 Open Access Article
Open Access ArticleDeconstruction of lignocellulosic biomass with ionic liquids
Agnieszka
Brandt
a,
John
Gräsvik
b,
Jason P.
Hallett
a and
Tom
Welton
*a
aDepartment of Chemistry, Imperial College London, London, SW7 2AZ, UK. E-mail: t.welton@imperial.ac.uk
bDepartment of Chemistry, Umeå University, Umeå, Sweden
First published on 19th December 2012
Abstract
This paper reviews the application of ionic liquids to the deconstruction and fractionation of lignocellulosic biomass, in a process step that is commonly called pretreatment. It is divided into four parts: the first gives background information on lignocellulosic biomass and ionic liquids; the second focuses on the solubility of lignocellulosic biomass (and the individual biopolymers within it) in ionic liquids; the third emphasises the deconstruction effects brought about by the use of ionic liquids as a solvent; the fourth part deals with practical considerations regarding the design of ionic liquid based deconstruction processes.
 Agnieszka Brandt | Agnieszka Brandt graduated from LMU Munich with a Bachelors in chemistry and biochemistry and a degree in chemistry. During her Masters, she investigated the production of n-butanol in bacteria. Her next research interest was the fractionation of lignocellulosic biomass with ionic liquids. In 2011, she completed her PhD at Imperial College London in this area under the joint supervision of Tom Welton, David Leak and Richard Murphy. Notably, she has investigated the impact of the ionic liquid anion on the effectiveness of the ionic liquid deconstruction process. Agi is currently the Business Manager of Imperial College spin-out Econic Technologies Ltd. |
 John Gräsvik | John Gräsvik was born in 1977 and in 2005 he obtained a Master of Science in Engineering with a major in chemistry from Umeå University. In 2008 he began his PhD studies at the same university and is expected to graduate in the summer of 2013. His research is focused on ionic liquids and in particular the pretreatment of biomass using ionic liquids. In 2011 he spent 6 months in Tom Welton’s research group at Imperial College London studying the physical properties of acids in ionic liquid. |
 Jason P. Hallett | Jason Hallett received his PhD in chemical engineering from the Georgia Institute of Technology. He joined Imperial College in 2006 with a Marshall-Sherfield Postdoctoral Fellowship in Sustainable Chemistry and is now a Research Lecturer in the Department of Chemistry. He has authored over 60 articles and holds 4 patents. He has a multidisciplinary background (organic chemistry/chemical engineering) and his research lies at the science–engineering interface. His current research interests involve the solvation behaviour of ionic liquids and the use of ionic liquids in biorefining, specifically the production of sustainable chemical feedstocks and lignocellulosic biofuels. |
 Tom Welton | Tom Welton received both his BSc and DPhil in chemistry from the University of Sussex. He later moved to the University of Exeter where he was the Demonstrator in Inorganic Chemistry. Upon being awarded a Lloyd’s of London Tercentenary Fellowship he moved again to Imperial College, where he has remained ever since and is now Professor of Sustainable Chemistry and head of the Chemistry Department. He has worked with ionic liquids since the mid-1980s, studying their structures, interactions with solutes, effects on reactivity and applications. |
1. Introduction
During the twentieth century, we came to rely on fossilised organic matter such as coal, gas and oil for the generation of energy and the production of chemical products. It is now clear that the carbon dioxide produced during combustion of fossil resources is causing significant climate change. In addition, the golden age of cheap and abundant supply, at least for petroleum, will soon pass. This has led to a growing interest in renewable technologies to replace fossil sources of carbon. One of these technologies is the conversion of biomass to fuels and chemicals in the so-called ‘Integrated Biorefinery’.1Currently, biofuels are the most significant biomass derived chemicals and are made from edible components of food crops, such as sucrose, starch and vegetable oils. Vegetable oils are converted to biodiesel, which can be used on its own or blended with other diesels. Sugars are converted to ethanol by microbial fermentation. Ethanol can be blended in any ratio with gasoline, being compatible with the existing fleet of cars at low concentrations (5–15%), while flex fuel cars are able to use any concentration. The fermentation of carbohydrates to ethanol is a truly ancient technology that has more recently been applied to the production of biofuels. It is relatively easy to bring sucrose and starch feedstocks into a form that can be fermented by microorganisms. Brazilian sugar cane ethanol can save 80% of greenhouse gas emissions (compared to gasoline) with current technology, while other options such as corn ethanol provide more modest savings due to the more energy-intensive cultivation of these crops.2 Concerns have been widely expressed that the production of fuels from edible biomass directly competes with food production and that CO2 emission savings obtained by replacing gasoline with bioethanol may be diminished by CO2 released as a consequence of land use change.3
Lignocellulosic biomass is the most abundant plant material on our planet and therefore available in much higher quantities (also due to higher yields per area of land) and at lower cost than starch and sucrose based materials.1c,4 It has been estimated that the US produces enough lignocellulosic feedstock today to substitute 30% of their liquid transport fuels with biofuels.4 Utilisation of lignocellulose as a biofuel feedstock is likely to provide much higher CO2 emission savings.2,5
Lignocellulose is the material that makes up the cell walls of woody plants such as trees, shrubs and grasses. It is a composite material, with three biopolymers, cellulose, hemicellulose and lignin, making up ca. 90% of the dry matter. It contains up to 60–70 wt% sugars/carbohydrates but is not utilised for food production. Lignocellulosic biomass for industrial use can come from various sources: agricultural and forest residues, municipal waste such as organic and paper waste and crops specifically grown for this purpose (dedicated biofuel crops). The use of agricultural residues, e.g. cereal straws and corn cobs, as biorefinery feedstock makes it possible to produce food and fuels using the same land.
A major obstacle to competitive lignocellulose utilisation is that cost-effective technologies for processing lignocellulosic material into fuels and chemicals are yet to be commercially developed.6 The increasing complexity of the biomass, from sucrose to starch to lignocellulose, leads to additional process steps (Fig. 1). Sucrose, extracted from sugar cane or sugar beet, can be directly utilised for yeast fermentation. The starch from corn or other grains is first hydrolysed (depolymerised) to glucose using enzymes. In order to access the carbohydrates in the biomass for biological conversion, an additional deconstruction step (also commonly called pretreatment or pre-treatment) is required to bring the sugar polymers into a form suitable for hydrolysis and subsequent fermentation.
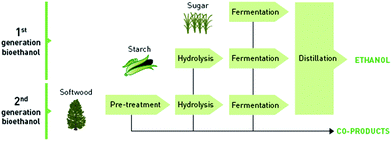 | ||
| Fig. 1 Conversion of first generation and second generation feedstocks into ethanol via the fermentation route. Adapted from ref. 7. | ||
This additional processing consists of a number of steps: feedstock comminution, the actual feedstock deconstruction, conditioning of the treated biomass and the hydrolysate (e.g. detoxification and neutralisation), hydrolysis/depolymerisation of the polysaccharides. The microbial fermentation of monosaccharides and product recovery (often by distillation) is similar to sucrose based processes. The deconstruction of lignocellulose is likely to account for an important fraction of the energy requirement for biomass processing.8 In addition, the utilisation of the non-carbohydrate fraction, lignin,9 is highly desirable for the economic viability of any future biorefinery. Therefore, optimisation of the deconstruction step is desired. This article comprehensively reviews the field of lignocellulose deconstruction with ionic liquids, one of several deconstruction options under development. The review covers the area from early development which has partly already been reviewed elsewhere10 and provides an update on recent advances.
1.1 The chemical composition of wood
Lignocellulose is a composite material synthesised by plant cells, consisting mainly of polymeric carbohydrates (cellulose and hemicelluloses) and the aromatic polymer lignin (Fig. 2). It also contains smaller amounts of pectins, inorganic compounds, proteins and extractives, such as waxes and lipids, which also have potential value. The exact composition of lignocellulose depends on the species, the plant tissue and the growth conditions.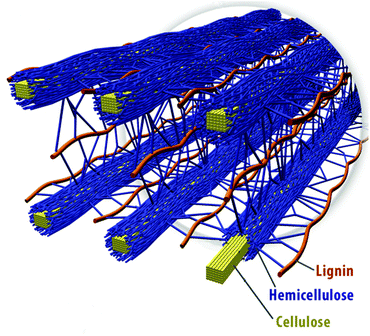 | ||
| Fig. 2 Spatial arrangement of cellulose hemicellulose and lignin in the cell walls of lignocellulosic biomass. Reproduced with permission from ref. 11. | ||
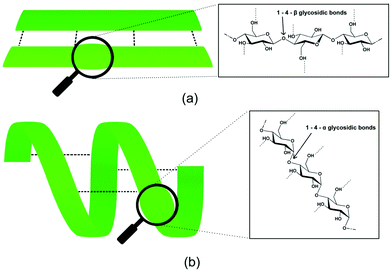 | ||
| Fig. 3 Impact of the geometry at the anomeric carbon on the polymer conformation. (a) A stretched chain is observed for cellulose with β-configuration the biological conformer (cellulose I) being a flat sheet composed of several strands linked by hydrogen-bonds (---) and (b) helical conformation of a starch molecule with an α-conformation. | ||
The linear conformation enables the packing of numerous cellulose strands into crystalline fibrils.12 In biosynthetic (native) cellulose (cellulose Ia and Ib), three hydrogen bonds per glucosyl unit occur: two intramolecular hydrogen bonds and one intermolecular hydrogen bond to a neighbouring cellulose molecule in the same sheet (Fig. 4b).13 The sheets are thought to interact mostly through van der Waals interactions which contribute significantly to the stabilisation of cellulose fibrils.14 Celluloses Ia and Ib can be transformed into cellulose II, a non-natural but thermodynamically more stable form of cellulose, by swelling (mercerization) and dissolution/regeneration;15 in this form, the crystal symmetry is changed and hydrogen-bonds between sheets occur (Fig. 4c).16 Cellulose has the highest degree of polymerisation among the lignocellulosic polymers. The number of glucosyl units in one polymer strand can be 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 or higher.17 Although the monomer (glucose) and short oligomers are water-soluble, cellulose is not. Reasons for this are the high molecular weight of cellulose (solubility is usually inversely related to polymer length) and the comparatively low flexibility of cellulose polymer chains.18 The intermolecular hydrogen-bonding, and the hydrophobic flat top and bottom surfaces enabling van der Waals interactions between sheets, allow intimate and ordered packing of cellulose strands and contribute to the polymer's insolubility in water and most solvents.
000 or higher.17 Although the monomer (glucose) and short oligomers are water-soluble, cellulose is not. Reasons for this are the high molecular weight of cellulose (solubility is usually inversely related to polymer length) and the comparatively low flexibility of cellulose polymer chains.18 The intermolecular hydrogen-bonding, and the hydrophobic flat top and bottom surfaces enabling van der Waals interactions between sheets, allow intimate and ordered packing of cellulose strands and contribute to the polymer's insolubility in water and most solvents.
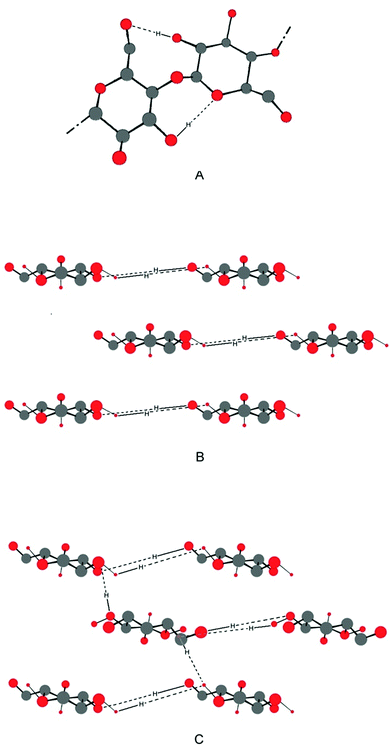 | ||
| Fig. 4 (A) Cellobiose, the repeating unit in crystalline cellulose I, with intramolecular hydrogen bonds shown. Axial cross sections of 3 sheets of (B) cellulose I and (C) cellulose II, with intermolecular hydrogen bonds shown. Cellulose strands are represented by cellobiose units and hydrogen atoms have been omitted for clarity unless involved in hydrogen-bonds. | ||
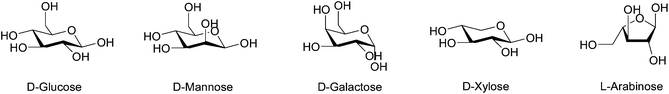 | ||
| Fig. 5 The hexoses and pentoses typically found in hemicellulose. | ||
Hemicellulose polymers can be branched and may be decorated with functionalities such as acetyl and methyl groups, cinnamic, glucuronic and galacturonic acids. For example, the main chain of galactoglucomannan, a branched hemicellulose found in softwood, is built from (1→4)-linked β-D-glucopyranosyl and (1→4)-linked β-D-mannopyranosyl units. The mannosyl units are also substituted to some extent by both acetyl groups in the C-2 and C-3 position and by a (1→6)-linked α-D-galactopyranosyl units (Fig. 6).20
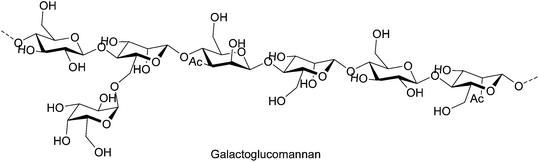 | ||
| Fig. 6 Galactoglucomannan, a branched hemicellulose found in softwood. | ||
Hemicellulose is thought to bind non-covalently to the surface of cellulose fibrils. It acts as an amorphous matrix material, holding the stiff cellulose fibrils in place. It has been suggested that the substitution with hydrophobic groups such as acetyl and methyl groups enhances the affinity of hemicellulose to lignin and thus aids the cohesion between the three major lignocellulosic polymers.21 The most common hemicellulose sugar in grasses and hardwood is xylose. In softwood, mannose is the major hemicellulose sugar.19 Due to its non-crystalline nature, hemicellulose is more susceptible to depolymerisation than cellulose (especially in acidic conditions), an aspect of its behaviour that is exploited by many deconstruction strategies.
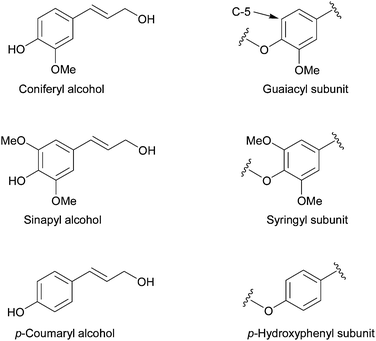 | ||
| Fig. 7 The three monolignols from which lignin is synthesised. The monomers vary in the substitution at the C-3 and C-5 ring positions. | ||
The composition of lignin differs between softwood, hardwood and grasses, with softwood consisting almost exclusively of guaiacyl units while hardwood also contains a large number of syringyl units. Grasses also contain minor amounts of p-hydroxyphenyl groups. This difference in composition has a great effect on the delignification chemistry and therefore on biomass deconstruction. Guaiacyl units are more likely to C–C cross-link at the C-5 position of the ring, these cross-links can form during lignification as well as during delignification.22 The C5 position is substituted in the syringyl unit, which therefore cannot participate in substitution reactions. The C–C cross-links cannot be hydrolysed by acid or base, making delignification of softwoods more difficult than for hardwoods and grasses.
The lignin polymer contains a wide range of linkages. The most common linkage is the β-O-4 ether bond. Roughly 50% of all inter-subunit bonds are of this type.23 The β-O-4 ether bonds lead to a linear elongation of the polymer. Other C–O and C–C linkages are present in lower abundance, and branching occurs when lignification is advanced. The most common linkages are depicted in Fig. 8.
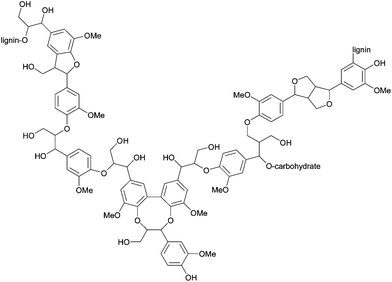 | ||
| Fig. 8 Lignin fragment with various C–O and C–C linkages typically present in native lignin. Reproduced with permission from ref. 24. | ||
The lignin crust has been identified as one of the major obstacles for an energy-efficient biomass deconstruction process.8 Native lignin not only prevents access of polysaccharide hydrolases to their substrates, but modified lignin adhering to the pulp after pretreatment also causes unproductive binding of hydrolases.25 This leads to the need for higher enzyme loadings in the enzymatic carbohydrate hydrolysis and prevents efficient enzyme recycling.17 Lignin can also be a source of compounds that inhibit hydrolases and fermentative organisms, including syringyl aldehyde and vanillic acid.26
Most chemical deconstruction methods modify lignin by hydrolysing its ether bonds, but only some remove it from the pulp (e.g. Organosolv pulping, some base treatments, sulfite pretreatment and pulping, Kraft pulping). The removal of lignin is usually a combination of chemical fragmentation and the ability of the liquor to solvate the modified lignin fragments. Lignin can become water-soluble, as in the case for sulfite pulping which produces lignosulfonates.27
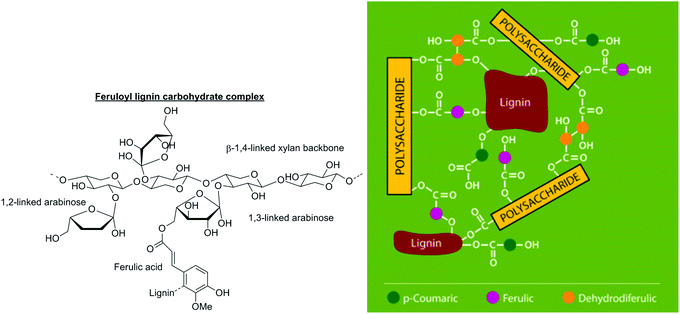 | ||
| Fig. 9 Grass lignin–carbohydrate complexes involving ferulic acid (left). The network that is formed, mediated by ferulic acid, is depicted on the right. Adapted with permission from ref. 30. | ||
Ferulic acid can also dimerise hemicellulose chains (Fig. 10). The extent of cross-linking via lignin–carbohydrate complexes has been correlated with increased cell wall rigidity and resistance to enzymatic digestion. Thus, these cross-links must be broken by chemically hydrolysing the ester bonds in order to obtain an effective deconstruction process.31 In softwood and hardwood, direct complexes between lignin and carbohydrates are present.32 It is thought that they are formed during lignification, when hydroxyl groups of carbohydrates react with electrophilic ketone methide intermediates of the growing lignin polymer chains.
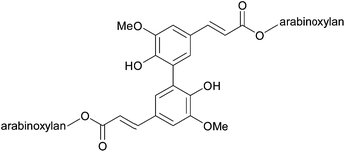 | ||
| Fig. 10 Ferulic acid dimer cross link. | ||
Between adjacent cell walls is a lumen called the middle lamella, whose content holds the cell walls together (Fig. 11). It is devoid of cellulose fibrils and therefore rich in hemicellulose. In mature woody tissue, the middle lamella becomes heavily encrusted with lignin. Most living plant cells disappear when they reach maturity, while their cell walls remain, sometimes for decades and centuries.
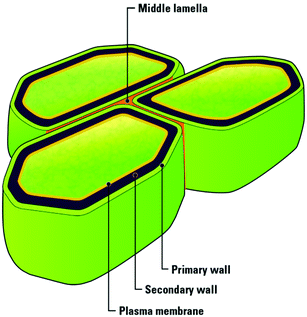 | ||
| Fig. 11 Porous structure of lignocellulosic tissue. Reproduced with permission from ref. 11. | ||
There are three major types of lignocellulosic biomass: softwood, hardwood and grasses. Each group contains promising candidates for future biorefinery feedstocks (Fig. 12a–c). The varying chemical composition (e.g. lignin composition and content) and structural differences (e.g. cell wall thickness and pore size) between the lignocellulose types affects their amenability to deconstruction, therefore the effects of each lignocellulose deconstruction method should be assessed on a number of feedstocks.
 | ||
| Fig. 12 Cultivation of lignocellulosic biomass for biorefinery application: (a) a softwood forest, (b) Miscanthus plantation and (c) a coppice of willows. | ||
1.1.5.1 Softwood. Fir, pine and spruce are softwoods and the dominant lignocellulosic feedstocks in the Northern hemisphere. Fast-growing and tall softwood species are among the most important commercial trees, grown in large plantations for their timber and wood-pulp. Unused softwood resources such as forest thinning, logging, timber mill and urban wood residues will all be part of the biorefinery feedstock base.4 Softwood has a largely uniform microscopic structure, due to the high abundance of a single cell type, the so-called tracheids, which are a narrow, thick-walled cell type. Although this feedstock is very abundant, it is also the most recalcitrant feedstock type. A relatively high lignin content combined with a high guaiacyl to syringyl ratio in the lignin is considered to be the reason for its higher resistance to delignification compared to grass or hardwood biomass. Therefore, harsher conditions are usually required and strong nucleophiles such as sulfide (S2−, in Kraft pulping) or sulfite ([HSO3]−) ions are added to aid lignin removal and prevent lignin recondensation.34 The main hemicellulose sugar in softwood is mannose, followed by xylose.
1.1.5.2 Hardwood. Willows and poplars are examples of potential hardwood biorefinery crops. Hardwoods have more complex structures than softwoods, containing large water-conducting pores or vessels that are surrounded by narrower fibre cells (Fig. 13). The lignin is made up of guaiacyl and syringyl units and the main hemicellulose sugar is not mannose but xylose.
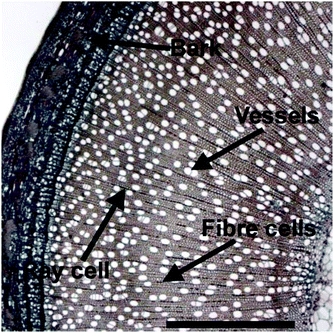 | ||
| Fig. 13 Cross section of a willow showing the bark, fibre cells, the vessels involved in nutrient transport and longitudinal ray cells. Adapted with permission from ref. 35. Scale bar = 1 mm. | ||
Willows (genus Salix) are a group of about 400 hardwood species and have been proposed as dedicated bioenergy crops.36 Willows take root from cuttings and grow quickly. Some species, especially S. viminalis and its hybrids, can be maintained as short rotation coppice from which branches are harvested every 3–5 years, resulting in high yields. Other fast-growing hardwood crops are poplars, with similar yields and harvest cycles.37
1.1.5.3 Perennial and non-perennial grasses. Both annual and perennial grasses are being proposed as sources of biofuels. Perennial bioenergy grasses such as Miscanthus and switchgrass have the advantage that high yields per area of land and year can be achieved. They are planted once and can be grown for many years with relatively low maintenance,38 but they are more resistant to lignocellulose deconstruction than annual grasses such as straws. Annual grass lignocelluloses, corn stalks, sugar cane bagasse, straws (e.g. wheat, rice or barley), are also under investigation as lignocellulosic feedstocks for fuels production and conveniently are by-products of food production.39
Grasses have a distinctively different pore structure compared to trees. Cross sections of Miscanthus reveal a rigid outer ring with thicker cell walls and softer pith consisting of thin cell walls (Fig. 14 left). The thin cell walls of the pith act as a foam core, while the outer ring gives stability against both compression and tension. The stems of other grasses, such as straws, can be hollow. Straws are usually, of all feedstocks, the least resistant to deconstruction. Grass lignin can usually be extracted by aqueous alkali, which is not effective on softwood. The major hemicellulose sugar is xylose.
 | ||
| Fig. 14 Biomass chips of Miscanthus, willow and pine. Scale bar = 10 mm. | ||
The focus of those working on biomass deconstruction processes is thus the preparation of carbohydrates (mainly cellulose) from lignocellulose in a form that can be readily metabolised by fermenting microorganisms. To achieve this, the structural and chemical obstacles that limit the release of carbohydrates must be overcome. The production of unwanted or downstream process inhibiting by-products must be limited. The initial focus of most deconstruction processes has been the cellulose portion, as it provides glucose which is a ready substrate for fermentation. To gain access to the cellulose the lignin–hemicellulose shield must be broken up (Fig. 15), usually by a chemical method at elevated temperature and often also elevated pressure. The exposed cellulose is subsequently hydrolysed in a separate saccharification step. The saccharification can be catalysed by chemicals41 or enzymes; enzymes are usually preferred due to their higher selectivity.17
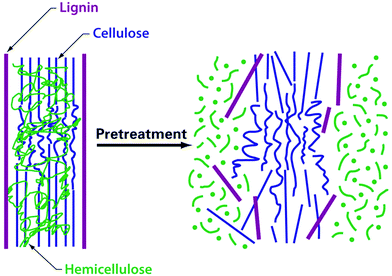 | ||
| Fig. 15 (Physico)chemical deconstruction disrupts the lignocellulose structure, so that the carbohydrates become accessible. Adapted with permission from ref. 8. | ||
Within the biorefinery, the focus must not only be upon obtaining the maximum amount of fermentable hexoses. The utilisation of the pentoses, particularly xylose, will improve the economics of biofuel production.42 In addition, the recovery of non-fermentable components as co-products, particularly lignin, will allow multiple revenue streams compared to a single fermentation product.1 There are a wide range of physical, chemical and combined approaches that are currently being explored to achieve these aims.43 One of these is the deconstruction of biomass with the aid of ionic liquids, which is the subject of this review.
Modern ionic liquids contain organic cations, usually quaternised aromatic or aliphatic ammonium ions. Alkylated phosphonium and occasionally sulfonium cations are also in use. A representative selection of common cations is shown in Fig. 16. Ionic liquid cations often have lengthy names, therefore a notation of the form [CnCm(CoCp)x] will be adopted throughout this article. C signifies an alkyl chain (up to 4 alkyl chains can be attached to the cation core), with subscripts denoting the chain length (number of methylene units plus the terminal methyl group) and x defines the cation core (e.g., N for ammonium, im for imidazolium, pyr for pyridinium and pyrr for pyrrolidinium salts). Ether functionalities within alkyl chains are denoted as CmOCn, where the subscripted ‘C’ describes the alkyl group on either side of the oxygen atom.
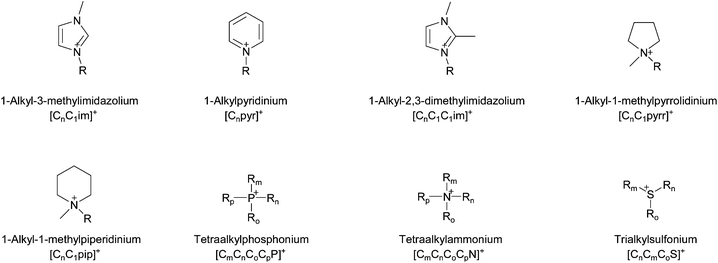 | ||
| Fig. 16 Common cations used in modern ionic liquids. | ||
Ionic liquid anions are either inorganic or organic (Fig. 17). With the exception of halide anions, they are usually polyatomic and the negative charge is distributed over several atoms. Many popular anions are substituted with electron-withdrawing fluorine atoms, such as trifluoromethanesulfonate or tetrafluoroborate, which aids delocalisation of the negative charge, but ionic liquids with non-halogenated anions have recently been developed due to their lower price and lower toxicity/environmental burden.
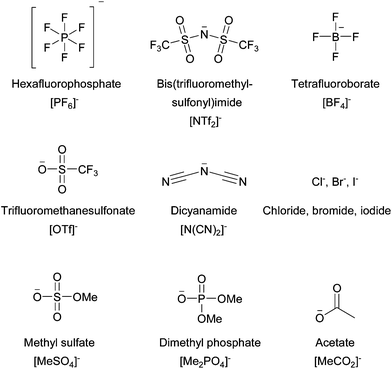 | ||
| Fig. 17 Selection of anions used in modern ionic liquids. | ||
The ionic liquids relevant to this article are listed in Table 1. References to their syntheses are supplied where these have been available in detail. Many of the ionic liquids are commercially available. Purity issues need to be considered for both synthesised and commercially sourced ionic liquids.
| Abbreviation | Structure | R | Name | Reference for synthesis |
|---|---|---|---|---|
| [C4C1im][ABS] |

|
n-C4H9 | 1-Butyl-3-methylimidazolium alkylbenzenesulfonate | 52 |
| [C2C1im][ABS] | n-C2H5 | 1-Ethyl-3-methylimidazolium alkylbenzenesulfonate | 52 | |
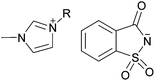
| [C4C1im][ace] |
|
n-C4H9 | 1-Butyl-3-methylimidazolium acesulfamate | 53 |
| [C2C1im][ace] | n-C2H5 | 1-Ethyl-3-methylimidazolium acesulfamate | 53 | |
| [C2pyr]Cl |
|
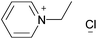
1-Ethylpyridinium chloride | 54 | |
| [C4Him][HSO4] |
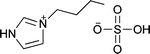
|
n-C4H9 | 1-Butylimidazolium hydrogen sulfate | 55 |
| [C4C1im][HSO4] |
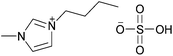
|
1-Butyl-3-methylimidazolium hydrogen sulfate | 55 | |
| [C4C1im][MeSO4] |

|
n-C4H9 | 1-Butyl-3-methylimidazolium methyl sulfate | 56 |
| [C1C1im][MeSO4] | CH3 | 1,3-Dimethylimidazolium methyl sulfate | Commercial | |
| [C4C1im][MeSO3] |
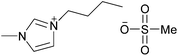
|
1-Butyl-3-methylimidazolium methanesulfonate | 56 | |
| [C2C1im][MeCO2] |

|
n-C2H5 | 1-Ethyl-3-methylimidazolium acetate | 45 |
| [C4C1im][MeCO2] | n-C4H9 | 1-Butyl-3-methylimidazolium acetate | 45 | |
| [C8C1im][MeCO2] | n-C8H17 | 1-Octyl-3-methylimidazolium acetate | 45 | |
| [(H(OC2)2)C1im][MeCO2] |
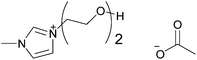
|
1-(2-(2-Hydroxy-ethoxy)ethyl)-imidazolium acetate | 57 | |
| [(C1(OC2)2)C2im][ MeCO2] |

|
1-(2-(2-Methoxy-ethoxy)ethyl)-3-ethylimidazolium acetate | 57 | |
| [(C1(OC2)4)C2im][MeCO2] | 1-(3,6,9,12-Tetraoxatridec-1-yl)-3-ethylimidazolium acetate | 57 | ||
| [(C1(OC2)7)C2im][MeCO2] | 1-(3,6,9,12,15,18,21-Heptaoxadocos-1-yl)-3-ethylimidazolium acetate | 57 | ||
| [(C1(OC2)2)-C2C2C2N][MeCO2] |

|
1-(2-(2-Methoxy-ethoxy)ethyl)-triethylammonium acetate | 57 | |
| [(HOC2)C1C1NH][MeCO2] |
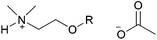
|
H | (2-Hydroxy-ethyl)-dimethylammonium acetate | 57 |
| [(C1OC2)C1C1NH][MeCO2] | CH3 | (2-Methoxyethyl)-dimethylammonium acetate | 57 | |
| [C1C1C1C1GH] [MeCO2] |
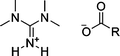
|
CH3 | Tetramethylguanidinium acetate | 58 |
| [C1C1C1C1GH] [EtCO2] | CH2CH3 | Tetramethylguanidinium propionate | 58 | |
| [C4C1im][HCOO] |

|
1-Butyl-3-methylimidazolium formate | 57 | |
| [(C4)4P][HCOO] |
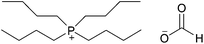
|
Tetrabutylphosphonium formate | 57 | |
| [(C4)4N][HCOO] |
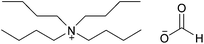
|
Tetrabutylammonium formate | 57 | |
| [C6C1im][OTf] |
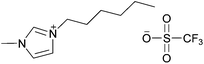
|
1-Hexyl-3-methylimidazolium trifluoromethanesulfonate | 59 | |
| [C4C1im]Cl |

|
n-C4H9 | 1-Butyl-3-methylimidazolium chloride | 56 |
| [C2C1im]Cl | n-C2H5 | 1-Ethyl-3-methylimidazolium chloride | 56 | |
| [C4C1im]Br |
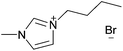
|
1-Butyl-3-methylimidazolium bromide | 56 | |
[C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C2C1im]Cl C2C1im]Cl |

|
1-Allyl-3-methylimidazolium chloride | 60 | |
| [C4C1C1im][BF4] |
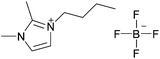
|
1-Butyl-2,3-dimethylimidazolium tetrafluoroborate | 56 | |
| [C4C1im][BF4] |

|
1-Butyl-3-methylimidazolium tetrafluoroborate | 56 | |
| [C4C1im][PF6] |
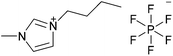
|
1-Butyl-3-methylimidazolium hexafluorophosphate | 56 | |
| [C4C1pyrr][PF6] |
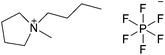
|
1-Butyl-1-methylpyrrolidinium hexafluorophosphate | 56 | |
| [C2C1im][C2C2PO4] |
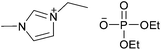
|
1-Ethyl-3-methylimidazolium diethyl phosphate | 61 | |
| [C2C1im][NO3] |
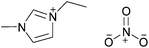
|
1-Ethyl-3-methylimidazolium nitrate | 57 | |
| [C1C1im][Me2PO4] |

|
CH3 | 1,3-Dimethylimidazolium dimethyl phosphate | Commercial |
| [C4C1im][Me2PO4] | n-C4H9 | 1-Butyl-3-methylimidazolium dimethyl phosphate | 56 | |
| [C1Him]Cl |
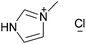
|
1-Methylimidazolium chloride | 62 |
1.2 The solubility of cellulose in ionic liquids
Cellulose is the most abundant polymer in lignocellulose (35–50%). Understanding its interactions with ionic liquids is thus important for the processing of lignocellulosic biomass in ionic liquids; therefore we will include a brief survey of this area here.Dissolution of cellulose is desired in biomass-to-fuels processing as well as for the production of man-made cellulose fibres. Only a few solvents are able to dissolve the crystalline polymer fibrils, e.g. N-methylmorpholine-N-oxide (NMO)63 or concentrated phosphoric acid,64 none of which are applied in lignocellulose processing. The Viscose and Lyocell processes are used for the commercial processing of pure cellulose (e.g. cotton). In the Viscose process, the cellulose is solubilised chemically by transforming the hydroxyl groups into xanthate esters using carbon disulfide (CS2), which makes the cellulose soluble in organic solvents.65 The cellulose is regenerated by hydrolysing these esters. This process uses toxic and highly flammable CS2 and produces large amounts of waste products, which has fuelled the search for a more benign replacement. The Lyocell process is a true dissolution process using NMO and an improvement in terms of health and environmental impact.66 Nevertheless, significant instability of the NMO/cellulose solution above 90 °C and redox activity of the NMO have been viewed as problems,67 which could be addressed by the use of more stable cellulose dissolving ionic liquids.
It has been known for some time that 1-ethylpyridiniumchloride, [C2pyr]Cl, can dissolve cellulose.68 More recently, it has been demonstrated that a range of ionic liquids, typically with 1,3-dialkylimidazolium cations, are also effective cellulose solvents.69 Clear viscous solutions are obtained, showing the typical behaviour of polymer solutions in general and solutions of cellulose in particular.70 This has sparked great interest. In the past few years, a considerable number of studies have been published on cellulose solubility in ionic liquids, patents have been granted for cellulose solubilisation using ionic liquids,71 and the literature has been reviewed several times.72 Many of these reviews contain comprehensive tables listing the substantial number of solubilisation studies for cellulose (and often other biopolymers), we therefore direct the reader to use these studies should they be interested in such information.
Cellulose dissolution is an industrially attractive application of ionic liquids, due to good solubilities (5–20 wt%, depending on the ionic liquid and the conditions), the competitive properties of cellulose reprecipitated from the ionic liquid solutions,73 the increased stability of ionic liquid cellulose solutions74 and the low toxicity of certain relevant ionic liquids (for example, [C2C1im][MeCO2] is classified as non-toxic and non-irritant). The dissolved cellulose can be modified in solution70,73b or regenerated (reprecipitated) by adding water, mixtures of water with organic solvents (e.g. acetone) or protic organic solvents, such as ethanol to form films and fibres.69a,75 The ordering of the regenerated cellulose is reduced compared to the initial state and it is transformed into cellulose II.75,76 This also results in significantly accelerated hydrolysis with cellulases compared to native cellulose,77 an effect that is very attractive in terms of the biorefinery and has sparked interest in the use of cellulose dissolving ionic liquids in lignocellulose deconstruction.
It has been observed that the ionic liquid anion plays an important role in determining an ionic liquid's ability to dissolve cellulose.69a Suitable ionic liquids identified to date contain anions that can form strong hydrogen bonds with hydroxyl groups, e.g. chloride,69a carboxylates (acetate, formate, propionate, lactate),78 dialkyl phosphates, dialkyl and trialkylphosphonates69d and amino acid anions.79 Ionic liquids with more exotic hydrogen-bond basic anions, such as phosphorthioates or phosphorselenoates, have proven to be less stable than the earlier discoveries.80 The dissolving power of these relevant ionic liquids has been typically attributed to strong hydrogen-bonding interactions between the anions and equatorial hydroxyl groups on the cellulose. This close association was confirmed by NMR studies on ionic liquid solutions of glucose and cellobiose81 and molecular dynamics studies of the interaction of [C2C1im][MeCO2]82 or [C4C1im]Cl83 with celloligomers. The 1H-NMR spectral shift of the ethanol hydroxyl proton peak in the presence of ionic liquids has been correlated with the cellulose solubilisation power of imidazolium ionic liquids.84
These findings also agree with empirical solvent polarity measurements, which are applied to predict solvent properties, among others, rate constants and solubility.46a Several empirical and semi-empirical polarity scales have been used to explain and predict solubility of biomolecules and biopolymers in solvents, for example the Hansen solubility parameters,21,85 COSMO-RS86 and the Kamlet–Taft polarity parameters.87 The empirical Kamlet–Taft model69c,d and the quantum mechanical COSMO-RS model88 have been used most frequently to predict or explain the solubility of cellulose (and other biopolymers) in ionic liquids, while few literature data exist for Hansen solubility parameters of ionic liquids.89
The Kamlet–Taft parameters are determined by measuring the UV-Vis spectra of dyes when dissolved in a solvent of interest. In the Kamlet–Taft system, the parameter describing the hydrogen-bond basicity is called β, while α is a measure of the solvents’ hydrogen-bond acidity and π* of its interactions through dipolarity and polarisability. In ionic liquids, particularly those with non-functionalised cations such as dialkylimidazolium, the hydrogen-bond basicity/value of β is primarily influenced by the anion.56 For cellulose solubility, it was shown that ionic liquids that dissolve cellulose are characterised by high values for parameters describing solvent hydrogen-bond basicity.78a
The importance of the anion's hydrogen-bond basicity is demonstrated in Fig. 18, where cellulose solubilities from a range of publications are plotted against the Kamlet–Taft β parameters of these liquids (only dialkylimidazolium cations were considered). Dialkylimidazolium chlorides usually have a β parameter of just above 0.80 and bromides around 0.75. Ionic liquids that have similar β parameters but different anions, such as [C4C1im][MeSO3] (β = 0.77)55 or [C4C1im][Me2PO3Se] (β = 0.82)84 do not dissolve cellulose. This suggests that, around the cut-off, secondary effects such as anion size and geometry play a role and may decide whether an ionic liquid solubilises cellulose or not. All empirical polarity scales have such limitations, they can only report on that which they measure.
Ionic liquids with imidazolium or phosphonium cations and amino acid anions have high hydrogen-bond basicity while being liquid at room temperature,90 but have not been investigated in detail for their ability to dissolve cellulose. Careful investigation of the stability of these ionic liquids under processing conditions is required, as it has been shown that high hydrogen bond basicity is associated with reduced ionic liquid stability.
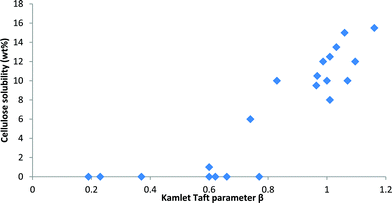 | ||
| Fig. 18 The solubility of cellulose in 1-ethyl-3-methylimidazolium and 1-butyl-3-methylimidazolium ionic liquids as a function of the Kamlet–Taft β value of the ionic liquid. The data were taken from a range of publications.55,69a,d,77,78a,113,114,124 Although the measurements were performed under a variety of conditions (e.g. different temperature, dissolution time, cellulose degree of polymerisation, moisture content, ionic liquid purity), which affect the maximum solubility, the importance of a high β value (>0.8) is apparent. | ||
The COSMO-RS predictions by Kahlen et al. also identify the anion as a major factor in the solubilisation power of ionic liquids.88b It has been suggested that it is not only β, but the combination of β and the difference between α and β can be used more generally to predict (based on the Kamlet–Taft solvent parameters) whether an ionic liquid can dissolve cellulose or not.
The choice of cation also affects solubility considerably. The effect of the cation on the solubility in ionic liquids with carboxylate anions (acetate and formate) is demonstrated in Table 2. Although data are not always consistent, several effects seem to play a role. Lengthening the alkyl (or glycol) chains on the cation progressively reduces cellulose solubility. Hydroxyl groups on the alkyl chains, functionalities that increase the hydrogen-bond acidity of the ionic liquid, also reduce the solubility of cellulose. The same effect is seen when a hydroxyl group is situated on the anion.78a A protic cation prevents cellulose solubilisation entirely in many cases.57,91 This could be due to stronger interactions between cations and anions, making the ionic liquid less able to dissolve cellulose. For cellulose solubility to be observed in protic ionic liquids, the cation must be based on a strong base such as tetramethylguanidine78b or 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)92 and a weak carboxylic acid such as acetic or propionic acid. These ionic liquids are characterised by high β values (1.1 or higher), a high π* and a moderate α value, in accordance with the rules laid out earlier.93 A relatively high α value seems to result in particularly high sensitivity to moisture; smaller amounts than for [C2C1im][MeCO2] were shown to inhibit solubilisation of cellulose entirely. Ionic liquids formed by proton transfer from an acid to a base have the advantage of simple preparation, low price and recovery by distillation.78b
| Ionic liquid | Solubility (wt%) | Comment |
|---|---|---|
| [C2C1im][MeCO2] | 15 | Highest solubility |
| [C8C1im][MeCO2] | <1 | Long alkyl chain reduces solubility |
| [C1(OC2)2C2im][MeCO2] | 12 | Effect of glycol chain |
| [C1(OC2)4C2im][MeCO2] | 10 | Effect of lengthening glycol chain |
| [C1(OC2)7C2im][MeCO2] | 3 | Effect of lengthening glycol chain |
| [H(OC2)2C1im][MeCO2] | 5 | Effect of –OH group on alkyl chain |
| [(C1OC2OC2)C2C2C2N][MeCO2] | 10 | Effect of changing cation core |
| [(C1OC2)C1C1NH][MeCO2] | <0.5 | Effect of substituting an alkyl group with H |
| [(HOC2)C1C1NH][MeCO2] | <0.5 | Effect of N–H group and OH group on alkyl chain |
| [C4C1im][HCOO] | 8 | Effect of changing cation core |
| [(C4)4P][HCOO] | 6 | Effect of changing cation core |
| [(C4)4N][HCOO] | 1.5 | Effect of changing cation core |
The strength and directionality of interactions between ionic liquid cations and cellulose is still the subject of debate. Most studies postulate significant van der Waals attractions and even weak hydrogen bonds between the two. Molecular simulation studies of glucose in [C1C1im]Cl found that cations are particularly likely to be found above and below the glucose ring.94 This is explained by the anisotropic molecular surface of the straight cellulose strands that have a hydrogen-bonding periphery but relatively hydrophobic top and bottom surfaces. Therefore, ionic liquids with relatively small, non-coordinating cations and small hydrogen-bonding anions may be particularly suitable solvents for cellulose due to their bi-functionality (amphiphilicity); the cations interact with the top and bottom surfaces through dispersion forces and hydrogen-bonds between the oxygen atoms in the glycosidic bonds and hydroxyl groups. This replaces the stacking interactions between the sheets in crystalline cellulose; the anions coordinate the equatorial hydroxyl groups, separating the strands from their lateral neighbours.
It must not be forgotten that dissolution of solutes in solvents is not only an enthalpic effect but also greatly affected by entropy;95 this is particularly important for large solutes such as polymers, but has only very recently been considered for ionic liquid cellulose solutions.96 The presence of solutes has a negative effect on the entropy of the solvent, as the solvent needs to order around the solute for it to be accommodated. The loss in solvent entropy is usually counteracted by increased entropy of the solute which gains translational, rotational and vibrational degrees of freedom upon dissolution. Since cellulose is a fairly rigid polymer, its ability to compensate for the loss of solvent entropy is limited, playing a large role in its insolubility.18 Therefore, a comparatively small loss of solvent entropy observed for ionic liquids when accommodating a solute83 is likely to contribute to the solubility of cellulose in ionic liquids.
In terms of Kamlet–Taft empirical solvent polarity, the optimum ionic liquids for cellulose dissolution should have high hydrogen-bond basicity and low hydrogen-bond acidity. Although the polarizability–dipolarity interactions of ionic liquids do not show the ranges seen for the other parameters, these should be higher, indicating that the ionic liquid cations and anions should not be decorated with long aliphatic alkyl chains. Similar conclusions can be drawn from COSMO-RS studies.88a,b
Mixtures of cellulose dissolving ionic liquids and polar organic co-solvents, such as dimethylformamide, dimethylsulfoxide and 1,3-dimethyl-2-imidazolidinone, have also been successfully applied.79,97 The great advantage of such mixtures is the lower viscosities of the cellulose solutions. This accelerates the process for dissolving cellulose from hours down to minutes.
The ionic liquid–organic solvent mixtures have been termed ‘organic electrolyte solutions’.97b For a co-solvent to be suitable for this application they are required to have high π* (>0.8), low hydrogen-bond acidity (α < 0.5) and a hydrogen-bond basicity that is moderate compared to ionic liquids but high for molecular solvents (β ≥ 0.4).97a Although Kamlet–Taft polarity strictly only applies to pure solvents, the methodology has been applied to ionic liquid–molecular solvent mixtures.97b,98 For many solutes, the local solvent environment is different from the bulk composition; this effect is called preferential solvation. Hence, the dye probe and solute of interest have different solvation environments (i.e. the probe is surrounded by a different mixture of solvent molecules–ions than the solute). Consequently, such studies should only be used as semi-quantitative guides to the solvation chemistry.
In addition to the ionic liquid structure, impurities can also affect cellulose solubility, the most prominent being water. Water can easily be absorbed by ionic liquids from air (Fig. 21) or introduced with wet cellulose/biomass and is a potent anti-solvent for cellulose. For example, a study by Mazza et al. of the precipitation of cellulose from [C2C1im]Cl showed the onset of cellulose precipitation occurring at ca. 0.15 wt% water and that it was essentially complete at ca. 0.25 wt% water;99 this is less than 3 mol% compared to the number of ionic liquid ions in the system and only 0.5 equivalents relative to the number of cellulose hydroxyl groups in solution. These results are interesting, as at the point of solubility inhibition, plenty of non-hydrated ions are still available. In the very first publication on cellulose solubility in ionic liquids by Swatloski et al., concentrations of 1 wt% water was stated as inhibitory,69a which is 5–10 times higher than that which Mazza et al. found in their study. Gericke et al. found that addition of 20 wt% water inhibited solubilisation of cellulose in all the ionic liquids that they investigated: [C4C1im]Cl, [CC2C1im]Cl and [C2C1im][MeCO2].100
Hauru et al. found that solutions of cellulose in [C2C1im][MeCO2] became turbid at 2–3 equivalents of water, which is equivalent to 20–25 wt% water content.93 This is substantially higher than has been found for chloride ionic liquids. The solution's viscoelastic properties also changed at 1.6 equivalents or 12 wt% water, suggesting the existence of a gel-like stage between the solution and turbid suspension stages. In the guanidinium based ionic liquids [C1C1C1C1GH][MeCO2] and [C1C1C1C1GH][EtCO2], water that was present before dissolution was inhibitory to cellulose solubilisation at <0.5 equivalents. If the water was added to an existing cellulose solution in the guanidinium ionic liquids more water was tolerated (up to 3 equivalents). Therefore it appears, as one might expect, that water tolerance is dependent on the nature of the ionic liquid.
The exact reason for the high sensitivity of cellulose solubility in ionic liquids to water remains to be fully explained. A possible reason is that water hydrogen-bonds strongly to the ionic liquid anions, so reducing their propensity to interact with the cellulose. Alternatively, water may hydrogen-bond to cellulose in such a manner that it prevents the coordination of ionic liquid anions to the cellulose. In both cases, the primary driving force (anion–cellulose hydrogen bonds) for the solubility of cellulose would be removed. The former explanation is supported by empirical solvent polarity measurements of wet ionic liquids, for which reduction in the measured Kamlet–Taft hydrogen-bond basicity has been observed (suggesting that the anion preferably interacts with water rather than a solute such as the Kamlet–Taft dye or cellulose).93,101 Changes in solvent entropy may also play a role. Finally, it is possible that different effects are contributing to this behaviour at different concentrations of water.
Interestingly, Ohno and co-workers recently reported the rapid dissolution (5 min) of 15 wt% cellulose in aqueous solutions of tetrabutylphosphonium hydroxide, [(C4)4P]OH, and tetrabutylammonium hydroxide, [(C4)4N]OH, containing 40–50% water by weight at room temperature.102 In both cases, even large quantities of water did not impair the solubility, as it typically does with other cellulose solubilising ionic liquids. The tolerance to water is important, as the water is essential for stabilising the organic hydroxide salt, which would decompose upon drying. The fast dissolution is due to the decreased viscosity of the resulting cellulose solutions. Cellulose solubility in cold (4 °C) aqueous hydroxide has been observed before.103 However, the organic hydroxide solution seems to work at temperatures that are more beneficial for processing and in a wider range of concentrations.
1.3 The solubility of lignin in ionic liquids
Lignin in its native form differs from the lignins extracted with common commercial methods. Despite this, lignin solubilities in ionic liquids have been typically tested with lignin preparations such as alkaline, Kraft or Organosolv lignin. Although solubility tests give valuable information, the solubility of such lignin preparations in an ionic liquid does not guarantee that the same ionic liquid will also excel at extraction of native lignin from a biomass substrate or at recovery of the solubilised lignin. Therefore, the ionic liquid solubility of lignin preparations obtained by extraction is discussed here, while the extraction and chemical modification of lignin by ionic liquids and lignin recovery are discussed in separate sections.The solubilities of lignin preparations in a range of dialkylimidazolium ionic liquids are listed in Table 3. They show a wide variation from virtually insoluble to 30 wt% solubility. As with cellulose, the solubility seems to be strongly affected by the choice of anion, although hydrogen-bond basicity does not need to be as high as for cellulose; some mid-range basic ionic liquids seem to be better solvents for lignin than their more hydrogen-bond basic relatives (Fig. 19). 1,3-Dialkylimidazolium ionic liquids with moderate to strong hydrogen-bonding anions such as trifluoromethanesulfonate (triflate, [OTf]−), methyl sulfate ([MeSO4]−), chloride, bromide and acetate anions exhibit high lignin solubility. Pu et al. reported a strong temperature dependence of the solubility for methyl sulfate and triflate ionic liquids. The solubility was 6 wt% in both [C1C1im][MeSO4] and [C4C1im][MeSO4] at 25 °C, but when the temperature was increased to 50 °C, solubility increased drastically to 26 wt%,104 with a similar temperature effect found for [C6C1im][OTf]. It has been shown that the methyl sulfate anions are reactive at elevated temperatures, particularly in the presence of catalytic amounts of acid, which promote ester hydrolysis (in the presence of water) and/or transesterification reactions with alcohols.55,105 Therefore, heating lignin in these ionic liquids is likely to be accompanied by condensation reactions between the ionic liquid and lignin and could explain the stark increase of lignin solubility with temperature in such ionic liquids.
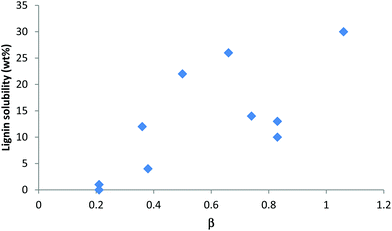 | ||
| Fig. 19 Solubilitiesof lignin in ionic liquids (Table 3) plotted against the ionic liquids’ Kamlet–Taft parameter for hydrogen-bond basicity (β).56,104 | ||
| Ionic liquid | Preparation | Solubility (wt/wt%) | Temperature | Reference |
|---|---|---|---|---|
| [C2C1im][MeCO2] | Kraft lignin (Indulin AT) | 30 | 90 °C | 108 |
| [C1C1im][MeSO4] | Residual softwood lignin | 26 | 50 °C | 104 |
| [C4C1im][MeSO4] | Residual softwood lignin | 26 | 50 °C | 104 |
| [C6C1im][OTf] | Residual softwood lignin | 22 | 70 °C | 104 |
| [C4C1im]Cl | Residual softwood lignin | 13 | 75 °C | 104 |
| [C4C1im]Cl | Kraft lignin (Indulin AT) | 10 | 90 °C | 109 |
| [C4C1im]Br | Residual softwood lignin | 14 | 75 °C | 104 |
| [C4C1C1im][BF4] | Residual softwood lignin | 12 | 100 °C | 104 |
| [C4C1im][BF4] | Kraft lignin (Indulin AT) | 4 | 90 °C | 109 |
| [C4C1im][PF6] | Kraft lignin (Indulin AT) | 1 | 90 °C | 109 |
| [C4C1im][PF6] | Residual softwood lignin | 0 | 120 °C | 104 |
| [C4C1pyrr][PF6] | Residual softwood lignin | 0 | 120 °C | 104 |
The ability of [C4C1im]Cl to extract and dissolve lignin has been recently employed in optical lignin quantification (although this ionic liquid does not show the best lignin solubilities).106 Generally, higher solubilities are possible for lignins than for cellulose and a larger number of ionic liquids are capable of dissolving lignin than are capable of dissolving cellulose. A theoretical model using dispersion corrected density functional theory found that imidazolium cations are particularly suitable for lignin solubilisation, as the cation can favourably interact with the lignin phenyl rings via the aromatic rings.107 However, due to the limited number of ionic liquids studied (essentially all being imidazolium ionic liquids) experimental validation of the relative importance of the imidazolium cation is still missing.
1.4 The solubility of lignocellulose in ionic liquids
Ionic liquids have also been used in attempts to dissolve the whole lignocellulosic biomass (Fig. 20). Most studies reporting solubilisation of lignocellulose have used ionic liquids with imidazolium cations, in particular dialkylimidazolium cations. The solubility of lignocellulosic material has already been reviewed.110 We will briefly recap the history and major insights and update to the current state of knowledge before concentrating on the aspects of lignocellulose solubility that are relevant to the deconstruction/fractionation of lignocellulose.![Dissolution of beech powder in [C2C1im][MeCO2]; left: ionic liquid, middle: beech powder, right: solution. Reproduced from ref. 114.](/image/article/2013/GC/c2gc36364j/c2gc36364j-f20.gif) | ||
| Fig. 20 Dissolution of beech powder in [C2C1im][MeCO2]; left: ionic liquid, middle: beech powder, right: solution. Reproduced from ref. 114. | ||
The dissolution of wood powder in 1-butyl-3-methylimidazolium chloride ([C4C1im]Cl) was first described by Rogers and co-workers in 2007.111 The dissolution of wood saw dust and thermomechanical wood pulp in [C4C1im]Cl and 1-allyl-3-methylimidazolium chloride ([(CC2)C1im]Cl) was reported shortly afterwards by Kilpeläinen et al.60 The study found that [(CC2)C1im]Cl was more effective than its dialkyl analogues under comparable conditions. Possible reasons for this are the reduced melting point and viscosity of [(CC2)C1im]Cl compared to [C4C1im]Cl. Another possible explanation is that the allyl side chain may be reactive under the high temperatures of the dissolution experiments, but this has not yet been investigated.
Further progress was made when Sun et al. reported the dissolution of wood particles in [C2C1im][MeCO2] for the first time.112 Under comparable conditions, this ionic liquid was also more effective than [C4C1im]Cl.113 Zavrel et al. measured light scattering to screen ionic liquids for partial or complete dissolution of lignocellulose (softwood and hardwood).114 The sufficiently large number of screened ionic liquids allowed them to establish a link between cellulose and lignocellulose solubility. This link was further strengthened by relating lignocellulose solubilities and Kamlet–Taft solvent polarities.101 It was shown that the hydrogen-bond basicity of the ionic liquid, controlled by the choice of anion, is crucial for obtaining an ionic liquid capable of dissolving or at least swelling lignocellulose.
Table 4 shows representative data for lignocellulose dissolution in ionic liquids. It provides evidence that, in most cases, a solid residue is left at the end of the dissolution experiment. Under the right conditions, this fraction is small. Even studies that report complete dissolution state that their solutions were hazy;111 thus, it appears to be difficult to achieve total dissolution of biomass. Ball milling for long periods of time is required to achieve completely uniform solutions of lignocellulosic biomass. Such ball milling is useful for analytical techniques,115 but uneconomical for an industrial process.
| Ionic liquid | Loading wt% (solubilisation %) | Feedstock | Particle size (mm) | Temperature and time | Reference |
|---|---|---|---|---|---|
| a Microwave irradiation. | |||||
| [C2C1im][MeCO2] | 5 (92.2) | Pine | <0.125 | 175 °C, 30 min | 119 |
| 5 (42.8) | Pine | <0.125 | 185 °C, 7 min | 119 | |
| 5 (93.5) | Pine | 0.250–0.500 | 110 °C, 16 h | 112 | |
| 5 (98.5) | Oak | 0.250–0.500 | 110 °C, 16 h | 112 | |
| 5 (75) | Beech | 0.100–0.500 | 115 °C, 24 h | 116 | |
| 5 (95) | Beech | 0.100–0.500 | 115 °C, 72 h | 116 | |
| 5 (40) | Spruce | 0.100–0.500 | 115 °C, 24 h | 116 | |
| 5 (75) | Spruce | 0.100–0.500 | 115 °C, 72 h | 116 | |
| [C4C1im]Cl | 5 (26.0) | Pine | 0.250–0.500 | 110 °C, 16 h | 112 |
| [C2C1im]Cl | 3 (95.0) | Beech | n.d. (wood flour) | 120 °C, 24 h | 120 |
| [CC2C1im]Cl |
8 (n.d.) | Spruce | 0.100–2.000 | 110 °C, 8 h | 111 |
| 5 (26) | Pine | 0.450–0.650 | 100 °C, 15 h | 121 | |
| 3 (50) | Pine | 0.450–0.650 | 110 °C,a 2 h | 121 | |
| 10 (100) | Spruce, Eucalyptus | Ball milled for 48 h | 75 °C, 48 h | 115 | |
| [C2C1im][C2C2PO4] | 4 (n.d.) | Wheat straw | <5 | 100 °C, 1 h | 122 |
The data in Table 4 show that a variety of factors influence the extent of lignocellulose solubilisation, for example, the choice of ionic liquid, feedstock type, feedstock particle size, dissolution time and temperature.112 In general, higher temperature or microwave irradiation accelerates the dissolution, but partial decomposition of feedstock and ionic liquid may be an issue under harsher conditions. Regarding the type of feedstocks, grass biomass is more easily solubilised, while the most difficult substrates are typically softwoods (pine and spruce). Table 4 also shows that most researchers choose a solid to liquid ratio around 5% (wt/wt) and good solubility is usually only observed for samples with less than 10 wt% solid loading.
The solubility of finely milled biomass is higher than that of coarser material. Sun et al. showed that particle size is one of the factors that influence the solubilisation of pine and oak wood in [C2C1im][MeCO2].112 The use of less finely ground feedstock (>0.5 mm) significantly reduced the solubilisation of the lignocellulosic substrates. Various studies reported that solubilisation of chipped feedstocks was incomplete in a reasonable time frame, even at temperatures over 100 °C.111,113 On the other hand, Viell and Marquardt reported the dissolution speed of air-dried spruce and beech wood chips (1 × 10 × 2 mm) to be similar to that of finer particles of the same feedstocks (0.1–0.5 mm),116 in contrast to the observations of others. Their study used a very long treatment period of 72 h to achieve satisfactory solubilisation. The similarity of dissolution kinetics was attributed to initial breaking up of the wood into individual cell walls, indicating that the middle lamella may dissolve faster than the walls. Fast dissolution of the middle lamella has also been reported for Organosolv treatment.117 Dispersion of the cell walls after middle lamella dissolution could also account for the occasional observation of lignocellulose solubility in non-cellulose dissolving ionic liquids such as [C4C1im][HSO4] or methyl sulfate based ionic liquids.118
There are some studies that report that they did not accomplish lignocellulose solubilisation in [C2C1im][MeCO2].109,123 These observations, contradicting reports of many other groups, may be due to very high biomass loadings (substantially exceeding the solubility limit for lignocellulose in this ionic liquid), the presence of inhibiting amounts of water in the ionic liquid or the biomass, temperatures that are too low to accomplish solubilisation, or combinations of these. It should be noted that, despite the impaired solubilisation, it is still possible that a deconstruction effect has been accomplished.
As just mentioned, the dissolution process is sensitive to water. Addition of 5–10% water to [C2C1im][MeCO2] and [C4C1im][MeCO2] prior to treatment of maple wood flour increased the crystallinity of cellulose in the treated wood compared to the cellulose treated with a dry treatment solvent.124 Another study found that 3–5 wt% of water will completely inhibit the solubilisation of Miscanthus in [C2C1im][MeCO2],118 while BASF claim in a patent application that the dissolution process using acetate anions tolerates up to 10% of water.125 The inhibitory effect of water on lignocellulose solubility is in agreement with the negative effect of water on the solubilisation of cellulose in ionic liquids. This sensitivity is posing a challenge for total biomass dissolution, as many hydrophilic ionic liquids have a high affinity towards water. These (hydrogen-bond basic) ionic liquids take up more than 1 equivalent of water at room temperature (Fig. 21). The uptake of 3.5 molecules of water per anion (or cation) was also reported by Troshenkova et al. for [C2C1im][MeCO2] (27 wt% which corresponds to 3.5 equivalents).126 A release of up to 11 kJ mol−1 heat upon mixing water and the dried ionic liquid was observed by the same study, demonstrating how strongly incorporation of water affects the solvent structure of such ionic liquids.
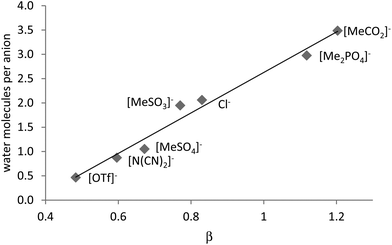 | ||
| Fig. 21 Correlation between the Kamlet–Taft β parameter of dry 1butyl3methylimidazolium ionic liquids with the number of water molecules per anion (or cation) when the ionic liquids are equilibrated with air.127 | ||
Since solubilisation of lignocellulose and cellulose seems to require similar ionic liquid properties, it comes as no surprise that dissolved lignocellulose can be regenerated by addition of an anti-solvent, just as for cellulose. The most widely used anti-solvent is water. The composition of the regenerated lignocellulose is often different from the original composition, which has triggered attempts to tune the regeneration solvent to fractionate lignocellulose, separating the lignin from the carbohydrates. Some researchers observed that the cellulose content in the precipitate can be enriched when organic solvent–water mixtures are used instead of pure water, in order to keep part of the lignin in solution when the pulp phase-separates. A 1:1 (vol%) acetone–water mixture has been used by the Rogers group,111,112 while Viell et al. used a 1:1 (vol%) ethanol–acetone mixture.116 Dibble et al. investigated the optimisation of wash solvent composition comprising ethanol and acetone to avoid gelling of cellulose.128
The crystallinity of the cellulose portion in regenerated lignocellulosic biomass has been reported to be reduced, usually based on studying the disappearance and/or shifting of signals in X-ray powder diffraction patterns.109 This has been attributed to the change from cellulose I to cellulose II76 as well as loss of the fibrillar ordering. Cheng et al. observed that the conversion of cellulose I into cellulose II during lignocellulose solubilisation occurred in the order of hours and progressed at different rates for different biomass types, in the order switchgrass > eucalyptus (hardwood) > pine (softwood).76b
In summary, a range of ionic liquids has been identified to date that can dissolve lignocellulosic biomass. The role of the workhorse ionic liquid has shifted from chloride based ionic liquids (notably [CnC1im]Cl and [(CC2)C1im]Cl) to ionic liquids with acetate anions, particularly [C2C1im][MeCO2]. The commercial availability of [C2C1im][MeCO2] in larger quantities and moderate prices has certainly contributed to allowing this change to happen, as well as observation of improved deconstruction effects (which will be discussed in the following part). The degree of solubilisation that can be achieved depends on many variables and finding conditions for optimum solubilisation while avoiding degradation reactions remains a challenge.
1.5 Lignocellulose deconstruction with ionic liquids
So far, we have discussed the solubilisation of lignocellulose and two of its major components, cellulose and lignin. From here on, the focus will be on the deconstruction of lignocellulosic biomass, so that it can be used as a feedstock for biofuel and chemicals production, and ionic liquids as fractionating solvents for the realisation of an ‘integrated biorefinery’. It should be noted that, in order to achieve this, solubilisation of all biomass components may be part of the deconstruction effort, but it is not essential (as solubilisation is not essential for other deconstruction options such as dilute acid or Organosolv treatment).Two distinct approaches to using ionic liquids for lignocellulose biomass deconstruction have emerged to date. The first and most widely studied approach places importance on solubilisation of the entire biomass composite. A variation of this seeks to concomitantly hydrolyse the carbohydrate polymers by using acidic or acidified lignocellulose dissolving ionic liquids.129 The second approach places importance on chemically disrupting the chemical lignocellulose composite without achieving complete biomass dissolution.109,123 This involves (partial) lignin and hemicellulose solubilisation while the cellulose fraction remains largely intact.55,130 We term these approaches the Dissolution Process and the Ionosolv Process. The latter derives its name from distinct similarities with the Organosolv Process.131
Various techniques can be applied to assess lignocellulose deconstruction on a laboratory scale. These can be used to assess the effect of ionic liquid treatment as well as treatment with other solvents:
• Solid yield (wt%) after treatment
• Saccharification yield (hydrolysis kinetics and/or maximum biomass digestibility)
• Composition of treated solid
• Cellulose crystallinity
• Chemical composition of lignin and carbohydrates (in treated solid or solubilised portion)
Cellulose crystallinity, often measured in connection with ionic liquid treatment, is of particular interest to the Dissolution Process, which aims to alter the structure of the native cellulose fibrils and thus accelerate the subsequent hydrolysis. Compositional analysis allows statements about the effect of the deconstruction experiment, e.g. whether lignin, hemicellulose and/or cellulose have been removed and to what extent. Typically, the quantities of the principal wood sugars (glucose, xylose, arabinose, mannose and galactose), lignin and an inorganic fraction (ash) are determined using standardised protocols, e.g. the protocols made publicly available by the US National Renewable Energy Laboratory (NREL).132 Untreated feedstock and solid biomass after deconstruction can be analysed for their composition.
Other analysis techniques can give insight into chemical changes in the lignin and carbohydrate fractions. 2D NMR techniques allow conclusions about chemical alterations in lignin and hemicellulose;133 gel permeation chromatography measures changes in polymer chain length134 and certain HPLC experiments can quantify the degradation products such as furfurals105 or lignin model compounds.135
| Feedstock | Temperature | Time | Glucose recovered (%) | Lignin removed (%) | Hemicellulose sugars removed (%) | Reference |
|---|---|---|---|---|---|---|
| Corn stover | 125 °C | 1 h | 83 | 44 | 34 | 120 |
| Energy cane | 120 °C | 30 min | 87 | 32 | 14 | 137 |
| Switchgrass | 120 °C | 3 h | 97 | 34 | 22 | 136 |
| 160 °C | 3 h | 79 | 65 | 83 | 136 | |
| 160 °C | 3 h | 85 | 69 | 77 | 138 | |
| Bagasse | 165 °C | 10 min | not meas. | 34 | not meas. | 119 |
| Maple wood | 130 °C | 1.5 h | 84 | 52 | 26 | 109 |
| Oak | 110 °C | 16 h | not meas. | 35 | not meas. | 112 |
| Pine | 110 °C | 16 h | not meas. | 26 | not meas. | 112 |
| Miscanthus (20% water) | 120 °C | 22 h | 96 | 56 | 37 | 55 |
| Willow (20% water) | 120 °C | 22 h | 79 | 18 | 44 | 55 |
| Pine (20% water) | 120 °C | 22 h | 88 | 17 | 0 | 55 |
| Triticale straw | 150 °C | 1.5 h | 86 | 64 | 76 | 139 |
| Triticale straw (50% water) | 150 °C | 1.5 h | 95 | 29 | 62 | 139 |
The glucan fraction, which is mainly cellulose, is usually almost completely recovered in the regenerated fraction (Table 4), although very harsh conditions do reduce cellulose recovery.136 Therefore, the ionic liquids treatments typically lead to enrichment of cellulose in the recovered solids. The dissolved fractions remain with the ionic liquid and are removed with the ionic liquid during separation of the liquor from the pretreated pulp. The subsequent separation of the solubilised biomass components from the ionic liquid and the associated challenges will be discussed later.
Wu et al. have investigated the effect of biomass loading on corn stover deconstruction using ionic liquids. They increased the loading stepwise from 4.8 wt% up to 50 wt% for [C2C1im][MeCO2] at 125 °C for 1 h and observed that lignin removal decreased substantially from 44% to 8%.123a Increasing the biomass loading is desirable from a processing point of view, as it improves the process economics. The cell walls swell rather than being solubilised; however, potential handling problems and reduction of yield during up-scaling beyond 15 wt% must be considered.17
Ionic liquids other than [C2C1im][MeCO2] can also remove lignin and hemicellulose from the feedstock (Table 6). Although chloride based ionic liquids have been used extensively for wood dissolution, it seems that they have not been investigated much for their impact on the feedstock composition. The reported delignification and hemicellulose removal are significantly lower than achieved with other ionic liquids under the conditions applied. These were coupled with very low glucose yields after enzymatic saccharification. Other ionic liquids, such as those containing hydrogen sulfate ions mixed with water, show more promise.55 For [C4C1im][HSO4], and also for its acid–base ionic liquid analogue [C4Him][HSO4], the delignification seems to be more extensive than with [C2C1im][MeCO2]. For the ionic liquid [C2C1im][ABS], data for delignification and hemicellulose removal have not been reported. However, recovery of only around 50% of the bagasse feedstock and production of a pulp that appears to be highly enriched in cellulose suggests that removal of both lignin and hemicellulose is considerable with this ionic liquid.52 Therefore sulfonate and sulfate containing ionic liquids appear to be the currently most potent ionic liquids in terms of separating cellulose from lignin (and hemicelluloses). As a potential drawback, the less basic or sometimes even acidic environment of non-acetate ionic liquids can cause more hemicellulose conversion to furfurals and/or humins, which is not seen for the acetate ionic liquids.55 Preservation of the hemicellulose (as the chemically most fragile component among the three main lignocellulosic polymers) by pre-extraction has been suggested for other deconstruction methods, mainly paper pulping,40 and may also be applicable to ionic liquid deconstruction.
| Ionic liquid | Biomass | Temperature | Time | Lignin removed (%) | Hemicellulose removed (%) | Reference |
|---|---|---|---|---|---|---|
| [C4C1im][MeSO4] (20% water) | Miscanthus | 120 °C | 2 h | 27 | 0 | 55 |
| [C4C1im][HSO4] (20% water) | 2 h | 44 | 51 | 55 | ||
| 22 h | 93 | 82 | 55 | |||
| [C4C1im][MeSO3] (20% water) | 68 | 73 | 55 | |||
| [C4Him][HSO4] (20% water) | 4 h | 81 | 84 | 55 | ||
| 20 h | 80 | 92 | 55 | |||
| [C4C1im][HSO4] (20% water) | Willow | 22h | 85 | 79 | 55 | |
| Pine | 65 | 66 | 55 | |||
| [C4C1im]Cl (20% water) | Miscanthus | 15 | 6 | 55 | ||
| [C4C1im]Cl (40% water) | Legume straw | 150 °C | 2 h | 30 | 9 | 140 |
| [C4C1im]Cl | Triticale straw | 90 °C | 24 h | 15 | 11 | 108 |
| [C4C1im][MeSO4] (7% H2SO4) | Sugarcane bagasse | 125 °C | 2 h | 26 | 88 | 130 |
A number of observations regarding the modification of lignin during ionic liquid deconstruction have been published. Tan et al. reported that lignin extracted with [C2C1im][ABS] has a lower molecular weight and a narrower polydispersity than a lignin obtained by aqueous auto-catalysed pretreatment.52 George et al. studied the impact of a range of ionic liquids on several commercial lignins and demonstrated a profound anion effect on the fragmentation mechanism and the degree of polymerisation, with liquids containing alkyl sulfate anions having the greatest ability to fragment the lignins and reduce polymer length.134 The order of molecular weight reduction was sulfates > lactate > acetate > chloride > phosphates. The functional group of the anion determined the effect rather than its size, while the cation did not play a significant role. The authors suggest that the more active anions act as nucleophiles during lignin depolymerisation. In support of this, an increased sulfur content of the lignin after treatment with ionic liquids with sulfur containing anions such as sulfonates and sulfamates (e.g. [ABS]− and acesulfamate)52,53 and sulfates ([MeSO4]− and [HSO4]−)55 has been reported. This may also explain why an attempt to design a ‘lignin friendly’ cation by adding an aromatic side chain had only moderate success.60
Analysis of the residual lignin in [C2C1im][MeCO2] pretreated maple wood with 2-dimensional NMR revealed a decrease of the β-O-4 aryl ether bond content as well as deacetylation of xylan.133 The effect of two lignocellulose-dissolving ionic liquids on a lignin model compound featuring a β-O-4 aryl ether linkage has been studied (Fig. 22).135 The compound was dissolved in [C2C1im]Cl or [C2C1im][MeCO2] at 120 °C. An α,β-dehydration reaction was reported for both ionic liquids. The dehydration was significantly faster in [C2C1im][MeCO2] than in [C2C1im]Cl, possibly reflecting the acetate's greater basicity and affinity towards water.141 The occurrence of such dehydration reactions has also been suggested by George et al. more recently.134
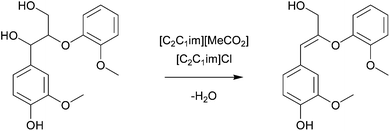 | ||
| Fig. 22 Dehydration of the lignin model compound guaiacylglycerol-β-guaiacyl ether in ionic liquids.135 | ||
Cleavage of the β-O-4 aryl ether bond in guaiacylglycerol-β-guaiacyl ether by hydrogen-bond acidic monoalkylimidazolium ionic liquids at 110–150 °C was investigated.142 Differences in reactivity depending on the anion were observed. More strongly hydrogen bond-basic anions (Cl−, Br− and [HSO4]−) resulted in higher yields of the cleavage products than weakly basic anions, as well as in different routes. The enol ether product in Fig. 23 (top) was observed as an intermediate when the ionic liquid contained a coordinating anion. Less coordinating anions such as [BF4]− resulted in elimination of the γ hydroxyl methylene group, resulting in formation of formaldehyde.
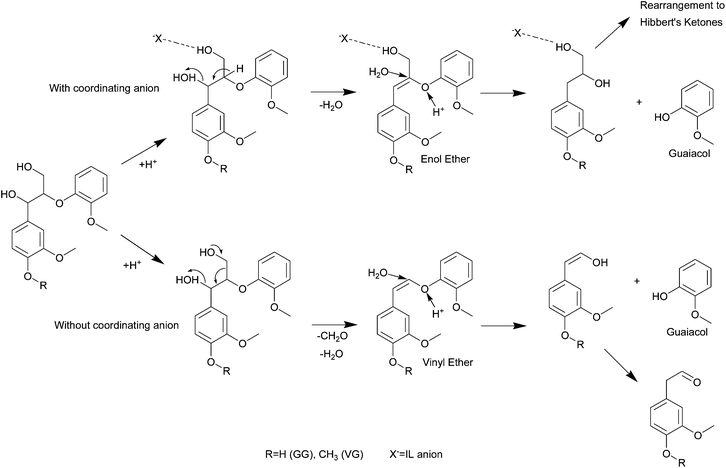 | ||
| Fig. 23 Mechanism of β-O-4 aryl ether bond cleavage in ionic liquids with coordinating (upper pathway) and non-coordinating anions. Redrawn with permission from ref. 142. | ||
Transformation of solubilised lignin in ionic liquids has attracted limited attention so far. The oxidation of Organosolv beech lignin in ionic liquids in the presence of transition metals and molecular oxygen has been demonstrated by Stärk et al.143 The main product was 2,6-dimethoxy-1,4-benzoquinone; vanillin, syringaldehyde and other less abundant oxidised aromatic fragments were also identified. The compounds were partially extracted into organic solvents.
Deliberate in situ hydrolysis of dissolved lignocellulose with acidified ionic liquids is also under investigation. There is now a commercialisation attempt in the US by start-up company Hyrax Energy.144 The hemicellulose and cellulose components are hydrolysed to soluble monomers and short oligomers. This deconstruction strategy may be considered a variation of the Dissolution Process, as complete solubilisation of the carbohydrate fraction is the goal, although there is typically no regeneration step and the enzymatic saccharification stage is not required. The simultaneous dissolution and saccharification process shares similarities with concentrated acid deconstruction (usually performed with sulfuric acid).145 Performing the (chemical) hydrolysis of cellulose and hemicellulose while dissolved in an ionic liquid has the advantage that the glycosidic bonds are more accessible in homogenous solution, allowing milder hydrolysis conditions. The chemical hydrolysis of polysaccharides has the advantage of being faster than an enzymatic hydrolysis step.
The in situ chemical hydrolysis of pure cellulose and the cellulose (and hemicellulose) in ionic liquids has been attempted by various researchers.146 In most studies, a (ligno)cellulose solubilising ionic liquid based on chloride, typically [C4C1im]Cl, and catalytic amounts of a strong acid were used. The use of Brønsted acidic ionic liquids, e.g. hydrogen sulfate containing ionic liquids, has been patented.146d,147
The most comprehensive study of the acid-catalysed hydrolysis of lignocellulosic carbohydrates has been conducted by Sievers et al. who hydrolysed pine flour in [C4C1im]Cl with added sulfuric acid at 100–150 °C.129a Although the solubilisation was close to 100% at 120 °C within 2 h, the yield of monomeric sugars was low. This is likely due to the limited amount of water available in the anhydrous system, halting the hydrolysis at the oligomer stage. The addition of small quantities of water enhanced the monomer yield, but larger amounts lowered it, leading to a maximum glucose yield of just over 10%. The decrease of the monomer yield at higher water content is probably due to decreased lignocellulose solubility. A secondary hydrolysis in diluted ionic liquid increased the monomer yield to up to 30%, supporting the explanation that the cellulose after acidic dissolution in ionic liquids is mostly oligomeric. Well-known carbohydrate degradation products obtained under acidic conditions are 2-furaldehyde (furfural, from pentoses), 5-hydroxymethyl furfural (HMF, from hexoses) and levulinic acid resulting from further HMF conversion,129a,148 as well as solid degradation products called humins.
The two-sided role of water (required for hydrolysis but reducing cellulose solubility), led to the application of a modified strategy by Binder and Raines,146a who dissolved cellulose and the carbohydrate fraction of corn stover with [C2C1im]Cl containing catalytic amounts of HCl. Water was added incrementally during the hydrolysis to maintain a balance between hydrolysis and solubility of the cellulose polymers (the shorter the polymer the more water was tolerated). Using corn stover as a substrate, the yield of glucose under optimised conditions was 70–80% and the HMF yield was 10% or less. Almost quantitative yield of glucose and cellobiose with very little HMF was achieved when hydrolysing pure cellulose in acidified [C4C1im]Cl with the incremental water method.149
The hydrolysis process has also been used to hydrolyse cellulose in an ‘organic electrolyte solution’ made up from [C4C1im]Cl and N-methylpyrrolidinone (NMP) with small amounts of water and sulfuric acid added.150 The hydrolysis temperature was only 70 °C and the presence of the co-solvent accelerated the dissolution substantially, although hydrolysis was slower. The product solution was not analysed for degradation products or lignin. The best conditions (using a 1:3 ration of NMP to ionic liquid) resulted in a 69% total reducing sugar yield and 39% glucose yield.
The choice of acid is important. Vanoye et al. and others correlated the (aqueous) pKa of various acids with the initial rates of cellobiose hydrolysis in [C2C1im]Cl.151 It was shown that only the use of acids with a pKa < 1.0 resulted in appreciable hydrolysis, showing that strong acids need to be used to achieve fast hydrolysis in chloride ionic liquids.
Solid acid catalysts have been used in ionic liquids as an alternative to soluble acids. Cellulose dissolved in [C4C1im]Cl was depolymerised in the presence of excess (24×) water.152 The use of [C4C1im][MeCO2] led to destruction of the heterogeneous catalysts. Similarly to soluble acids, the production of short cellulose oligomers was observed rather than complete hydrolysis to monomers. It was concluded that macroreticulated resins functionalized with sulfonic groups (–SO3H) such as Amberlyst 15DRY, which have a large surface area, are compatible with viscous solutions and sufficiently acidic for the reaction to occur. This study suggested that the cellulose hydrolysis proceeds on the surface of the resin, while subsequent studies have shown that the acidic protons contained in the solid catalyst are released into the ionic liquid and that the hydrolysis rate is proportional to the H+ concentration in the liquid phase.153
A two-stage process combining simultaneous dissolution and saccharification and enzymatic saccharification to shorten the over-all time needed for the saccharification step has also been suggested.154 Initially, high molecular weight cellulose is broken down to oligomers using a short simultaneous dissolution and saccharification step in [C4C1im]Cl, and, after precipitation, a subsequent enzymatic saccharification which hydrolysed the regenerated oligomers to glucose. The total time requirement was 3.5–5 h (0.5–1 h for the dissolution/acid hydrolysis and 3–4 h for the enzymatic hydrolysis) which is significantly shorter than enzymatic saccharification alone but with a similar selectivity. Application to lignocellulosic biomass, a more challenging substrate due to the presence of lignin and more labile hemicellulose, needs to be demonstrated as well as the economical advantage of running a shorter two-stage process compared to a longer one-stage saccharification.
The influence of the anion on the sugar monomer yield during chemical saccharification of cellulose in ionic liquids was investigated by Binder and Raines.146a While several ionic liquids containing chloride anions were effective in generating glucose from cellulose, the ionic liquids [C2C1im][NO3], [C4C1im][BF4], [C1C1im][Me2PO4] and [C2C1im][MeCO2] did not result in the formation of detectable amounts of glucose. For the first two ionic liquids, it is suggested that their inability to dissolve the polymer inhibits access of the acid to the glycosidic bonds and thus prevents hydrolysis. The latter two ionic liquids can dissolve cellulose, but the relatively high basicity of their anions, which are both counter-ions of weak acids (with pKa values of 1.29155 for dimethyl phosphoric acid and 4.76 for acetic acid) reduces the acidity of these solutions so much that glucose production cannot be observed. A different study came to a similar conclusion when investigating hydrolysis of the glycosidic bond in cellobiose in [C2C1im][MeCO2] with added acid.153
When acidic protons are present in a mixture of proton acceptors the weaker acid always forms. This must be born in mind when dealing with the ionic liquids whose anions are comparatively good proton acceptors. This gives rise to ‘solvent levelling’ of the acidity, with the ionic liquid anion playing the role of the solvent's basic functional group. This is found for all solutions of strong acids. The adverse effect of high anion basicity on the proton activity and thus on the rates of acid-catalysed reactions has been observed elsewhere.156 Imidazole impurities, e.g., 1-methylimidazole stemming from the synthesis, in chloride ionic liquids have also been shown to inhibit in situ polysaccharide hydrolysis, which is again due to a relatively high pKa of alkylimidazole of around 7.2.153b
Although the simultaneous dissolution and hydrolysis of lignocellulosic biomass seems attractive, more research is needed. The cost-effective separation of monomeric sugars from ionic liquids needs to be investigated. From a processing point of view, the separation of sugars from the ionic liquid and the recycling of the ionic liquid are issues that have the potential to be process limiting problems.
The optimisation of sugar yields in balance with the formation of furfural, HMF, levulinic acid, formic acid and insoluble polymeric humins is an important challenge. In scenarios where maximum sugar yield is desired, the suppression of side reactions giving rise to other products is paramount. However, the carbohydrate dehydration products have the potential to raise revenue, as they could be isolated from the non-volatile ionic liquid by distillation – together with other volatile compounds. The production of furfural and HMF as main products could also be desirable. A range of investigations has been conducted into the selective dehydration of glucose (and fructose as an intermediate of cellulose to HMF conversion) in ionic liquids.157 The selective production of HMF in an aqueous environment is difficult, because it is a reactive intermediate on the way to more stable levulinic and formic acid. Ionic liquids could provide an anhydrous environment for this reaction, resulting in higher HMF yields, as the formation of the acids requires water. Recently, very good conversion of fructose to HMF in 1-methylimidazolium chloride, [C1Him]Cl, has been reported.158 The high yields are ascribed to the high solubility of fructose in this ionic liquid and the water-free environment. The reported HMF yield was high, but not quantitative, probably due to the release of water during formation of the HMF, which is then consumed to form levulinic and formic acid.
The optimal raw material for HMF synthesis would be glucose, which can be obtained from abundant lignocellulosic biomass. To address this, a chromium-catalysed, high-yielding transformation of glucose into HMF in ionic liquids has been reported.159 The CrCl2 catalyst showed superior selectivity compared to various other metal and protic acid catalysts. It is proposed that the CrCl2 facilitates the isomerisation of aldoses into ketoses, while the fairly anhydrous conditions in the ionic liquid stabilise the HMF. In a second study, HMF could be generated directly from cellulose or even from lignocellulosic biomass, using catalytic quantities of CrCl2 in [C2C1im]Cl to achieve yields of up to 54%.160
Representative examples of glucose and xylose yields after enzymatic saccharification of ionic-liquid treated and washed biomass are listed in Table 7. In general, treatments with ionic liquids that are able to remove lignin and hemicellulose also increase the accessibility of the cellulose fraction to hydrolytic enzymes. Both grasses and hardwoods have been pretreated effectively this way. Data for softwood are scarcer; it seems that it can be pretreated but may require harsher conditions.
Saccharification yields after [C4C1im][MeCO2] pretreatment for various lengths of time have been correlated with the crystallinity index of the cellulose in the pulp and the degree of delignification.109,124 The lower the crystallinity index and the lower the lignin content, the higher was the glucose yield. Enhanced absorption of cellulases onto regenerated cellulose was observed, which indicates that the surface area available to the enzymes is increased.77 The increased accessible surface area may explain why cellulose pulp re-precipitated from cellulose-dissolving ionic liquids can be hydrolysed faster by cellulases than crystalline cellulose.77,162 Doherty et al. also found a positive correlation between the saccharification yield and the acetate content in [C4C1im][MeSO4]/[MeCO2] mixtures.124 The ionic liquid [C4C1im][MeSO4] alone did not affect the crystallinity. Addition of water to [C4C1im][MeCO2] or [C2C1im][MeCO2] also had a negative effect on the glucose yield.
| Biomass | Ionic liquid (additives in wt%) | Pretreatment conditions | Glu yield | Xyl yield | Reference |
|---|---|---|---|---|---|
| Maple | [C4C1im][MeCO2] | 90 °C, 24 h | 74% | 64% | 124 |
| [C4C1im][MeCO2] (10% H2O) | 59% | 52% | 124 | ||
| [C2C1im][MeCO2] | 70% | 64% | 124 | ||
| [C4C1im][MeSO4] | 1% | 5% | 124 | ||
| [C4C1im][MeSO4] (10% H2O) | ca. 7% | ca. 7% | 124 | ||
| Eucalyptus | [CC2C1im]Cl |
120 °C, 5 h | 15% | n.d. | 129b |
| Miscanthus | [C2C1im][MeCO2] (20% H2O) | 120 °C, 22 h | 73% | 38% | 55 |
| [C4C1im][HSO4] (20% H2O) | 91% | 21% | 55 | ||
| [C4C1im][MeSO4] (20% H2O) | 89% | 21% | 55 | ||
| [C4C1im][MeSO3] (20% H2O) | 98% | 24% | 55 | ||
| [C4Him][HSO4] (1 mol% H2SO4/20% H2O) | 75% | 3% | 55 | ||
| Willow | [C4C1im][HSO4] (20% H2O) | 81% | 16% | 55 | |
| Bagasse | [C4C1im][MeSO4] (7%H2SO4) | 125, 120 min | 77% | n.d. | 130 |
While there is a some variability in the saccharification yields after [CnC1im][MeCO2] treatment, depending on the study, results for the use of [MeSO4]− based ionic liquids are in stark contrast. Brandt et al. reported glucose yields close to 90% for [C4C1im][MeSO4]/water mixtures55 and Diedericks et al. found 77% glucose release using [C4C1im][MeSO4]/7 wt% H2SO4,130 while Doherty et al. found very low saccharification yields, close to those obtained from untreated maple wood for neat ionic liquid as well as with 10% water added.124 An explanation for this could be that the [MeSO4]− anion requires (partial) hydrolysis to hydrogen sulfate before it is active, with the water either added separately or introduced with the ionic liquid or biomass. The hydrolysis is initiated by adding acid or a sufficiently high treatment temperature, inducing autohydrolysis. Sufficient water must, of course, be present for this hydrolysis reaction to occur.
A few studies have compared ionic liquid deconstruction with other treatment methods. Li et al. compared deconstruction of corn stover with [C2C1im][MeCO2] with ammonia fibre expansion (AFEX) treatment at selected conditions and found higher final glucose yield after the ionic liquid treatment, but lower hemicellulose recovery from the solids due to substantial solubilisation of the hemicellulose by the ionic liquid. Li et al. compared dilute sulfuric acid treatment and the [C2C1im][MeCO2] Dissolution Processing of switchgrass.138 The treatments were conducted at the same temperature (160 °C) but the treatment time was different (20 min for acid treatment and 3 h for treatment with ionic liquid). The saccharification of ionic liquid treated switchgrass was substantially faster than of the acid-treated biomass. This was attributed to the reduced ordering of regenerated cellulose and the more extensive lignin removal by ionic liquid treatment. However, at such a high temperature, it is likely that some of the ionic liquid degraded during the treatment. Sathitsuksanoh et al. compared the effect of concentrated phosphoric acid (COSLIF, 85%) and [C4C1im]Cl on the saccharification yield of both pure cellulose and corn stover. The COSLIF process was run at only 50 °C and was shown to result in higher saccharification yields than the ionic liquid pretreatment.163
Ionic liquids can be inhibitory to the saccharification enzymes. Spiess et al. thoroughly studied cellulase activity in the presence of a range of cellulose-dissolving ionic liquids.162 They showed destabilising effects of small amounts of regularly used ionic liquids such as [C2C1im][MeCO2] or [C4C1im]Cl. For example, adding 10% (by volume) ionic liquid to water decreased cellulase activity by 70–85%. The ionic liquids studied all contained dialkylimidazolium cations ([CnC1im]; n = 1, 2 or 4, also CC2) and little cation effect was discovered. The anion effects on cellulase activity were much more noticeable, with [C1C1im][Me2PO4] showing the highest activity (30% relative to buffer), followed by [CC2C1im]Cl (22%), [C4C1im]Cl (18%) and [C2C1im][MeCO2] (15%). In all cases, the activity decreased rapidly with increasing ionic liquid concentration. The authors were able to separate the various effects and determine that viscosity was having the greatest negative impact, followed by ionic strength and finally pH. However, there is some residual chemical effect still attributable to the ionic liquid itself, as even when a buffer solution was corrected for viscosity, ionic strength and pH the activity was more than twice as high as for a comparable ionic liquid concentration. Interestingly, while the cellulose activity was decreased in the presence of ionic liquid, the stability (determined by incubation with ionic liquid) was not affected (Fig. 24), suggesting that the ionic liquids are inhibiting enzymatic activity rather than irreversibly denaturing the enzymes. All of these ionic liquid inhibition effects strongly suggest that careful washing of cellulose pulps to remove residual ionic liquid contaminants is a vital process consideration.
![Activity of celluclast cellulase preparation in number of ionic liquids (NaAc, sodium acetate, [MMIM][DMP] = dimethylimidazolium dimethyl phosphate, [AMIM]Cl = 1-allyl-3-methylimidazolium chloride, [BMIM]Cl = 1-butyl-3-methylimidazolium chloride, [EMIM][Ac] = 1-ethyl-3-methylimidazolium acetate). Reproduced with permission from ref. 162.](/image/article/2013/GC/c2gc36364j/c2gc36364j-f24.gif) | ||
| Fig. 24 Activity of celluclast cellulase preparation in number of ionic liquids (NaAc, sodium acetate, [MMIM][DMP] = dimethylimidazolium dimethyl phosphate, [AMIM]Cl = 1-allyl-3-methylimidazolium chloride, [BMIM]Cl = 1-butyl-3-methylimidazolium chloride, [EMIM][Ac] = 1-ethyl-3-methylimidazolium acetate). Reproduced with permission from ref. 162. | ||
Nevertheless, one-pot processes including a pretreatment step and, upon substantial dilution, an enzymatic saccharification have been carried out. They have used pure cellulose rather than real lignocellulosic substrates.164 Although saccharification has been achieved, the high energy demand for evaporating water which makes up 90% of the mixture upon dilution seems prohibitive. Perhaps alternative reconcentration methods can be devised, which would be beneficial for any ionic liquid deconstruction process that requires dilution with water.
| Ionic liquid (water content) | Biomass | Temp. | Time | Precipitation solvent | Quantity of precipitate | Reference |
|---|---|---|---|---|---|---|
| a [MeSO4]− anions partially hydrolysed to [HSO4]−. | ||||||
| [C2C1im][MeCO2] | Pine | 110 °C | 16 h | Water | 10% | 112 |
| Spruce | 115 °C | 72 h | Water | 36% | 116 | |
| Beech | 115 °C | 72 h | Water | 43% | 116 | |
| [C2C1im][MeCO2] (20% water) | Miscanthus | 120 °C | 22 h | Water | 18% | 55 |
| [C4C1im][HSO4] (20% water) | 120 °C | 22 h | Water | 47% | 55 | |
| [C4C1im][MeSO4]a (20% water) | 120 °C | 26 h | Water | 43% | 55 | |
| [C4C1im][MeSO3] (20% water) | 120 °C | 22 h | Water | 30% | 55 | |
| [C4Him][HSO4] (1 mol% H2SO4 20% water) | 120 °C | 24 h | Water | 99% | 55 | |
| [C2C1im][ABS] | Sugarcane bagasse | 190 °C | 30 min | Water, pH 2 | 67% | 52 |
| 190 °C | 60 min | Water, pH 2 | 97% | 52 | ||
| 190 °C | 90 min | Water, pH 2 | 118% | 52 | ||
| [C4C1im][Ace] | Eucalyptus | 100 °C | 2 h | Acetone | 38% | 53 |
| Pine | 100 °C | 2 h | Acetone | 38% | 53 | |
| 140 °C | 4 h | Acetone | 88% | 53 | ||
In the case of the ionic liquid Dissolution Process, a two-stage process appears most promising (Fig. 25). Firstly, cellulose is precipitated while the lignin remains in solution. The lignin is precipitated in a second step. This two-stage process is popular because several studies observed that better separation can be achieved when the cellulose is precipitated with an organic solvent mixture or an organic solvent–water mixture. Protic solvents or mixtures of solvents, that contain at least one protic component, are generally preferred. An example is the study by Sun et al.112,113 After treatment of pine with [C2C1im][MeCO2] and precipitation with acetone–water, the acetone was evaporated. This resulted in a second precipitation that allowed recovery of 31% of the lignin (although the yield would have been lower if the liquor had not been acidified during precipitation). The acidification is performed to reduce the basicity of the acetate containing ionic liquids, thus lowering lignin solubility. However, the acid will accumulate in the ionic liquid when it is recycled. While there is no chemical obstacle to neutralising the added acid during the recycling process step, this is highly questionable from an economic and environmental viewpoint. An alternative is extraction of lignin from the ionic liquid into a separate organic phase without previous acidification.165
 | ||
| Fig. 25 Strategies to separate lignin and cellulose and ionic liquids from homogenous lignocellulose ionic liquid solution. (A) The ionic liquid is diluted with a water–organic solvent mixture (e.g. water–acetone),113 the more volatile organic solvent is then evaporated which leads to precipitation of lignin. (B) The cellulose is precipitated upon dilution with a protic organic solvent or organic solvent mixture. The solvent is removed and a lignin-precipitating solvent added. The lignin yield is occasionally increased by adding acid to the liquor. | ||
The Ionosolv approach uses ionic liquids with neutral or acidic anions to dissolve the lignin while the cellulose remains undissolved throughout the treatment. Tan et al. have demonstrated lignin recovery from sugar cane bagasse after treatment with 1-ethyl-3-methylimidazolium alkylbenzenesulfonate [C2C1im][ABS] followed by a precipitation step, which also involved acidification of the liquor during dilution with water.52 This study reported precipitate recovery between 67–118% after extraction with [C2C1im][ABS], depending on the process time and the temperature. Lignin recovery has also been achieved with ionic liquids combining the [C2C1im]+or [C4C1im]+ cation with the food additive derived acesulfamate anion.53 Lignin was precipitated upon dilution with water (without addition of acid) from [HSO4]− containing ionic liquid liquors by Brandt et al.55 They reported a precipitate yield of over 60 wt% of the lignin content of untreated Miscanthus after pretreatment with [C4C1im][HSO4] water mixtures. Acidification of the liquor was not necessary to obtain reasonable yields, but they also found that the more acidic ionic liquid liquor [C4Him][HSO4] + 1 mol% sulfuric acid generated yields close to 100%, or above, relative to the lignin content of the untreated biomass. Yields exceeding the original lignin content suggest that the precipitates contain other substances, for instance residual ionic liquid (possibly incorporated into the lignin) or insoluble carbohydrate degradation products (e.g. humins). The precipitate could also contain oligomeric carbohydrates that are soluble in concentrated ionic liquid but not in dilute aqueous solutions, particularly when the precipitation solvent has a low solubility for carbohydrates. IR spectra are often used to identify the precipitate as lignin. However, minor differences between spectra of reference and extracted lignins are frequently observed, which can be due to differing subunit content, extraction method and conditions, but also due to above mentioned contaminants. The presence of humins in their precipitate was confirmed by Tan et al. using solid state NMR.52 Wei et al. found that the precipitate obtained after Dissolution treatment of legume straw with [C4C1im]Cl water mixtures at 150 °C for 2 h contained 35% cellulose, 64% lignin and 1% ash.140 More details about the chemical alterations of lignin by the ionic liquids can be found in Section 1.5.2.
Interestingly, ionic liquids designed for selective lignin dissolution seem to be more tolerant to moisture52 or even require certain levels of water to function optimally.55 This is probably due to the fact that lignin solubilisation does not require as strictly anhydrous conditions as does cellulose, while the hydrolysis of ether bonds in lignin and in lignin–carbohydrate complexes is favoured in the presence of water, so aiding separation.
1.6 Challenges for an ionic liquids based biorefinery
While they have shown promise, ionic liquid based lignocellulose deconstruction processes are currently not at a stage where any are commercialised. In this section, we first describe how possible ionic liquid deconstruction/fractionation processes might look and also note the main obstacles that need to be overcome for these to be realised. There has been a great deal of research into biorefinery applications of ionic liquids. Many slightly differing approaches are under investigation, e.g. pre-extraction of hemicellulose with ionic liquids,84,166 which we won't deal with in detail here. Similar to other deconstruction processes, ionic liquids will only be applied commercially if they provide the most economically and environmentally sustainable processes.We have identified two distinct types of processes employing ionic liquids for deconstruction and named these the Dissolution Process and the Ionosolv Process. They mainly differ in the treatment of the cellulose fraction. During the Dissolution Process, the cellulose is dissolved together with the rest of the biomass or at least its ordering is substantially reduced. In the Ionosolv Process, the structure of cellulose is not changed. The next three sections highlight progress made on important process related aspects of lignocellulose deconstruction with ionic liquids, before the two processes are discussed in more detail.
To do this, the stability of compounds is often characterised by thermogravimetric analysis (TGA), a technique that correlates sample weight loss with temperature. During a typical TGA scan, the temperature is increased at a fixed rate. Under such circumstances, the decomposition/weight loss is not instantaneous; therefore the data are interpreted by fitting two tangents to the weight loss curve. This step-tangent method yields the onset temperature of decomposition, Tonset.168Tonset is suitable for comparing the relative stability of different ionic liquids. It has been found that the type of both cation and anion influences the temperature of decomposition,169 but there is a strong effect of the anion. It has been shown that anions that are very nucleophilic, such as chloride and acetate, can lower Tonset to around 200 °C, while dissolved cellulose appears to have no effect on the IL decomposition temperature, even up to 12% cellulose.74
If the stability of an ionic liquid under process conditions is of interest, Tonset is only an indicator, as significant mass loss occurs below Tonset (Fig. 26).170 This partial degradation affects the recycling efficiency as well as the properties of the ionic liquid as degradation products accumulate. It has been shown that the economic viability of ionic liquid deconstruction is very sensitive to the efficiency of ionic liquid recycling and therefore decomposition must be avoided.171
![Long-term stability of ionic liquids at 120 °C. Weight loss after 60 h from [C4C1im][MeCO2] was 10%, while [C4C1pyrr][MeCO2] containing an ammonium cation decomposed entirely.174](/image/article/2013/GC/c2gc36364j/c2gc36364j-f26.gif) | ||
| Fig. 26 Long-term stability of ionic liquids at 120 °C. Weight loss after 60 h from [C4C1im][MeCO2] was 10%, while [C4C1pyrr][MeCO2] containing an ammonium cation decomposed entirely.174 | ||
In order to find the temperature at which the ionic liquid decomposition is negligible, Baranyi et al. suggested to use the temperature at which the ionic liquid decomposes by 1% within 10 h (T0.01/10 h) as a suitable measure.172 Wooster et al. also suggested a maximum stable operating temperature for ionic liquid based process to be 10 °C lower than T0.01/10 h.173 Due to the lack of data in the literature, we measured T0.01/10 h for [C4C1im][MeCO2] and determined it as 102 °C.174 Since the anion and the cation core are very similar for [C4C1im][MeCO2] and [C2C1im][MeCO2], the T0.01/10 h for [C2C1im][MeCO2] is anticipated to be very similar. This would result in a maximum process temperature of ca. 90 °C for both of these very commonly used ionic liquids. It is important to note that the majority of biomass deconstruction experiments using these ionic liquids have been performed at temperatures greatly in excess of this limit. Optimisation of the carbohydrate yield after deconstructing wheat straw with a [C2C1im][MeCO2] process using the Design of Experiment (DOE) approach resulted in an optimal process temperature of 158 °C.175 This was at 50.5 wt% water content, where the effect of water on the ionic liquid is likely to be considerable. The effect of water on the stability of ionic liquids used for biomass processing should be investigated. [C2C1im]Cl and [C4C1im]Cl are long-term stable at 120 °C and show signs of decomposition at 140 °C.176 The same study showed methanesulfonate based ionic liquids [C2C1im][MeSO3] and [C4C1im][MeSO3] to be stable at 140 °C and to only slowly degrade at 200 °C.
Data from deconstruction experiments support this: Rogers and co-workers have observed 15% weight loss within 10 min when treating bagasse and pine with [C2C1im][MeCO2] at 185 °C.119 The decomposition is much slower at 60 °C below Tonset, but still detectable (Fig. 26). Decomposition of [C2C1im][MeCO2] and [C2C1im]Cl at 100–130 °C was almost complete within 4 months.177 When designing the ionic liquid, cation effects are also important. The ionic liquid [C4C1pyrr][MeCO2], based on an aliphatic (cyclic) ammonium cation, which usually show lower resistance to decomposition than imidazolium cations, decomposed completely within just 30 h at 120 °C.
Wang et al. reused [CC2C1im]Cl once after a dissolution treatment of 5 wt% pine and did not detect any reduction in performance.121 Although this is a start, one repeat is not sufficient to assess performance on an industrial scale where many cycles will be necessary to make an ionic liquid deconstruction process viable. Li et al. reused [CC2C1im]Cl three times for deconstructing 8 wt% Eucalyptus using the Dissolution Process and found that the saccharification yields decreased progressively.129b In addition, higher pulp recoveries were observed when the ionic liquid was reused, which could be due to less extensive lignin removal. Increasing quantities of carboxylic acid and hydroxyl groups (both phenolic and aliphatic) in the ionic liquid were also observed, indicating that both carbohydrates and lignin fragments accumulate in the liquor.
Another study reused [C4C1im]Cl 7 times for fractionation of legume straw with ionic liquid water mixtures.140 The recovery procedure was simple removal of water. An increase of the amount of recovered pulp was observed after the 4th cycle; the lignin content in the recovered solid remained constant over the course of the experiment.
Brandt et al. detected varying amounts of monomeric hemicellulose and furfurals in liquors of [C4C1im][MeSO4], [C4C1im][HSO4] and [C4C1im][MeSO3] after treatment of Miscanthus with fresh ionic liquid, the quantities detected depending on the ionic liquid acidity and the treatment length.55 Wu et al. reused [C2C1im][MeCO2] 10 times at 4.8 wt% corn stover loading at 125 °C. The saccharification yield was unaffected, as was the decrystallisation of the cellulose, but less lignin was extracted in the last cycle compared to the first one (44% reduced to 20%). Rogers and co-workers reused [C2C1im][MeCO2] once after treatment of 4.8 wt% bagasse at 185 °C for 10 min;119 the delignification efficiency dropped from 63% to 38%. Lan et al. fractionated bagasse into cellulose, hemicelluloses and lignin four times with the same batch of [C4C1im]Cl.178 The delignification efficiency dropped slightly, the saccharification yield was not investigated. These studies show results for the changing performance of the ionic liquids in the process step in which they were used, not the condition of the ionic liquid solution that was being recycled. These solutions may contain ionic liquid decomposition products, biomass decomposition products and most likely both. These need to be known before any improvements in performance can be achieved.
The preliminary observations that fractionation and, in some cases, the saccharification are somewhat adversely affected by repeated use of the ionic liquid suggests that a clean-up step is likely to be necessary. Separation of [C2C1im][MeCO2] from non-volatile sugars with ion exclusion chromatography has been suggested.179 Francisco et al. found that adsorption of glucose onto zeolites from ionic liquid water mixtures and subsequent desorption into water could be used for removing sugars from the ionic liquid liquor.180 Shill et al. have attempted to lower the energy use for drying the ionic liquid after dissolution of Miscanthus by re-concentrating diluted ionic liquids with the inorganic salt potassium phosphate (K3PO4), but saw a negative effect on saccharification yields after reuse.181 Brennan et al. have devised an extraction method for glucose and xylose recovery from [C2C1im][MeCO2] using boronates such as naphthalene-2-boronate. The sugars are extracted from the basic ionic liquid solution into an organic phase by forming a complex with the boronate and are stripped from the organic solvent into dilute acidic aqueous solution.182
The particle size reduction process is an important target for optimisation, especially in the case of woody biomass. A recent study shows that ionic liquids could help to reduce the energy input for grinding wood-derived lignocellulose. The non-baseline power consumption for grinding pine chips was significantly reduced (up to 75%), while a finer powder was obtained, by adding the ionic liquid before the grinding.187 The savings were shown to be due to the lubricating properties of ionic liquids, rather than partial modification of the lignocellulose matrix. This is particularly interesting for non-dissolving ionic liquid deconstruction efforts. As an alternative, Viell and Marquardt reported the breaking up of air-dried beech wood chips into individual cell walls under dissolving conditions;116 this was accomplished by vigorous stirring for 2 h. Although grinding costs are saved, the energy required for drying harvested biomass to 7–10% moisture, and thoroughly drying the ionic liquid, must be accounted for.
1.7 Biomass processing with ionic liquids: the Ionosolv and the Dissolution Processes
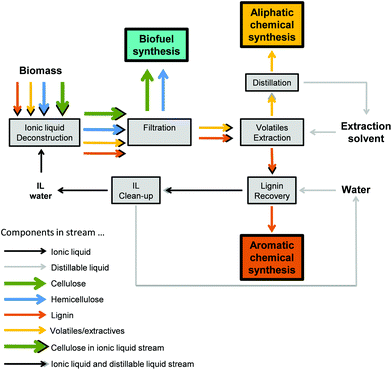 | ||
| Fig. 27 Potential process flow scheme for the Ionosolv Process. | ||
The small number of papers in the literature that use the Ionosolv Process approach indicates that it is unlikely to have yet been optimised. In spite of this, almost quantitative removal of lignin has been achieved.55 This is likely due to the presence of mid-range nucleophilic, neutral or acidic anions in the ionic liquids employed by the Ionosolv Process; these seem to act as catalytic delignifiers and possibly reagents.
For the Ionosolv, as well as the Dissolution Process, it is important that the cellulose fraction be as free of ionic liquid as possible to avoid unwanted deactivation of the saccharification enzymes162 or toxicity to the fermenting microorganisms.188 This restraint places great importance on the efficiency of the cellulose washing step. This can be approached in two ways. The first is the engineering design of the wash process itself – solvent(s) used, temperature, time, mixing etc.; the second approach is to design the ionic liquid to be more easily removed from the cellulose during washing. Since the fractionation ability of an ionic liquid is dominated by the nature of the anion, it is likely that this will determine the anion selection. Therefore, it is by cation design that an ionic liquid could be optimised for washing.
Regardless of how successfully the ionic liquid is separated from the cellulose, it is likely that some of the ionic liquid will pass into subsequent process operations. Hence, another approach is to design the ionic liquid to be less hostile to the process enzymes77 and fermenting microorganisms.188 Of course, it is also possible to develop these enzymes189,190 and microorganisms to be more resilient to the presence of small amounts of ionic liquid.
The filtrate will contain lignin and probably some hemicelluloses. It is also likely that this is where the extractable organic small molecules will be found (oils, acetic acid, ferulic acid, vanillin, other small lignin fragments, furfurals, levulinic acid etc.). These compounds can be removed with this stream. This is likely to be achieved through extraction using either a non-polar organic solvent,165 supercritical fluid extraction191 or distillation. This organic extract stream could be further processed to aliphatic chemical products and organic fuels, such as 5-hydroxymethylfuran.159
Water can be added to the ionic liquid phase to precipitate the dissolved lignin for further processing or incineration.52,53,55 This is made possible by the relatively acidic nature of the ionic liquid, which keeps the dissolved lignin in fully protonated form (phenol groups rather than phenolates). Lignin precipitation requires more water than is generally used to precipitate cellulose from the Dissolution Process solutions. For the Ionosolv Process to be viable, it will be necessary to reconcentrate the ionic liquid for reuse. Although the recovered ionic liquid does not need to be fully dried (as ca. 20% water is required for the pretreatment step), this is still likely to be a highly energy intensive step and has the potential to be a process-limiting problem.
An alternative route is to regard the lignin solution in the ionic liquid as the starting point for further chemical conversion of lignin to high-value aromatic products, such as vanillin or other aromatic derivatives.143 Although there has very recently been an increased interest in the conversion of lignin into chemicals,9 to date there are no available commercial processes for this upgrading. This could lead to, for example, low molecular weight compounds that could be separated without the addition of water. This concept does not remove the need for ionic liquid remediation for recycling. However, it does have the potential to avoid an energy intensive drying step. Of course, if this method is to be used, the reactivity of ionic liquids towards the major lignocellulose components must be investigated in more detail and the inertness of the ionic liquid during catalysis needs to be ensured. For example, the incorporation of ionic liquid components into lignin or cellulose pulp could hamper the economic viability of ionic liquid deconstruction by reducing the maximum recovery as well as the value of the recovered pulp and the lignin.
Ionic liquids are the most expensive solvents that are currently under investigation for lignocellulose deconstruction. This is an oft-stated criticism of any proposal to apply ionic liquids as process solvents. In spite of this complaint, for other applications, ionic liquids have been applied as process solvents on a large scale.51 Notwithstanding this, reduction in the price of ionic liquids used for deconstruction of biomass is essential if this is to become commercial reality. Careful analysis of the ionic liquids that are actually used in the chemicals processing industry on a large scale shows that they are mostly based on protic cations, such as [C1Him]+, usually combined with a mineral acid derived anion, such as Cl−. This makes these ionic liquids much cheaper than those with fully alkylated cations (e.g. [C2C1im]+ in [C2C1im][MeCO2]) used in most academic laboratories. The protic ionic liquids are also distillable and hence much more easily recycled than the ionic liquids with fully alkylated cations. Unfortunately, some ionic liquids with protic cations may not be applicable in the Dissolution Process due to their increased hydrogen-bond acidity, although others have been shown to be good cellulose solvents.78b
During recycling, the ionic liquid is likely to experience a build-up of carbohydrate monomers and oligomers, furfurals, organic acids, lignin fragments, inorganic salts and other solutes that are released during the pretreatment. The use of distillable acid–base ionic liquids may be a solution to this problem, although distillation of the ionic liquid after each cycle is likely to be uneconomical. Therefore, other separation procedures, such as solvent extraction, will probably be used to minimize solute accumulation and to ensure the ionic liquid remains sufficiently clean for repeated recycling, with distillation only being used when this cleaning fails. It is possible that some form of base wash could be used to liberate the more expensive amine-based component in manner similar to the regeneration of 1-methylimidazole in BASF's BASIL™ process.192 However, until the chemistries for lignin upgrading are known, this remains mere speculation.
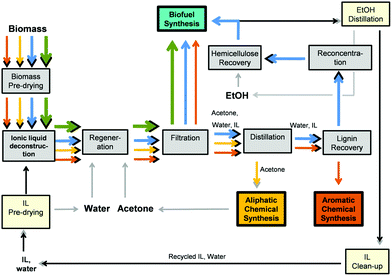 | ||
| Fig. 28 Potential process flow diagram for the Dissolution Process based on ref. 194. | ||
After pretreatment, the ionic liquid-biomass solution is diluted with an antisolvent in order to precipitate a cellulose-enriched pulp, which is filtered and washed to remove residual ionic liquid (and the solubilised part of the biomass). The pulp, containing amorphous cellulose, is slurried with water for depolymerisation by cellulose enzymes and subsequent fermentation for conversion to biofuels.
As has been noted above, the solubility of cellulose in the ionic liquids used in the Dissolution Process is sensitive to the presence of water. Cellulose has extremely low water solubility, and the water content of acetate and chloride based ionic liquids has been observed to have the single greatest impact on the cellulose solubility. The ionic liquids used for biomass dissolution are highly hydroscopic (Fig. 21). Raw biomass is wet, typically containing 50% water and even thoroughly air-dried biomass typically contains around 5–10 wt% residual moisture. Unless drying is a prominent feature of the process, the build-up of water in the ionic liquid will lead to levels that will prevent cellulose dissolution and hence stop the process. The energy costs of this drying have to be assessed and have the potential to be a process-limiting problem. Since good cellulose solubility (Fig. 18) has been related to the ionic liquid anion's β value in the same way as its hydrophilicity (Fig. 21), we cannot see a way to address this problem through ionic liquid design. It may be possible to partially circumvent this problem by basing the pretreatment on the swelling of the biomass without dissolving the cellulose.109,123 Promising results have been obtained by allowing for a higher biomass loading, but these need to be further verified.123a
An advantage of the Dissolution Process is the recovery of the majority of the hemicelluloses with the pulp, although along with some lignin, as the delignification in this process is not complete.109,136 The lignin remaining after saccharification could be further processed for chemicals production or burned, depending on the purity.
As with the Ionosolv Process, it is important that the ionic liquid is separated from the precipitated cellulose to prevent enzyme deactivation or toxicity to the fermenting microorganisms in subsequent process steps. This problem will need to be tackled in the same ways as described above (engineering design, ionic liquid design and enzyme development) for the Ionosolv Process. There is also a particular problem regarding the use of acetate ionic liquids. It is essential that significant concentrations of ionic liquid ions in the fermentation step are avoided. Acetate is a proven metabolic inhibitor of most yeast systems. As little as 0.25% residual [C2C1im][MeCO2] in the fermentation broth will completely inhibit yeast growth and biofuel production.188 The ionic liquid [C2C1im]Cl also affects growth of the yeast, but to a lesser extent.188 The ionic liquid cation was identified as the main cause of cell toxicity, as control experiments with inorganic acetate and chloride salts did not lead to the same level of inhibition. The chloride ions also inhibit enzymatic activity in concentrations of less than 1%.195 The study found that water was more effective in removing residual ionic liquid than either pure ethanol or acetone–ethanol mixtures. The design of less inhibitory ionic liquids could be beneficial, as extensive washing will worsen the process economics due to a large, possibly environmentally harmful waste water stream.
The filtrate (precipitation solvent + ionic liquid) contains dissolved lignin, some hemicelluloses and the organic extractives. No reported work has considered the direct use of this solution. The solubilised sugars and other organic compounds released during pretreatment are also valuable and should be retrieved. This is another reason to improve the separation of ionic liquid from dissolved components.
2. Conclusion – why use ionic liquids?
Ionic liquids are being investigated as lignocellulose deconstruction solvents for a number of reasons. As already mentioned, a major and widely highlighted advantage of the ionic liquid Dissolution Process compared to other pretreatment options is its ability to decrystallise the cellulose portion of lignocellulosic biomass and simultaneously disrupt the lignin and hemicellulose network. While the latter is a prerequisite for enzymatic saccharification to occur to significant extent, the changed crystallinity of the cellulose also has a measurable positive impact on the speed of saccharification.76a The possibility of removing lignin with the ionic liquid and recovering a separate, possibly more valuable lignin fraction is also an attractive feature. A less frequently mentioned advantage of ionic liquids is their low volatility, making it possible to pretreat biomass at atmospheric pressure even at temperatures surpassing the boiling point of water, as well as the ability to handle a non-odorous and relatively safe liquor compared to volatile solvents or sulfide containing aqueous solutions.However, deconstruction with ionic liquid will only be viable if its advantages outweigh the drawbacks of ionic liquids, most prominently the cost of ionic liquids relative to the value of the substrate processed by them. Modelling of the process is required to estimate energy requirements and cost, so more informed comparison with other pretreatment operations will be possible. The first techno-economical analysis modelling of ionic liquid deconstruction has determined that among the investigated variables the order of importance/sensitivity is ionic liquid price > biomass loading > recycling rate.171 In addition, the ability to sell the lignin and/or its products will heavily impact on the process costs.
The following list, whilst not exhaustive, contains aspects of ionic liquid pretreatment that should receive attention in the future. Further investigations will help to determine whether the identified challenges of ionic liquid processing of lignocellulose are inherent disadvantages or can be improved upon by optimisation or technical innovation. These future research targets are:
• Low cost ionic liquids and co-solvents (ideally ≤$2.50 kg−1)
• Increased biomass loading
• High yield and high selectivity fractionation
• Short pretreatment time
• Applicability to a wide range of feedstocks (biomass type, geographical location and time of year)
• Increased moisture tolerance; reduced energy input during drying
• Reduced ionic liquid losses; optimised recycling
• End of use ionic liquid recovery
• Reduced impact of residual ionic liquid on downstream processing (toxicity on fermenting organisms and inhibition of enzymes)
• ‘Greenness’ of ionic liquid and co-solvent (health and environmental impact).
References
- (a) A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484 CrossRef CAS; (b) J. H. Clark, F. E. I. Deswarte and T. J. Farmer, Biofuels Bioprod. Biorefin., 2009, 3, 72 CrossRef CAS; (c) J. Pickett, Sustainable Biofuels: Prospects and Challenge, The Royal Society, 2008 Search PubMed.
- Food and Agricultural Organisation of the United Nations, The State of Food and Agriculture 2008, Rome, 2008. Available at http://www.fao.org/ Search PubMed.
- T. Searchinger, R. Heimlich, R. A. Houghton, F. Dong, A. Elobeid, J. Fabiosa, S. Tokgoz, D. Hayes and T.-H. Yu, Science, 2008, 319, 1238 CrossRef CAS.
- R. D. Perlack, L. L. Wright, A. F. Turhollow, R. L. Graham, B. J. Stokes and D. C. Erbach, Biomass as a Feedstock for a Bioenergy and Bioproducts Industry: the Technical Feasibility of a Billion-ton Annual Supply, U.S. Department of Energy and U.S. Department of Agriculture, 2005 Search PubMed.
- M. R. Schmer, K. P. Vogel, R. B. Mitchell and R. K. Perrin, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 464 CrossRef CAS.
- U.S. DOE, Breaking the Biological Barriers to Cellulosic Ethanol: A Joint Research Agenda, DOE/SC-0095, 2006.
- The NILE project 2005–2009, Advances in lignocellulosic ethanol, project supported by the 6th RTD Framework Programme of the European Commission, brochure published January 2008.
- N. Mosier, C. Wyman, B. Dale, R. Elander, Y. Y. Lee, M. Holtzapple and M. Ladisch, Bioresour. Technol., 2005, 96, 673 CrossRef CAS.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS.
- (a) A. Stark, Energy Environ. Sci., 2011, 4, 19 RSC; (b) H. Tadesse and R. Luque, Energy Environ. Sci., 2011, 4, 3913 RSC; (c) M. FitzPatrick, P. Champagne, M. F. Cunningham and R. A. Whitney, Bioresour. Technol., 2010, 101, 8915 CrossRef CAS; (d) C.-Z. Liu, F. Wang, A. R. Stiles and C. Guo, Appl. Energy, 2012, 92, 406 CrossRef CAS; (e) T. Vancov, A.-S. Alston, T. Brown and S. McIntosh, Renew. Energy, 2012, 45, 1 CrossRef CAS.
- U.S Department of Energy Genome Programs image gallery, http://genomics.energy.gov (accessed 22 February 2011).
- A. C. O'Sullivan, Cellulose, 1997, 4, 173 CrossRef CAS.
- X. Qian, S.-Y. Ding, M. R. Nimlos, D. K. Johnson and M. E. Himmel, Macromolecules, 2005, 38, 10580 CrossRef CAS.
- Y. Nishiyama, P. Langan and H. Chanzy, J. Am. Chem. Soc., 2002, 124, 9074 CrossRef CAS.
- E. Dinand, M. Vignon, H. Chanzy and L. Heux, Cellulose, 2002, 9, 7 CrossRef CAS.
- F. J. Kolpak and J. Blackwell, Macromolecules, 1976, 9, 273 CrossRef CAS.
- H. Jørgensen, J. Kristensen and C. Felby, Biofuels Bioprod. Biorefin., 2007, 1, 119 CrossRef.
- C. D. Armeniades and E. Baer, in Introduction to Polymer Science and Technology, ed. H. S. Kaufman and J. J. Falcetta, Wiley, New York, 1977 Search PubMed.
- T. E. Timell, Wood Sci. Technol., 1967, 1, 45 CrossRef CAS.
- S. Willför, K. Sundberg, M. Tenkanen and B. Holmbom, Carbohydr. Polym., 2008, 72, 197 CrossRef.
- C. M. Hansen and A. Björkman, Holzforschung, 1998, 52, 335 CrossRef CAS.
- W. Boerjan, J. Ralph and M. Baucher, Annu. Rev. Plant Biol., 2003, 54, 519 CrossRef CAS.
- R. El Hage, N. Brosse, L. Chrusciel, C. Sanchez, P. Sannigrahi and A. Ragauskas, Polym. Degrad. Stab., 2009, 94, 1632 CrossRef CAS.
- Wikipedia, Structure of Lignin, http://en.wikipedia.org/wiki/File:LigninStructure.png (accessed 20 July 2011).
- X. J. Pan, D. Xie, N. Gilkes, D. J. Gregg and J. N. Saddler, Appl. Biochem. Biotechnol., 2005, 121, 1069 CrossRef.
- A. Berlin, M. Balakshin, N. Gilkes, J. Kadla, V. Maximenko, S. Kubo and J. Saddler, J. Biotechnol., 2006, 125, 198 CrossRef CAS.
- A. P. Marques, D. V. Evtuguin, S. Magina and A. Prates, J. Wood Chem. Technol., 2009, 29, 322 CrossRef CAS.
- K. E. Myton and S. C. Fry, Planta, 1994, 193, 326 CrossRef CAS.
- J. Ralph, J. H. Grabber and R. D. Hatfield, Carbohydr. Res., 1995, 275, 167 CrossRef CAS.
- U.S. Department of Energy, Breaking the Biological Barriers to Cellulosic Ethanol, A Joint Research Agend, DOE/SC-0095, 2006 Search PubMed.
- J. H. Grabber, R. D. Hatfield and J. Ralph, J. Sci. Food Agric., 1998, 77, 193 CrossRef CAS.
- M. Lawoko, G. Henriksson and G. Gellerstedt, Holzforschung, 2006, 60, 156 CAS.
- C. J. Jacobs-Young, R. A. Venditti and T. W. Joyce, Effect of Enzymatic Pretreatment on the Diffusion of Sodium Hydroxide in Wood, TAPPI, Norcross, GA, USA, 1998 Search PubMed.
- J. Zhu, W. Zhu, P. Obryan, B. Dien, S. Tian, R. Gleisner and X. Pan, Appl. Microbiol. Biotechnol., 2010, 86, 1355 CrossRef CAS.
- N. J. B. Brereton, F. E. Pitre, M. J. Ray, A. Karp and R. J. Murphy, Biotechnol. Biofuels, 2011, 4, 13 CrossRef CAS.
- A. Karp, S. J. Hanley, S. O. Trybush, W. Macalpine, M. Pei and I. Shield, J. Integr. Plant Biol., 2012, 53, 151 CrossRef.
- (a) P. Sannigrahi, A. J. Ragauskas and G. A. Tuskan, Biofuels Bioprod. Biorefin., 2010, 4, 209 CrossRef CAS; (b) M. J. Aylott, E. Casella, I. Tubby, N. R. Street, P. Smith and G. Taylor, New Phytol., 2008, 178, 358 CrossRef.
- R. Samson, S. Mani, R. Boddey, S. Sokhansanj, D. Quesada, S. Urquiaga, V. Reis and C. Ho Lem, Crit. Rev. Plant Sci., 2005, 24, 461 CrossRef.
- R.-C. Sun, Cereal Straw as a Resource for Sustainable Biomaterials and Biofuels, Elsevier, Oxford, 2010 Search PubMed.
- A. J. Ragauskas, M. Nagy, D. H. Kim, C. A. Eckert, J. P. Hallett and C. L. Liotta, Ind. Biotechnol., 2006, 2, 55 CrossRef CAS.
- J. F. Saeman, Ind. Eng. Chem., 1945, 37, 43 CrossRef CAS.
- Y. H. Zhang, J. Ind. Microbiol. Biotechnol., 2008, 35, 367 CrossRef CAS.
- (a) L. da Costa Sousa, S. P. S. Chundawat, V. Balan and B. E. Dale, Curr. Opin. Biotechnol., 2009, 20, 339 CrossRef; (b) J. Zhu, X. Pan and R. Zalesny, Appl. Microbiol. Biotechnol., 2010, 87, 847 CrossRef CAS; (c) S. P. S. Chundawat, G. T. Beckham, M. E. Himmel and B. E. Dale, Annu. Rev. Chem. Biomol. Eng., 2011, 2, 121 CrossRef CAS.
- (a) Ionic Liquids in Synthesis, ed. P. Wasserscheid and T. Welton, VCH Wiley, Weinheim, 2nd edn, 2008 Search PubMed; (b) Ionic Liquids, ed. B. Kirchner, Top. Curr. Chem., 2009, p. 290 Search PubMed.
- J. S. Wilkes and M. J. Zaworotko, J. Chem. Soc., Chem. Commun., 1992, 965 RSC.
- (a) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS; (b) T. Welton, Chem. Rev., 1999, 99, 2071 CrossRef CAS.
- (a) V. I. Parvulescu and C. Hardacre, Chem. Rev., 2007, 107, 2615 CrossRef CAS; (b) T. Welton, Coord. Chem. Rev., 2004, 248, 2459 CrossRef CAS; (c) J. Dupont, R. F. de Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS; (d) P. Wasserscheid and W. Keim, Angew. Chem., Int. Ed., 2000, 39, 3772 CrossRef CAS.
- (a) F. van Rantwijk and R. A. Sheldon, Chem. Rev., 2007, 107, 2757 CrossRef CAS; (b) U. Kragl, M. Eckstein and N. Kaftzik, Curr. Opin. Biotechnol., 2002, 13, 565 CrossRef CAS; (c) S. Muginova, A. Galimova, A. Polyakov and T. Shekhovtsova, J. Anal. Chem., 2010, 65, 331 CrossRef CAS.
- R. Hagiwara and J. S. Lee, Electrochem., 2007, 75, 23 CrossRef CAS.
- H. Zhao, Chem. Eng. Commun., 2006, 193, 1660 CrossRef CAS.
- N. V. Plechkova and K. R. Seddon, Chem. Soc. Rev., 2008, 37, 123 RSC.
- S. S. Y. Tan, D. R. MacFarlane, J. Upfal, L. A. Edye, W. O. S. Doherty, A. F. Patti, J. M. Pringle and J. L. Scott, Green Chem., 2009, 11, 339 RSC.
- A. Pinkert, D. F. Goeke, K. N. Marsh and S. Pang, Green Chem., 2011, 13, 3124 RSC.
- J. C. Nardi, C. L. Hussey, L. A. King and R. A. Carpio, US Patent, 4,115,390, 1978 Search PubMed.
- A. Brandt, M. J. Ray, T. Q. To, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2011, 13, 2489 RSC.
- M. A. Ab Rani, A. Brandt, L. Crowhurst, A. Dolan, J. P. Hallett, N. H. Hassan, P. A. Hunt, M. Lui, H. Niedermeyer, J. M. Perez-Arlandis, M. Schrems, T. Q. To, T. Welton and R. Wilding, Phys. Chem. Chem. Phys., 2011, 13, 16831 RSC.
- H. Zhao, G. A. Baker, Z. Y. Song, O. Olubajo, T. Crittle and D. Peters, Green Chem., 2008, 10, 696 RSC.
- A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301 CrossRef CAS.
- H. Mehdi, A. Bodor, D. Lantos, I. T. Horváth, D. E. De Vos and K. Binnemans, J. Org. Chem., 2006, 72, 517 CrossRef.
- I. Kilpeläinen, H. Xie, A. King, M. Granstrom, S. Heikkinen and D. S. Argyropoulos, J. Agric. Food Chem., 2007, 55, 9142 CrossRef.
- E. Kuhlmann, S. Himmler, H. Giebelhaus and P. Wasserscheid, Green Chem., 2007, 9, 233 RSC.
- M. Hirao, K. Ito and H. Ohno, Electrochim. Acta, 2000, 45, 1291 CrossRef CAS.
- C. H. Kuo and C. K. Lee, Bioresour. Technol., 2009, 100, 866 CrossRef CAS.
- Y. H. P. Zhang, S. Y. Ding, J. R. Mielenz, J. B. Cui, R. T. Elander, M. Laser, M. E. Himmel, J. R. McMillan and L. R. Lynd, Biotechnol. Bioeng., 2007, 97, 214 CrossRef CAS.
- T. Liebert, Cellulose solvents – remarkable history, bright future, ACS Symp. Ser., 2010, 1033, 3 CrossRef CAS.
- H. P. Fink, P. Weigel, H. J. Purz and J. Ganster, Prog. Polym. Sci., 2001, 26, 1473 CrossRef CAS.
- S. Dorn, F. Wendler, F. Meister and T. Heinze, Macromol. Mater. Eng., 2008, 293, 907 CrossRef CAS.
- C. Gränacher, U.S. Patent, 1,943,176, 1934 Search PubMed.
- (a) R. P. Swatloski, S. K. Spear, J. D. Holbrey and R. D. Rogers, J. Am. Chem. Soc., 2002, 124, 4974 CrossRef CAS; (b) J. Wu, J. Zhang, H. Zhang, J. S. He, Q. Ren and M. Guo, Biomacromolecules, 2004, 5, 266 CrossRef CAS; (c) Y. Fukaya, A. Sugimoto and H. Ohno, Biomacromolecules, 2006, 7, 3295 CrossRef CAS; (d) Y. Fukaya, K. Hayashi, M. Wada and H. Ohno, Green Chem., 2008, 10, 44 RSC; (e) S. Köhler, T. Liebert, M. Schöbitz, J. Schaller, F. Meister, W. Günther and T. Heinze, Macromol. Rapid Commun., 2007, 28, 2311 CrossRef.
- M. Gericke, K. Schlufter, T. Liebert, T. Heinze and T. Budtova, Biomacromolecules, 2009, 10, 1188 CrossRef CAS.
- R. P. Swatloski, R. D. Rogers and J. D. Holbrey, US Patent, 6824599 B2, 2004 Search PubMed.
- (a) S. Zhu, Y. Wu, Q. Chen, Z. Yu, C. Wang, S. Jin, Y. Ding and G. Wu, Green Chem., 2006, 8, 325 RSC; (b) A. Pinkert, K. N. Marsh, S. Pang and M. P. Staiger, Chem. Rev., 2009, 109, 6712 CrossRef CAS; (c) M. E. Zakrzewska, E. Bogel-Łukasik and R. Bogel-Łukasik, Energy Fuels, 2010, 24, 737 CrossRef CAS; (d) Z. Meng, X. Zheng, K. Tang, J. Liu and S. Qin, e-Polymers, 2012, 028, 1 Search PubMed; (e) H. Wang, G. Gurau and R. D. Rogers, Chem. Soc. Rev., 2012, 41, 1519 RSC.
- (a) B. Kosan, C. Michels and F. Meister, Cellulose, 2008, 15, 59 CrossRef CAS; (b) R. Sescousse, K. Le, M. E. Riesand and T. Budtova, J. Phys. Chem. B, 2010, 114, 7222 CrossRef CAS.
- F. Wendler, L.-N. Todi and F. Meister, Thermochim. Acta, 2012, 528, 76 CrossRef CAS.
- M. G. Freire, A. R. R. Teles, R. A. S. Ferreira, L. D. Carlos, J. A. Lopes-da-Silva and J. A. P. Coutinho, Green Chem., 2011, 13, 3173 RSC.
- (a) A. P. Dadi, S. Varanasi and C. A. Schall, Biotechnol. Bioeng., 2006, 95, 904 CrossRef CAS; (b) G. Cheng, P. Varanasi, C. Li, H. Liu, Y. B. Melnichenko, B. A. Simmons, M. S. Kent and S. Singh, Biomacromolecules, 2011, 12, 933 CrossRef CAS.
- (a) H. Zhao, C. I. L. Jones, G. A. Baker, S. Xia, O. Olubajo and V. N. Person, J. Biotechnol., 2009, 139, 47 CrossRef CAS; (b) X.-F. Tian, Z. Fang, D. Jiang and X.-Y. Sun, Biotechnol. Biofuels, 2011, 4, 1 CrossRef.
- (a) A. R. Xu, J. J. Wang and H. Y. Wang, Green Chem., 2010, 12, 268 RSC; (b) A. W. T. King, J. Asikkala, I. Mutikainen, P. Järvi and I. Kilpeläinen, Angew. Chem., Int. Ed., 2011, 50, 6301 CrossRef CAS.
- K. Ohira, Y. Abe, M. Kawatsura, K. Suzuki, M. Mizuno, Y. Amano and T. Itoh, ChemSusChem, 2012, 5, 388 CrossRef CAS.
- M. Hummel, C. Froschauer, G. Laus, T. Roder, H. Kopacka, L. K. J. Hauru, H. Weber, H. Sixta and H. Schottenberger, Green Chem., 2011, 13, 2507 RSC.
- R. C. Remsing, G. Hernandez, R. P. Swatloski, W. W. Massefski, R. D. Rogers and G. Moyna, J. Phys. Chem. B, 2008, 112, 11071 CrossRef CAS.
- H. Liu, K. L. Sale, B. M. Holmes, B. A. Simmons and S. Singh, J. Phys. Chem. B, 2010, 114, 4293 CrossRef CAS.
- A. S. Gross, A. T. Bell and J.-W. Chu, J. Phys. Chem. B, 2011, 115, 13433 CrossRef CAS.
- C. Froschauer, M. Hummel, G. Laus, H. Schottenberger, H. Sixta, H. K. Weber and G. Zuckerstätter, Biomacromolecules, 2012, 13, 1973 CrossRef CAS.
- (a) Hansen Solubility Parameters: A User's Handbook, CRC Press, Inc., Boca Raton, FL, 2007. ISBN: 9780849372483 Search PubMed; (b) http://www.hansen-solubility.com/, accessed 11 November 2012; (c) C. M. Hansen and B. H. Andersen, Am. Ind. Hyg. Assoc. J., 1988, 49, 301 CrossRef CAS.
- (a) A. Klamt, J. Phys. Chem., 1995, 99, 2224 CrossRef CAS; (b) F. Eckert and A. Klamt, AIChE J., 2002, 48, 369 CrossRef CAS.
- S. Spange, D. Keutel and F. Simon, J. Chim. Phys. Phys.–Chim. Biol., 1992, 89, 1615 CAS.
- (a) Z. Guo, B.-M. Lue, K. Thomasen, A. S. Meyer and X. Xu, Green Chem., 2007, 9, 1362 RSC; (b) J. Kahlen, K. Masuch and K. Leonhard, Green Chem., 2010, 12, 2172 RSC; (c) S. Padmanabhan, M. Kim, H. W. Blanch and J. M. Prausnitz, Fluid Phase Equilib., 2011, 309, 89 CrossRef CAS.
- M. Mora-Pale, L. Meli, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 1229 CrossRef CAS.
- H. Ohno and K. Fukumoto, Acc. Chem. Res., 2007, 40, 1122 CrossRef CAS.
- A. Pinkert, K. N. Marsh and S. Pang, Ind. Eng. Chem. Res., 2010, 49, 11809 CrossRef CAS.
- G. D'Andola, L. Szarvas, K. Massone and V. Stegmann, US Patent, 2010/0048829, 2010 Search PubMed.
- L. K. J. Hauru, M. Hummel, A. W. T. King, I. Kilpeläinen and H. Sixta, Biomacromolecules, 2012, 13, 2896 CrossRef CAS.
- T. G. A. Youngs, C. Hardacre and J. D. Holbrey, J. Phys. Chem. B, 2007, 111, 13765 CrossRef CAS.
- (a) B. Lindman, G. Karlström and L. Stigsson, J. Mol. Liq., 2010, 156, 76 CrossRef CAS; (b) B. Medronho, A. Romano, M. Miguel, L. Stigsson and B. Lindman, Cellulose, 2012, 19, 581 CrossRef CAS; (c) W. G. Glasser, R. H. Atalla, J. Blackwell, R. M. Brown, W. Burchard, A. D. French, D. O. Klemm and Y. Nishiyama, Cellulose, 2012, 19, 589 CrossRef CAS.
- A. S. Gross, A. T. Bell and J.-W. Chu, Phys. Chem. Chem. Phys., 2012, 14, 8425 RSC.
- (a) M. Gericke, T. Liebert, O. A. E. Seoud and T. Heinze, Macromol. Mater. Eng., 2011, 296, 483 CrossRef CAS; (b) R. Rinaldi, Chem. Commun., 2011, 47, 511 RSC.
- (a) A. Pinkert, J. Chem. Eng. Data, 2012, 57, 1338 CrossRef CAS; (b) R. Rinaldi, J. Chem. Eng. Data, 2012, 57, 1341 CrossRef CAS.
- M. Mazza, D. A. Catana, C. Vaca-Garcia and C. Cecutti, Cellulose, 2009, 16, 207 CrossRef CAS.
- M. Gericke, T. Liebert, O. A. E. Seoud and T. Heinze, Macromol. Mater. Eng., 2011, 296, 483 CrossRef CAS.
- A. Brandt, J. P. Hallett, D. J. Leak, R. J. Murphy and T. Welton, Green Chem., 2010, 12, 672 RSC.
- M. Abe, Y. Fukaya and H. Ohno, Chem. Commun., 2012, 48, 1808 RSC.
- K. Kamida, K. Okajima, T. Matsui and K. Kowsaka, Polym. J., 1984, 16, 857 CrossRef.
- Y. Q. Pu, N. Jiang and A. J. Ragauskas, J. Wood Chem. Technol., 2007, 27, 23 CrossRef CAS.
- S. Himmler, S. Hormann, R. van Hal, P. S. Schulz and P. Wasserscheid, Green Chem., 2006, 8, 887 RSC.
- L. M. Kline, D. G. Hayes, A. R. Womac and N. Labbe, BioResources, 2010, 5, 1366 CAS.
- B. G. Janesko, Phys. Chem. Chem. Phys., 2011, 13, 11393 RSC.
- D. Fu, G. Mazza and Y. Tamaki, J. Agric. Food Chem., 2010, 58, 2915 CrossRef CAS.
- S. H. Lee, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2009, 102, 1368 CrossRef CAS.
- (a) J. Wang, Y. Zhen and S. Zhang, in Clean Energy Systems and Experiences, ed. K. Eguchi, Sciyo, 2010 Search PubMed; (b) P. Mäki-Arvela, I. Anugwom, P. Virtanen and J.-P. Mikkola, Ind. Crops Prod., 2010, 32, 175 CrossRef.
- D. A. Fort, R. C. Remsing, R. P. Swatloski, P. Moyna, G. Moyna and R. D. Rogers, Green Chem., 2007, 9, 63 RSC.
- N. Sun, M. Rahman, Y. Qin, M. L. Maxim, H. Rodriguez and R. D. Rogers, Green Chem., 2009, 11, 646 RSC.
- S. Padmanabhan, M. Kim, H. W. Blanch and J. M. Prausnitz, Fluid Phase Equilib., 2011, 309, 89 CrossRef CAS.
- M. Zavrel, D. Bross, M. Funke, J. Buchs and A. C. Spiess, Bioresour. Technol., 2009, 100, 2580 CrossRef CAS.
- (a) L. Zoia, A. W. T. King and D. S. Argyropoulos, J. Agric. Food Chem., 2011, 59, 829 CrossRef CAS; (b) T. Leskinen, A. W. T. King, I. Kilpeläinen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2011, 50, 12349 CrossRef CAS.
- J. Viell and W. Marquardt, Holzforschung, 2011, 65, 519 CrossRef CAS.
- B. B. Hallac, M. Ray, R. J. Murphy and A. J. Ragauskas, Biotechnol. Bioeng., 2010, 107, 795 CrossRef CAS.
- S. Padmanabhan, M. Kim, H. W. Blanch and J. M. Prausnitz, Fluid Phase Equilib., 2011, 309, 89 CrossRef CAS.
- W. Li, N. Sun, B. Stoner, X. Jiang, X. Lu and R. D. Rogers, Green Chem., 2011, 13, 2038 RSC.
- H. Miyafuji, K. Miyata, S. Saka, F. Ueda and M. Mori, J. Wood Sci., 2009, 55, 215 CrossRef CAS.
- X. Wang, H. Li, Y. Cao and Q. Tang, Bioresour. Technol., 2011, 102, 7959 CrossRef CAS.
- Q. Li, Y.-C. He, M. Xian, G. Jun, X. Xu, J.-M. Yang and L.-Z. Li, Bioresour. Technol., 2009, 100, 3570 CrossRef CAS.
- (a) H. Wu, M. Mora-Pale, J. Miao, T. V. Doherty, R. J. Linhardt and J. S. Dordick, Biotechnol. Bioeng., 2011, 108, 2865 CrossRef CAS; (b) N. Labbé, L. M. Kline, L. Moens, K. Kim, P. C. Kim and D. G. Hayes, Bioresour. Technol., 2012, 104, 701 CrossRef; (c) K. M. Torr, K. T. Love, O. P. Cetinkol, L. A. Donaldson, A. George, B. M. Holmes and B. A. Simmons, Green Chem., 2012, 14, 778 RSC.
- T. V. Doherty, M. Mora-Pale, S. E. Foley, R. J. Linhardt and J. S. Dordick, Green Chem., 2010, 12, 1967 RSC.
- T. Balensiefer, H. Schröder, S. Freyer, G. D'Andola and K. Masonne, US Patent, 20100081798, 2010 Search PubMed.
- S. Troshenkova, E. Sashina, N. Novoselov, K. Arndt and S. Jankowsky, Russ. J. Gen. Chem., 2010, 80, 106 CrossRef CAS.
- A. Brandt, PhD Thesis, Ionic liquid pretreatment of lignocellulosic biomass. Imperial College London, London, 2012 Search PubMed.
- D. C. Dibble, C. Li, L. Sun, A. George, A. Cheng, O. P. Çetinkol, P. Benke, B. M. Holmes, S. Singh and B. A. Simmons, Green Chem., 2011, 13, 3255 RSC.
- (a) C. Sievers, M. B. Valenzuela-Olarte, T. Marzialetti, D. Musin, P. K. Agrawal and C. W. Jones, Ind. Eng. Chem. Res., 2009, 48, 1277 CrossRef CAS; (b) B. Li, J. Asikkala, I. Filpponen and D. S. Argyropoulos, Ind. Eng. Chem. Res., 2010, 49, 2477 CrossRef CAS.
- D. Diedericks, E. V. Rensburg, M. del Prado García-Aparicio and J. F. Görgens, Biotechnol. Prog., 2012, 28, 76 CrossRef CAS.
- (a) C. Arato, E. Pye and G. Gjennestad, Appl. Biochem. Biotechnol., 2005, 123, 871 CrossRef; (b) T. J. McDonough, Tappi J., 1993, 186 CAS; (c) X. Pan, N. Gilkes, J. Kadla, K. Pye, S. Saka, D. Gregg, K. Ehara, D. Xie, D. Lam and J. Saddler, Biotechnol. Bioeng., 2006, 94, 851 CrossRef CAS.
- (a) A. Sluiter, B. Hames, R. Ruiz, C. Scarlata, J. Sluiter, D. Templeton and D. Crocker, Determination of Structural Carbohydrates and Lignin in Biomass; NREL/TP-510-42618, US National Renewable Energy Laboratory, Golden, Colorado, 2008 Search PubMed; (b) A. Sluiter, R. Ruiz, C. Scarlata, J. Sluiter and D. Templeton, Determination of Extractives in Biomass, NREL/TP-510-42619, US National Renewable Energy Laboratory, Golden, Colorado, 2008 Search PubMed.
- Ö. P. Çetinkol, D. C. Dibble, G. Cheng, M. S. Kent, B. Knierim, M. Auer, D. E. Wemmer, J. G. Pelton, Y. B. Melnichenko, J. Ralph, B. A. Simmons and B. M. Holmes, Biofuels, 2010, 1, 33 CrossRef.
- A. George, K. Tran, T. J. Morgan, P. I. Benke, C. Berrueco, E. Lorente, B. C. Wu, J. D. Keasling, B. A. Simmons and B. M. Holmes, Green Chem., 2011, 13, 3375 RSC.
- S. Kubo, K. Hashida, T. Yamada, S. Hishiyama, K. Magara, M. Kishino, H. Ohno and S. Hosoya, J. Wood Chem. Technol., 2008, 28, 84 CrossRef CAS.
- R. Arora, C. Manisseri, C. Li, M. Ong, H. Scheller, K. Vogel, B. Simmons and S. Singh, BioEnergy Res., 2010, 3, 134 CrossRef.
- Z. Qiu, G. M. Aita and M. S. Walker, Bioresour. Technol., 2012, 117, 251 CrossRef CAS.
- C. Li, B. Knierim, C. Manisseri, R. Arora, H. V. Scheller, M. Auer, K. P. Vogel, B. A. Simmons and S. Singh, Bioresour. Technol., 2010, 101, 4900 CrossRef CAS.
- D. Fu and G. Mazza, Bioresour. Technol., 2011, 102, 7008 CrossRef CAS.
- L. Wei, K. Li, Y. Ma and X. Hou, Ind. Crops Prod., 2012, 37, 227 CrossRef CAS.
- L. Cammarata, S. G. Kazarian, P. A. Salter and T. Welton, Phys. Chem. Chem. Phys., 2001, 3, 5192 RSC.
- B. J. Cox, S. Jia, Z. C. Zhang and J. G. Ekerdt, Polym. Degrad. Stab., 2011, 96, 426 CrossRef CAS.
- K. Stärk, N. Taccardi, A. Bösmann and P. Wasserscheid, ChemSusChem, 2010, 3, 719 CrossRef.
- http://hyraxenergy.com/, accessed on 4 November 2012.
- M. J. Taherzadeh and K. Karimi, BioResources, 2007, 2, 472 CAS.
- (a) J. B. Binder and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4516 CrossRef CAS; (b) C. Li, Q. Wang and Z. K. Zhao, Green Chem., 2008, 10, 177 RSC; (c) K. Massonne, G. D'Andola, V. Stegmann, W. Mormann, M. Wezstein and W. Leng, WO2007/101812 A1, 2007 Search PubMed.
- M. Fanselow, J. Holbrey, K. R. Seddon, L. Vanoye and A. Zheng, WO2009/030949 A1, 2009 Search PubMed.
- C. Sievers, I. Musin, T. Marzialetti, M. Olarte, P. Agrawal and C. Jones, ChemSusChem, 2009, 2, 665 CrossRef CAS.
- S. Morales-delaRosa, J. M. Campos-Martin and J. L. G. Fierro, Chem. Eng. J., 2012, 181–182, 538 CrossRef CAS.
- Z. Zhang, B. Liu and Z. Zhao, Polym. Degrad. Stab., 2012, 97, 573 CrossRef CAS.
- L. Vanoye, M. Fanselow, J. D. Holbrey, M. P. Atkins and K. R. Seddon, Green Chem., 2009, 11, 390 RSC.
- R. Rinaldi, R. Palkovits and F. Schüth, Angew. Chem., Int. Ed., 2008, 47, 8047 CrossRef CAS.
- (a) A. A. Dwiatmoko, J. W. Choi, D. J. Suh, Y.-W. Suh and H. H. Kung, Appl. Catal., A, 2010, 387, 209 CrossRef CAS; (b) R. Rinaldi, N. Meine, J. vom Stein, R. Palkovits and F. Schüth, ChemSusChem, 2010, 3, 266 CrossRef CAS.
- R. Rinaldi, P. Engel, J. Büchs, A. C. Spiess and F. Schüth, ChemSusChem, 2010, 3, 1151 CrossRef CAS.
- W. D. Kumler and J. J. Eiler, J. Am. Chem. Soc., 1943, 65, 2355 CrossRef CAS.
- T. P. Wells, J. P. Hallett, C. K. Williams and T. Welton, J. Org. Chem., 2008, 73, 5585 CrossRef CAS.
- P. Mäki-Arvela, E. Salminen, T. Riittonen, P. Virtanen, N. Kumar and J.-P. Mikkola, Int. J. Chem. Eng., 2012, 674761 Search PubMed.
- C. Moreau, A. Finiels and L. Vanoye, J. Mol. Catal. A: Chem., 2006, 253, 165 CrossRef CAS.
- H. Zhao, J. E. Holladay, H. Brown and Z. C. Zhang, Science, 2007, 316, 1597 CrossRef CAS.
- Y. Su, H. M. Brown, X. Huang, X.-d. Zhou, J. E. Amonette and Z. C. Zhang, Appl. Catal., A, 2009, 361, 117 CrossRef CAS.
- J. H. Van Vleet and T. W. Jeffries, Curr. Opin. Biotechnol., 2009, 20, 300 CrossRef CAS.
- P. Engel, R. Mladenov, H. Wulfhorst, G. Jager and A. C. Spiess, Green Chem., 2010, 12, 1959 RSC.
- N. Sathitsuksanoh, Z. Zhu and Y. H. P. Zhang, Cellulose, 2012, 19, 1161 CrossRef CAS.
- (a) T. Auxenfans, S. Buchoux, K. Djellab, C. Avondo, E. Husson and C. Sarazin, Carbohydr. Polym., 2012, 90, 805 CrossRef CAS; (b) N. Kamiya, Y. Matsushita, M. Hanaki, K. Nakashima, M. Narita, M. Goto and H. Takahashi, Biotechnol. Lett., 2008, 30, 1037 CrossRef CAS.
- Q. Xin, K. Pfeiffer, J. M. Prausnitz, D. S. Clark and H. W. Blanch, Biotechnol. Bioeng., 2012, 109, 346 CrossRef CAS.
- (a) I. Anugwom, P. Mäki-Arvela, P. Virtanen, R. Sjöholm, S. Willför and J.-P. Mikkola, Carbohydr. Polym., 2012, 87, 2005 CrossRef CAS; (b) N. Sun, H. Rodriguez, M. Rahman and R. D. Rogers, Chem. Commun., 2011, 47, 1405 RSC.
- A. W. T. King, A. Parviainen, P. Karhunen, J. Matikainen, L. K. J. Hauru, H. Sixta and I. Kilpelainen, RSC Adv., 2012, 2, 8020 RSC.
- A. Seeberger, A.-K. Andresen and A. Jess, Phys. Chem. Chem. Phys., 2009, 11, 9375 RSC.
- W. H. Awad, J. W. Gilman, M. Nyden, R. H. Harris, T. E. Sutto, J. Callahan, P. C. Trulove, H. C. DeLong and D. M. Fox, Thermochim. Acta, 2004, 409, 3 CrossRef CAS.
- D. M. Fox, W. H. Awad, J. W. Gilman, P. H. Maupin, H. C. De Long and P. C. Trulove, Green Chem., 2003, 5, 724 RSC.
- D. Klein-Marcuschamer, B. A. Simmons and H. W. Blanch, Biofuels Bioprod. Biorefin., 2011, 5, 562 CrossRef CAS.
- K. J. Baranyai, G. B. Deacon, D. R. MacFarlane, J. M. Pringle and J. L. Scott, Aust. J. Chem., 2004, 57, 145 CrossRef CAS.
- T. J. Wooster, K. M. Johanson, K. J. Fraser, D. R. MacFarlane and J. L. Scott, Green Chem., 2006, 8, 691 RSC.
- A. Brandt, M. Clough, J. Hallett and T. Welton, unpublished results.
- D. B. Fu and G. Mazza, Bioresour. Technol., 2011, 102, 8003 CrossRef CAS.
- N. Meine, F. Benedito and R. Rinaldi, Green Chem., 2010, 12, 1711 RSC.
- P. Keil, M. Kick and A. König, Chem. Ing. Tech., 2012, 84, 859 CAS.
- W. Lan, C.-F. Liu and R.-C. Sun, J. Agric. Food Chem., 2011, 59, 8691 CrossRef CAS.
- N. L. Mai, N. T. Nguyen, J. I. Kim, H. M. Park, S. K. Lee and Y. M. Koo, J. Chromatogr., A, 2012, 1227, 67 CrossRef CAS.
- M. Francisco, A. N. Mlinar, B. Yoo, A. T. Bell and J. M. Prausnitz, Chem. Eng. J., 2011, 172, 184 CrossRef CAS.
- K. Shill, S. Padmanabhan, Q. Xin, J. M. Prausnitz, D. S. Clark and H. W. Blanch, Biotechnol. Bioeng., 2011, 108, 511 CrossRef CAS.
- T. C. Brennan, S. Datta, H. W. Blanch, B. A. Simmons and B. M. Holmes, BioEnergy Res., 2010, 3, 123 CrossRef.
- L. Cadoche and G. D. López, Biol. Wastes, 1989, 30, 153 CrossRef CAS.
- Z. Miao, T. E. Grift, A. C. Hansen and K. C. Ting, Ind. Crops Prod., 2011, 33, 504 CrossRef CAS.
- S. Sokhansanj, A. Kumar and A. F. Turhollow, Biomass Bioenergy, 2006, 30, 838 CrossRef.
- S. Mani, L. G. Tabil and S. Sokhansanj, Biomass Bioenergy, 2004, 27, 339 CrossRef.
- A. Brandt, J. K. Erickson, J. P. Hallett, R. J. Murphy, A. Potthast, M. J. Ray, T. Rosenau, M. Schrems and T. Welton, Green Chem., 2012, 14, 1079 RSC.
- M. Ouellet, S. Datta, D. C. Dibble, P. R. Tamrakar, P. I. Benke, C. Li, S. Singh, K. L. Sale, P. D. Adams, J. D. Keasling, B. A. Simmons, B. M. Holmes and A. Mukhopadhyay, Green Chem., 2011, 13, 2743 RSC.
- S. Datta, B. Holmes, J. I. Park, Z. Chen, D. C. Dibble, M. Hadi, H. W. Blanch, B. A. Simmons and R. Sapra, Green Chem., 2010, 12, 338 RSC.
- J. Pottkämper, P. Barthen, N. Ilmberger, U. Schwaneberg, A. Schenk, M. Schulte, N. Ignatiev and W. R. Streit, Green Chem., 2009, 11, 957 RSC.
- M. A. McHugh, Supercritical Fluid Extraction [Electronic Resource]: Principles and Practice, Butterworth-Heinemann, 2nd edn, 1994 Search PubMed.
- M. Maase, K. Massonne, K. Halbritter, R. Noe, M. Bartsch, W. Siegel, V. Stegmann, M. Flores, O. Huttenloch and M. Becker, WO2003 062171, 2003 Search PubMed.
- H. Xie, H. Shen, Z. Gong, Q. Wang, Z. K. Zhao and F. Bai, Green Chem., 2012, 14, 1202 RSC.
- M. Rahman, Y. Qin, M. L. Maxim and R. D. Rogers, WO2010/056790 A1, 2010 Search PubMed.
- M. B. Turner, S. K. Spear, J. G. Huddleston, J. D. Holbrey and R. D. Rogers, Green Chem., 2003, 5, 443 RSC.
| This journal is © The Royal Society of Chemistry 2013 |