suitable crystals for X-ray crystallography and thermal polymerization.
:5 v/v) gave suitable crystals for X-ray crystallography and thermal polymerization.
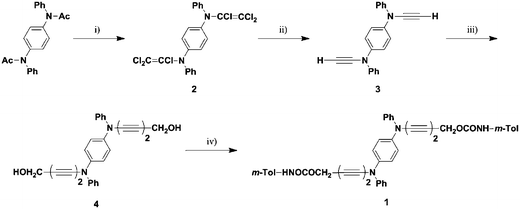 | ||
| Scheme 1 Preparation of the monomer 1. (i) PCl5, toluene, 55% (ii) n-BuLi, THF, 50% (iii) 2-propyn-1-ol, O2, CuCl, TMEDA, acetone, 50% (iv) m-TolNCO, THF, 61%. | ||
Fig. 3 shows the crystal structure of the monomer 1. The crystal data were listed in Table 1. The selected bond distances and angles were summarized in Table 2. The crystal consists of two crystallographically independent molecules, A (Fig. 3(a)) and B (Fig. 3(b)), where each molecule has an inversion center on their phenylene rings. The diacetylene groups curve slightly and clear bond alternations were recognized in both molecules. The bond distances of the ynamine moieties, N(1A)–C(1A) and N(1B)–C(1B), are 1.345(3) Å and 1.347(3) Å, respectively. These distances are consistent with those reported in the literatures.29,35–37 The structures around N(1A) and N(1B) are planar, because the sum of the angles around N(1A) and N(1B) are 359.8° and 359.6°, respectively. The dihedral angles of plane A (around N(1A)) and plane B (around N(1B)) with phenylene ring are 37.9° and 35.6°, respectively. These dihedral angles support that phenylenediamine linker connects two π-conjugated systems of the diacetylene groups effectively. The structures of urethane groups are also planar in both molecules. However, significant difference was observed in the dihedral angles with m-tolyl groups, where the angles are 35.9° in molecule A and 65.5° in molecule B, respectively. The difference of orientation in m-tolyl groups affords a herring-bone type interaction within the columnar structure (vide infra).
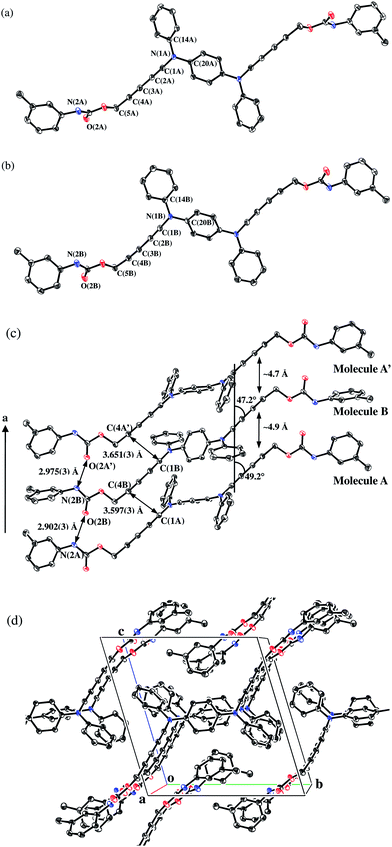 | ||
| Fig. 3 Crystal structure of 1 with 50% probability level.† Hydrogen atoms were omitted for clarity: (a) Molecule A; (b) Molecule B; (c) Molecular stacking viewed along the a axis; (d) Molecular packing viewed from the a axis. | ||
| Compound | 1 |
|---|---|
| Empirical formula | C44H34N4O4 |
| Formula weight | 682.78 |
| Temperature/K | 93 |
| Crystal system | Triclinic |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (#2) (#2) |
| a/Å | 9.589(3) |
| b/Å | 13.452(4) |
| c/Å | 14.158(4) |
| α/° | 106.847(3) |
| β/° | 91.096(5) |
| γ/° | 100.771(4) |
| V/Å3 | 1711.9(9) |
| Z | 2 |
| D c/g cm−3 | 1.325 |
| R 1(I > 2.00σ(I)) | 0.0654 |
| wR2 (I > 1.50σ(I)) | 0.1771 |
| Goodness of Fit | 1.004 |
| Molecule A | Molecule B | ||
|---|---|---|---|
| Selected bond distances/Å | |||
| N(1A)–C(14A) | 1.442(3) | N(1B)–C(14B) | 1.436(3) |
| N(1A)–C(20A) | 1.430(3) | N(1B)–C(20B) | 1.433(3) |
| N(1A)–C(1A) | 1.345(3) | N(1B)–C(1B) | 1.347(3) |
| C(1A)–C(2A) | 1.203(3) | C(1B)–C(2B) | 1.200(3) |
| C(2A)–C(3A) | 1.371(3) | C(2B)–C(3B) | 1.375(3) |
| C(3A)–C(4A) | 1.206(3) | C(3B)–C(4B) | 1.203(3) |
| C(4A)–C(5A) | 1.453(3) | C(4B)–C(5B) | 1.460(3) |
| Selected bond angles/° | |||
| C(1A)–N(1A)–C(14A) | 118.68(17) | C(1B)–N(1B)–C(14B) | 118.47(17) |
| C(1A)–N(1A)–C(20A) | 118.90(17) | C(1B)–N(1B)–C(20B) | 118.45(17) |
| C(14A)–N(1A)–C(20A) | 122.17(15) | C(14B)–N(1B)–C(20B) | 122.63(16) |
| N(1A)–C(1A)–C(2A) | 176.8(3) | N(1B)–C(1B)–C(2B) | 177.2(3) |
| C(1A)–C(2A)–C(3A) | 178.17(18) | C(1B)–C(2B)–C(3B) | 176.85(19) |
| C(2A)–C(3A)–C(4A) | 179.57(19) | C(2B)–C(3B)–C(4B) | 176.6(3) |
| C(3A)–C(4A)–C(5A) | 177.17(18) | C(3B)–C(4B)–C(5B) | 173.00(19) |
The stacking structure of the monomer along the a axis was shown in Fig. 3(c). Fig. 3(d) also shows the packing structure viewed from the a axis. Molecules A and B stack alternately to form a columnar structure, where the phenylenediamine, the phenyl and the m-tolyl groups make a herring-bone type of stacking structure by CH⋯π interaction. There are also one-dimensional intermolecular hydrogen bonds between the urethane groups, where the distances of N(2A)–O(2B) and N(2B)–O(2A′) are 2.902(3) Å and 2.975(3) Å, respectively.
The diacetylene moieties stack alternately along the a axis where the averaged stacking intervals are ca. 4.7 Å and 4.9 Å. The inclination angles with the a axis are ca. 49.2° (molecule A) and ca. 47.3° (molecule B), respectively. Intermolecular distances of the potentially polymerizing carbons are 3.597(3) Å and 3.651(3) Å. This molecular arrangement is thought to be suitable for solid-state polymerization of diacetylenes,10 although the stacking is alternated.13,38
Polymerization of the monomer 1 was found to proceed by thermal annealing above 80 °C and the color of the sample changed from pale purple to black. The polymerization could be monitored by the intensities of νC≡C (2169 cm−1, 2246 cm−1) of the monomers. The decay trace of the monomers at 100 °C is shown in Fig. 4. The polymerization proceeded gradually without a marked induction period and the decay trace showed an exponential curve. The polymerization was found to be fully completed within 400 h judging by the disappearance of the signals of the monomers. DSC peaks were not observed in the annealed sample up to 180 °C. This result indicated that there were no remaining monomers and that the obtained polymer had thermal stability up to at least 180 °C. The obtained polymer gave a novel signal at 2110 cm−1, which was assigned to the stretching of PDA backbone.
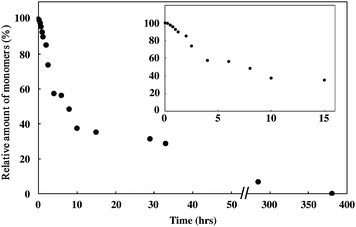 | ||
| Fig. 4 The decay trace of the monomer annealed at 100 °C under an argon atmosphere. The inset is the enlargement of the initial stage of polymerization. | ||
The obtained polymer was insoluble in common organic solvents and was a crystalline solid judged by the SEM image and the powder X-ray diffraction (PXRD) pattern. Fig. 5 shows the SEM image of the polymer 1. A bundle of fiber-like solids could be seen in the image, which is a typical shape for a low-dimensional solid such as PDAs. A diameter of microcrystalline fibers was estimated to be 1 μm. The direction of the fibers was thought to be the a axis, because the polymerization proceeds along this axis. In this polymerization, the shape of the crystals was maintained, suggesting maintenance of the crystal lattices. PXRD results (Fig. 6) also support the maintenance of the crystal lattice during thermal polymerization because the polymer 1 gave sharp diffraction lines.

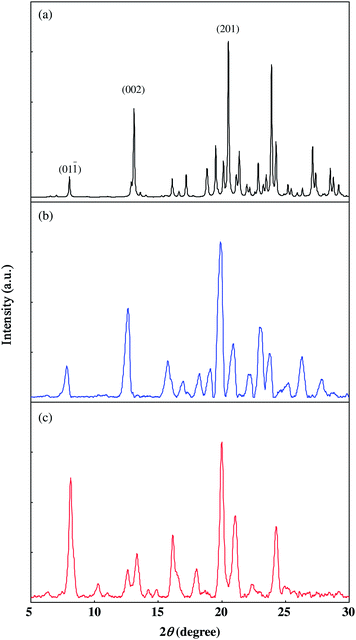 | ||
| Fig. 6 Powder X-ray diffraction (PXRD) patterns: (a) Simulated PXRD pattern of the monomer 1 based on the crystal structure; (b) PXRD pattern of the monomer 1; (c) PXRD pattern of the polymer 1. | ||
PXRD charts of both the monomer (Fig. 6(b)) and the polymer (Fig. 6(c)) gave strong diffraction lines around 2θ = 20°. The simulation of the PXRD pattern of 1 (Fig. 6(a)) based on the crystal structure afforded almost the same patterns to that of the monomer, although a slight difference in the angles was recognized because of the difference in observed temperature and experimental error. The index of the strong peak around 2θ = 19.9° was assigned to (2 0 1), the spacing of which was d = 4.46 Å. The diffraction was assigned to the spacing of stackings of m-tolyl groups. The result that the strong diffraction was still observed at the polymer suggested that the stacking structures of the m-tolyl groups were kept even after polymerization. The frequencies of νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and νN–H in the polymer were observed at 1700 cm−1 and 3288 cm−1, respectively. Although the frequencies were slightly shifted to a lower region compared with those of the monomer, the IR results indicated intermolecular N–H⋯OC hydrogen bonds were maintained even in the polymer as well as the stacking structures of the tolyl groups.
O and νN–H in the polymer were observed at 1700 cm−1 and 3288 cm−1, respectively. Although the frequencies were slightly shifted to a lower region compared with those of the monomer, the IR results indicated intermolecular N–H⋯OC hydrogen bonds were maintained even in the polymer as well as the stacking structures of the tolyl groups.
In many cases thermally polymerized polymers are disturbed structurally, however this two-legged ladder polymer keeps high regularity. This is presumably because the framework of the ladder polymer and the intermolecular hydrogen bonds maintained the local arrangement.39
The polymerization showed two unique characteristics compared with usual solid-state-polymerization of diacetylenes. One is the absence of a marked induction period and the other is little correlation in polymerization between the two diacetylenic units which are connected to π-conjugated linker. These behaviors might be explained by alternate stacks of PDs and/or PD itself that reduce the lattice strain caused by the polymerization.
Fig. 7 shows the absorption spectra of the monomer and the polymer. The polymer showed a broad band from the visible to the near IR region, and the absorption edge reached 1000 nm. The band gap of the ladder polymer was estimated to be ca. 1 eV. Since band gaps of usual one-leg PDAs were estimated to be ca. 2.5 eV,40–42 the gap of the ladder polymer was found to be very small. The observed red shift of the ladder polymer was considered to originate from the expansion of the π-conjugated system from a one-dimensional line to a ladder. The high regularity of the polymer was thought to contribute effectively to the expansion of the π-system. Such a red shift was also observed in a PDA-ladder system prepared from hexaacetylenes.23,27,43 The red shift originating in the expansion of π-system was also predicted theoretically.44
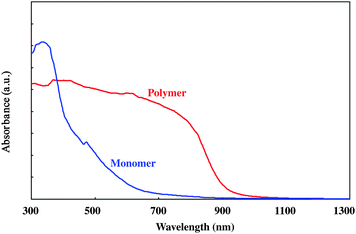 | ||
| Fig. 7 UV-Vis-NIR spectra of the monomer 1 (blue line) and the polymer 1 (red line). | ||
Although an excitonic transition was usually observed in PDAs around 2 eV,41,42 the lowest absorption band was assigned to an interband transition. The reasons for the assignment are: (i) the band gap estimated theoretically is accordance with the optically observed one;44 and (ii) the absence of the transition was reported in the case where nitrogen atoms were attached directly to PDAs.45
Conductivities of the polymer measured by the 2 probe methods in compressed pellets were summarized in Table 3. Before doping by iodine, conductivities of the polymer was σRT = 5 × 10−12 S cm−1 which was a usual value for PDAs without doping.46 When the sample was placed under iodine vapor at room temperature, its conductivity was increased by six order to reach σRT = 1.2 × 10−5. Judging from the crystal structure (Fig. 3(d)) and the SEM images, iodines were suggested to penetrate into crystalline fibers from the a axis.
| T/°Ca | p I2/torrb | σ T/S cm−1c | σ RT/S cm−1d | E a/meVe |
|---|---|---|---|---|
| a Doping temperature. b Vapor pressure of iodine. c Conductivities monitored during doping. d Conductivities at room temperature after 15 min. e E a was estimated by Arrhenius plot. f Unable to estimate because of cracks owing to rapid cooling to −30 °C. | ||||
| 20 | 0.2 | 1.2 × 10−5 | 1.2 × 10−5 | 360 |
| 40 | 1.0 | 5.7 × 10−5 | 1.3 × 10−5 | 470 |
| 60 | 4.3 | 2.3 × 10−4 | 1.6 × 10−5 | 500 |
| 80 | 15 | 1.9 × 10−3 | 8.4 × 10−5 | —f |
| 100 | 46 | 2.3 × 10−1 | 3.7 × 10−2 | —f |
The conductivity at room temperature decreased gradually to the original value when the sample was placed in air. This is presumably because iodine within the sample escaped outside. When the sample was doped by iodine again, the conductivity recovered to the former value of σRT = 1.2 × 10−5. This reversible feature indicated that iodine did not react with the PDA backbone. This reversibility was also recognized even at higher temperature.
The conductivity depended heavily on a vapor pressure of iodine. It increased with increasing doping temperature to σ333K = 2.3 × 10−4 S cm−1 at 60 °C. The conductivity finally reached σ373K = 2.3 × 10−1 S cm−1 at 100 °C. After doping at 100 °C, the conductivity of the sample at room temperature decreased to σ293K = 3.7 × 10−2 S cm−1. This is three thousand times as large as that doped at room temperature and is significantly bigger than those reported in the literature.18,19 The record for conductivity of chemically doped PDAs was 3 × 10−2 S cm−1 at 70 °C, which was realized by grinding the single crystals into small particles.18 Our results surpass the reported data even at room temperature. Such a high conductivity is thought to be derived from both the expansion of π-conjugate system and the decrease of ionization potential by introduction of 1,4-phenylenediamine moiety as a conjugated linker. The amount of iodine within the polymer was, unfortunately, unable to be estimated because of the deposition of iodine crystals.
The conductivity of the doped polymer was also measured at the lower temperature region of −40 °C to 0 °C. In this region, the conductivities showed reversible temperature dependence. This is presumably because iodine in the polymer is trapped tightly and cannot escape outside the sample. Fig. 8 shows the temperature dependence of the conductivities, where the ln σT was plotted as a function of T−1. Linear relationship was recognized between ln σT and T−1 for all samples. When the temperature dependence was assumed to obey Arrhenius's eqn (1), activation energies were estimated by the slope to be 360 meV (doped at 20 °C), 470 meV (doped at 40 °C) and 500 meV (doped at 60 °C) (Table 3).
| σT = σ exp(−Ea/RT) | (1) |
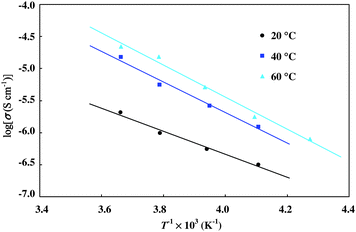 | ||
| Fig. 8 The Arrhenius plot of conductivities of the polymer 1 after I2 doping. | ||
The reports for the activation energy of PDAs are limited to a few studies. Sakamoto et al. reported the energies of PTS (poly[2,4-hexadiyne-1,6-diol-bis-(p-toluene sulfonate)]) and DCH (poly[2,4-hexadiyne-1,6-di(N-carbazolyl)]) in single crystals to be 200 meV and 20 meV by implantation of 75As.47 The energy of chemically doped DCH in a single crystal was estimated to be 200 meV.48 The activation energy of the ladder polymer was comparable with those of PDA single crystals, although the samples were compacted pellets.
Conclusion
In conclusion, we succeeded in providing a novel two-legged conjugated ladder polymer based on PDAs, where phenylenediamine moieties were introduced as a linker. The obtained polymer was crystalline solid. This is presumably because the regularity of the polymer was maintained by the framework of the ladder system. The band gap of the ladder polymer was estimated to be ca. 1.0 eV, which was a very small value compared with that of the reported PDAs. This tendency was also explained by the expansion of the π-conjugated system of the polymer.The conductivity of the polymer changed significantly by I2 doping from insulator (σ293K = 5 × 10−12 S cm−1) to semiconductor (1.2 × 10−5 S cm−1). The conductivity depended heavily on a pressure of iodine and reached σ373K = 2.3 × 10−1 S cm−1 at 100 °C. Such high regularity and conductivity were unusual for PDAs. The methodology, introduction of a linker to connect one-dimensional π-systems, is found to be useful for developing organic conducting polymers.
Experimental
1. General procedure
All chemicals were purchased from Kanto Chemical Co. Ltd. or Sigma Aldrich Co. Ltd. and used without further purification. Gel permeation chromatography (GPC) was performed on a JAI LC-918 equipped with JAIGEL -1H and -2H columns. 1H and 13C NMR spectra were recorded on a JEOL JNM-ECA-400 spectrometer in deuterated solvents (chloroform-d or acetone-d6) with tetramethylsilane as an internal standard. All 13C NMR spectra were obtained with complete proton decoupling. IR spectra were recorded on a JASCO FT/IR-420 spectrometer by using a KBr pellet. UV-Vis spectrum was measured on a HITACHI U-2010 spectrometer. UV-Vis-NIR spectrum in solid state was measured on a SHIMADZU UV-3100PC spectrometer equipped with an ISR-3100 integrating sphere attachment. Elemental analysis was recorded on a J-SCEINCE LAB MICRO CORDER JM10.X-Ray crystallographic data were obtained by a RIGAKU Saturn 724 + CCD device with a monochromatic Mo-Kα radiation at −180 °C. The structure was solved by a direct method (SIR 92), and refined by a full-matrix least-squares method. The positions of atoms were obtained from differential Fourier maps. Non-hydrogen atoms were treated anisotropically. Hydrogen atoms were treated isotropically and were not refined. Powder X-ray diffraction (XRD) was recorded on a RIGAKU MiniFlex II diffractometer with a monochromatic Cu-Kα radiation at ambient temperature. Theoretical calculations were performed on Spartan 03 software (Wavefunction, Inc.) with DFT 6-31G* level.
Polymerization of the monomer 1 was performed in an electric oven. The powdered monomer was sealed into glass tubes with exchange gas of argon. The tubes were annealed at 100 °C.
Scanning electric microscope (SEM) image was taken by JEOL JSM-6390 for pelletized sample, where the accelerating voltage was 20 kV. Differential scanning calorimetry (DSC) was performed on SHIMADZU DSC-50 calorimeter.
The conductivity of the polymer was measured by 2-probe method. The compressed polymer, sealed with excess amounts of iodine, was placed in an electric oven. The size of the pellets was ca. in 100 μm thickness and ca. 200 × 200 μm2 in area.
2. Materials
NMR data: δH (400 MHz; chloroform-d; TMS) 7.07 (4H, s), 7.08 (4H, dd, J = 8.5, 1.2 Hz), 7.12 (2H, tt, J = 7.5, 1.2 Hz), 7.33 (4H, dd, J = 8.5, 7.5 Hz).
NMR data: δH (400 MHz; chloroform-d; TMS) 2.90 (2H, s), 7.10 (2H, tt, J = 7.1, 1.2 Hz), 7.28 (4H, dd, J = 8.6, 1.2 Hz), 7.28 (4H, s), 7.34 (4H, dd, J = 8.6, 7.1 Hz). δC (100 MHz; chloroform-d; TMS) 55.06, 81.90, 120.27, 122.48, 123.95, 129.27, 139.27, 143.02.
Elemental analysis (Found: C, 80.41; H, 4.88; N, 6.83. Calc. for C28H20N2O2: C, 80.75; H, 4.84; N, 6.73%). IR absorption (KBr): (νmax/cm−1) 3331 (O–H), 2241 and 2165 (C≡C). NMR data: δH (400 MHz; acetone-d6; TMS) 2.93 (2H, br s), 4.33(4H, s), 7.24 (2H, tt, J = 7.4, 0.9 Hz), 7.31 (4H, dd, J = 8.7, 0.9 Hz), 7.36 (4H, s), 7.46 (4H, dd, J = 8.7, 7.4 Hz). δC (100 MHz; acetone-d6; TMS) 51.19, 55.52, 69.76, 76.83, 83.43, 121.67, 123.44, 126.04, 130.61, 140.19, 143.22.
Elemental analysis (Found: C, 77.05; H, 5.39; N, 7.96. Calc. for C44H34N4O4: C, 77.40; H, 5.02; N, 8.21%). UV-Vis absorption: λmax(CHCl3)/nm 296 (log ε/dm3 mol−1 cm−1 4.6). IR absorption (KBr): (νmax/cm−1) 3312 (NH), 2246 and 2169 (C≡C), 1705 (CO). NMR data: δH (400 MHz; acetone-d6; TMS) 2.30 (6H, s), 4.93 (4H, s), 6.86 (2H, d, J = 7.7 Hz), 7.17 (2H, dd, J = 7.8, 7.7 Hz), 7.25 (2H, tt, J = 7.4, 1.1 Hz), 7.31 (4H, dd, J = 8.6, 1.1 Hz), 7.35 (2H, d, J = 7.8 Hz), 7.36 (4H, s), 7.40 (2H, s), 7.46 (4H, dd, J = 8.6, 7.4 Hz), 8.78 (2H, br s). δC (100 MHz; acetone-d6; TMS) 21.59, 53.61, 55.41, 71.89, 77.97, 78.88, 116.44, 119.82, 121.80, 123.50, 124.47, 126.25, 129.53, 130.69, 139.31, 139.75, 140.09, 142.94, 153.56.
Acknowledgements
This work was supported by Research for Promoting Technological Seeds from Japan Science and Technology Agency (JST).References
- H. Usta, A. Facchetti and T. J. Marks, Acc. Chem. Res., 2011, 44, 501–510 CrossRef CAS.
- T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011 DOI:10.1021/cr100428a.
- P. M. Beaujuge and J. R. Reynolds, Chem. Rev., 2010, 110, 268–320 CrossRef CAS.
- A. C. Grimsdale, K. L. Chan, R. E. Martin, P. G. Jokisz and A. B. Holmes, Chem. Rev., 2009, 109, 897–1091 CrossRef CAS.
- G. Wegner, Z. Naturforsch., B: J. Chem. Sci., 1969, B24, 824–832 Search PubMed.
- D. Ahn and J. Kim, Acc. Chem. Res., 2008, 41, 805–816 CrossRef CAS.
- T. Manaka, H. Kohn, Y. Ohshima, E. Lim and M. Iwamoto, Appl. Phys. Lett., 2007, 90, 171119 CrossRef.
- T. Sakanoue, M. Yahiro, C. Adachi, K. Takimiya and A. Toshimitsu, J. Appl. Phys., 2008, 103, 094509 CrossRef.
- T. Koyanagi, M. Muratsubaki, Y. Hosoi, T. Shibata, K. Tsutsui, Y. Wada and Y. Furukawa, Chem. Lett., 2006, 35, 20–21 CrossRef CAS.
- R. H. Baughman, J. Polym. Sci., Polym. Phys. Ed., 1974, 12, 1511–1535 CrossRef CAS.
- V. Enkelmann, in Advances in Polymer Science, ed. H. J. Cantow, Springer-Verlag, Berlin, 1984, vol. 63, pp. 91–136 Search PubMed.
- Y. Okawa and M. Aono, Nature, 2001, 409, 683–684 CrossRef CAS.
- Y. Okawa and M. Aono, J. Chem. Phys., 2001, 115, 2317–2322 CrossRef CAS.
- Y. Okawa, S. K. Mandal, C. Hu, Y. Tateyama, S. Goedecker, S. Tsukamoto, T. Hasegawa, J. K. Gimzewski and M. Aono, J. Am. Chem. Soc., 2011, 133, 8227–8233 CrossRef CAS.
- K. Takami, J. Mizuno, M. Akai-kasaya, A. Saito, M. Aono and Y. Kuwahara, J. Phys. Chem. B, 2004, 108, 16353–16356 CrossRef CAS.
- K. Takami, Y. Kuwahara, T. Ishii, M. Akai-Kasaya, A. Saito and M. Aono, Surf. Sci., 2005, 591, L273–L279 CrossRef CAS.
- H. Nakanishi, F. Mizutani, M. Kato and K. Hasumi, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 983–987 CrossRef CAS.
- V. A. Marikhin, E. G. Guk and L. P. Myasnikova, Phys. Solid State, 1997, 39, 686–689 CrossRef.
- H. Matsuda, H. Nakanishi, S. Kato and M. Kato, J. Polym. Sci., Part A: Polym. Chem., 1987, 25, 1663–1669 CrossRef CAS.
- H. Matsuda, H. Nakanishi, N. Minami and M. Kato, Mol. Cryst. Liq. Cryst., 1988, 160, 241–251 CrossRef.
- A. Sarkar, S. Okada, H. Nakanishi and H. Matsuda, Macromolecules, 1998, 31, 9174–9180 CrossRef CAS.
- G. Wegner, J. Polym. Sci., Part B: Polym. Lett., 1971, 9, 133–144 CrossRef CAS.
- A. Sarkar, S. Okada, H. Matsuzawa, H. Matsuda and H. Nakanishi, J. Mater. Chem., 2000, 10, 819–828 RSC.
- S. Okada, H. Matsuda, A. Masaki, H. Nakanishi and K. Hayamizu, Chem. Lett., 1990, 19, 2213–2216 CrossRef.
- K. Hayamizu, S. Okada, S. Tsuzuki, H. Matsuda, A. Masaki and H. Nakanishi, Bull. Chem. Soc. Jpn., 1994, 67, 342–345 CrossRef CAS.
- A. Sarkar, S. Okada, K. Komatsu and H. Nakanishi, Macromolecules, 1998, 31, 5624–5630 CrossRef CAS.
- S. Okada, K. Hayamizu, H. Matsuda, A. Masaki, N. Minami and H. Nakanishi, Macromolecules, 1994, 27, 6259–6266 CrossRef CAS.
- H. Matsuzawa, S. Okada, A. Sarkar, H. Matsuda and H. Nakanishi, Polym. J., 2001, 33, 182–189 CrossRef CAS.
- T. Okuno, S. Ikeda, N. Kubo and D. J. Sandman, Mol. Cryst. Liq. Cryst., 2006, 456, 35–44 CAS.
- Y. Yamashita, T. Suzuki, G. Saito and T. Mukai, Chem. Lett., 1985, 14, 1759–1762 CrossRef.
- J. W. Lauher, F. W. Fowler and N. S. Goroff, Acc. Chem. Res., 2008, 41, 1215–1229 CrossRef CAS.
- Y. Okamoto and S. K. Kundu, J. Org. Chem., 1970, 35, 4250–4252 CrossRef CAS.
- C. Cuniberti, G. Dellepiane, P. Piaggio, R. Franco, G. F. Musso, C. Dell'Erba and G. Garbarino, Chem. Mater., 1996, 8, 708–713 CrossRef CAS.
- A. S. Hay, J. Org. Chem., 1962, 27, 3320–3321 CrossRef CAS.
- J. J. Mayerle and M. A. Flandera, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1978, 34, 1374–1376 CrossRef.
- R. Galli, M. Neuenschwander and P. Engel, Helv. Chim. Acta, 1988, 71, 1914–1923 CrossRef CAS.
- R. Galli, M. Neuenschwander and P. Engel, Helv. Chim. Acta, 1989, 72, 1324–1336 CrossRef CAS.
- R. Xu, V. Gramlich and H. Frauenrath, J. Am. Chem. Soc., 2006, 128, 5541–5547 CrossRef CAS.
- Y. Xu, M. D. Smith, M. F. Geer, P. J. Pellechia, J. C. Brown, A. C. Wibowo and L. S. Shimizu, J. Am. Chem. Soc., 2010, 132, 5334–5335 CrossRef CAS.
- H. Bässler, in Advances in Polymer Science, ed. H. J. Cantow, Springer-Verlag, Berlin, 1984, vol. 63, pp. 1–48 Search PubMed.
- N. S. Sariciftci, B. Kraabel, C. H. Lee, K. Pakbaz, A. J. Heeger and D. J. Sandman, Phys. Rev. B: Condens. Matter, 1994, 50, 12044–12051 CrossRef CAS.
- G. Barcza, Ö. Legeza, F. Gebhard and R. M. Noack, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 045103 CrossRef.
- S. Inayama, Y. Tatewaki and S. Okada, Polym. J., 2010, 42, 201–207 CrossRef CAS.
- S. Yang and M. Kertesz, Macromolecules, 2007, 40, 6740–6747 CrossRef CAS.
- K. Ichimura, H. Matsuda, H. Nakanishi and T. Kobayashi, Phys. Rev. B: Condens. Matter, 1993, 47, 6250–6255 CrossRef CAS.
- G. Wegner and W. Schermann, Colloid Polym. Sci., 1974, 252, 655–672 CAS.
- M. Sakamoto, B. Wasserman, M. S. Dresselhaus, G. E. Wnek, B. S. Elman and D. J. Sandman, J. Appl. Phys., 1986, 60, 2788–2796 CrossRef CAS.
- F. Ebisawa, T. Kurihara and H. Tabei, Synth. Met., 1987, 18, 431–436 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures; NMR spectra; X-ray crystallographic data for 1. CCDC reference number 838856. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1jm13896k |
| This journal is © The Royal Society of Chemistry 2012 |