Efficient synthesis of cyanohydrin trimethylsilyl ethers via 1,2-chemoselective cyanosilylation of carbonyls†
Giacomo
Strappaveccia
a,
Daniela
Lanari
*b,
Dmitri
Gelman
c,
Ferdinando
Pizzo
a,
Ornelio
Rosati
b,
Massimo
Curini
b and
Luigi
Vaccaro
*a
aLaboratory of Green Synthetic Organic Chemistry, CEMIN – Dipartimento di Chimica, Università di Perugia, Via Elce di Sotto, 8, Perugia, Italy. E-mail: luigi@unipg.it; Fax: +39 075 5855560; Tel: +39 075 5855541
bDipartimento di Chimica e Tecnologia del Farmaco, Università di Perugia, Via del Liceo, 06123 Perugia, Italy
cInstitute of Chemistry, The Hebrew University, Edmond Safra Campus, Givat Ram, 91904 Jerusalem, Israel
First published on 2nd November 2012
Abstract
Here we report a sustainable protocol for the cyanosilylation of carbonyl compounds 1a–g and 3a–m using trimethylsilyl cyanide and triphenylphosphine supported on polystyrene as a catalyst under solvent-free conditions. It has been shown that a small amount of the catalyst allows the chemoselective 1,2-addition of trimethylsilyl cyanide to α,β-unsaturated carbonyls 1a–g (5 mol%) and to saturated carbonyls 3a–m (2 mol%). The preparation of cyanohydrin trimethylsilyl ethers 2a–g and 4a–m has been accomplished in good yields (72–99%) and very low E-factor values (5–10). Finally, efficiency has been further improved by setting two different flow procedures that have allowed us to perform the representative preparation of cyanohydrin trimethylsilyl ether 4a on a large scale and with the E-factor of 0.16 or 0.47 consisting in a reduction of 90 or 72% of waste compared to our batch conditions.
Introduction
Cyanohydrin trimethylsilyl ethers are key intermediates for the synthesis of many valuable molecules such as α-hydroxy acids, α-hydroxy aldehydes and β-amino alcohols.1 A large variety of synthetic procedures have been reported in the literature utilizing different cyanide sources such as HCN and its related salts,2 acyl cyanides,3 alkyl cyanoformates4 and trialkylsilyl cyanides.5 The utilization of the latter is particularly convenient as it allows us to obtain silyl protected products in high yields and readily accessible for further manipulations. However, the use of silyl cyanides requires activation by different catalysts. For example, Lewis acids,6 amines,7 phosphines7,8 and phosphazanes9 have been employed for this purpose. Furthermore, in the effort to develop sustainable processes a wide range of heterogeneous organocatalysts have been introduced.10Lately we have devoted our research to the optimization of synthetic procedures by employing eco-friendly reaction conditions such as the use of polymer supported organocatalysts,11 use of water as a reaction medium12 or application of solvent-free reaction conditions (SolFC).13 In addition, batch procedures have been further optimized by setting up single and multi-step flow reactors. These innovative approaches allowed us to develop a large number of eco-friendly synthetic procedures for many chemical transformations with a significant minimization of waste production (that can be trivially measured using the Environmental Factor, i.e. E-factor = kg of waste/kg of desired product).14 We have also previously shown that the combination polymer-supported organocatalyst under SolFC conditions is the best choice in terms of sustainability and of chemical efficiency.
In this paper we present a new optimized protocol for the synthesis of a variety of cyanohydrin trimethylsilyl ethers by the chemoselective 1,2-cyanosilylation of carbonyl compounds. Following our approach we have investigated the development of an efficient procedure involving the use of trimethylsilyl cyanide (TMSCN) as a cyanide source and polystyrene-supported triphenylphosphine (PS-TPP) under SolFC.
Results and discussion
Preliminary studies on the cyanosilylation have been devoted to the identification of the most efficient supported catalyst. The screening was performed using (E)-hept-3-en-2-one (1a) as a reference compound and different solid catalysts bearing an amine moiety such as 4-dimethylamino pyridine (PS-DMAP), trimethylamine (PS-TMA) or 1,5,7-triazabicyclo[4.4.0]dec-5-ene (PS-TBD) and those bearing a phosphine moiety such as triphenylphosphine (PS-TPP). The results are shown in Table 1.|
|
|||
|---|---|---|---|
| Entry | Catalyst | Solvent (M) | Conversiona (%) |
| a Conversion measured by GC analyses. b Reaction time 4 h. c Reaction performed in 12 h with 1.1 eq. of TMSCN. d Reaction performed in 4 h and 5 mol% catalyst. e Reaction performed in 4 h and 2 mol% catalyst. | |||
| 1 | PS-DMAP | — | 75 |
| 2 | PS-TMA | — | 88 |
| 3 | PS-TBD | — | 74 |
| 4 | PS-TPP | — | 93 |
| 5 | PS-TPP | H2O (0.5 M) | 0 |
| 6 | PS-TPP | CH2Cl2 (0.5 M) | 0 |
| 7 | PS-TPP | CH3CN (0.5 M) | 0 |
| 8 | PS-TPP | — | 100b |
| 9 | PS-TPP | — | 90c |
| 10 | PS-TPP | — | 100d |
| 11 | PS-TPP | — | 80e |

The best catalytic efficiency was observed for PS-TPP (Table 1, entry 4), which gave complete conversion in 4 h under SolFC (Table 1, entry 8). Remarkably, no reaction occurred when a reaction medium (organic or aqueous) was employed (Table 1, entries 5–7) stressing again the importance of SolFC to increase the efficiency of a catalyst. When the amount of catalyst was decreased from 10% to 5% no loss in reactivity was observed (Table 1, entry 10). It should be noticed that TMSCN is partially hydrolyzed under the reaction conditions with the formation of hexamethyldisiloxane. A slight excess was therefore used to achieve the complete conversion of 1a.
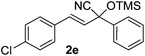
The optimized conditions reported herein were applied to the α,β-unsaturated carbonyl compounds 1b–g and chemoselectively led to the formation of the sole cyanohydrin trimethylsilyl ethers in excellent yields (90–99%) and with low E-factors (5–10) (Table 2).







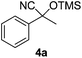
In order to extend the optimized protocol to a wider range of compounds, we tested its applicability to cyanation of saturated carbonyls as shown in Table 3. Also in the case of these substrates good results have been consistently obtained with the use of just 2 mol% of the catalyst and 1.1 eq. of TMSCN.
|
|
||||
|---|---|---|---|---|
| Entry | Product | t (h) | Yielda (%) | E-factor |
| a Isolated yield of the pure product without further purification. b Reaction performed with 5 mol% of catalyst. c Reaction performed at 30 °C. | ||||
| 1 |
|
3 | 97 | 7 |
| 2 |

|
7 | 96b | 6 |
| 3 |

|
2.5 | 96 | 7 |
| 4 |
|
1 | 98 | 5 |
| 5 |

|
8 | 72 | 10 |
| 6 |

|
4.5 | 96 | 6 |
| 7 |

|
1 | 83c | 8 |
| 8 |

|
1 | 85c | 8 |
| 9 |

|
1.5 | 95c | 6 |
| 10 |

|
3.5 | 96c | 6 |
| 11 |

|
3.5 | 95c | 6 |
| 12 |

|
2 | 98c | 6 |
| 13 |
|
7 | 98b,c | 7 |

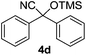
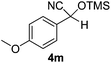
E-factor values calculated for the preparation of cyanohydrin trimethylsilyl ethers 4a–m ranged from 5 to 10 (see the Experimental section for calculation details).
After having optimized the batch protocol for the efficient synthesis of cyanohydrin trimethylsilyl ethers, in the case of the representative substrate 3a the protocol was also performed on a larger scale and the recycling of the catalyst was investigated to further evaluate the sustainability of the process.
On the 20 mmol scale the E-factor was reduced to 1.7 (see the Experimental section for calculation details) and similar results were obtained also on the 50 mmol scale. Not completely satisfactory results were instead obtained in our attempts to recycle the PS-TPP. In the test reaction of acetophenone (3a) to yield 4a in batch the catalyst remained active even after four repetitive runs but longer reaction times were required to achieve complete conversion (Table 4).
|
|
||
|---|---|---|
| Run | t (h) | Yield (%) |
| 1st | 3 | 97 |
| 2nd | 3 | 94 |
| 3rd | 7 | 96 |
| 4th | 18 | 95 |

In order to minimize waste production and to highlight the efficiency of the use of a heterogeneous recoverable catalyst under SolFC, the reaction was also performed in flow on the representative substrate 3a.
We have investigated two different flow procedures, one operating in a cyclic mode reproducing the batch procedure but in flow (Fig. 1) and the other operating in a continuous-flow manner which required a specific optimization of the flow rate and the catalyst packing (Fig. 2). Below the operating details and results obtained are described.
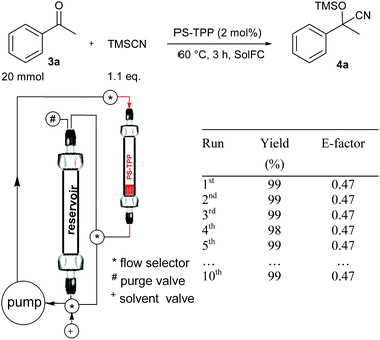 | ||
| Fig. 1 Cyanosilylation of acetophenone 3a using a cyclic-mode flow reactor* (*a thermostated chamber was used but is not shown for clarity). | ||
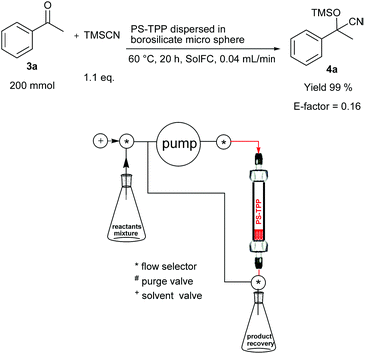 | ||
| Fig. 2 Cyanosilylation of acetophenone (3a) using a continuous flow reactor* (*a thermostated chamber was used but is not shown for clarity). | ||
The cyclic-mode flow procedure is basically a batch-like procedure where stirring of the reaction mixture through the solid catalyst is performed by using a pump instead of a magnetic stirrer.13 Therefore to set the cyclic-mode flow protocol the same optimized data were used.
Acetophenone (3a) and TMSCN were charged into a glass column functioning as a reservoir and PS-TPP (0.4 mmol, 2 mol% measured on the ketone 3a) was charged into a glass column (labelled as PS-TPP). The reaction mixture was continuously pumped through the catalysts for 3 h (the same conditions reported in Table 3, entry 1) until complete conversion to 4a was achieved.
At this point the pump was left to run in order to recover the reaction mixture into the reservoir. Then EtOAc (2 × 1 mL) was introduced into the system and cyclically pumped (10 min) to wash the catalyst and then collected in the reservoir. After the evaporation of the solvent product 4a was obtained in pure form and in 99% yield and with a very low E-factor of 0.47 (for calculation details see the Experimental section).
According to this procedure, the catalyst is safely conserved in the glass column and could be reused without loss in activity keeping the reaction time as short as 3 hours (see the results in Fig. 1 and ESI† for the recycling procedure).
It should be noticed that by using this type of flow condition, similar and often better results in terms of the E-factor have been obtained by increasing the reaction scale. Therefore, we consider that 20 mmol is the minimal reaction scale where the reported E-factor values can be obtained.
For comparison the representative reaction of 3a with TMSCN was also performed in a continuous-flow procedure on the 200 mmol scale (Fig. 2) after a further specific optimization of the reaction conditions. PS-TPP (0.2 mol% measured on the ketone 3a) was dispersed in a 45–60 mesh borosilicate microsphere. In this case the reactants were premixed, pumped through the catalyst (best results were obtained using a 0.04 mL min−1 flow rate) and the resulting mixture was collected in a recovery flask. When the reactants mixture was consumed, EtOAc (2.5 mL × 2) was cyclically pumped through the catalyst, the system was washed and the product 4a was recovered in 99% yield and with a very low 0.16 E-factor value (Fig. 2) (see the Experimental section for calculation details). Depending on the reaction scale adopted, a partial recovery of the solvent can be also defined further reducing the waste production.
Conclusions
In conclusion we have developed a new efficient and sustainable procedure for the chemoselective cyanosilylation of a large variety of carbonyl compounds. Our protocol employed PS-TPP as a solid-supported catalyst and TMSCN as a cyanide source. The solvent-free environment dramatically enhanced the system efficiency while consistently resulting in excellent yields. We set up two flow procedures operating in cyclic- or continuous-mode that allowed optimal recovery and reuse of the catalyst. Using the flow approach, the sustainability of the protocol was greatly increased as proved by the 72–90% reduction of the waste produced compared to our batch conditions.Experimental section
Unless otherwise stated, all chemicals were purchased and used without any further purification. GC analyses were performed by using a Hewlett-Packard HP 5890A equipped with a capillary column DB-35MS (30 m, 0.53 mm), a FID detector and helium as the gas carrier. GC-EIMS analyses were carried out by using a Hewlett-Packard HP 6890N Network GC system/5975 Mass Selective Detector equipped with an electron impact ionizer at 70 eV.All 1H NMR and 13C NMR spectra were recorded at 200 MHz or 400 MHz, and at 50.3 or 100.6 MHz respectively, using a Bruker DRX-ADVANCE 200 MHz spectrometer and a Bruker DRX-ADVANCE 400 MHz spectrometer. The deuterated solvent used was CDCl3, and TMS was employed as the internal standard. Chemical shifts were reported in ppm and coupling constants in hertz. Elemental analyses were realized by using a FISONS instrument EA 1108 CHN.
Compounds 2a,152b,162c,172d,5c2f,182g,194a,5c4b,5c4c,204d,5c4e,214f,224g,234h,234i,10b4j,10b4k,5i4l,194m10j are known compounds. Compound 2e is an unknown compound.
Characterization data and copies of the 1H and 13C NMR spectra for compounds 2a–g and 4a–m are reported in ESI.†
Representative experimental procedure
In a screw capped vial equipped with a magnetic stirrer PS-TPP (0.008 g, 0.025 mmol, 3.2 mmol g−1), (E)-hept-3-en-2-one (1a) (0.065 mL, 0.5 mmol), and TMSCN (0.075 mL, 0.6 mmol) were consecutively added and the resulting mixture was left under stirring at 60 °C. After 4 hours Et2O (1 mL) was added and the catalyst was filtered off and the solvent was removed under vacuum. Pure (E)-2-methyl-2-((trimethylsilyl)oxy)hept-3-enenitrile (2a) was obtained as an oil (99% yield, 0.104 g).Representative E-factor calculation
–Batch (0.5 mmol):(Acetophenone (60 mg) + TMSCN (54 mg) + Washing EtO2 (710 mg) + PS-TPP (3 mg) − Product mass (106 mg))/Product mass (106 mg) = 7
–Batch (20 mmol):
(Acetophenone (2.4 g) + TMSCN (2.18 g) + Washing EtOAc (7.2 g) − Product mass (4.34 g))/Product mass (4.34 g) = 1.7
–Cyclic-mode flow reactor (20 mmol) (1st run):
(Acetophenone (2.4 g) + TMSCN (2.18 g) + Washing EtOAc (1.8 g) − Product mass (4.34 g))/Product mass (4.34 g) = 0.47
–Continuous flow reactor (200 mmol):
(Acetophenone (24 g) + TMSCN (21.8 g) + Washing EtOAc (4.5 g) − Product mass (43.4 g))/Product mass (43.4 g) = 0.16/Product mass (43.4 g) = 0.16
Acknowledgements
We gratefully acknowledge the Ministero dell'Istruzione, dell'Università e della Ricerca (MIUR) within the projects PRIN 2008 and “Firb-Futuro in Ricerca”, and the Università degli Studi di Perugia for financial support. This work was also supported by the Israel–Italy Joint Innovation Program for Scientific and Technological Cooperation in R&D 2012–2014.Notes and references
- (a) J.-M. Brunel and I. P. Holmes, Angew. Chem., Int. Ed., 2004, 43, 2752–2778 CrossRef CAS; (b) R. J. H. Gregory, Chem. Rev., 1999, 99, 3649–3682 CrossRef CAS.
- HCN and its salts (a) T. S. A. Heugebaert, B. I. Roman, A. De Blieck and C. V. Stevens, Tetrahedron Lett., 2010, 51, 4189–4191 CrossRef CAS; (b) F. L. Cabirol, A. E. C. Lim, U. Hanefeld, R. A. Sheldon and I. M. Lyapkalo, J. Org. Chem., 2008, 73, 2446–2449 CrossRef CAS; (c) I. V. P. Raj and A. Sudalai, Tetrahedron Lett., 2005, 46, 8303–8306 CrossRef CAS; (d) F. Effenberg, Tetrahedron Lett., 1998, 32, 2605–2608 CrossRef.
- Alkylcyanoformates (a) K. Motokura, M. Tadaand and Y. Iwasawa, J. Am. Chem. Soc., 2007, 129, 9540–9541 CrossRef CAS; (b) S. Aoki, S. Kotani, M. Sugiura and M. Nakajima, Tetrahedron Lett., 2010, 51, 3547–3549 CrossRef CAS.
- Acylcyanides (a) T. Watahiki, S. Ohba and T. Oriyama, Org. Lett., 2003, 5, 2679–2681 CrossRef CAS; (b) W. Zhang and M. Shi, Org. Biomol. Chem., 2006, 4, 1671–1674 RSC; (c) F. Li, K. Widyan, E. Wingstrand and C. Moberg, Eur. J. Org. Chem., 2009, 3917–3922 CrossRef CAS.
- TMSCN: (a) Y. Kikukawa, K. Suzuki, M. Sugawa, T. Hirano, K. Kamata, K. Yamakughi and N. Mizuno, Angew. Chem., Int. Ed., 2012, 51, 1–6 CrossRef; (b) G. M. Dekamin, R. Alizadeh and M. R. Naimi-Jamal, Appl. Organomet. Chem., 2010, 24, 229–235 CrossRef; (c) Z. Shen and S. Ji, Synth. Commun., 2009, 39, 775–791 CrossRef CAS; (d) Y. Ogasawara, S. Uchida, K. Yamaguchi and N. Mizuno, Chem.–Eur. J., 2009, 15, 4343–4349 CrossRef CAS; (e) J. Gawronski, N. Wascinska and J. Gajewy, Chem. Rev., 2008, 108, 5227–5252 CrossRef CAS; (f) N. H. Khan, S. Agrawal, R. I. Kureshy, S. H. R. Abdi, S. Singh and R. V. Jasra, J. Organomet. Chem., 2007, 692, 4361–4366 CrossRef CAS; (g) L. Wang, X. Huang, J. Jiang, X. Liu and X. Feng, Tetrahedron Lett., 2006, 47, 1581–1584 CrossRef CAS; (h) J. J. Song, F. Gallou, J. T. Reeves, Z. Tan, N. K. Yee and C. H. Senanayake, J. Org. Chem., 2006, 71, 1273–1276 CrossRef CAS; (i) L. Minuti, A. Taticchi, A. Marrocchi, S. Landi and E. Gacs-Baitz, Tetrahedron Lett., 2005, 46, 5735–5737 CrossRef CAS; (j) N. Kurono, M. Yamaguchi, K. Suzuki and T. Ohkuma, J. Org. Chem., 2005, 70, 6530–6532 CrossRef CAS , and ref. 6–10.
- (a) B. Y. Park, K. Y. Ryu, J. H. Park and S.-g. Lee, Green Chem., 2009, 11, 946–948 RSC; (b) J. Wang, Y. Masui, K. Watanabe and M. Onaka, Adv. Synth. Catal., 2009, 351, 553–557 CrossRef CAS; (c) B. Karimi and L. Ma'Mani, Org. Lett., 2004, 6, 4813–4815 CrossRef CAS; (d) P. Saravanan, R. V. Anand and V. K. Singh, Tetrahedron Lett., 1998, 39, 3823–3824 CrossRef CAS.
- S. E. Denmark and W. Chung, J. Org. Chem., 2006, 71, 4002–4005 CrossRef CAS , and citations therein.
- (a) D. A. Evans and R. Y. Wong, J. Org. Chem., 1977, 42, 350–352 CrossRef CAS; (b) S. Kobayashi, Y. Tsuchiya and T. Mukaiyama, Chem. Lett., 1991, 537–540 CrossRef CAS; (c) S. Matsukawa, I. Sekine and A. Iitsuka, Molecules, 2009, 14, 3353–3359 CrossRef CAS.
- Z. Wang, B. Fetterly and J. G. Verkade, J. Organomet. Chem., 2002, 646, 161–166 CrossRef CAS.
- (a) S. Matsukawa and S. Fujikawa, Tetrahedron Lett., 2012, 53, 1075–1077 CrossRef CAS; (b) Y. Teng and P. H. Toy, Synlett, 2011, 551–554 CAS; (c) M.-A. Lacour, N. J. Rahier and M. Taillefer, Chem.–Eur. J., 2011, 17, 12276–12279 CrossRef CAS; (d) S. A. Pourmousavi and H. Salahshornia, Bull. Korean Chem. Soc., 2011, 32, 1575–1578 CrossRef CAS; (e) G. M. Pawara and M. R. Buchmeisera, Adv. Synth. Catal., 2010, 352, 917–928 CrossRef; (f) B. Thirupathi, M. K. Patil and B. M. Reddy, Appl. Catal. A, General, 2010, 384, 147–153 CrossRef CAS; (g) K. Iwanami, J.-C. Choi, B. Lu, T. Sakakura and H. Yasuda, Chem. Commun., 2008, 1002–1004 RSC; (h) W. K. Cho, S. M. Kang, A. K. Medda, J. K. Lee, I. S. Choi and H.-S. Lee, Synthesis, 2008, 507–510 CrossRef CAS; (i) S. T. Kadam, Bull. Korean Chem. Soc., 2008, 29, 1320–1322 CrossRef CAS; (j) A. Procopio, G. Das, M. Nardi, M. Oliverio and L. Pasqua, ChemSusChem, 2008, 1, 916–919 CrossRef CAS; (k) M. L. Kantam, K. Mahendar, B. Sreedhar, K. V. Kumar and B. M. Choudary, Synth. Commun., 2008, 38, 3919–3936 CrossRef CAS; (l) K. Yamaguchi, T. Imago, Y. Ogasawara, J. Kasai, M. Kotani and N. Mizuno, Adv. Synth. Catal., 2006, 348, 1516–1520 CrossRef CAS; (m) M. L. Kantam, P. Sreekanth and P. L. Santhi, Green Chem., 2000, 2, 47–48 RSC; (n) M. Curini, F. Epifano, M. C. Marcotullio, O. Rosati and M. Rossi, Synlett, 1999, 315–316 CrossRef CAS; (o) M. Onaka, K. Higuchi, K. Sugita and Y. Izumi, Chem. Lett., 1989, 1393–1396 CrossRef CAS.
- (a) S. Bonollo, D. Lanari, J. M. Longo and L. Vaccaro, Green Chem., 2012, 14, 164–169 RSC; (b) S. Bonollo, D. Lanari, T. Angelini, F. Pizzo and L. Vaccaro, J. Catal., 2012, 285, 216–222 CrossRef CAS; (c) K. Oded, S. Mussa, D. Gelman and J. Blum, Catal. Commun., 2012, 20, 68–70 CrossRef CAS; (d) P. Wolfer, M. L. Santarelli, L. Vaccaro, L. Yu, T. D. Anthopulos, P. Smith, N. Stingelin-Stutzmann and A. Marrocchi, Org. Electron., 2011, 12, 1886–1892 CrossRef CAS.
- (a) T. Angelini, D. Lanari, R. Maggi, F. Pizzo, G. Sartori and L. Vaccaro, Adv. Synth. Catal., 2012, 354, 908–916 CrossRef CAS; (b) D. Lanari, O. Piermatti, F. Pizzo and L. Vaccaro, Synthesis, 2012, 2181–2184 Search PubMed; (c) S. Calogero, D. Lanari, M. Orrù, O. Piermatti, F. Pizzo and L. Vaccaro, J. Catal., 2011, 282, 112–119 CrossRef CAS; (d) S. Bonollo, D. Lanari, F. Pizzo and L. Vaccaro, Org. Lett., 2011, 13, 2150–2152 CrossRef CAS; (e) S. Bonollo, D. Lanari and L. Vaccaro, Eur. J. Org. Chem., 2011, 2587–2598 CrossRef CAS; (f) S. Bonollo, F. Fringuelli, F. Pizzo and L. Vaccaro, Synlett, 2007, 2683–2686 CAS; (g) F. Fringuelli, F. Pizzo and L. Vaccaro, J. Org. Chem., 2001, 66, 4719–4722 CrossRef CAS.
- (a) D. Lanari, R. Ballini, A. Palmieri, F. Pizzo and L. Vaccaro, Eur. J. Org. Chem., 2011, 2874–2884 CrossRef CAS; (b) F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Green Chem., 2010, 12, 1301–1305 RSC; (c) A. Zvagulis, S. Bonollo, D. Lanari, F. Pizzo and L. Vaccaro, Adv. Synth. Catal., 2010, 352, 2489–2496 CrossRef CAS; (d) R. Ballini, L. Barboni, L. Castrica, F. Fringuelli, D. Lanari, F. Pizzo and L. Vaccaro, Adv. Synth. Catal., 2008, 350, 1218–1224 CrossRef CAS; (e) F. Fringuelli, R. Girotti, F. Pizzo and L. Vaccaro, Org. Lett., 2006, 8, 2487–2489 CrossRef CAS; (f) D. Amantini, F. Fringuelli, O. Piermatti, F. Pizzo, E. Zunino and L. Vaccaro, J. Org. Chem., 2005, 70, 6526–6529 CrossRef CAS.
- (a) R. A. Sheldon, Chem. Ind. (London, U.K.), 1997, 12–15 CAS; (b) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 RSC; (c) J. Augé, Green Chem., 2008, 10, 225–231 RSC; (d) R. A. Sheldon, Chem. Commun., 2008, 3352–3365 RSC.
- J. Cao, F. Zhou and J. Zhou, Angew. Chem., Int. Ed., 2010, 49, 4976–4980 CrossRef CAS.
- H. Deng, M. P. Isler, M. L. Snapper and A. H. Hoveyda, Angew. Chem., Int. Ed., 2002, 41, 1009–1012 CrossRef CAS.
- Y. Hirata, T. Yukawa, N. Kashihara, Y. Nakao and T. Hiyama, J. Am. Chem. Soc., 2009, 131, 10964–10973 CrossRef CAS.
- S. S. Kim, G. Rajagopal and S. C. George, Appl. Organomet. Chem., 2007, 21, 368–372 CrossRef CAS.
- S. M. Raders and J. G. Verkade, Tetrahedron Lett., 2009, 50, 5317–5321 CrossRef CAS.
- S. J. Zuend and E. N. Jacobsen, J. Am. Chem. Soc., 2007, 129, 15872–15883 CrossRef CAS.
- X. Wang and S. Tian, Tetrahedron Lett., 2007, 48, 6010–6013 CrossRef CAS.
- B. M. Fetterly and J. G. Verkade, Tetrahedron Lett., 2005, 46, 8061–8066 CrossRef CAS.
- S. C. George, S. S. Kim and S. T. Kadam, Appl. Organomet. Chem., 2007, 21, 994–998 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Characterization data and copies of the 1H and 13C NMR spectra for compounds 2a–g and 4a–m. See DOI: 10.1039/c2gc36442e |
| This journal is © The Royal Society of Chemistry 2013 |