Elemental mass spectrometry for Se-dependent glutathione peroxidase determination in red blood cells as oxidative stress biomarker
Juan
Gómez-Espina
,
Elisa
Blanco-González
*,
Maria
Montes-Bayón
and
Alfredo
Sanz-Medel
*
Department of Physical and Analytical Chemistry, Faculty of Chemistry, University of Oviedo, C/ Julián Clavería, 33006 Oviedo, Spain. E-mail: asm@uniovi.es; eblancog@uniovi.es
First published on 1st October 2012
Abstract
Glutathione Peroxidase-1 (GPx1) is an enzyme playing an important role in the defense against oxidative stress which is associated with many pathological conditions. Thus, changes in the expression of this enzyme in different human tissues and fluids could be an indicator used for oxidative status assessment. Since most analytical methods for GPx1 determination are based on relative activity measurements, here we propose the absolute quantification of this protein in red blood cells through the measurement of Se present as selenocysteine in its structure. For this purpose, the sample is treated for hemoglobin precipitation and further fractionated by size-exclusion liquid chromatography (SEC) with ICP-MS for final detection. Purity of the species in the fraction obtained by SEC was assessed by orthogonal chromatographic separation based on a reversed phase mechanism (RP) of the peptides obtained by tryptic digestion of the corresponding fraction. The use of ICP-MS detection after such capillary-RP-HPLC permitted us to detect a single Se-containing peptide that was further confirmed by electrospray ionization mass spectrometric detection (ESI-MS) to belong to GPx1. Once the purity of such fraction was addressed, the quantitative analysis of GPx1 was conducted by Se post-column isotope dilution analysis after SEC separation in different samples of human red blood cells. When the concentration results obtained via SEC-ICP-MS for different protein standards are plotted versus the activity measurements (using the spectrophotometric H2O2/NADPH/GR method) a good correlation curve is obtained. Such results permit us, from ICP-MS measurements, to obtain simultaneously the GPx1 absolute concentration as well as the activity by interpolation in the previously obtained curve.
Introduction
Glutathione Peroxidases (GPx, EC 1.11.1.9) represent a major class of enzymes in the human body for defense against oxidant species.1 The most predominant and important GPx is the classical/cellular GPx1, which is located in the cytoplasm and is found in most parenchymal organs and peripheral blood cells, particularly erythrocytes.2,3 GPx1 is a tetrameric protein with four identical 22–23 kDa subunits, each of which contains one selenocysteine residue at the catalytic site.4 This enzyme catalyzes the reduction of harmful peroxides using cellular glutathione as a reducing agent.5,6 Therefore, GPx1 plays a crucial role in protecting cells from oxidative damage,1,7 a condition known as oxidative stress (disturbance in the pro-oxidant–antioxidant balance in favor of the former).8 Accordingly, impairment of the expression of this antioxidant enzyme in human tissues and in blood has been related to the development of several pathologies associated with oxidative stress, such as Parkinson's, Alzheimer's and Huntington's diseases; premature aging; cancer; diabetes and atherosclerosis.9–13 Thus, there is a need for the accurate measurement of GPx1 in blood and tissues under different patho-physiological conditions.Currently, enzymatic activity measurements using spectrophotometric assays are the method of choice for detection of changes in GPx1 activity. However, these assays are prone to interferences due to the presence of non-selenium dependent GPx-like enzymes with a cysteine residue instead of selenocysteine at the catalytic center which shows some GPx1 activity.14,15 The family of enzymes known as glutathione S-transferases can also interfere since they promote reduction of organic hydroperoxides just as GPx does.16 In addition, inter-laboratory variation in GPx1 activity values is large which precludes setting generally applicable reference values for optimum activity.17
Additionally, activity measurements provide relative values that have to be referred to the specific assay through which they have been obtained.
In contrast to the intense application of measurements of GPx activity changes, only a few experiments have studied changes in the absolute concentration of the enzyme. For this purpose, enzyme-linked immunosorbent assay (ELISA) methods have been developed for the determination of GPx in human erythrocytes,18,19 rat liver20 and human plasma.17 Unfortunately, the levels of immunoreactive GPx detected by ELISA appear to be significantly higher than the true GPx levels.17,19 Moreover, the results obtained for ELISA measurement of plasma GPx seem to be considerably inconsistent. Plasma levels have been reported to be 50–60 ng mL−1 in healthy US adults21 and 6 mg mL−1 in healthy control patients in Poland.22 Therefore more robust methods, other than immunoreactive assays, for accurate quantification of GPx in biological samples are still needed.
Nowadays, inductively coupled plasma mass spectrometry (ICP-MS) coupled with chromatography or electrophoresis (capillary and gel) has become a valuable technique for absolute quantification of heteroatom-containing proteins with high sensitivity and accuracy.23 In fact, affinity high-performance liquid chromatography (AF-HPLC)-ICP-MS has been proven to be a reliable approach to the speciation analysis of selenoproteins including GPx in human serum.24–26 In a previous work27 we have demonstrated that the use of HPLC-ICP-MS allows to obtain simultaneously the concentration and the enzymatic activity of Cu,Zn-SuperOxide Dismutase (Cu,Zn-SOD) in real samples by monitoring the Cu present in the catalytic center. Thus, in the present work this possibility has been explored for GPx1 assessment in human red blood cells by measuring the selenium present in the protein using post-column isotope dilution analysis (IDA)-HPLC-ICP-MS strategies.27,28
Experimental
Reagents, materials and samples
Analytical reagent grade chemicals were used throughout unless otherwise stated. All solutions and dilutions were made with high-purity deionized water (>18 MΩ Milli-Q water, Millipore, Bedford, MA, USA). Glutathione Peroxidase-1 (GPx1) from bovine erythrocytes was purchased from Sigma-Aldrich (St. Louis, MO, USA). The standards used to size-calibrate the size exclusion column were ferritin (440 kDa), transferrin (80 kDa), α-lactoalbumin (14 kDa) and vitamin B12 (1.4 kDa), all from Sigma-Aldrich.For protein digestion, trypsin from bovine pancreas (Sigma-Aldrich), dithiothreitol (DTT) (Sigma-Aldrich), iodoacetamide (Sigma-Aldrich), ammonium bicarbonate (Merck, Darmstadt, Germany) and acetic acid (Merck) were used. HPLC grade ethanol and chloroform for hemoglobin precipitation were supplied by Teknocroma S.L. (Barcelona, Spain). Cut-off membrane filters (10 kDa) were from Amicon-Ultra (Millipore, Iberica, Madrid).
The mobile phase for HPLC containing 50 mM ammonium acetate (Merck), pH 7.4, was prepared by diluting the solid salts with ultrapure water (Millipore). Acetonitrile for capillary HPLC was obtained from VWR International SAS (Fontenay-sous-Bois, France) and formic acid was purchased from Merck.
Isotopically enriched Se samples with relative abundances of 98.7955% 74Se, 0.1391% 76Se, 0.0779% 77Se, 0.2619% 78Se, 0.5821% 80Se and 0.1435% 82Se were obtained from Cambridge Isotope Laboratories as elemental Se powder and dissolved in a minimum volume of sub-boiled nitric acid and then diluted with ultrapure water, as required. The concentration of Se in these solutions was established by reverse isotope dilution analysis using a natural abundance Merck certified standard.
The glutathione peroxidase cellular activity assay kit composed of potassium phosphate, EDTA, sodium azide, glutathione reductase, reduced glutathione and NADPH was purchased from Sigma-Aldrich. Hydrogen peroxide used as the substrate was also from Sigma-Aldrich.
The blood samples from healthy volunteers were kindly provided by the Hospital Central of Asturias, Laboratory for Biochemical Analysis (Oviedo, Spain). Samples were anonymous and collected in accordance with protocols approved by the relevant institutional review boards and with the Declaration of Helsinki. All blood samples were collected using BD Vacutainer® tubes containing gel for serum separation. Red blood cells were aliquoted in 2 mL tubes and frozen at −20 °C until analysis. The samples were thawed before analysis at room temperature and were not frozen again.
SEC-ICP-MS
HPLC separations were carried out using a dual-piston liquid chromatographic pump (Shimadzu LC-10AD, Shimadzu Corporation, Kyoto, Japan) equipped with a sample injection valve from Rheodyne, model 7125 (Cotati, CA, USA), fitted with a 50 μL injection loop, a size exclusion chromatography column Superdex™ 200 10/300 GL (300 mm × 10 mm i.d., GE Healthcare Bio-Sciences, Sweden) and a diode array detector (DAD) from Agilent Technologies (1100 Series, Waldbronn, Germany). A 50 μL aliquot of the sample was injected into the Superdex 200 column (fractionation range 10–600 kDa) and eluted under isocratic conditions with 50 mM ammonium acetate buffer as the mobile phase (pH 7.4) at a flow rate of 0.6 mL min−1. The Superdex column was calibrated using different proteins with known molecular weights ranging from 1.4 to 450 kDa.Specific atomic detection of Se in the column effluent was performed using an ICP-MS model 7500 from Agilent Technologies (Agilent, Tokyo, Japan) equipped with a collision cell system (ICP-(ORS)-MS) using H2 as the reaction gas at 4 mL min−1. Details of the instrumental operating conditions are given in Table 1.
| Size exclusion chromatography | ||
| Column | Superdex™ 200 (10 × 300) | |
| Mobile phases | 50 mM ammonium acetate, pH = 7.4 | |
| Injection volume | 50 μL | |
| Flow rate | 0.6 mL min−1 | |
| Capillary reversed phase chromatography | ||
| Column | Zorbax SB-C18 | |
| Mobile phases | (A) 0.1% formic acid in water | |
| (B) 0.1% formic acid in ACN | ||
| Injection volume | 4 μL | |
| Flow rate | 8 μL min−1 | |
| Gradient conditions | Time | % B |
| 0 | 3 | |
| 11 | 3 | |
| 51 | 60 | |
| 56 | 80 | |
| 58 | 80 | |
| 60 | 3 | |
| ICP-MS parameters | ||
| RF power | 1500 W | |
| Carrier gas flow rate | 1.12 L min−1 | |
| Collision/reaction gas | H2 | |
| Gas flow | 4 mL min−1 | |
| Cell entrance | −40.0 V | |
| Cell exit | −76.0 V | |
| Isotopes monitored | 80Se, 78Se, 74Se | |
Capillary-RP-HPLC-ICP-MS
These separations were performed with an Agilent 1100 series capillary liquid chromatography system consisting of a four channel on-line de-gasser, a capillary pump and a micro-well plate autosampler. Capillary-RP-HPLC analysis was performed on an Agilent Zorbax SB-C18 column (150 mm × 0.3 mm, 5 μm particle size) (Agilent Technologies).The fraction corresponding to GPx1 elution from the SEC column was collected, desalted using 10 kDa cutoff filters and preconcentrated by freeze-drying. After reconstitution in water (preconcentration factor of approximately 6 calculated for individual samples by weight), the fraction was digested to peptides using trypsin and then injected into the cap-C18 column (injection volume 4 μL). Separation was performed by means of a five step gradient of 3 to 80% acetonitrile in 60 min at a flow rate of 8 μL min−1. A micro-nebuliser, based on a modified CEI 100 nebuliser from CETAC (CETAC, Omaha, NE, USA), and a quartz injector tubing extension, which allows the direct connection of the nebulizer and the torch maintaining the lowest possible internal volume, were used as an interface between capHPLC and the ICP-MS system. Operating conditions are given in Table 1.
Capillary-RP-HPLC-ESI-Q-TOF
The ESI-Q-TOF instrument used for this study was a QStar XL model (Applied Biosystems, Darmstadt, Germany) equipped with an ion-spray source and using N2 as the nebulization gas. The capillary column outlet was directly connected to the nanospray ion source, which was operated at 2.8 kV at a flow rate of 8 μL min−1. The selected mass range for peptides was between 400 and 4000 Da in the TOF-MS mode. A reserpine standard was used for external calibration and the instrument was operated in the positive mode.Procedures
Sample treatment
The red blood cells were hemolysed by adding ice-cold distilled water (4.5 mL per 0.5 mL of cells) and shaking it for 3 min using a vortex mixer at room temperature. For haemoglobin precipitation, a published procedure was adopted with some modifications.29 In this case, an ethanol–chloroform mixture (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added to the cell lysate (1 mL per 4 mL of lysate). After manual shaking for 5 min, the mixture was centrifuged (12000g, 4 °C, 1 h), and the supernatant was divided into two aliquots: one was directly used to conduct the activity measurements and the second one was preconcentrated (10-fold) using a Speed-Vac Concentrator 5301 (Eppendorf AG, Hamburg, Germany) for further HPLC-ICP-MS measurements.
:1) was added to the cell lysate (1 mL per 4 mL of lysate). After manual shaking for 5 min, the mixture was centrifuged (12000g, 4 °C, 1 h), and the supernatant was divided into two aliquots: one was directly used to conduct the activity measurements and the second one was preconcentrated (10-fold) using a Speed-Vac Concentrator 5301 (Eppendorf AG, Hamburg, Germany) for further HPLC-ICP-MS measurements.
The recovery of the sample treatment procedure described was evaluated by measuring the specific activity of GPx in the erythrocyte lysate before and after the treatment. Recoveries of about 90 ± 20% (mean ± SD, n = 3) were obtained. Since the GPx enzymatic activity depends on the presence of Se in the molecule, quantitative recoveries calculated through activity measurements imply the stability of species throughout the precipitation step.
Tryptic digestion
The trypsin digestion procedure was taken from the literature and modified slightly. In brief, 10 μL aliquots of both the GPx1 standard solution and the fraction collected after SEC separation were incubated in 25 μL of 50 mM ammonium bicarbonate solution containing 1.5 μL of 100 mM DTT for 5 min at 95 °C. Then, after allowing the sample to reach room temperature, 3 μL of 100 mM iodoacetamide solution was added and was left to stand for 20 min to let the alkylation reaction proceed. After that, a 3 μL aliquot of 1 mg mL−1 trypsin solution was added and the digestion was carried out overnight at 37 °C. The reaction was stopped by adding 1 μL acetic acid.Activity measurement
The activity of GPx1 was determined by a modification of the method of Paglia and Valentine.30 This method is based on the oxidation of reduced glutathione (GSH) to oxidized glutathione (GSSG) catalyzed by GPx1, which is then coupled to the recycling of GSSG back to GSH using glutathione reductase (GR) and β-nicotine adenine dinucleotide phosphate reduced (NADPH). The decrease in absorbance at 340 nm due to the oxidation of NADPH to NADP+ is indicative of GPx1 activity, since GPx1 is the rate limiting factor for the coupled reactions. The assay was carried out in a 1000 μL reaction mixture containing 50 mM potassium phosphate (pH 7.4), 1 mM EDTA, 1 mM NaN3, 0.2 mM NADPH, 1 unit per mL GR, 1 mM GSH and 20–50 μL of the enzyme (0.25 U per mL). The reaction was started by the addition of 0.25 mM H2O2. The change in absorbance was determined using a UV/VIS spectrophotometer model Genesys 10S (Thermo scientific, Bremen, Germany).Results and discussion
Separation and characterization of Glutathione Peroxidase-1
In order to separate GPx1 from other Se-proteins in the sample we used a SEC column (Superdex 200). For this purpose, 50 mM ammonium acetate solution was chosen as an eluent as it provides adequate compatibility with ICP-MS detection and the separation was carried out at the physiological pH of 7.4. The chromatograms obtained with UV/VIS detection (280 nm) and ICP-MS detection of Se for a GPx1 standard from bovine erythrocytes are shown in Fig. 1A. The separation reveals the presence of a main peak at about 22 minutes showing UV absorption and Se intensity. The UV signal at 280 nm shows also the presence of different peaks, besides GPx1, that absorb at this wavelength confirming that the commercial standards are not really pure. However, the elemental detection by ICP-MS shows a main Se-containing peak (tR = 22 min) that fits to the molecular mass of GPx1, according to the column calibration (∼90 kDa). This selenium peak was also observed in human red blood cells (after the sample processing described in the Experimental section), as can be seen in Fig. 1B. In this case, no other detectable Se-signals were observed in the chromatogram except for a broad peak at about 9 minutes that could correspond to larger proteins in which Se is present, not necessarily Se-proteins (UV trace can also be seen in black). In order to confirm that there is no coelution of GPx1 and other Se containing species possibly present in the red blood cells extract at 22 minutes, it was necessary to use an orthogonal chromatographic approach, typical in elemental speciation of biomolecules.31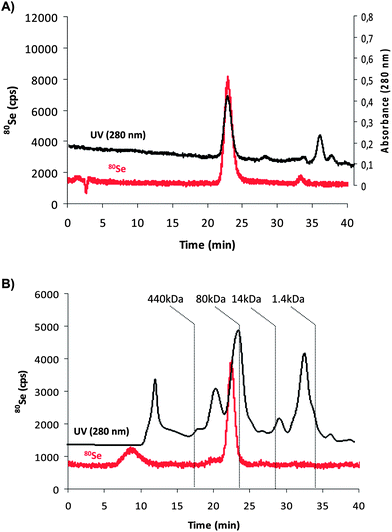 | ||
| Fig. 1 Chromatograms obtained by size exclusion chromatography (SEC) using (A) UV-Vis detection (absorbance at 280 nm, black trace) and ICP-MS detection (80Se, red trace) for the GPx1 standard from bovine erythrocytes and (B) ICP-MS trace of 80Se extracted from the red blood cell sample. | ||
For this purpose, reversed phase HPLC was used as a second dimension to determine the peak purity of the SEC fraction. Since Se is part of the backbone of the protein structure as selenocysteine, it is possible to conduct species identification at the peptide level. Thus, as proved by other authors,32 Se can be monitored as an heteroatom in the peptides obtained by tryptic digestion of GPx1 using ICP-MS. Additionally by using the same chromatographic separation with ESI-MS detection it is possible to simplify the search for the Se-isotope pattern necessary for species identification. With this aim, the fraction eluting from the SEC column at 22 min (see Fig. 1) was collected, desalted and preconcentrated by freeze-drying (in both GPx standard and sample). After reconstitution in water, the fraction was trypsin digested into the corresponding peptides. The separation of the obtained peptides was done by capillary reverse phase high performance liquid chromatography (capRP-HPLC) with ICP-MS for Se detection and with ESI-MS for molecular identification.
Fig. 2 shows the chromatogram, obtained by capRP-HPLC-ICP-MS, for the fraction of SEC corresponding to GPx1 in the standard (Fig. 2A) and in the red blood cells samples (Fig. 2B). In both cases, a major Se-containing peak was observed at approximately 42 minutes; some other traces of minor Se species were also apparent. The identity of this predominant Se peak was investigated by capRP-HPLC coupled to ESI-Q-TOF. The Se-isotope pattern was observed only in the m/z 896.9741, as shown in Fig. 3A. Such molecular mass corresponds to the doubly charged ion of the peptide with the sequence VLLIENVASLZGTTVR where Z denotes selenocysteine and that belongs to the peptide resulting from tryptic digestion of GPx1 (theoretical monoisotopic mass 896.9579 Da) after carboxymethylation of Se. The fitting for the theoretical and obtained patterns was very good as can be also observed when comparing the obtained isotopic pattern (Fig. 3A) and the calculated one for the selenopeptide (Fig. 3B).
 | ||
| Fig. 2 Selenium profile obtained by capillary reversed phase liquid chromatography (RPHPLC) with ICP-MS detection (80Se) of the digested fraction (22 min in Fig. 1) for: (A) a GPx1 standard from bovine erythrocytes and (B) a red blood cell sample extract. | ||
 | ||
| Fig. 3 Mass spectrum obtained by ESI-Q-TOF corresponding to the doubly charged peptide (monoisotopic m/z 896.9741) with the sequence VLLIENVASLZGTTVR where Z corresponds to Se-cysteine. The profiles belong to the obtained (A) and calculated (B) peptide isotope patterns. | ||
These results demonstrate not only the identity but also the peak purity of the Se peak first obtained by SEC at 22 minutes (since the calculated recovery for capRP-HPLC and trypsin digestion, including desalting, is about 74% for the GPx standard). Therefore further quantitative studies of GPx1 can be done directly after SEC separation. For this quantification purpose, however, it was necessary to ensure that the recovery of Se with the SEC separation procedure is quantitative as well. To evaluate this recovery, the eluate from the entire chromatographic run after injection of the GPx1 standard was collected (n = 3) and Se was quantified by ICP-MS using a calibration graph obtained with Se standard solutions of 0–20 ng mL−1 (7 calibration points) and Ge as the internal standard. The total concentration of Se found was compared with that measured in the standard before HPLC injection. The results of this mass balance for Se turned out to be quantitative (99 ± 10%) and allowed the use Se post-column IDA for quantification of GPx1.
GPx1 quantitative analysis by isotope dilution analysis
After the assessment of the species identity in the samples, quantitative studies were undertaken. For this purpose, an online isotope dilution method was applied and a post-column spike of isotopically enriched Se (74Se, 98.7955%) was continuously pumped by an arm of a T-piece and mixed with the eluent coming from the chromatographic column. After calculating the isotope ratio at each point of the chromatogram and applying the online isotope dilution equation it is possible to calculate the total Se at each peak of the chromatogram.28 Taking into account the Se/protein stoichiometry in GPx1 (4:1), the final protein concentration was calculated from the Se measurements (calculated detection limit 1.5 ng mL−1 of Se). Such online isotope dilution method was applied for the determination of total Se and protein concentration, both in the bovine GPx1 standard and in the red blood cells (after the sample treatment described before). The obtained results for the analyzed samples (n = 3) turned out to be about 5.11 ± 0.21 mg L−1 for GPx1 in red blood cells of healthy individuals, as can be seen in Table 2 (1 mol GPx1 is equivalent to 4 mol Se).
| Se (ng mL−1) (HPLC-IDA-ICP-MS) | GPx (μg mL−1) (HPLC-IDA-ICP-MS) | Activity measureda (U per mL) | Activity calculateda (U per mL) |
|---|---|---|---|
| a Corrected by the corresponding dilution factor. | |||
| 18.0 ± 0.36 | 5.06 ± 0.20 | 3.07 ± 0.57 | 2.84 ± 0.48 |
| 17.9 ± 0.35 | 5.03 ± 0.20 | 3.77 ± 0.61 | 3.01 ± 0.51 |
| 18.7 ± 0.38 | 5.25 ± 0.22 | 3.48 ± 0.57 | 3.08 ± 0.52 |
Of course, since the enzymatic activity of GPx1 is only driven by the presence of selenocysteine in the catalytic site of the enzyme, only if Se is retained within the protein structure of the molecule all along the proposed chromatographic separation, the enzymatic activity will be preserved. If this is so, a linear relationship between the Se concentration found in the enzyme and its activity should be expected. Thus, a single SEC-ICP-MS measurement should be enough to obtain, simultaneously, GPx1 absolute concentration and activity, as previously demonstrated for Cu,Zn-SOD.27 For this purpose, Se in the GPx1 peak was quantified in 5 different standards by post-column IDA after SEC separation as previously described. Additionally, these standard solutions were diluted to 1:1000 with the activity assay buffer and its activity was measured by monitoring the change in absorbance at 340 nm due to the oxidation of NADPH (for the standard method for determining GPx activity, see procedures). Both sets of results were plotted against each other obtaining a linear correlation, as can be observed in Fig. 4. Thus from the quantitative Se data obtained for the analyzed samples (shown in the first column of Table 2), the enzyme activity was obtained by interpolating the straight line in Fig. 4. Additionally, direct measurements of the GPx1 activity with the proposed enzymatic assay were also conducted (after diluting the cell lysate to 1:100 as recommended by the activity assay). The last two columns of Table 2 compare the results observed for both sets of results proving that they are statistically indistinguishable and validating, in this way, the proposed ICP-MS based methodology for enzyme activity measurements.
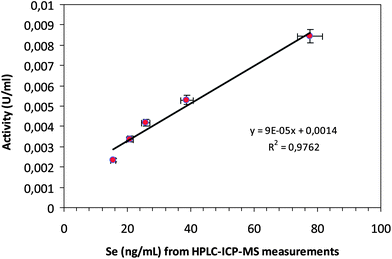 | ||
| Fig. 4 Linear correlation between Se concentration (measured by SEC-ICP-MS) and enzyme activity (measured by the Paglia and Valentine method) for GPx1 standards from bovine erythrocytes. | ||
Conclusions
A new method for GPx1 absolute quantitative determination has been developed based on a postcolumn IDA approach with SEC-ICP-MS measurement of selenium. After optimization of the sample treatment and the separation conditions, it has been demonstrated that Se can be quantitatively recovered in association with GPx1 after the chromatographic separation. Since the protein catalytic activity is related to the presence of Se, our results indicate that the SEC separation seems not to affect the enzymatic activity of the GPx1 protein. Such conclusion has been validated by measuring the activity of the protein by the spectrophotometric method of Paglia and Valentine and comparing the results with the GPx1 concentration obtained by SEC-ICP-MS experiments for a number of standards, obtaining a good linear relationship between both sets of data. Furthermore, such a linear correlation line could be used to extract activity data directly from the ICP-MS absolute concentration. This concept has been proved by analyzing GPx1 in red blood cells.References
- M. Raes, C. Michiels and J. Remacle, Free Radical Biol. Med., 1987, 3, 3–7 CrossRef CAS.
- J. R. Arthur, Cell. Mol. Life Sci., 2000, 57, 1825–1835 CrossRef CAS.
- X. G. Lei, W. H. Cheng and J. P. McClung, Annu. Rev. Nutr., 2007, 27, 41–61 CrossRef CAS.
- O. Epp, R. Ladenstein and A. Wendel, Eur. J. Biochem., 1983, 133, 51–69 CrossRef CAS.
- C. Rocher, J. L. Lalanne and J. Chaudiere, Eur. J. Biochem., 1992, 205, 955–960 CrossRef CAS.
- T. Finkel and N. J. Holbrook, Nature, 2000, 408, 239–247 CrossRef CAS.
- H. Steinbrenner and H. Sies, Biochim. Biophys. Acta, Gen. Subj., 2009, 1790, 1478–1485 CrossRef CAS.
- B. Halliwell and M. Whiteman, Br. J. Pharmacol., 2004, 142, 231–255 CrossRef CAS.
- W. Droge and H. M. Schipper, Aging Cell, 2007, 6, 361–370 CrossRef CAS.
- M. T. Lin and M. F. Beal, Nature, 2006, 443, 787–795 CrossRef CAS.
- P. Newsholme, E. P. Haber, S. M. Hirabara, E. L. Rebelato, J. Procopio, D. Morgan, H. C. Oliveira-Emilio, A. R. Carpinelli and R. Curi, J. Physiol., 2007, 583, 9–24 CAS.
- P. Storz, Front. Biosci., 2005, 10, 1881–1896 CrossRef CAS.
- D. Harrison, K. K. Griendling, U. Landmesser, B. Hornig and H. Drexler, Am. J. Cardiol., 2003, 91, 7A–11A CrossRef CAS.
- N. B. Ghyselinck, I. Dufaure, J. J. Lareyre, N. Rigaudiere, M. G. Mattei and J. P. Dufaure, Mol. Endocrinol., 1993, 7, 258–272 CrossRef CAS.
- S. Herbette, P. Roeckel-Drevet1 and J. R. Drevet, FEBS J., 2007, 274, 2163–2180 CrossRef CAS.
- D. E. Ullrey, J. Anim. Sci., 1987, 65, 1712–1726 CAS.
- G. A. Jacobson, C. Narkowicz, Y. C. Tong and G. M. Peterson, Clin. Chim. Acta, 2006, 369, 100–103 CrossRef CAS.
- K. Takahaswi, P. E. Newburger and H. J. Cohen, J. Clin. Invest., 1986, 77, 1402–1404 CrossRef.
- H. Suemizu, S. Yoshimura, N. Toda, K. Watanabe and T. Moriuchi, Hybridoma, 1992, 11, 795 CrossRef CAS.
- S. A. B. Knight and R. A. Sunde, J. Nutr., 1987, 4, 732–738 Search PubMed.
- C. R. McGill, N. R. Green, M. C. Meadows and S. S. Gropper, J. Nutr. Biochem., 2003, 14, 656–662 CrossRef CAS.
- H. Donica, Renal Failure, 2001, 23, 231–238 CrossRef CAS.
- A. Sanz-Medel, M. Montes-Bayón, M. R. Fernández de la Campa, J. Ruiz Encinar and J. Bettmer, Anal. Bioanal. Chem., 2008, 390, 3–16 CrossRef CAS.
- P. Jitaru, G. Cozzi, A. Gambaro, P. Cescon and C. Barbante, Anal. Bioanal. Chem., 2008, 391, 661–669 CrossRef CAS.
- L. Hinojosa-Reyes, J. M. Marchante-Gayon, J. I. Garcia Alonso and A. Sanz-Medel, J. Anal. At. Spectrom., 2003, 18, 1210–1216 RSC.
- K. Shigeta, K. Sato and N. Furuta, J. Anal. At. Spectrom., 2007, 22, 911–916 RSC.
- Y. N. Ordoñez, M. Montes-Bayon, E. Blanco-Gonzalez and A. Sanz-Medel, Anal. Chem., 2010, 82, 2387–2394 CrossRef.
- P. Rodriguez-Gonzalez, J. M. Marchante-Gayon, J. I. García Alonso and A. Sanz-Medel, Spectrochim. Acta, Part B, 2005, 60, 151–207 CrossRef.
- A. Deisseroth and A. L. Dounce, Physiol. Rev., 1970, 50, 319–3759 CAS.
- D. E. Paglia and W. N. Valentine, J. Lab. Clin. Med., 1967, 70, 158–169 CAS.
- A. Sanz-Medel, Spectrochim. Acta, Part B, 1998, 53, 197–211 CrossRef.
- G. Ballihaut, S. Mounicou and R. Lobinski, Anal. Bioanal. Chem., 2007, 388, 585–591 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |