Size and elemental composition of nanoparticles using ion mobility spectrometry with inductively coupled plasma mass spectrometry†
Efthymios A.
Kapellios
and
Spiros A.
Pergantis
*
Environmental Chemical Processes Laboratory, Department of Chemistry, University of Crete, Voutes Campus, Heraklion, 71003, Greece. E-mail: spergantis@chemistry.uoc.gr; Fax: +30 2810 545001; Tel: +30 2810 545084
First published on 28th October 2011
Abstract
The combined use of nanoelectrospray ion mobility spectrometry (IMS) with off-line inductively coupled plasma mass spectrometry (ICP-MS) for determining engineered nanoparticles (NPs) in aqueous solution is demonstrated here for the first time. The resulting analytical technique exhibits capabilities for determining the size and metal content of NPs and can also in principle be used for determining NP concentration. The resolving power exhibited by IMS along with the high sensitivity offered by single particle mode ICP-MS are the main features that make this combination attractive for analysing NPs.
Over a decade ago nanoelectrospray (nES)-IMS with condensed particle counting (CPC) detection (a technique referred to as GEMMA) was first introduced1 and shown to offer unique capabilities for sizing large biomolecules, protein complexes and various other types of NPs having diameters in the range of 3 to 100 nm.2 However, one of the technique's disadvantages is the lack of chemical information that is provided by the CPC detector. This extremely sensitive detector does not provide information regarding the elemental composition of the NPs detected. In an effort to overcome this shortcoming we recently demonstrated the first coupling of nES-IMS on-line with ICP-MS.3 In particular, when electrospraying solutions with high concentrations of various salts, i.e. of CsI, the system provided information regarding the size of the resulting aggregates along with their elemental composition. The aggregates in this case did not pre-exist in solution but were formed as a result of the nES process. Unfortunately, however, the system was not sufficiently sensitive to simultaneously determine the elemental composition of metal containing proteins or other metal containing NPs, while accurately determining their size. Using higher concentrations did not solve the problem as nES-induced aggregates are formed, giving inaccurate size measurements.
In our continuing effort to improve the sensitivity of ICP-MS to function as a detector for nES-IMS we investigated the possibility of their off-line coupling. The approach described in this manuscript involves the use of a NP collection system.4 NPs of a specific size, selected by appropriately setting the IMS voltage, are collected in a metal cup to which a high voltage has been applied. Following collection, the NPs of each fraction are dispersed in water and subsequently analyzed via pneumatic aspiration ICP-MS. However, in order to achieve adequate sensitivity and to obtain information on the metal content of the NPs and their concentration it was imperative for the ICP-MS to be operated in the single particle mode (SPM).
It is now well established that SPM-ICP-MS can be used for the sensitive detection of various types of NPs in environmental and biological samples.5 The advantages of this approach being that it analyzes individual NPs, providing information about their metal content as well as their concentration, with extremely high sensitivities.6
The off-line approach was investigated using a modified NP collection device (Nanometer Aerosol Sampler, Model 3089, TSI inc.) which was installed between the IMS (which is a differential mobility analyzer) and the CPC detector. In order to have the NP collector function efficiently for the purpose of this study two modifications were made to it. First its pumping system was turned off and second the two outlets of the NP collection chamber were connected to a T-piece which via a single piece of non-conducting tubing was connected to the CPC detector. This allowed for the CPC unit, functioning at an air intake rate of 1.5 L min−1, to dictate the air flow going through the NP collection device. In order to trap the incoming NPs a voltage is applied to the collector electrode upon which a metal collection cup is placed. Tape is used to keep the collection cup facing the incoming monodisperse aerosol flow. Voltages up to −10 kV can be applied to the collector electrode. A schematic of the system configuration is presented in Fig. 1 (for photographs see ESI†, Fig. S1 and S2). Its NP trapping efficiency is demonstrated by flowing 60 nm Au-NPs through the NP collector and counting how many reach the CPC, without and with a voltage applied to the collector electrode (Fig. 2). From this it is clearly observed that almost all Au-NPs are removed from the gas flow by the collector device. Since the surface of the collection electrode is covered by the metal cup it is expected that most Au-NPs are captured within it. This behavior was observed for all types of NPs examined, i.e. metal NPs, biomolecules, salt clusters, etc. Smaller diameter NPs required lower voltages for efficient trapping, whereas larger NPs required higher voltages.4
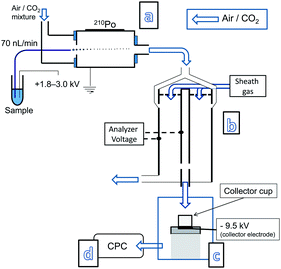 | ||
| Fig. 1 Nanoelectrospray (a) with an ion mobility system (b) on-line with a NP collection device (c) installed between the outlet of an IMS and the inlet of a CPC detector (d). | ||
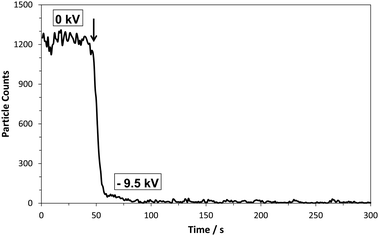 | ||
| Fig. 2 Au NPs (60 nm) counted by the CPC detector without collector voltage (0 kV) and with collector voltage (−9.5 kV). Arrow indicates the voltage onset. | ||
To test the capability of ICP-MS to detect the collected NPs, solutions of Au-NPs with a nominal size of 60 nm, obtained from NIST,7 were analyzed following sample preparation.8 Initially, a CPC electropherogram of such an analysis was recorded (Fig. 3), in which case the collector device was off-line.9 Two main peaks were detected. The peak at 9.1 nm is believed to correspond to residual salts present in the analyzed Au-NP solution. Whereas the peak around 60 nm is believed to correspond to the Au-NPs. However, these tentative assignments highlight the disadvantage of the CPC detector not being able to provide information regarding the elemental composition of the detected NPs.
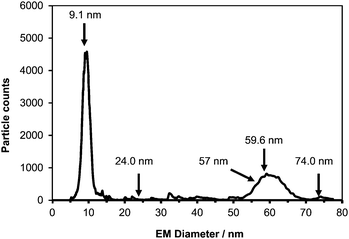 | ||
| Fig. 3 CPC electropherogram of 60 nm Au NPs from NIST. Arrows indicate the sizes at which 30 min fractions were collected for subsequent SPM-ICP-MS analysis (120 s scan time). | ||
Subsequently, the NP collection device was put in-series between the IMS and the CPC, and the IMS was set to allow specific size NPs through to the collector device. In this mode NPs were collected for 30 min time intervals and the resulting fractions were analyzed for their metal content. The sizes of the five collected fractions are shown in Fig. 3 with arrows. Following the completion of NP collection 1 ml of de-ionised water was added to each fraction in order to have the NPs dispersed in solution. The resulting dispersions were then transferred via a pipette to Eppendorf tubes from where they were analyzed by using ICP-MS in the single particle mode. In all cases the collection voltage was set to −9.5 kV.
The NP samples were analyzed using SPM ICP-MS by setting the mass analyzer to monitor a single isotope, i.e.197Au, with a 10 ms dwell time. Each NP entering the plasma gives an ion plume which is counted by the detector. Using short enough dwell times, i.e. 10 ms, allows for the recording of the entire plume of ions as a single event. In the case of the 60 nm Au NP approximately 100 ions are counted for each NP (the rest of the ions entering the interface are lost in the spectrometer). Detection of such NP single events results in signal pulses or spikes. The number of ions detected in each pulse is directly proportional to the gold mass in the corresponding single nanoparticle. The resulting spike graphs for two of the collected fractions are shown in Fig. 4. More specifically, for the 59.6 nm fraction the resulting spike graph (Fig. 4A) shows a series of intensity spikes each one corresponding to the Au ion plume resulting from a single Au-NP. In which case 22 individual NPs were detected, whereas in 3 cases double and triple NPs were detected within the 10 ms dwell time (the probability of having multiple NP entering the plasma within the same 10 ms dwell time cannot be totally excluded, even though it is reduced substantially upon dilution). The average intensity of the resulting spikes (93 ± 18 counts) matched well with the average intensity recorded for a solution of 60 nm Au-NPs (97 ± 33 counts) at a Au concentration of 50 ng L−1 (see ESI†, Fig. S3). This confirmed that each detected NP with a size of 59.6 nm (as determined by the IMS) has the same amount of Au as the NIST 60 nm Au NPs. The IMS technique also provides size information, i.e. an electrophoretic mobility diameter of 59.6 nm (not corrected for the thickness of any nonvolatile salts encrusted on the surface of the particles) which agrees well with the values provided by NIST ranging from 54.9 to 56.6 nm, obtained using a variety of independent analytical techniques.7
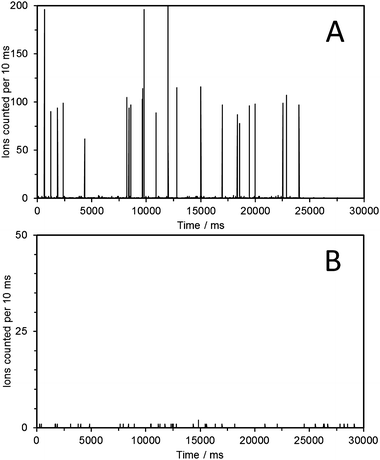 | ||
| Fig. 4 Single particle mode ICP-MS analysis (m/z 197, dwell time 10 ms) of the fraction collected at 59.6 nm following the electrospraying of 60 nm Au NPs (A). The fraction collected at 24 nm showed no spikes, indicating the absence of Au containing NPs (B). | ||
Determining NP numbers or NP concentrations in the analyzed solution is a somewhat more complicated procedure because the combined nES and NP collecting efficiency needs to be determined. This of course is possible by analyzing the initial Au-NP solution (prior to it being electrosprayed) by using SPM ICP-MS and comparing the number of NPs counted with the number of NPs counted in the collected fraction(s). However, because numerous experimental parameters (collection time, collector voltage, IMS flow rate and nES spray conditions) affect the combined nES and NP collection efficiency substantially more work is required.
The second fraction that was analyzed had been collected at 24 nm (Fig. 4B). In this case it is clearly observed that no Au-NPs are detected as no intensity spikes are recorded. As expected, similar behavior was observed for the fractions collected at 9.1 nm and 74 nm. However, it is possible to occasionally detect a single NP of the correct Au mass content. This we attribute to NPs possibly entrapped within the system that are later released. However, it is so infrequent that it does not affect the overall profiles obtained. The fraction collected at the front of the Au-NP peak (57 nm) also gave rise to intensity spikes of the correct intensity for the 60 nm Au-NPs. See ESI†, Fig. S4–S6, for SPM-ICP-MS analysis of all additional fractions collected. It should also be kept in mind that a dimer of a 60 nm Au spherical NP should appear at about 1.24 × 60 nm = 72.4 nm, according to Pease III et al.,10 which is close to the 74 nm peak for which a sample was collected. However, in this study no convincing evidence for its presence was obtained.
In conclusion, the fact that nES-IMS is suitable for sizing a variety of NPs has encouraged us to investigate its potential to be coupled off-line with ICP-MS in an effort to determine the NP metal composition. The analytical approach described here is a relatively straightforward off-line technique making use of a NP collection device and the operation of an ICP-MS in the recently described extremely sensitive single particle mode. Coupling these unique pieces of instrumentation along with newly developed NP monitoring procedures makes the described approach highly attractive for further investigation into its application for ultratrace detection of metal-containing NPs, determining their size (approximately 20–100 nm), their metal content and their concentration in environmental and biological samples.
This work was supported by the project “IRAKLITOS II—University of Crete” of the Operational Programme for Education and Lifelong Learning 2007–2013 (E.P.E.D.V.M.) of the NSRF (2007–2013), which is co-funded by the European Union (European Social Fund) and National Resources.
Notes and references
- S. L. Kaufman, J. W. Skogen, F. D. Dorman, F. Zarrin and K. Lewis, Anal. Chem., 1996, 68, 1895 CrossRef CAS; S. L. Kaufman, A. R. Kuchumov, M. Kazakevich and S. N. Vinogradov, Anal. Biochem., 1998, 259, 195 CrossRef.
- G. Bacher, W. W. Szymanski, S. L. Kaufman, P. Zollner, D. Blaas and G. Allmaier, J. Mass Spectrom., 2001, 36, 1038 CrossRef CAS; J. F. de la Mora, S. Ude and B. A. Thomson, Biotechnol. J., 2006, 1, 988 CrossRef; C. S. Kaddis and J. A. Loo, Anal. Chem., 2007, 79, 1778 CrossRef; G. Allmaier, C. Laschober and W. W. Szymanski, J. Am. Soc. Mass Spectrom., 2008, 19, 1062 CrossRef; C. Laschober, J. Wruss, D. Blaas, W. W. Szymanski and G. Allmaier, Anal. Chem., 2008, 80, 2261 CrossRef; L. F. Pease, III, D.-H. Tsai, K. A. Brorson, S. Guha, M. R. Zachariah and M. J. Tarlov, Anal. Chem., 2011, 83, 1753 CrossRef.
- C. Carazzone, R. Raml and S. A. Pergantis, Anal. Chem., 2008, 80, 5812 CrossRef CAS.
- J. Dixkens and H. Fissan, Aerosol Sci. Technol., 1999, 30, 438 CAS.
- C. Degueldre, P. Y. Favarger and C. Bitea, Anal. Chim. Acta, 2004, 518, 137 CrossRef CAS; C. Degueldre and P. Y. Favarger, Talanta, 2004, 62, 1051 CrossRef; C. Degueldre, P. Y. Favarger and S. Wold, Anal. Chim. Acta, 2006, 555, 263 CrossRef; S. Hu, R. Liu, S. Zhang, Z. Huang, Z. Xing and X. Zhanga, J. Am. Soc. Mass Spectrom., 2009, 20, 1096 CrossRef; K. S. Ho and W. T. Chan, J. Anal. At. Spectrom., 2010, 25, 1114 RSC; Y. Suzuki, H. Sato, S. Hikida, K. Nishiguchi and N. Furuta, J. Anal. At. Spectrom., 2010, 25, 947 RSC.
- F. Laborda, J. Jiménez-Lamana, E. Bolea and J. R. Castillo, J. Anal. At. Spectrom., 2011, 26, 1362 RSC; E. M. Heithmar and S. A. Pergantis, U.S. Environmental Protection Agency, Washington, DC, EPA/600/R-10/117, 2010 RSC; S. Gschwind, L. Flamigni, J. Koch, O. Borovinskaya, S. Groh, K. Niemax and D. Günther, J. Anal. At. Spectrom., 2011, 26, 1166 RSC.
- National Institute of Standards & Technology, Report of Investigation for Reference Material 8013, Gold Nanoparticles, 2007 Search PubMed.
- Aqueous suspensions of citrate-stabilized 60 nm Au nanoparticles (RM 8013) were acquired from NIST. From this native suspension, 900 μL were transferred to a low-binding Eppendorf tube and centrifuged (MiniSpin centrifuge, Eppendorf) for 12 min at 7.6 krpm. The visually clear supernatant was removed (typically 850 μL) and replaced with 25 μL of 2 mmol L−1ammonium acetate solution at pH 8 (puriss. p.a, Reag. ACS, Sigma-Aldrich), the sample was re-homogenized and inserted into the nES-IMS for analysis. According to the NIST RM 8013 report of investigation (ref. 7) and the above sample preparation procedure the solution that was nES sprayed had a Au concentration of 622 μg mL−1, whereas the total amount of Au sprayed during the 30 minute collection period was 1.3 μg.
- A nES-IMS instrument with a CPC detector from TSI Inc. (St Paul, MN, USA) was used in this study. The instrument configuration consisted of a nES source unit (model 3480C) equipped with a neutralizing chamber (210Po α-source; 5 mCi, model P-2042 Nucleospot local air ionizer; NRD, Grand Island, NY, USA), whereas the ion mobility spectrometer used for the IMS separation was a differential mobility analyzer (TSI Inc., macroIMS, series 3080C). Detection was achieved using a butanol-based Ultrafine CPC (TSI Inc., series 3025A). IMS version 2.0.1.0 (TSI Inc.) software was used for data acquisition and data analysis. NP sample solutions in Eppendorf tubes were placed into the nES unit. One end of a 25 cm long, 25 μm i.d. (150 μm o.d.) polyimide-coated fused silica capillary was immersed into the solution, while the other end having a conical shape was located within the nES chamber. The spray was achieved by introducing the sample into the capillary by applying an air pressure of 3.7 bar and a voltage between 1.8 and 3.0 kV to the Pt wire dipped in the sample solution. This resulted in a nES flow rate of ∼70 nL min−1. To operate nES in stable cone-jet mode a mixture of CO2 (0.2 L min−1) and clean air (1.0 L min−1) was used. The sheath/carrier gas flow rate was 11 L min−1 and the flow entering the particle counter was 1.5 L min−1. When operating in the NP collection mode a NP collector (Nanometer Aerosol Sampler, Model 3089, TSI Inc.) was installed between the IMS analyzer and the CPC detector. NPs were collected in aluminium collection cups (1.2 cm i.d. × 1.1 cm height; Aluminum Shell Crowns, Prestige Dental Products, Inc., Anaheim, California USA) by applying a voltage of −9.5 kV. An X-series ICP-MS (Thermo Scientific) was used in the single particle mode throughout the study.
- L. F. Pease, III, D.-H. Tsai, J. L. Hertz, R. A. Zangmeister, M. R. Zachariah and M. J. Tarlov, Langmuir, 2010, 26, 11384 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) is available. See DOI: 10.1039/c1ja10237k |
| This journal is © The Royal Society of Chemistry 2012 |