Cu Kβ2,5 X-ray emission spectroscopy as a tool for characterization of monovalent copper compounds
J. R.
Vegelius
a,
K. O.
Kvashnina
b,
M.
Klintenberg
a,
I. L.
Soroka
c and
S. M.
Butorin
*a
aDepartment of Physics and Astronomy, Uppsala University, Box 516, S-751 20 Uppsala, Sweden. E-mail: sergei.butorin@physics.uu.se
bID26, European Synchrotron Radiation Facility, BP220, F-38043, Grenoble Cedex, France
cNuclear Chemistry, KTH Chemical Science and Engineering, Royal Institute of Technology, SE-100 44 Stockholm, Sweden
First published on 22nd August 2012
Abstract
Cu Kβ2,5 X-ray emission and resonant inelastic X-ray scattering measurements were performed on monovalent and divalent copper compounds. The data were compared with the results of local-density-approximation calculations. The methods were found to be efficient tools for studies of Cu 4p states in the valence band and for distinguishing between different monovalent copper compounds. This is of particular importance for the debate concerning copper corrosion in oxygen-free water.
I. Introduction
The properties of a material are closely connected to its electronic structure. In this work we focus on copper which is of significant industrial importance. Especially since the concept of a deep-underground repository for spent nuclear fuel in copper canisters has been planned particularly in Sweden and Finland.1 Copper corrosion has been studied for over 100 years and lately the debate has escalated further due to the issue concerning copper corrosion in oxygen free environments.2,3 Resonant inelastic X-ray scattering (RIXS) spectroscopy at the Cu L3 (2p3/2–3d, 4s transitions) edge has been shown to be a particularly useful tool in studies of the electronic structure of Cu compounds. It can be used to distinguish between different species even with the same oxidation state for the element in question. This was particularly shown for divalent Cu systems with a pronounced 3d9 character.4,5In Cu L3 RIXS, Cu 3d and 4s states in the valence band are probed due to dipole selection rules. Thus, the method is useful for characterization of systems with an unfilled 3d shell. However, monovalent Cu compounds have mainly a 3d10 character so that mostly Cu s, p states are involved in the chemical bonding. For this reason Cu L3 RIXS is expected to be less sensitive to the states responsible for chemical bonding because the spectra are dominated by the 3d contribution and the 4s contribution is significantly smaller.
X-ray emission spectroscopy at the Cu K (1s–4p transitions) edge, on the other hand, probes Cu 4p states rather than 3d states. This technique can therefore be expected to serve as a better tool for characterization of the electronic structure in monovalent copper systems. In this respect emission spectroscopies at the L3 and K edges can be considered as complementary.
It is a challenging task to probe both occupied and unoccupied states within the same high-energy spectroscopic experiment. For example, it is difficult to combine X-ray photoemission and inverse photoemission techniques in the study of states in valence and conduction bands, respectively, during the same round of measurements. An X-ray absorption spectrum as a probe of unoccupied density of states can be affected by the core-hole present in the final state of the spectroscopic process. The resolution of non-resonant X-ray emission spectroscopy (XES) has a fundamental restriction due to a significant core-hole lifetime broadening. We show here that the valence-to-core RIXS spectra at the Cu K edge of monovalent Cu compounds can be interpreted in simple terms as a convolution of unoccupied and occupied Cu 4p densities of states (DOSs), thus making the technique sensitive to changes in the electronic structure both in the valence and conduction bands.
In this study we present Cu Kβ2,5 XES and RIXS data measured for divalent and monovalent Cu compounds. While the spectra of divalent Cu systems do not significantly differ from each other within the applied experimental resolution the spectra of monovalent Cu compounds appreciably change on going from one compound to another. This makes the technique particularly useful in studies of problems such as the possible formation of monovalent Cu hydroxide6 as a main product of Cu corrosion in “pure” water.2,3,7,8
II. Experimental details
The copper hydride (CuH) was synthesized by a wet-chemistry method by the reaction of aqueous copper sulphate with hypophosphorous acid. The details of synthesis can be found in ref. 9. The obtained powder was packed and sealed in a glass tube. The procedure was fulfilled under a nitrogen protective atmosphere in the glove-box. The diameter of the glass tube was 0.7 mm. The thickness of the walls was 10 μm, which is transparent for the X-ray beam. Other copper compounds were purchased from Alfa Aesar. The samples studied were in powder form.The measurements were performed at beamline ID26 of the European Synchrotron Radiation Facility (ESRF) in Grenoble.10 The incident energy was selected using the 〈311〉 reflection from a double Si crystal monochromator. Rejection of higher harmonics was achieved by three Cr–Pd mirrors at an angle of 2.5 mrad relative to the incident beam. Cu Kβ2,5 (mainly 4p–1s transitions) XES spectra were measured using an X-ray emission spectrometer.11 At an incident photon beam energy of 9.1 keV, the sample, analyzer crystal and photon detector (silicon drift diode) were arranged in a vertical Rowland geometry. The emission energy was selected using the 〈800〉 reflection of two spherically bent Ge crystal analyzers (with 1 m bending radius) aligned at 77° Bragg angle. The intensity was normalized to the incident flux. A combined (incident convoluted with emitted) energy resolution of 0.9 eV was obtained as determined by measuring the full width at half-maximum (FWHM) of the elastic peak.
The full RIXS map around the Cu Kβ2,5 line was also measured by scanning the incident energy at different emission energies, i.e., at each emission energy the monochromator was scanned throughout the desired energy interval. The procedure resulted in a two-dimensional RIXS map with incident energy and energy of transfer on the x- and y-axes respectively. The total energy resolution was estimated to be around 0.8 eV.
The radiation damage of the samples was monitored by recording the 1 min X-ray absorption (XAS) scans immediately after the insertion of the sample into the X-ray beam and after the measurement of the XES scan (30 min duration). No changes in the shape of XAS spectra were observed. Furthermore, the possible degradation of halide samples upon exposure to X-ray beam in air is expected to be in the form of oxidation and therefore should also lead to changes in the XES spectra characteristic for oxides. This was not observed.
III. Calculations
The electronic structures in this work were calculated as part of the Electronic Structure Project (ESP).12An all electron full-potential linear muffin-tin orbital (FP-LMTO) method13,14 within the local density approximation (LDA) was used for all calculations. All electrons reflect that we explicitly include the core electrons (the Dirac equation is solved) in the self-consistent cycle as opposed to pseudopotential implementations such as VASP. The full potential part indicates that we put no geometric constraints on the expansion of the density and potential.
A solution within the density functional theory (DFT) framework means obtaining the Kohn–Sham (KS) eigenvectors and eigenvalues. Considering the approximations intrinsic to DFT it is not obvious that the KS solution is a valid model for the electronic structure. However, for a ground state system physical properties related to the electronic structure can often be reproduced within 5–10% of experiment but at the same time it is not surprising that for example some features in the XAS spectrum are not well reproduced.
A more complete description of the computational method used can be found in ref. 13 and 14 but it can be noted that an extended basis set has been used in order to remove the tail energy dependence in the solution and that parameters such as the k-point grid and fast Fourier transform (FFT) mesh were set to give a converged electronic structure. The muffin-tin radii were optimized to cover 90% of the nearest neighbor distance and the unit cells are those of ref. 15–29.
IV. Results and discussion
A. XES
Spectra in Fig. 1 and 2 represent the result of Cu Kβ2,5 XES measurements for divalent and monovalent Cu compounds, respectively. As can be seen in the figures the differences between the spectra of monovalent compounds are more prominent than those between divalent compounds. | ||
| Fig. 1 Cu Kβ2,5 X-ray emission spectra of divalent Cu compounds. The spectra were normalized to the maximum intensities. | ||
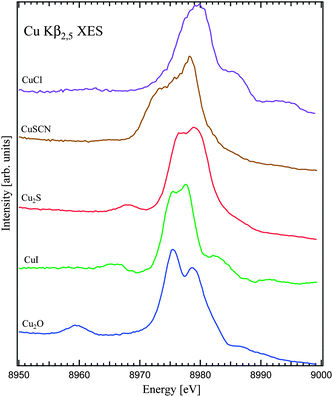 | ||
| Fig. 2 Non-resonant X-ray emission spectra of monovalent Cu compounds. The spectra were normalized to the maximum intensities. | ||
In the case of divalent Cu compounds (Fig. 1) the electronic configuration of Cu is best described as a 3d9 configuration. Due to an unfilled 3d shell the 3d states are most important for the chemical bonding. The distribution of Cu 4p states is strongly affected by the Cu 3d–ligand p state hybridization and correspondingly by the Cu 3d distribution.
Within the experimental resolution, the major Cu Kβ2,5 spectral structures which mostly represent the occupied Cu 4p states look somewhat alike (except for CuO) with the main line split into two intense peaks. The structure around 8959 eV in the spectrum of CuO is called Kβ′′ and corresponds to Cu 4p states hybridized with O 2s states.11,30 Similar structures in the spectra of other divalent Cu compounds in Fig. 1 somewhat differ in energy position, intensity and shape as a result of hybridization with the 2s states of oxygen atoms from different chemical groups. On the other hand, it is not easy to identify the contributions of Cu states hybridized with Cl 3s states.
Interestingly, however, the presence of CO32− groups seems to lead to a widening and intensity gain of the low-energy peak at ∼8975 eV in the main Kβ2,5 line (see for example the spectrum of CuCO3) and to an increased spectral weight in the energy range around 8970 eV of the main Kβ2,5 line.
The electronic configuration of Cu in monovalent Cu compounds is mainly 3d10. Since the 3d shell is filled the Cu chemical bonding is mainly defined by the states of 4s and 4p character. The Kβ2,5 XES measurements (Fig. 2) probe Cu 4p states in the valence band and the spectra vary significantly in terms of their shapes and energy position of the main Kβ2,5 structures.
The position and intensity of the Kβ′′ line also change significantly between different monovalent Cu compounds. It is strongest in Cu2O, becomes less intense in Cu2S and CuI and essentially smeared out in CuSCN (see the structure around 8960 eV in the spectrum of Cu2O in Fig. 2). At the same time it is difficult to identify the Kβ′′ contribution in the spectrum of CuCl because Kβ′′ is expected to be closer to the Kβ2,5 main line. The energy positions of Kβ′′ are defined by the energies of O 2s in Cu2O, I 5s in CuI, S 3s in Cu2S, and S 3s, N 2s, C 2s in CuSCN.
The features on the high-energy side of the main Kβ2,5 line, especially pronounced in CuCl and CuI (Fig. 2), are due to contribution of the multiple ionization satellites.11,31 The strength of these satellites depends on the degree of ionicity of the chemical bonding and the size of the band gap. The satellite appearance is also affected by the profile of the Cu 4p DOS.
In Fig. 3 and 4 the measured Cu Kβ2,5 spectra of divalent and monovalent Cu compounds, respectively, are compared with the results of LDA calculations. The calculated spectra were obtained by broadening of the occupied Cu 4p-projected DOS to account for the 1s core-hole lifetime-broadening and the experimental resolution. Since the Cu 1s X-ray photoelectron spectra of copper compounds were not measured and the experimental 1s binding energies were not established, we took advantage of the results of LDA calculations and used relative changes in calculated energies of Cu 1s core levels on going from one compound to another to align the data on the binding energy scale in Fig. 3 and 4.
 | ||
| Fig. 3 Comparison of measured (dashed curves) and LDA-calculated (full curves) Cu Kβ2,5 X-ray emission spectra of divalent Cu compounds. | ||
 | ||
| Fig. 4 Comparison of measured (dashed curves) and LDA-calculated (full curves) Cu Kβ2,5 X-ray emission spectra of monovalent Cu compounds. | ||
The calculations help to identify the character of the structures in the Kβ2,5 spectra. For divalent Cu compounds, the double peak structure of the main Kβ2,5 line mainly originates from the contribution of π-bonding (low-energy peak, e.g., at 8975 eV in Cu(OH)2) and π-anti-bonding (high-energy peak at 8979 eV) states as a result of the Cu 3d–ligand p–Cu 4p hybridization. On the other hand, for monovalent Cu compounds, such as Cu2O, the low-energy peak at ∼8976 eV of the Kβ2,5 line consists of contribution of the σ bonding states.
The energy positions of the Kβ′′ are not accurately described in calculations for divalent Cu compounds while they were found to be in better agreement with the experiment for monovalent Cu compounds.
As to Kβ2,5, some differences between experimental data and theoretical results in Fig. 3 and 4 are observed which can have different origins.
For Cu(II) compounds in general, the effect of the on-site Coulomb interaction U between Cu 3d electrons is underestimated in the conventional LDA formalism. This interaction also influences to some extent the distribution of fairly delocalized O 2p and Cu 4p states through their admixture to Cu 3d states, although the LDA results still remain overall useful for the description of spectra probing such delocalized states as shown for O Kα X-ray emission of cuprates.32 The so-called LDA + U and LDA + DMFT (dynamical mean-field theory) calculational schemes usually provide better results in the description of electron-correlated systems but require an introduction of the U-parameter and are not ab initio methods in this sense.
For atacamite and malachite in particular, we noticed a significant sensitivity of the results of calculations to the type of distortions in the crystal structure (orthorombic, monoclinic or trigonal). Although, the right types of the crystallographic distortions were used (in accord with established in the scientific literature) for the calculated spectra of atacamite and malachite in Fig. 3, the results of calculations can be possibly improved by further optimization of crystal structures (unit-cells) to have them as close as possible to those of particular mineral-samples studied here.
Furthermore, the LDA-formalism is usually not the best method for the calculations of the electronic structure of complex molecular-like systems such as CuSCN. In turn, for CuI with the relatively heavy iodine anion, the inclusion of the relativistic effects in calculations is important which was not done in our case.
We can speculate that calculations using the general gradient approximation (GGA) formalism would probably provide a rather better platform for the description of the electronic structure in the case of evaluation of variety of compounds with different characters and properties.
Despite some shortcomings, the predictive ability of the LDA calculations as an ab initio, self-consistent method can be still used to evaluate the behavior of the Cu Kβ2,5 spectra in various copper systems, especially in systems with Cu(I).
In debates concerning copper corrosion in oxygen-free water2,3,7,8 intermediate Cu–O–H phases with Cu(I) are discussed. The importance of methods for distinguishing between different monovalent copper compounds has, hence, been brought up. LDA-based calculations by Korzhavyi et al.6 for CuH, Cu2O and CuOH indicate significant differences in the distribution of the DOS in the valence band, thus suggesting the possibility to distinguish between these species by using Cu Kβ2,5 spectroscopy. Our results are pointing in the same direction. In Fig. 5 our calculations are presented showing the total DOS, O 2p states of Cu2O, H 1s states of CuH and Cu 4p and 3d states. In Cu2O, the hybridization between O 2p and Cu 3d states gives rise to two groups of structures in the occupied Cu 4p DOS at ∼−1 eV and ∼−7 eV. In CuH, the hybridization between H 1s and Cu 3d states also generates two groups of structures in the occupied Cu 4p DOS at ∼−2 eV and ∼−8 eV with different relative weight distributions compared to Cu2O.
 | ||
| Fig. 5 Total and partial densities of states derived from LDA calculations for CuH and Cu2O. | ||
The recorded Cu Kβ2,5 XES data shown in Fig. 6 reveal a similar trend as in the predictions by the calculations with respect to the intensity ratio between the two main structures of Cu2O and CuH, i.e., the low-energy structure is more pronounced in Cu2O compared to that in CuH. CuH is not a very stable compound in air at room temperature. It loses hydrogen and turns into metallic copper. That is why the sample was sealed into the glass capsule for our experiment and was transported to the beamline in the cooled box. In Fig. 6 we also show the Kβ2,5 spectrum of Cu metal for comparison which confirms the survival of the CuH sample during the measurements. Furthermore, the recorded Kβ1 (3p–1s transitions) spectrum of CuH revealed the chemical shift characteristic for Cu(I).
 | ||
| Fig. 6 Cu Kβ2,5 X-ray emission spectra of Cu2O, CuH and Cu metal. At the bottom, broadened Cu p DOSs of Cu2O and CuH are shown. | ||
The survival of the CuH sample sealed in the glass tube over a period of time longer than the duration of our XES measurement was also verified by X-ray diffraction characterization. The radiation damage during the experiment was monitored by recording the 1 min XAS scans before and after the XES measurements (30 min duration) and no changes in the shape of XAS spectra were observed. Nevertheless, we cannot entirely rule out some H leaching out of the sample. Such leaching may lead to a minor contribution of metallic Cu to the XES spectrum.
The LDA calculations by Korzhavyi et al.6 suggest the possible existence of three main structures in the Cu Kβ2,5 spectrum (for the experimental resolution used) of the Cu–O–H phase of Cu(I) as a result of the Cu 4s,p–O 2p–H 1s hybridization. This makes it possible to detect Cu–O–H formation if it is formed throughout the corrosion process in oxygen-free water. Unfortunately, we had difficulties in obtaining a sample of the Cu–O–H phase in the form suitable for our measurements. Therefore, theoretical findings are yet to be confirmed.
B. RIXS
In the present study the core-to-valence RIXS was measured which directly involves valence electrons; the energy transfer is of only a few tens of electron volts as shown in Fig. 7 and 8. RIXS data are shown here as a contour map in a plane of incident and transferred photon energies, where the vertical axis represents the energy difference between the incident and emitted energies. Variations of the colour on the plot correspond to the different scattering intensities. | ||
| Fig. 7 Measured (left) and calculated (right) RIXS maps of Cu2O. | ||
 | ||
| Fig. 8 Measured (left) and calculated (right) RIXS maps of Cu2S. | ||
Core-to-valence RIXS calculations at the Cu K edge for Cu2O and Cu2S were performed by introducing the Cu 4p projected DOS into the Kramers–Heisenberg equation:
 | (1) |
Following the treatment by Tulkki and Åberg34 of continuous excitations at the K edge for the description of resonant X-ray Raman scattering, eqn (1) was derived by Jimenez-Mier et al.35 from the Kramers–Heisenberg equation. They called the derived equation as “the expression for Raman scattering cross-section” which was obtained in the presence of an excited spectator electron. Such an approach for the description of the RIXS process was later supported by further theoretical work (see e.g.ref. 36) and experimental data (e.g.ref. 37).
In this case the results of the RIXS process at the Cu K edge can be viewed as a convolution of the occupied and unoccupied Cu 4p DOSs. Here we used the projected DOS obtained in FEFF8.4 calculations. The reason for not using the LDA-calculated DOSs was the necessity to calculate spectra for a quite extended range above the Fermi level. The accuracy of the LDA calculations in the description of the DOS distribution decreases at such an extended range.
Detailed analysis of eqn (1) shows that the energy difference between the occupied and unoccupied Cu p states will correspond to the energy transfer value of the observed RIXS transitions. Experimental RIXS data shown in Fig. 7 and 8 demonstrate the splitting of the RIXS features in the valence band at ∼5, ∼7, and ∼15 eV in energy transfer for Cu2S and ∼5, ∼9 and ∼25 eV in energy transfer for Cu2O. We find a reasonably good agreement between theoretical and experimental core-to-valence RIXS results at the Cu K edge for both Cu systems, with respect to features such as shapes, positions and relative intensities.
The requirement for RIXS structures/resonances to have constant energy transfer for at/above core threshold excitations is usually applied in the case of localized states and many-body approach. Even then, it was shown for CoO38,39 and NiO40 that RIXS resonances at the metal 2p edge do not necessarily follow the elastic peak within the width of the valence band and therefore can be confused with characteristic X-ray fluorescence.
On the other hand, in one-electron approach (which is adopted here) the radiative (RIXS) transitions as a result of core excitations at the corresponding absorption edge are rather decoupled from the increasing energy of incident X-rays and rather depend on the dispersion of the involved bands (see e.g.ref. 41) in the momentum space (or the gerade–ungerade rule for molecules). The RIXS transitions are restricted from following the elastic peak by the width of the occupied part of the valence band (see e.g. corresponding discussion and Fig. 1 in ref. 38).
RIXS provides potential advantages compared to other X-ray spectroscopic methods on the Cu K edge. In RIXS occupied and unoccupied DOSs are multiplied. Thus, RIXS is sensitive to occupied and unoccupied DOSs and differences in either or both can be visible in experiments. Hence, conditions for distinguishing between different Cu compounds are extended. While non-resonant XES provides only one 1D-spectrum, for RIXS map there is a possibility to make one-dimensional cuts along the energy transfer axis at a number of incident photon energies, in particular at those energies where the difference between compounds can be emphasized the most (such as between spectra of Cu2O and Cu2S on the XAS resonance/peak at around 8981.5 eV, 7 eV and 10 eV above it). Another advantage is that the resolution of the RIXS process is not limited by the core-hole lifetime broadening so that only the experimental resolution defines the total resolution.
V. Conclusion
Cu Kβ2,5 XES and RIXS were found to be efficient tools for characterizing the electronic structure and the chemical bonding (associated with the Cu atoms) and consequently speciation in monovalent Cu compounds. These spectroscopies applied at the Cu K edge mainly provide information about Cu 4s, p states which govern the chemical bonding in monovalent Cu compounds. XES spectra of divalent Cu compounds, however, were found to be fairly similar to each other.Acknowledgements
We acknowledge Rolf Berger for providing the samples. This work was supported by the Swedish Research Council and the Goran Gustafsson Foundation for Research in Natural Science and Medicine. The authors would like to thank the technical support staff at the ESRF for the assistance during the experiment. I.L.S. acknowledges the financial support from Swedish Nuclear Fuel and Waste Management Company (SKB).References
- L. Werme, Technical report TR 98-08, Design Premises for Canister for Spent Nuclear Fuel, SKB, 1998, Wendy Barnaby, Nature, 1978, vol. 274, p. 6 Search PubMed.
- G. Hultquist, M. J. Graham, P. Szakalos, G. I. Sproule, A. Rosengren and L. Gråsjö, Corros. Sci., 2011, 53, 310 CrossRef CAS.
- G. Hultquist, Corros. Sci., 1986, 26, 173 CrossRef CAS.
- K. O. Kvashnina, S. M. Butorin, A. Modin, I. Soroka, M. Marcellini, J.-H. Guo, L. Werme and J. Nordgren, J. Phys.: Condens. Matter, 2007, 19, 226002 CrossRef.
- K. O. Kvashnina, S. M. Butorin, J.-H. Guo, R. Berger, L. Werme and J. Nordgren, Phys. B Condens. Matter, 2009, 404, 3559 CrossRef CAS.
- P. A. Korzhavyi, I. L. Soroka, E. I. Isaev, C. Lilja and B. Johansson, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 686 CrossRef CAS PubMed.
- J. P. Simpson and R. Schenk, Corros. Sci., 1987, 27, 1365 CrossRef CAS.
- T. E. Eriksen, P. Ndalamba and I. Grenthe, Corros. Sci., 1989, 29, 1241 CrossRef CAS.
- P. A. Korzhavyi, I. Soroka, M. Boman and B. Johansson, Solid State Phenom., 2011, 172–174, 973 CrossRef CAS.
- C. Gauthier, V. A. Solé, R. Signorato, J. Goulon and E. Moguiline, J. Synchrotron Radiat., 1999, 6, 164 CrossRef CAS PubMed.
- P. Glatzel and U. Bergmann, Coord. Chem. Rev., 2005, 249, 65–95 CrossRef CAS.
- C. Ortiz, O. Eriksson and M. Klintenberg, Comput. Mater. Sci., 2009, 44, 1042 CrossRef CAS , http://gurka.fysik.uu.se/esp.
- J. M. Wills, O. Eriksson, M. Alouani and D. L. Price, in Lecture Notes in Physics, Electronic Structure and Physical Properties of Solids: The Uses of the LMTO Method, ed. H. Dreysse, Springer-Verlag, Berlin, 2000, vol. 535 Search PubMed.
- J. M. Wills, M. Alouani, P. Andersson, A. Delin, O. Eriksson and O. Grechnyev, Full-Potential Electronic Structure Method: Energy and Force Calculations with Density Functional and Dynamical Mean Field Theory, Springer-Verlag, Berlin, 2010 Search PubMed.
- M. Kabesova, M. Dunaj Jurco, M. Serator and J. Gazo, Inorg. Chim. Acta, 1976, 17, 161 CrossRef CAS.
- P. Suesse, Acta Crystallogr., 1967, 22, 146 CrossRef CAS.
- E. L. Belokoneva, Yu. K. Gubina and J. B. Forsyth, Phys. Chem. Miner., 2001, 28, 498 CrossRef CAS.
- S. Asbrink and L. J. Norrby, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1970, 26, 8 CrossRef CAS.
- N. V. Belov and V. P. Butuzov, Dokl. Akad. Nauk SSSR, 1946, 54, 717 CAS.
- R. W. G. Wyckoff and E. Posnjak, J. Am. Chem. Soc., 1922, 44, 30 CrossRef CAS.
- S. W. Peterson and H. A. J. Levy, Chem. Phys., 1957, 26, 220 CAS.
- E. A. Owen and E. L. Yates, Philos. Mag., 1933, 15, 472 CrossRef CAS.
- H. Mueller and A. J. Bradley, Proc. K. Ned. Akad. Wet., 1926, 25, 27 Search PubMed.
- L. Vegard and G. Skofteland, Arch. Math. Naturvidensk., 1942, 45, 163 CAS.
- J. B. Parise and B. G. Hyde, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1986, 42, 1277 CrossRef.
- H. Seidel, K. Viswanathan, W. Johannes and H. Ehrhardt, Z. Anorg. Allg. Chem., 1974, 410, 138 CrossRef CAS.
- P. C. Burns and F. C. Hawthorne, Am. Mineral., 1993, 78, 187 CAS.
- A. Kirfel and K. D. Eichhorn, Acta Crystallogr., Sect. B: Struct. Sci., 1990, 46, 271 Search PubMed.
- H. R. Oswald, A. Reller, H. W. Schmalle and E. Dubler, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1990, 46, 2279 CrossRef.
- V. I. Anisimov, V. A. Gubanov and E. Z. Kurmaev, J. Struct. Chem., 1980, 21, 291 CrossRef.
- J. Bremer and H. Sorum, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 2749 CrossRef CAS.
- S. M. Butorin, et al. , J. Electron Spectrosc. Relat. Phenom., 2000, 110–111, 235 CrossRef CAS.
- J. C. Fuggle and J. E. Inglesfield, Top. Appl. Phys., 1992, 69, 1–23 CrossRef CAS.
- J. Tulkki and T. Åberg, J. Phys. B: At. Mol. Phys., 1982, 15, L435 CrossRef CAS.
- J. J. Jimenez-Mier, J. van Ek and D. L. Ederer, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 2649 CrossRef CAS.
- J. J. Kas, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 235114 CrossRef.
- N. Smolentsev, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 235113 CrossRef.
- S. M. Butorin, J. Electron Spectrosc. Relat. Phenom., 2000, 110–111, 213 CrossRef CAS.
- M. Magnuson, et al. , Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 205106 CrossRef.
- M. Matsubara, et al. , J. Phys. Soc. Jpn., 2005, 74, 2052 CrossRef CAS.
- Y. Ma, et al. , Phys. Rev. Lett., 1992, 69, 2598 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2012 |