
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCu-catalyzed C(sp2)–N-bond coupling of boronic acids and cyclic imides†
Linn Neerbye
Berntsen
 ,
Thomas Nordbø
Solvi
,
Kristian
Sørnes
,
David S.
Wragg
and
Alexander H.
Sandtorv
*
,
Thomas Nordbø
Solvi
,
Kristian
Sørnes
,
David S.
Wragg
and
Alexander H.
Sandtorv
*
Department of Chemistry, University of Oslo, Oslo N-0315, Norway. E-mail: a.h.sandtorv@kjemi.uio.no
First published on 19th October 2021
Abstract
A general Cu-catalyzed strategy for coupling cyclic imides and alkenylboronic acids by forming C(sp2)–N-bonds is reported. The method enables the practical and mild preparation of (E)-enimides. A large range of cyclic imides are allowed, and di- and tri-substituted alkenylboronic acids can be used. Full retention was observed in the configuration of the alkene double bond in the coupled products. The method is also applicable for preparing N-arylimides, using arylboronic acids as coupling partners. The usefulness of this strategy is exemplified by the convenient derivatization of the chemotherapy medication 5-flurouracil, the nucleoside uridine and the anti-epileptic drug phenytoin.
Enimides (Fig. 1) are functional groups found in N-sulfonylurea isosteres,1 biologically active structures,2,3 functional materials,4 and natural products such as the parazoanthines A–E.5 They are also building blocks in synthesis of complex structures,6 polycyclic architecture,7 and β-2-amino acid derivatives.8
 | ||
| Fig. 1 Some examples of cyclic enimides. | ||
The growing interest in the enimide moiety has catalyzed a recent spurt of attention for methodology appropriate for its construction (Fig. 2).9 In reactions where the enimidic C(sp2)–N-bond is formed, only a few strategies are known. The main access point is the Ru-catalyzed hydroimidation strategy, wherein imides and alkynes are condensed (Fig. 2, strategy 1).10 Drawbacks include the use of an expensive Ru-catalysts, and the structural limitations imposed. A second approach involves the Cu-mediated coupling of imides and vinylic halides (Fig. 2, strategy 2).11 This strategy is only applicable to phthalimide, and therefore specialized. Other examples are substrate specific.12,13
 | ||
| Fig. 2 Prior developments in the area of C(sp2)–N-bond formation. | ||
The Chan–Lam14 inspired Cu-catalyzed process using alkenylic boron coupling partners, is an attractive route to enimides for several reasons: (i) the availability of alkenylboronic reagents15 provide synthetic flexibility, (ii) the use of an inexpensive Cu-catalyst is attractive, and (iii) a potentially larger structural diversity of enimides is conceivable, compared to Ru-methods currently used. Thus far, only highly substrate dependent examples are known,16 but a general method has not been reported before now.
Our endeavors were initiated as shown in Table 1. Due to our ongoing interest17 in the pharmaceutically relevant hydantoin framework,18 hydantoin 1a was used as a model substrate, with styrylboronic acid 2a as reagent. The optimal conditions (entry #1) involved the use of excess reagent 2a (3.0 equiv.) with copper(II)triflate and pyridine in ethanol at 25 °C. Less expensive Cu-salts may also be effective (#2 and 3). The process was also effective using 2.0 equiv. of the reagent (entry #4), so less reagent can be employed if a slight reduction in yield is acceptable. Our investigations uncovered that the process was inefficient in aprotic solvents such as DMF and toluene (entries #6 and 7), and that the use of base/ligand was of paramount importance. Surprisingly, triethylamine (entry #8) performed poorly compared to pyridine. Strong, non-nucleophilic bases (entries #9 and 11) were also ineffective. The complete optimization study can be found in the ESI.†
|
|
||||||
|---|---|---|---|---|---|---|
| # | Catalyst | 2a (equiv.) | Additive/base (equiv.) | Solvent | Time (h) | Yielda (%) |
| Conditions: N-Methylhydantoin 1a (0.20 mmol, 1.0 equiv.), boronic acid 2a (as specified), catalyst (0.010 mmol, 0.050 equiv.), additive/base (as specified) in solvent (as specified, 1 mL).a 1H NMR yield using mesitylene as internal standard.b Isolated yield.c The (hemi)pentahydrate salt was used.d Reaction performed at 40 °C. | ||||||
| 1 | Cu(OTf)2 | 3.0 | Py (1.0) | EtOH | 9/15 | 98/97b |
| 2 | Cu(NO3)2c | 3.0 | Py (1.0) | EtOH | 24 | 96 |
| 3 | CuCl | 3.0 | Py (1.0) | EtOH | 24 | 95 |
| 4 | Cu(OTf)2 | 2.0 | Py (1.0) | EtOH | 24 | 94 |
| 5d | Cu(OTf)2 | 1.0 | Py (1.0) | EtOH | 24 | 71 |
| 6 | Cu(OTf)2 | 3.0 | Py (1.0) | DMF | 24 | 26 |
| 7 | Cu(OTf)2 | 3.0 | Py (1.0) | PhMe | 24 | 0 |
| 8 | Cu(OTf)2 | 3.0 | Et3N (1.0) | EtOH | 24 | 8 |
| 9 | Cu(OTf)2 | 3.0 | t-BuOK (1.0) | EtOH | 18 | 22 |
| 10 | — | 3.0 | t-BuOK (2.0) | EtOH | 24 | 0 |
| 11 | Cu(OTf)2 | 3.0 | LiHMDS (1.0) | EtOH | 18 | 3 |

We next investigated the scope and limitations of the method, Scheme 1. Complete retention of the configuration of the alkene double bond was observed in all cases. The anti-epileptic drug phenytoin 1c was conveniently derivatized in excellent yield. Hydantoin 1e was also smoothly alkenylated. The aldol adduct 1h was smoothly N-3-alkenylated and not O-alkenylated, in excellent yield. We typically observed N-1,N-3-bisalkenylated hydantoins as byproducts in these reactions, such as the disubstituted hydantoin 3j′. Phtalimide 3m and glutarimide 3p were obtained in excellent yields. Uracils are privileged structures in drug discovery.19 Uracil 1q, the chemotherapy medication 5-fluorouracil 1r and the nucleoside uridine 1s were bis-alkenylated to 3q, 3r and 3s, respectively. These results show that the method can conjugate drugs and nucleosides and provide uracils cumbersome to access.20 Lastly, 2,4-thiazolidinedione 1n was unreactive, but the ferrocenyl derivative 3f was obtained in excellent yield.
 | ||
Scheme 1 Imide scope of the reaction. Conditions: imide 1a–1u (0.40 mmol, 1.0 equiv.), boronic acid 2a (1.2 mmol, 3.0 equiv.), Cu(OTf)2 (0.020 mmol, 0.050 equiv.), pyridine (0.40 mmol, 1.0 equiv.) and EtOH (2 mL). a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) (E)-Styryl-9-BBN was used as coupling partner. bCuCl (5.0 mol%) was used as catalyst. c2.0 equiv. boronic acid 2a was used. dReaction performed at 1.5 mmol scale. eReaction performed at 0.40 and 1.0 mmol scale. f4.0 equiv. boronic acid 2a was used. (E)-Styryl-9-BBN was used as coupling partner. bCuCl (5.0 mol%) was used as catalyst. c2.0 equiv. boronic acid 2a was used. dReaction performed at 1.5 mmol scale. eReaction performed at 0.40 and 1.0 mmol scale. f4.0 equiv. boronic acid 2a was used. | ||
The method is applicable to cyclic imides, and not linear imides, as imide 1o failed to react under our conditions. As suggested by Wasielewski et al.21 the carbonyl groups can adopt a parallel, coplanar conformation22 and may chelate to Cu(II)-ions. The chelation is likely detrimental for the coupling reaction.
A selection of alkenylboronic acids 2b–2f were next investigated with some imides, Scheme 2. The performance of the coupling reaction varied. The 1-pentenyl reagent 2b transferred in good to moderate yields, depending on the imide substrate. The cyclopentenyl coupling partner 2c was challenging, likely imposing a high steric demand in relevant Cu-species in the catalytic cycle. The coupled product 4g was obtained in good yield, but the six membered product 4f was not detected. The 1,1,2-trisubstituted alkenes obtained, are not currently accessible employing the Ru-catalyzed methods mentioned in the introduction.
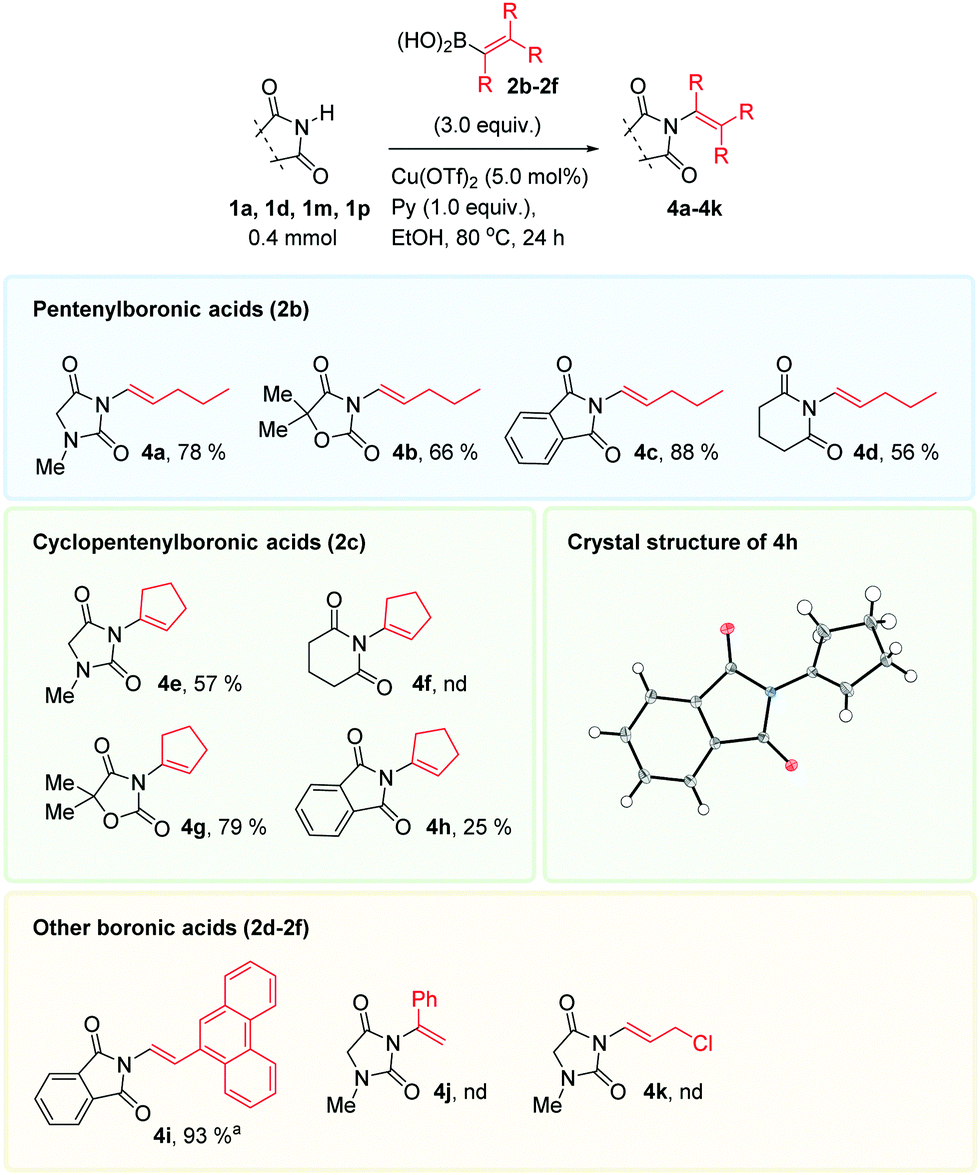 | ||
| Scheme 2 Alkenylboronic acid scope. Conditions: imide 1a, 1d, 1m or 1p (0.40 mmol, 1.0 equiv.), boronic acid 2b–2f (1.2 mmol, 3.0 equiv.), Cu(OTf)2 (0.020 mmol, 0.050 equiv.), pyridine (0.40 mmol, 1.0 equiv.) and EtOH (2 mL). aReaction performed at 0.18 mmol scale at 25 °C. | ||
Our earlier work17 spurred an interest in employing arylboronic acids for N-arylation. Although the Cu-catalyzed or -mediated arylation of imides has been documented earlier,23 the use of such conditions is sparingly described24 using the pharmaceutically relevant hydantoin, 2,4-oxazolidinedione and related frameworks as substrates.
The method was high yielding and tolerable to diverse aryl groups, Scheme 3. The reaction performance varied if the arylboronic acid (method 1), or the corresponding anhydride (triarylboroxine, method 2) was employed. It is not possible to draw a conclusion as to which method operates best with which substrate. The pyridine likely has several key functions, such as being a Cu-ligand, acting as a base, and stabilizing boroxines in situ.25
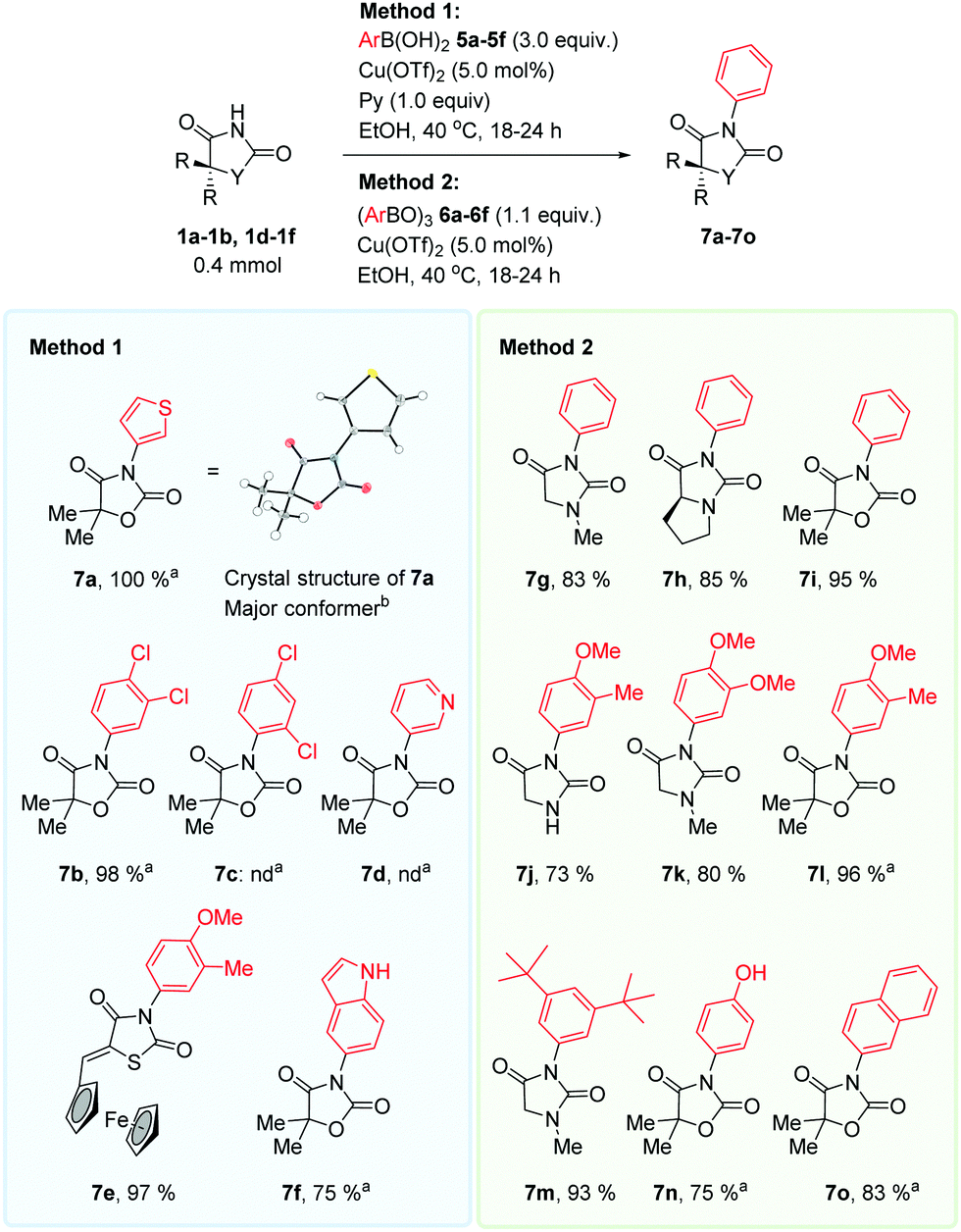 | ||
| Scheme 3 Scope of N-arylation. Method 1: hydantoin 1a, b or 1d–1f (0.40 mmol, 1.0 equiv.), boronic acid 5a–5f (1.2 mmol, 3.0 equiv.), Cu(OTf)2 (0.020 mmol, 0.050 equiv.), pyridine (0.020 mmol, 1.0 equiv.) in EtOH (2 mL). Method 2: hydantoin 1a, b or 1d–1f (0.40 mmol, 1.0 equiv.), boroxine 6a–6f (0.44 mmol, 1.1 equiv.), Cu(OTf)2 (0.020 mmol, 0.050 equiv.). aReaction performed on 0.14 mmol scale. bOnly the major conformer of the thiophene-ring is shown. | ||
The method smoothly transferred electron-rich aryl groups to form products such as 7j, 7k, 7l, 7m and 7n in mostly excellent yields. Electron-rich heterocyclic fragments were also efficiently coupled. The thiophenyl product 7a was obtained in excellent yield, and the indolyl product 7f was obtained in good yield. The pyridinyl product 7d was not obtained. The electron-poor 3,4-dichlorophenyl group coupled to afford product 7b in excellent yield, whereas the 2,4-dichlorophenyl group did not couple. We attribute this to the steric hinderance of the chlorine atom in the 2-position of the aryl group. Electronically neutral phenyl and naphthyl groups coupled in good yields.
L. N. B. and T. N. S. acknowledge the Department of Chemistry at the University of Oslo for funding their PhD fellowships. George Peletis is ackowledged for performing initial experiments on the N-arylation chemistry of hydantoin. The project has received support from UiO: Life Science and was partially supported by the Research Council of Norway through the Norwegian NMR Platform, NNP (226244/F50). We acknowledge the use of the Norwegian national infrastructure for X-ray diffraction and scattering (RECX, Research Council of Norway project number 208896).
Conflicts of interest
There are no conflicts to declare.Notes and references
- B. Metayer, A. Angeli, A. Mingot, K. Jouvin, G. Evano, C. T. Supuran and S. Thibaudeau, J. Enzyme Inhib. Med. Chem., 2018, 33, 804–808 CrossRef CAS PubMed.
- H. Uhr, C. Boie, H. Rieck, B.-W. Krueger, U. Heinemann, R. Market, M. Vaupel, M. Kugler, K. Stenzel, U. Wachendorff-Neumann, A. Mauler-Machnik, K.-H. Kuck, P. Loesel and S.-I. Narabu, Preparation of aryloxydithiazole and aryloxydithiazine dioxides as pesticides and fungicides, Patent DE 19918294, 2000.
- (a) A. D. Brown, M. E. Bunnage, C. A. L. Lane, R. A. Lewthwaite, P. A. Glossop, K. James and D. A. Price, Preparation of formylamino hydroxyphenyl phenylacetamide derivatives as β2 adrenoceptor agonists, Patent US 20050215590 A1, 2005; (b) D. L. Kirkpatrick and M. Indarte, Preparation of N-substituted thiazolecarboxamides for inhibiting CNKSR1, Patent US 20160304533 A1, 2016; (c) S. Louzoun Zada, K. D. Green, S. K. Shrestha, I. M. Herzog, S. Garneau-Tsodikova and M. Fridman, ACS Infect. Dis., 2018, 4, 1121–1129 CrossRef CAS PubMed.
- (a) T. Ogawa and H. Yamada, Preparation of N-styrylphthalimide derivatives as nonlinear organic optical materials, Patent JP 08176107, 1996; (b) S. Otsu, K. Ofuku and N. Kagawa, Semiconductor for photoelectric conversion material, photoelectric converter, and photoelectrochemical cell, Patent, JP 2005019124, 2003.
- N. Cachet, G. Genta-Jouve, E. L. Regalado, R. Mokrini, P. Amade, G. Culioli and O. P. J. Thomas, J. Nat. Prod., 2009, 72(9), 1612–1615 CrossRef CAS PubMed.
- (a) E. Alacid and C. Najera, Adv. Synth. Catal., 2008, 350, 1316–1322 CrossRef CAS; (b) W. Susanto, C.-Y. Chu, W. J. Ang, T.-C. Chou, L.-C. Lo and Y. Lam, J. Org. Chem., 2012, 77, 2729–2742 CrossRef CAS PubMed; (c) F. de Nanteuil and J. Waser, Angew. Chem., Int. Ed., 2013, 52, 9009–9013 CrossRef CAS PubMed; (d) J. Szudkowska-Frątczak, G. Hreczycho and P. Pawluć, Org. Chem. Front., 2015, 2, 730–738 RSC; (e) E. Landagaray, M. Ettaoussi, M. Rami, J. A. Boutin, D.-H. Caignard, P. Delagrange, P. Melynyk, P. Berthelot and S. Yous, Eur. J. Med. Chem., 2017, 127, 621–631 CrossRef CAS PubMed.
- (a) S. K. Murphy, M. Zeng and S. B. Herzon, Science, 2017, 356, 956–959 CrossRef CAS PubMed; (b) P. Kramer, J. Schonfeld, M. Bolte and G. Manolikakes, Org. Lett., 2018, 20, 178–181 CrossRef CAS PubMed.
- J. Dai, W. Ren, W. Chang, P. Zhang and Y. Shi, Org. Chem. Front., 2017, 4, 297–302 RSC.
- (a) Z. J. Song, M. D. Tellers, P. G. Dormer, D. Zewge, J. M. Janey, A. Nolting, D. Steinhuebel, S. Oliver, P. N. Devine and D. M. Tschaen, Org. Process Res. Dev., 2014, 18, 423–430 CrossRef CAS; (b) H. Lingua, F. Vibert, D. Mouysset, D. Siri, M. P. Bertrand and L. Feray, Tetrahedron, 2017, 73, 3415–3422 CrossRef CAS; (c) W.-M. Cheng, R. Shang and Y. Fu, Nat. Commun., 2018, 9, 5215–5223 CrossRef PubMed; (d) W. Lin, W. Li, D. Lu, F. Su, T.-B. Wen and H.-J. Zhang, ACS Catal., 2018, 8, 8070–8076 CrossRef CAS.
- (a) L. J. Goossen, M. Blanchot, C. Brinkmann, K. Goossen, R. Karch and A. J. Rivas-Nass, Org. Chem., 2006, 71, 9506–9509 CrossRef CAS PubMed; (b) M. Arndt, K. S. M. Salih, A. Fromm, L. J. Goossen, F. Menges and G. Niedner-Schatteburg, J. Am. Chem. Soc., 2011, 133, 7428–7449 CrossRef CAS PubMed; (c) A. E. Buba, M. Arndt and L. J. Goossen, J. Organomet. Chem., 2011, 696, 170–178 CrossRef CAS; (d) E. Semina, P. Tuzina, F. Bienewald, A. S. K. Hashmi and T. Schaub, Chem. Commun., 2020, 56, 5977–5980 RSC.
- (a) R. G. R. Bacon and A. Karim, J. Chem. Soc., Perkin Trans. 1, 1973, 278–280 RSC; (b) T. Ogawa, T. Kiji, K. Hayami and H. Suzuki, Chem. Lett., 1991, 1443–1446 CrossRef CAS.
- D. G. Kohler, S. N. Gockel, J. L. Kennemur, P. J. Waller and K. L. Hull, Nat. Chem., 2018, 10, 333–340 CrossRef CAS PubMed.
- (a) L. V. Desai and M. S. Sanford, Angew. Chem., Int. Ed., 2007, 46, 5737–5740 CrossRef CAS PubMed; (b) Z. Song and W. Yi, Adv. Synth. Catal., 2016, 358, 2727–2732 CrossRef CAS.
- J.-Q. Chen, J.-H. Li and Z.-B. Dong, Adv. Synth. Catal., 2020, 362, 3311–3331 CrossRef CAS.
- (a) R. J. Perner, C.-H. Lee, M. Jiang, Y.-G. Gu, S. DiDomenico, E. K. Bayburt, K. M. Alexander, K. L. Kohlhaas, M. F. Jarvis, E. L. Kowaluk and S. S. Bhagwat, Bioorg. Med. Chem. Lett., 2005, 14, 2803–2807 CrossRef PubMed; (b) G. A. Molander, L. N. Cavalcanti, B. Canturk, P.-S. Pan and L. E. Kennedy, J. Org. Chem., 2009, 74, 7364–7369 CrossRef CAS PubMed; (c) J. Hu, B. Cheng, X. Yang and T.-P. Loh, Adv. Synth. Catal., 2019, 361, 4902–4908 CrossRef CAS.
- (a) Y. Bolshan and R. A. Batey, Angew. Chem., Int. Ed., 2008, 47, 2109–2112 CrossRef CAS PubMed; (b) J.-W. Jiao, H.-Y. Bi, P.-S. Zou, Z.-X. Wang, C. Liang and D.-L. Mo, Adv. Synth. Catal., 2018, 360, 3254–3259 CrossRef CAS; (c) L. Steemers, L. Wijsman and J. H. van Maarseveen, Adv. Synth. Catal., 2018, 360, 4241–4245 CrossRef CAS; (d) J. Li, Z. Zhang, L. Wu, W. Zhang, P. Chen, Z. Lin and G. Liu, Nature, 2019, 574, 516–521 CrossRef CAS PubMed.
- L. Neerbye Berntsen, A. Nova, D. S. Wragg and A. H. Sandtorv, Org. Lett., 2020, 22, 2687–2691 CrossRef CAS PubMed.
- (a) L. Konnert, F. Lamaty, J. Martinez and E. Colacino, Chem. Rev., 2017, 117, 13757–13809 CrossRef CAS PubMed; (b) P. Thilmany, P. Gerard, A. Vanoost, C. Deldaele, L. Petit and G. Evano, J. Org. Chem., 2019, 84, 394–400 CrossRef PubMed.
- A. Palasz and D. Ciez, Eur. J. Med. Chem., 2015, 97, 582–611 CrossRef CAS PubMed.
- R. Dalpozzo, A. De Nino, L. Maiuolo, A. Procopio, R. Romeo and G. Sindona, Synthesis, 2002, 172–174 CAS.
- E. T. Chernick, M. J. Ahrens, K. A. Scheidt and M. R. Wasielewski, J. Org. Chem., 2005, 70, 1486–1489 CrossRef CAS PubMed.
- C. S. Kraihanzel and S. C. Grenda, Inorg. Chem., 1965, 4, 1037–1042 CrossRef CAS.
- Z.-G. Zheng, J. Wen, N. Wang, B. Wu and X. Q. Yu, Beilstein J. Org. Chem., 2008, 4, 40 Search PubMed.
- H. M. Hügel, C. J. Rix and K. Fleck, Synlett, 2006, 2290–2292 CrossRef.
- A. L. Korich and P. M. Iovine, Dalton Trans., 2010, 39, 1423–1431 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2099681, 2099682, 2099683, 2099684 and 2099686. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1cc04356k |
| This journal is © The Royal Society of Chemistry 2021 |