 Open Access Article
Open Access ArticleThe synthesis of N-heterocycles via copper/TEMPO catalysed aerobic oxidation of amino alcohols†
James C. A.
Flanagan
,
Laura M.
Dornan
,
Mark G.
McLaughlin
,
Niall G.
McCreanor
,
Matthew J.
Cook
* and
Mark J.
Muldoon
*
School of Chemistry and Chemical Engineering, Queen's University Belfast, David Keir Building, Stranmillis Road, Belfast, BT9 5AG, Northern Ireland. E-mail: m.cook@qub.ac.uk; Tel: +44 (0) 28 9097 4682m.j.muldoon@qub.ac.uk; Tel: +44 (0) 28 9097 4420
First published on 28th February 2012
Abstract
N-Heterocycles can be prepared using alcohol oxidation as a key synthetic step. Herein we report studies exploring the potential of Cu/TEMPO as an aerobic oxidation catalyst for the synthesis of substituted indoles and quinolines.
N-Heterocycles are important scaffolds for many drugs and natural products, and substituted quinolines and indoles are the basis of many top selling pharmaceuticals; for example, Singulair® and Maxalt®. The synthesis of such substituted N-heterocycles can be challenging with many traditional approaches requiring harsh conditions and delivering poor selectivity.1–3
An attractive route for the synthesis of N-heterocycles is to utilize alcohols as substrates as they are readily available and easy to handle. There are numerous examples where catalytic transfer hydrogenations or “hydrogen borrowing” methods have been used to prepare N-heterocycles from alcohols.4 A particularly desirable route for the selective synthesis of substituted N-heterocycles is the intramolecular oxidative cyclization of amino alcohols. Watanabe and co-workers previously employed this route for the synthesis of indoles, using a RuCl2(PPh3)3 catalyst.5 More recently, heterogeneous ruthenium catalysts (Ru/CeO2 and Ru/ZrO2) were used to prepare indole using the same route.6 Fujita et al. reported the use of a [Cp*IrCl2]2 catalyst for the synthesis of indoles, 1,2,3,4-tetrahydroquinolines and 2,3,4,5-tetrahydro-1-benzazepine.7 For the same starting materials, it was found that when the catalyst was switched to [Cp*RhCl2]2 it produced the corresponding five-, six-, and seven-membered ring lactams.8
This approach would be greatly improved if we could move away from expensive precious metals and employ more earth abundant metals. Given that oxidation of an alcohol to an aldehyde is the key step in this route, the Cu/TEMPO/O2 system is an attractive alternative.9 This biomimetic system employs a Cu(II) or Cu(I)9e salt complexed by a ligand such as 2,2-bipyridine, the stable free radical TEMPO (2,2,6,6-tetramethylpiperidinyloxyl), a base and dioxygen as the terminal oxidant. Along with the fact that it employs copper, this system was appealing because it is known to operate under mild conditions and is selective for the oxidation of primary alcohols to aldehydes.9 This feature would increase the scope of this oxidative intramolecular oxidative cyclization and allow us to prepare N-heterocycles using substrates containing secondary alcohol functionality. Furthermore, because it is an aerobic approach we envisaged that this would allow us to obtain quinolines as opposed to tetrahydroquinolines7 and lactams8 which were obtained using the Ir and Rh catalysts mentioned earlier. While there have been numerous studies exploring Cu/TEMPO for catalytic alcohol oxidation, these studies have primarily focused on simple model substrates;9 the catalyst had not been exploited for the synthesis of N-heterocycles.10
Indole synthesis
We began by exploring the synthesis of indole from the commercially available starting material 2-aminophenethyl alcohol. We explored a range of different reaction conditions and Table 1 highlights some selected examples.|
|
||||
|---|---|---|---|---|
| Entry | Cu source (mol%) | Co-catalyst (mol%) | Temperature (time, h) | Yield (%) |
| a Ligand = 2′2-bipyridine, base = 3 mol% NMI and 3 mol% DBU, solvent = acetonitrile, 3 Å molecular sieves present. b Aqueous biphasic system with decalin as the organic solvent, ligand = 2′2-bipyridine, base = 3 mol% NMI and 3 mol% DBU. c Ligand = 1,10-phenanthroline, base = 2 equivalent K2CO3, solvent = toluene. | ||||
| 1a | Cu(OTf)2 (3) | TEMPO (3) | 60 °C (4) | 46 |
| 2a | Cu(OTf)2 (3) | TEMPO (3) | 60 °C (24) | 30 |
| 3a | Cu(OTf)2 (9) | TEMPO (9) | 60 °C (4) | 90 |
| 4b | Cu(OTf)2 (3) | HO-TEMPO (3) | 90 °C (18) | 39 |
| 5c | CuCl (5) | DBAD (5) | 90 °C (4) | 33 |

The catalyst system which we mainly used was previously optimised for alcohol oxidation by Kumpulainen and Koskinen9d and consisted of Cu(OTf)2/2,2′-bipyridine/TEMPO, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), N-methylimidazole (NMI), 3 Å molecular sieves and acetonitrile. In our studies, it was found that longer reaction times led to lower yields (cf. entries 1 and 2 in Table 1), suggesting that the product is undergoing decomposition. This was further confirmed by subjecting pure indole to our reaction conditions, resulting in the recovery of 82% of indole (isolated yield). Additionally, we followed this product inhibition by 1H NMR using methyl benzoate as an internal standard and we clearly observed the disappearance of indole over time. So far we have been unable to identify the by-products from the reaction between the catalyst and indole, but one possible explanation is that indole is reacting with the Cu(II) via the nucleophilic C3-position. Work by Gaunt and co-workers has demonstrated that Cu(OTf)2 can be used to catalyse the arylation of indoles at this position.11 Further evidence that the product is reacting with the catalyst in this manner was obtained when we subjected 3-methylindole to standard reaction conditions. In this case, we could only recover and isolate 26% of the original 3-methylindole, with many unidentified by-products produced. We believe this greater reactivity can be rationalised by the fact that 3-methylindole is more nucleophilic at the 3-position than indole.
The fact that we can obtain a 90% isolated yield for the synthesis of indole (entry 3 in Table 1) indicates that the catalyst will preferentially oxidize the substrate over this unwanted reaction with the product. As shown in Fig. 1, when we monitored the reaction by 1H NMR of 1a, we observed smooth kinetics over the course of the reaction (until we obtained a yield of ∼80%).
 | ||
| Fig. 1 Kinetic study for the oxidation of 1a, monitored by 1H NMR using PhCO2Me as an internal standard in CD3CN. | ||
The results indicate that if we had a method of removing the product this could lead to greater efficiency and catalyst stability. We attempted to address the product inhibition problem by exploring the use of aqueous biphasic systems. In these cases, we employed either 4-hydroxy-TEMPO or 4-carboxy-TEMPO to ensure that both the copper complex and TEMPO were retained in the aqueous phase.† It was hoped that if the product was extracted into the upper organic phase (we explored the use of toluene, xylene, decalin and nonane) it would reduce product decomposition. Unfortunately, it was found that the reactions were very slow and the highest yield obtained was 39% (entry 3 in Table 1). As part of our optimisation studies we also investigated the use of copper(I)/di-tert-butyl-azodicarboxylate (DBAD) or its corresponding hydrazine (DBADH2) for the synthesis of indole. This catalyst system was reported by Markó as an effective method for alcohol oxidation,12 however we found that it was not as effective as Cu/TEMPO and a maximum yield of 33% was obtained for these catalysts (entry 4 in Table 1).
Taking our optimised conditions that allowed us to obtain 90% isolated yield of indole, we then applied these conditions for the synthesis of some substituted indoles. As can be seen in Table 2, these could be obtained in moderate to good (isolated) yields.

|
The oxidation of 3 to 4 is an impressive illustration of the ability of the Cu/TEMPO system to selectively oxidize the primary alcohol to an aldehyde whilst leaving the secondary alcohol unaffected (eqn (1)). The carbonyl group formed is attacked by the N atom of the amino group to afford the indole in 58% isolated yield with a secondary alcohol still intact.
 | (1) |
Quinoline synthesis
We began studying the synthesis of quinolines by once again looking at the simplest product, quinoline itself. The results of optimisation experiments for this procedure once again suggested some mechanistic details. Firstly, the issues of product inhibition experienced with the indole experiments do not seem to exist (at least to the same extent) for quinoline synthesis. However, it was found that a significant amount of 1,2,3,4-tetrahydroquinolin-2-one was produced in each experiment. The amount of this minor product increased further if less rigorously anhydrous conditions were used. This observation leads to the suggestion that once aldehyde oxidation is complete there are two competing slow steps: a dehydration to give the 3,4-dihydroquinoline and a further alcohol oxidation to give a 1,2,3,4-tetrahydroquinoline. The removal of water (by sieves and drying tube) clearly helps to drive the reaction towards quinoline production.
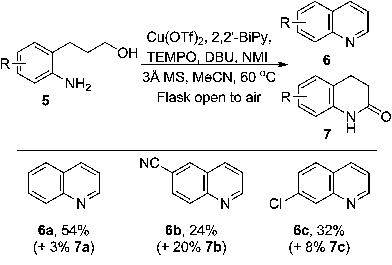
|
Conclusions
The study indicates that the Cu/TEMPO oxidation catalyst has potential for the synthesis of N-heterocycles. In the case of indole synthesis, the issue of product inhibition needs to be further resolved. The kinetics suggest that formation of the product is faster than the inhibition reaction that leads to catalyst deactivation. If it was possible to immobilise the catalyst, continuous flow operation could allow the product to be removed and improve the efficiency of the reaction. There is much room for expanding the substrate scope, especially with those containing both primary and secondary alcohols. We also aim to explore the use of Cu/TEMPO for synthesis of other N-heterocycles.Experimental
General (optimised) procedure: Copper(II) trifluoromethanesulfonate (Cu(OTf)2) (9 mol%), 2,2′-bipyridyl (2,2′-bipy) (9 mol%) and 2,2,6,6-tetramethylpiperidinyloxyl (TEMPO) (9 mol%) were added to acetonitrile (1.5 cm3) in an oven-dried round-bottomed flask to give a green solution. The starting amino alcohol (1 equivalent, typically 0.5–1 mmol), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (3 mol%) N-methylimidazole (NMI) (3 mol%), and 3 Å molecular sieves (0.5 g) were then successively added. The solution was stirred at 60 °C in a round-bottomed flask with a reflux condenser attached. The reflux condenser had a drying tube fitted to the top and was open to the air. In case of indoles reactions were stopped after 4 h while 24 h were required for quinolines. The mixture was then placed in a separating funnel and extracted three times with 15 ml diethyl ether and 15 ml water. The organic layers were then combined, dried with anhydrous magnesium sulfate, filtered, evaporated in vacuo and purified by flash column chromatography (EtOAc–hexane).†Acknowledgements
We are grateful to the RCUK, the Nuffield Foundation and Almac Discovery for funding. We also gratefully acknowledge GlaxoSmithKline for the donation of laboratory equipment.Notes and references
- Reviews of indole synthesis: R. B. Van Order and H. G. Lindwall, Chem. Rev., 1942, 30, 69 CrossRef CAS; G. W. Gribble, J. Chem. Soc., Perkin Trans. 1, 2000, 1045 RSC; G. R. Humphrey and J. T. Kuethe, Chem. Rev., 2006, 106, 2875 CrossRef; S. Cacchi and G. Fabrizi, Chem. Rev., 2005, 105, 2873 CrossRef.
- Reviews of quinoline synthesis: V. V. Kouznetsov, L. Y. Vargas Méndez and C. M. Meléndez Gómez, Curr. Org. Chem., 2005, 9, 141 CrossRef CAS; S. Madapa, Z. Tusi and S. Batra, Curr. Org. Chem., 2008, 12, 1116 CrossRef.
- J. A. Joule and K. Mills, Heterocyclic Chemistry, Wiley-Blackwell, 5th edn, 2010, ch. 9, 20 Search PubMed.
- Reviews: A. C. Marr, Catal. Sci. Technol., 2012, 2, 279 Search PubMed; M. H. S. A. Hamid, P. A. Slatford and J. M. J. Williams, Adv. Synth. Catal., 2007, 349, 1555 CrossRef CAS.
- Y. Tsuji, S. Kotachi, K. Huh and Y. Watanabe, J. Org. Chem., 1990, 55, 580 CrossRef CAS.
- S. Shimura, H. Miura, K. Wada, S. Hosokawa, S. Yamazoe and M. Inoue, Catal. Sci. Technol., 2011, 1, 1340 CAS.
- K. Fujita, K. Yamamoto and R. Yamaguchi, Org. Lett., 2002, 4, 2691 CrossRef CAS.
- K. Fujita, Y. Takahashi, M. Owaki, K. Yamamoto and R. Yamaguchi, Org. Lett., 2004, 6, 2785 CrossRef CAS.
- Examples of key papers: (a) M. F. Semmelhack, C. R. Schmid, D. A. Cortes and C. S. Chou, J. Am. Chem. Soc., 1984, 106, 3374 CrossRef CAS; (b) P. Gamez, I. W. C. E. Arends, J. Reedijk and R. A. Sheldon, Chem. Commun., 2003, 2414 RSC; (c) P. Gamez, I. W. C. E. Arends, R. A. Sheldon and J. Reedijk, Adv. Synth. Catal., 2004, 346, 805 CrossRef CAS; (d) E. T. Kumpulainen and A. M. Koskinen, Chem.–Eur. J., 2009, 15, 10901 CrossRef CAS; (e) J. M. Hoover and S. S. Stahl, J. Am. Chem. Soc., 2011, 133, 16901 CrossRef CAS.
- During the preparation of this manuscript a report describing the use of copper/4-HO-TEMPO for the synthesis of 2-substituted quinazolines and 4H-3,1-benzoxazines was published. In this case the catalyst does not perform an alcohol oxidation but oxidises in-situ prepared aminals and hemi-aminals. B. Han, X.-Y. Long, C. Wang, Y.-W. Bai, T.-C. Pan, X. Chen and W. Yu, J. Org. Chem., 2012, 77, 1136 CrossRef CAS.
- R. J. Phipps, N. P. Grimster and M. J. Gaunt, J. Am. Chem. Soc., 2008, 130, 8172 CrossRef CAS.
- (a) I. E. Markó, P. R. Giles, M. Tsukazaki, S. M. Brown and C. J. Urch, Science, 1996, 274, 2044 CrossRef; (b) I. E. Markó, A. Gautier, I. Chellé-Regnaut, P. R. Giles, M. Tsukazaki, C. J. Urch and S. M. Brown, J. Org. Chem., 1998, 63, 7576 CrossRef; (c) I. E. Markó, A. Gautier, R. Dumeunierl, K. Dodal, F. Philippart, S. M. Brown and C. J. Urch, Angew. Chem., Int. Ed., 2004, 43, 1588 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Further details of experiments and synthesis of starting materials. See DOI: 10.1039/c2gc35062a |
| This journal is © The Royal Society of Chemistry 2012 |