Abundance and sources of hydrophilic and hydrophobic water-soluble organic carbon at an urban site in Korea in summer
Seung Shik
Park
*,
Ja-Hyun
Kim
and
Jae-Uk
Jeong
Department of Environmental Engineering, Chonnam National University, 77 Yongbong-Ro, Buk-ku, Gwangju, 500-757, Republic of Korea. E-mail: park8162@chonnam.ac.kr; Fax: +82-62-530-1859; Tel: +82-62-530-1863
First published on 14th November 2011
Abstract
In this study, the characteristics of total water-soluble organic carbon (WSOC) and isolated WSOC fractions were examined to gain a better understanding of the pathway of organic aerosol production. 24 h PM2.5 samples were collected during the summer (July 28–August 28, 2009) at an urban site in Korea. A glass column filled with XAD7HP resin was used to separate the filtered extracts into hydrophilic (WSOCHPI) and hydrophobic (WSOCHPO) fractions. The origins of air mass pathways arriving at the sampling site were mostly classified into three types, those originating over the East Sea of Korea that passed over the eastern inland urban and industrial regions (type I); those from the marine (western/southwestern/southern marine) and passed over the national industrial complex regions (type II); and those from northeastern China that passed through North Korea and metropolitan areas of South Korea (type III). Measurements showed an increase in the average WSOC fraction of total OC from the type II to III air mass (53 to 64%) periods. Also, higher SO42−/SOx (=SO2 + SO42−) was observed in the type III air mass (0.70) than those in the types I (0.49) and II (0.43). According to the average values of WSOC/OC and SO42−/SOx, measurements suggest that the aerosols collected during the type III air mass period were more aged or photo-chemically processed than those during the types I and II air mass periods. The relationship between the SO42−/SOx and WSOC/OC (R2 = 0.64) suggests that a significant fraction of the observed WSOC at the site could be formed by an oxidation process similar to SO42− aerosols, probably the oxidation process using OH radicals, or in-cloud processing. The photochemical production of WSOCHPO was also observed to significantly contribute to the total OC.
Environmental impactThe water-soluble organic carbon (WSOC) fraction of the OC could provide an important clue to the composition and chemical processes of organic aerosols. This study revealed that the WSOC fraction of total OC and SO42−/(SO2 + SO42−) ratio increased when the air masses arriving at the receptor site originated from the northeastern polluted areas of China. Our results also indicated that the photochemical production of the hydrophobic WSOC fraction was a large contributor to the OC during the summer at the site. |
Introduction
Organic aerosol particles in ambient air are a highly complex mixture composed of oxygenated carbon functional groups;1 thus, making the comprehensive chemical characterization of aerosol organic carbon difficult using gas chromatography coupled with mass spectrometry (GC-MS).2 One approach has focused on the isolation of the water-soluble organic carbon (WSOC) fraction from organic aerosols into broad and comprehensive chemical fractions. WSOC constitutes a significant fraction of particulate organic aerosols, ranging from 10 to 90% of organic carbon mass depending on the locations.3–10 The WSOC particles play an important role in global climate change by altering the hygroscopicity of atmospheric aerosols.11,12WSOC is also of interest because it may possess important physical properties. For example, when inhaled, these compounds could produce an adverse health effect.13 These water-soluble organic compounds have both primary and secondary sources. Because one of the main sources of WSOC is secondary organic aerosol (SOA) formed through the photo-oxidation of precursor organics,14–16 the study of WSOC is one method for investigating SOA. Various methods have been applied to separate and quantify the WSOC component of ambient aerosol particles into neutral, basic and acid functional groups.12,17–23 Some studies have also shown that WSOC is composed of compounds, such as aliphatic acids, carbonyls, aromatic acids, saccharides, polyols, phenols, organic nitrates, and amines.20,22,24 These compounds can be broadly classified into hydrophilic and hydrophobic fractions. The hydrophilic WSOC fraction tends to be highly soluble in water and includes compounds with low molecular weights, such as aliphatic carboxylic acids and carbonyls (<4 carbons), saccharides and amines, while the hydrophobic WSOC fraction tends to be less hygroscopic, including aliphatic carboxylic acids and carbonyls (>3 to 4 carbons), aromatic acids, phenols, organic nitrates, cyclic acids, and Suwannee River fulvic acids.An XAD resin coupled with a TOC (total organic carbon) analyzer has been used to isolate the WSOC components into hydrophilic and hydrophobic fractions.20,22–25 Urban measurements from Atlanta during summer indicated that the hydrophilic WSOC fraction accounted for an average of 61% of the total WSOC, suggesting that the production processes of secondary organic aerosol produce significant amounts of hydrophilic compounds.9 However, Miyazaki et al.20 showed that the hydrophobic WSOC fraction at a rural site in the Pearl River Delta, China, during summer 2006, accounted for an average of 60% of the total WSOC mass, with increases in both the hydrophilic and hydrophobic fractions with photochemical aging. Jeong et al.25 also reported that the hydrophilic and hydrophobic fractions of WSOC averaged 30.5 and 69.5%, respectively, of the total WSOC in PM2.5 measured between May and September 2010, at an urban site in Seoul, Korea. Humic-like substances (HULIS) in the WSOC aerosol have been reported to apparently be associated with hydrophobic WSOC.2,18,19 These HULIS compounds have unique properties, including light absorption and strong surface activity,2,26 which could decrease the surface tension of aerosol and fog water.27,28
Despite the much improved available information on the composition, physical and chemical properties, sources and transformation characteristics of organic aerosols over recent years, information relating to northeastern Asia is still rather limited. Therefore, a comprehensive study is required to better understand the sources and atmospheric processing of organic aerosol particles at an urban airshed by isolating organic compounds in aqueous solutions by their chemical functional groups. In this paper, the results of group separation of WSOC samples from an urban site in Gwangju, Korea, using PM2.5 filter samples collected during summer, are presented. XAD7HP resin, with TOC detection, was used to separate the WSOC into hydrophilic and hydrophobic fractions.
Experimental
PM2.5 measurements
24 h integrated PM2.5 samples, starting at about 09:00 a.m., were collected between 28 July and 28 August 2009 from the roof of a three-story building (54.3 m above sea level) at the university campus (35°11′N, 126°54′E) at the Gwangju metropolitan area, Korea. The sampling site was approximately 150 m from a two-lane road carrying heavy traffic during rush-hour, and located within an urban airshed that was greatly influenced by regionally produced pollution aerosols.9,29 The site was also appropriate for the investigation of the Asian continental impact of aerosol outflows prior to their affecting the northern Pacific and North America. A detailed description of the regional characteristics of the measurement site can be found in our previous study.30Three PM2.5 cyclone samplers were used to collect ambient air particles. The PM2.5 inlet cyclone sampler draws ambient air at a flow rate of 16.7 l min−1. In the first and second samplers, the PM2.5 samples were collected on pre-fired 47 mm quartz filters for analyses of the organic and elemental carbon contents (OC and EC), total water-soluble OC (WSOC), and fractionated WSOC. A carbon impregnated filter (CIF), multi-channel, parallel plate denuder was placed upstream of the filter-pack to remove semi-volatile organic vapors. All quartz fibers used to collect the carbonaceous particles were pre-cleaned by baking at 500 °C for 10 h. Aerosol samples in the third sampler were collected on 47 mm Teflon filters (Zefluor, 2 μm pore size, Gelman Science) behind an annular denuder and used for determining the ionic species as well as the PM2.5 mass. Field blank samples for the quartz and Teflon filters during the entire measurement period were collected three times by loading the filter into the sampler without the vacuum pump turned on. Hourly average concentrations of five ambient air pollutants, including sulfur dioxide (SO2), ozone (O3), carbon monoxide (CO), and nitrogen oxides (NOx), were also measured using an ambient air monitoring system from the Ministry of the Environment, at a location about 2.0 km from the aerosol sampling site.
Classification of air mass pathways
Even though the sampling site was located within an urban area, where the influence of traffic emissions was significant, the site has been reported to be greatly impacted by regionally produced aerosols.9,29,30 Isentropic four-day back trajectories were calculated using the Hybrid Single-Particle Lagrangian Integrated Trajectories (HYSPLIT 4.5) model31 to examine the origins of the air masses arriving at the sampling site, at 0000 UTC time (09:00 local time) each day, at three altitudes (500, 1000 and 1500 m) above ground level (AGL). The origins of the calculated air mass trajectories were mostly classified into three types, as shown in Fig. 1. The first type represented air masses originating over the East Sea of Korea, passing over the eastern inland urban and industrial regions of Korea prior to reaching the sampling site (type I). This type of air mass was observed during 29.6% of the entire sampling period (8 times) and may contain a mixture of marine, local and transported pollution. Originating from the East China or Yellow Seas, or southern marine and east Pacific Ocean, the second type of air mass (type II) passed over large-scale industrial complex regions, located about 80 km southeast of the site, and may include a mixture of marine, local anthropogenic emissions and transported pollutants emitted or produced from the Korean Peninsula. The pathway of the second type of air mass was observed during 44.4% (12 times) of the entire sampling period. The third type of air mass, originating from regions of Southern Russia or Mongolia, passed over northeastern regions of China, North Korea and the Korean peninsula (Seoul, Inchon, etc.) prior to reaching the sampling site. This type of air mass accounted for 25.9% (7 times) during the entire period, which indicated the large influence of anthropogenic emissions from the northeastern or eastern regions of China and heavily polluted metropolitan regions of South Korea. | ||
| Fig. 1 Fig. 1Four-day back trajectory plots representing three different categories of (a) East Sea of Korea—Korean peninsula, (b) marine (China Sea/Yellow Sea/Pacific Ocean), and (c) northeastern China–North Korea. | ||
Chemical analyses of OC, EC, total WSOC, and ionic species
Typically, the quartz filter from the first PM2.5 sampler was analyzed to determine the amounts of OC and EC using a thermal–optical transmittance (TOT) standard method,32 at Sunset Laboratory Inc. (NC office, USA). The OC and EC were determined as follows: OC was evolved under a stream of ultrahigh purity He (99.999% minimum), while heating the sample in four temperature steps to a final temperature of 870 °C; the temperature program consisted of 310 °C for 60 s, 475 °C for 60 s, 615 °C for 60 s and 870 °C for 90 s. The OC evolved during these four steps was operationally defined as volatile OC. During the initial temperature ramp, a fraction of the OC is pyrolytically converted to EC carbon due to the charring of OC. Any charring of OC results in increased absorbance of the laser, which lowers the OC measurement and increases the original EC measurement. To evolve EC and pyrolyzed OC, the sample was first cooled to 550 °C and then heated under a mixture of 2% O2 + 98% He. The ECOC analyzer utilized laser transmission to correct for sample charring. The EC was determined as the carbon evolved after the filter transmittance returned to its initial value. In this study, the method detection limit of a substance was calculated as the average blank value of the substance plus three times the standard deviation of the blanks. The values of the average blank and standard deviation of blanks used in the calculation are 0.32 and 0.04 μg C cm−2 for the OC, and 0.01 and 0.02 μg C cm−2 for the EC, respectively. The OC and EC detection limits for this sample set were 0.23 and 0.03 μg C m−3, respectively. The OC and EC measurements had precisions of 1.3 and 2.5%, respectively.Quartz filter samples from the second sampler were extracted in 40 mL of ultrapure distilled de-ionized water (DDW), produced using an ultrapure water purification system (Barnstead Nanopure, #D11901, Thermo Scientific, USA), viaultrasonication for 60 min. The water extracts were filtered using a syringe membrane filter (Millipore 0.45 μm) to remove the insoluble particles and filtered scraps. The filtrate was then analyzed to determine the WSOC content using a total organic carbon analyzer (TOC, Sievers 5310C, USA). A detailed description about the TOC analysis can be found elsewhere.33 The WSOC detection limit was measured to be 0.22 μg C m−3. The WSOC measurement showed a precision of <± 5%. The remaining extracts were used for the group separation of bulk WSOC into hydrophilic and hydrophobic fractions.
The Teflon filters were weighed before and after sample collections with a micro-balance of 1 μg sensitivity and analyzed for eight ionic species. The filters were conditioned for about 48 h in a clean chamber maintained at a relative humidity of 40% and a temperature of 20 °C. In order to extract the ionic species from the Teflon filters, each filter was put into a 20 mL vial. The Teflon filter, with the sampled aerosols, was first wetted with 20 mL of DDW, and then extracted using ultrasonication for 60 min. The water extracts were filtered using a syringe membrane filter (Millipore 0.45 μm) to remove the insoluble particles and then analyzed to determine amounts of ionic species such as chloride (Cl−), nitrate (NO3−), sulfate (SO42−), sodium (Na+), ammonium (NH4+), potassium (K+), magnesium (Mg2+) and calcium (Ca2+) using ion chromatography (IC). The IC system consisted of an anion IC (Metrohm model 861 with a suppressor, equipped with a Metrohm Metrosep A Supp-5, 4 × 150 mm, anion column) and a cation IC (Metrohm model 861 without a suppressor, equipped with a Metrohm Metrosep C4, 4 × 150 mm, cation column). The eluants were 3.2 mM sodium carbonate (Na2CO3)/1.0 mM sodium bicarbonate (NaHCO3) for the anion IC and 1.7 mM nitric acid (HNO3)/0.7 mM dipicolinic acid for the cation IC. The eluant flow rate for the anion and cation was maintained at 0.7 and 1.0 mL min−1, respectively. The detection limits for NO3−, SO42−, and NH4+ were measured to be 0.08, 0.17, and 0.05 μg m−3, respectively.
Group separation of WSOC fractions using XAD resin column
In order to separate the total WSOC in the filter samples from the second sampler into hydrophilic and hydrophobic fractions, a XAD7HP resin (Rohm & Haas France S.A.S) was used. Some previous studies have used XAD-8, DAX-8, or OASIS HLB resin to isolate WSOC hydrophobic fractions from ambient air samples.8,22,23,34 Because XAD-8 resin is no longer commercially available, a substitute resin, XAD7HP, was used in this study. The XAD7HP is a polymeric adsorbent available as white insoluble beads. It is a nonionic, aliphatic acrylic crosslinked polymer which derives its adsorptive properties from its macroreticular structure, high surface area and the aliphatic nature of its surface. The organic compounds that passed through the XAD7HP column are the more hydrophilic fractions, while those retained are strongly related to the hydrophobic WSOC components. The XAD7HP adsorbent was cleaned prior to use until the organic compounds and salts had been removed. In this study, the resin was primarily pre-treated with methanol and acetone for 48 h each using a Soxhlet extraction apparatus. This primary washing cycle was repeated three times. After the Soxhlet treatments the resin was washed with DDW for 2.1 h to eliminate the residual solvent. For the group separation of the WSOC, a 6 mm ID × 10 cm long aqueous chromatography column (Spectrum Laboratories, Inc., Houston, TX, USA), hand-packed with the resin, was used with TOC detection. Before the liquid extracts were continuously pumped onto the XAD7HP resin, the washing cycle of the resin column consisted of alternating 0.1 M sodium hydroxide (NaOH) and 0.1 M hydrogen chloride (HCl) for 20 min each, all at flow rates of 2.0 mL min−1 by a multi-channel peristaltic pump (Ismatec IPC-N-16). An intermediate washing step with DDW was carried out between alkali and acid treatments. This NaOH–HCl cycle was repeated three times. After all the washing procedures were completed, the resin column was tested three times with DDW. The DDW passing through the column was analyzed using the TOC analyzer to check the quality of the washing. The TOC levels before (“total WSOC”) and after (“hydrophilic WSOC”) passing through the resin column were found to be 10–15 and 7–10 ppb C, respectively, which may be negligible, implying that the washed quality of the resin was fairly good. The average blank values were used as the background for subtraction from each filter sample. Prior to loading, the aqueous sample solution was adjusted to pH 2 with HCl, and then introduced onto the XAD7HP resin column at a rate of 2.0 mL min−1. The flow rate over the column is changed from the cleaning phase to sample loading. The sample flow rate through the column was maintained at 1.3 mL min−1 for 20 min. In this study, the organic compounds penetrating the XAD7HP column were measured using the TOC analyzer, and referred to as the hydrophilic fraction. Recovery experiments were conducted to extract the hydrophobic fraction of WSOC for the resin column, but smaller amounts were quantified than real amounts of the hydrophobic WSOC fraction (=total WSOC − hydrophilic WSOC). It varied with the sample extracts, but approximately 15% or more could be unrecovered in the pH 13 eluent. Similar results were also obtained by other researchers.20,22 Thus, the difference between the total and hydrophilic WSOC was defined as the hydrophobic WSOC fraction.The penetration efficiencies of the XAD7HP resin column were examined using a variety of water-soluble organic compounds relevant to atmospheric aerosols. These experiments were performed for a few organic standard solution concentrations. The results of the organic compounds penetration tests can be found elsewhere.35 Briefly, the experimental results showed that aliphatic dicarboxylic acids and carbonyls (<4 carbons), amines and saccharides were hydrophilic WSOC. The hydrophobic WSOC included aliphatic dicarboxylic acids (>4–5 carbons), phenols, aromatic acids and cyclic acid, as well as Suwannee River fulvic acid. Since the hydrophobic WSOC was calculated from the total and the hydrophilic WSOC contents, some uncertainty may exist in the determination of the hydrophobic WSOC fraction. The detection limits for the hydrophilic and hydrophobic WSOC measurements made with the field blanks were estimated to be 0.10 and 0.08 μg C m−3, respectively.
Results and discussion
General characteristics of PM2.5 and its chemical species
A summary of the statistics for PM2.5, OC, EC, total WSOC, hydrophilic and hydrophobic WSOC fractions, OC/EC, WSOC/OC, and major ionic components are presented in Table 1. A summary of the PM2.5 and its chemical species for the pathways of the different air mass types is also presented in Table 1. Fig. 2 shows temporal profiles of PM2.5, OC, EC, NO3−, SO42−, and NH4+ concentrations over the study period. Time-series plots of total WSOC and hydrophilic WSOC concentrations are shown in Fig. 3. The observed OC and EC concentrations were 4.4 (2.1–6.9 μg C m−3) and 1.4 μg C m−3 (0.6–2.1 μg C m−3), respectively. The OC/EC values ranged from 2.0 to 4.7, with an average of 3.3. The contributions of organic matter (OM = 1.6 × OC)36 and EC were 27.2 ± 8.4 (range: 11.4–44.0%) and 5.6 ± 2.4% (range: 2.1–11.8%) of the PM2.5 mass, respectively. The total average fraction of the sum of three major inorganic water-soluble components (NO3−, SO42− and NH4+) was 40.9% (9.1–63.9%) of the PM2.5, with SO42− particles being the most dominant, accounting for 26.9% (6.5–44.0%) of the PM2.5 mass concentration. | ||
| Fig. 2 Temporal profiles of PM2.5 and its major chemical species concentrations. | ||
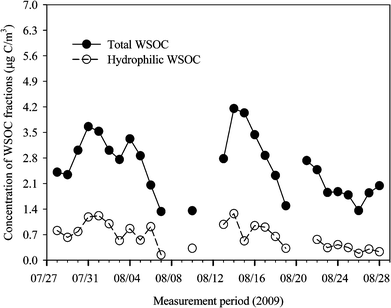 | ||
| Fig. 3 Temporal plot of total WSOC and hydrophilic WSOC concentrations. | ||
| Parameter | Entire data set | Air mass type | ||||
|---|---|---|---|---|---|---|
| Unit | Mean | Range | I | II | III | |
| PM2.5 | μg m−3 | 26.5 | 10.5–59.6 | 24.8 ± 5.5 | 22.3 ± 7.8 | 35.6 ± 13.7 |
| OC | μg C m−3 | 4.4 | 2.1–6.9 | 4.5 ± 0.6 | 3.9 ± 1.1 | 5.0 ± 2.2 |
| EC | μg C m−3 | 1.4 | 0.6–2.1 | 1.3 ± 0.4 | 1.3 ± 0.3 | 1.5 ± 0.6 |
| WSOC | μg C m−3 | 2.7 | 1.3–4.2 | 2.8 ± 0.5 | 2.2 ± 0.7 | 3.2 ± 1.3 |
| Hydrophilic | μg C m−3 | 0.7 | 0.2–1.3 | 0.8 ± 0.2 | 0.5 ± 0.3 | 0.7 ± 0.4 |
| Hydrophobic | μg C m−3 | 2.0 | 1.0–3.5 | 2.0 ± 0.3 | 1.6 ± 0.5 | 2.5 ± 0.9 |
| OC/EC | — | 3.3 | 2.0–4.7 | 3.5 ± 0.9 | 3.2 ± 0.8 | 3.3 ± 0.5 |
| WSOC/OC | — | 0.61 | 0.36–0.77 | 0.57 ± 0.09 | 0.53 ± 0.08 | 0.64 ± 0.06 |
| NO3− | μg m−3 | 1.2 | 0.1–5.1 | 1.5 ± 0.9 | 0.6 ± 0.3 | 1.9 ± 2.0 |
| SO42− | μg m−3 | 7.8 | 0.7–26.2 | 6.0 ± 2.2 | 5.8 ± 3.9 | 13.4 ± 7.0 |
| NH4+ | μg m−3 | 2.8 | 0.1–9.3 | 2.5 ± 0.9 | 2.0 ± 1.4 | 4.6 ± 2.3 |
| SO42−/PM2.5 | % | 26.9 | 6.5–44.0 | 23.8 ± 6.9 | 23.6 ± 8.8 | 36.1 ± 6.3 |
| Σsecondary ions/PM2.5 | % | 40.9 | 9.1–63.9 | 39.5 ± 8.8 | 34.4 ± 12.6 | 53.7 ± 7.2 |
| SO42−/(SO2 + SO42−) | — | 0.51 | 0.12–0.79 | 0.49 | 0.43 | 0.70 |
As indicated in Table 1, the average contributions of carbonaceous species (OM, total WSOC, and EC) to the PM2.5 mass were higher in the types I and II than the type III air mass periods when the air masses had originated from polluted northeastern regions of China and were transported over metropolitan areas of Korea. The PM2.5 mass concentration observed for the type III air mass period (August 13–15, 19, 24, 27–28) was dominated by secondary inorganic species (46.6–63.9%, mean: 53.7%), especially SO42− (27.6–44.0%; mean: 36.1%), as shown in Fig. 2. The contribution of the secondary ionic species was about 20% higher in the type III air mass than in the types I and II. For the types I and II air masses, the average contributions (35.3–36.3%) of carbonaceous materials (OM + EC) to the PM2.5 mass were comparable to, or slightly lower than those of the sum of the secondary inorganic components, while the contribution of carbonaceous species for the type III air mass was about 2.1 times lower than that of the secondary ionic species (53.7%). The SO42−/SOx(=SO2 + SO42−) ratio has been reported to increase with aging of the air mass due to the chemical conversion of SO2 to SO42− and faster removal of SO2 from the atmosphere.37 In the present study, SO42−/SOx for the type III air mass period was 0.70, which was relatively high compared to those for types I and II air masses (0.43–0.49). This suggests that the chemical components of the observed PM2.5 during the type III period were further photo-chemically processed, rather than significantly influenced by primary emissions from urban sources. The OC to EC ratio was within the range 3.1–3.5 for the types I, II, and III air masses. No significant difference in the OC/EC was observed among the air mass types.
Investigation of chemical evolution of OC using bulk WSOC and other parameters
The WSOC fraction in the OC could provide an important key to the composition and chemical processes of organic aerosols. The WSOC concentration ranged from 1.3 to 4.2 μg C m−3, with a mean of 2.7 μg C m−3. The highest WSOC concentration (based on carbon mass), observed on August 14th, accounted for 62.0% of the measured OC. As shown in Table 1, the daily average WSOC/OC value was 0.61, ranging from 0.36 to 0.77, which was very similar to that found in the study previously conducted at the same sampling site during summer.9,10 Based on the classified air mass types, the average WSOC/OC value was highest (0.64) for the type III period when the air masses had originated from the northeastern polluted areas of China and transported over the Korean peninsula, with a slow air mass transport velocity. For the types I and II air masses, the WSOC/OC was within the range of 0.53–0.57. The high WSOC/OC value observed during the type III air mass period might be attributable to further atmospheric transformation processing of volatile organic species during the transport of air masses. Evidence for this result can be observed by the high SO42−/SOx during the type III air mass period. The result from a regression analysis between the WSOC and OC is shown in Fig. 4. The close correlation between the WSOC and OC concentrations (R2 = 0.86, p < 0.01) suggested that the OC and WSOC at the site had very similar chemical characteristics. The water-insoluble OC (WIOC) concentrations ranged from 0.7 to 2.9 μg C m−3, with a mean value of 1.6 μg C m−3, accounting for 23 to 64% (mean 41%) of the OC. It has been suggested that the WIOC in urban areas is most likely formed via incomplete combustion of fossil fuels.38 As shown in Fig. 4, the WIOC was correlated with the EC, which is known as a tracer of incomplete combustion of fossil fuels, with a R2 value of 0.51 and a slope of 1.01. The relationship between the EC and WIOC suggested that some of the observed WIOC was associated with primary combustion emissions.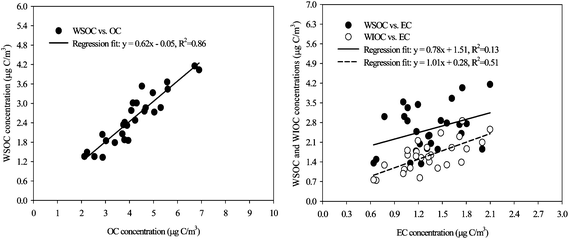 | ||
| Fig. 4 Regression relationships of WSOCversusOC and of WSOC and WIOCversus EC. | ||
To investigate the chemical evolution of OC, the relationships between the WSOC and other parameters were examined, the results of which are also shown in Fig. 4. The WSOC and EC concentration was poorly correlated, with a R2 value of 0.13, suggesting that significant amounts of the observed WSOC were unlikely to be due to primary combustion emissions. Several previous studies have also indicated that primary urban emissions of WSOC were negligible.16,39 Therefore, several possibilities could be considered to account for the high WSOC fractions of the total OC at the site. Firstly, some studies have revealed that the photochemistry of O3 was positively associated with the WSOC concentration.4,40 The average OC/EC of 3.3 (2.0–4.7) in this study was greater than the OC/EC of primary emissions (1.5–1.7) reported for an urban site in Gwangju, Korea.41 An enhanced OC/EC during the summer may reflect enhanced secondary organic aerosol production due to the more favorable conditions for gas/particle conversion of volatile organic compounds as a result of photochemical activity. In this study, the OC/EC, WSOC/OC, OC and WSOC were regressed as a function of the sum of O3 and NO2 concentrations. The reason we used the sum of O3 and NO2 as an indicator for photochemical processing is that in urban areas, the O3 may be not a good indicator for photochemical processing due to the fast reaction of NO + O3 ↔ NO2 and there being a significant amount of NO present. However, the 24 h average O3 + NO2 concentration observed at the site was poorly correlated with the OC/EC (R2 = 0.13), WSOC/OC (R2 = 0.06), OC (R2 = 0.13) and WSOC (R2 = 0.15) (not shown). These poor correlations may suggest that a significant fraction of the observed WSOC was associated with aged secondary products formed through atmospheric processing of primary organic species during the transport of air masses, rather than through local photochemical processes, which are linked to O3 production. Secondly, previous studies have suggested that heterogeneous acid-catalyzed reactions could enhance the production of secondary organic aerosol (SOA) in acidic aerosol environments.42–47 In this study, in situ aerosol properties such as acidity and the liquid water content of the aerosols were evaluated to investigate the aerosol acidity characteristics and the role of heterogeneous acid-catalyzed reactions in SOA formation. The aerosol acidity of PM2.5 ([H+]total) was estimated using the following relationship: [H+]total = 2[SO42−] + [NO3−] − [NH4+]. The in situ acidity ([H+]in situ) and aerosol water content were estimated using a thermodynamic model (Aerosol Inorganic Mode: E-AIM; http:///www.aim.env.uea.ac.uk/aim/aim.php)48 described elsewhere.47,49 Aerosol acidity was in the range of −1 to 192 nmol m−3 with a mean of 24 nmol m−3. The average aerosol acidity levels determined in this study were lower than those in aerosol samples collected at four major cities in China (Beijing, Shanghai, Guangzhou, and Lanzhou).49 The in situ acidity was on average 10 nmol m−3, accounting for 42% of the aerosol acidity due to the aerosol water content in the filter samples. However, the in situ acidity indicated weak correlations with the WSOC and WSOC/OC, with R2 values of 0.01 and 0.01, respectively. This result suggests that acid-catalyzed heterogeneous reactions were also unlikely to be a possible pathway for the formation of WSOC enhancements in the acidic aerosols. Therefore, it is not certain, but it can be suggested that the atmospheric transformation processes during the transport of air masses over distances could be a possible pathway for the formation of the observed WSOC (or OC) at the site.
To further demonstrate the production processes of WSOC particles, the correlations between the WSOC and OC/EC, and the WSOC/OC and SO42−/SOx were calculated, the results of which are shown in Fig. 5. As seen in Fig. 5, the total WSOC mass concentrations during the summer study period tended to increase with increasing OC/EC (R2 = 0.61, p < 0.05). Because the SO42−/SOx can be used as an indicator of the formation process of SO42− aerosol,37,50 the chemical transformation processes of the WSOC can presumably be evaluated from the relationship between the WSOC and SO42−/SOx. The WSOC/OC was highly correlated with the SO42−/SOx, with a R2 of 0.64. This would suggest that a significant fraction of the observed WSOC was formed in chemical processes similar to SO42− aerosols, which are produced by in-cloud aqueous-phase oxidation, or photochemical oxidation of SO2 by OH radicals.51 Also this result could explain why there was a weak correlation of the OC/EC, WSOC/OC, OC, and WSOC with O3 + NO2, as discussed above.
 | ||
| Fig. 5 Relationships between WSOC and OC/EC, and between WSOC/OC and SO42−/SOx. | ||
Investigation of chemical evolution of OC using isolated WSOC fractions
In the previous section, it was examined that a significant fraction of the observed WSOC could be largely produced via atmospheric transformation processes. To further examine the WSOC production processes, the characteristics of the fractionated WSOC (hydrophilic and hydrophobic WSOC) are discussed below. As shown in Table 1, the hydrophobic WSOC concentration was observed to be greater than the hydrophilic fraction. The average hydrophilic (WSOCHPI) and hydrophobic WSOC (WSOCHPO) concentrations were 0.7 (0.2–1.3) and 2.0 (1.0–3.5) μg C m−3, respectively. The average mass concentration of WSOCHPO accounted for 75.3% of the total WSOC, about three times more than that of the WSOCHPI (24.7%). The relationships between the isolated WSOC fractions and total WSOC (not shown) indicated that the 24 h averaged variability in WSOC concentration was more highly associated with the WSOCHPO fraction (WSOCHPO = 0.63WSOC + 0.26, R2 = 0.92) than the WSOCHPI fraction (WSOCHPI = 0.37WSOC – 0.26, R2 = 0.79). Similarly, when compared to OC, the R2 value for the WSOCHPO (WSOCHPO = 0.45OC + 0.04, R2 = 0.83) was greater than that for the WSOCHPI (WSOCHPI = 0.19OC – 0.12, R2 = 0.46). These results suggest that the hydrophobic WSOC mass was an important factor that could contribute to the OC mass of fine particles at the site. The results from our sampling location were the inverse of those from an urban Atlanta site, USA, where the hydrophilic WSOC fractions in the PM2.5 accounted for ∼60% of the total WSOC and dominated throughout the summer.22 The observed contrasts between the two urban sites could have been due to several factors, such as the different atmospheric chemical and meteorological processes, regional characteristics and local emissions. As discussed in the Experimental section, our sampling site was frequently affected by Asian continental outbreaks of polluted air and anthropogenic aerosols from polluted regions of the Korean peninsula. The results from the study of Miyazaki et al.,20 which was conducted at a rural site in PRD, China, during summer, suggested that significant amounts of hydrophobic WSOC compounds, containing large carbon numbers (>C4), were produced by photochemical processing. Also, previous studies from urban sites or a rural site in China, and the Gosan background site, Korea, indicated that significant fractions of the WSOC were highly refractory compounds related to hydrophobic WSOC.40,52,53As with the relationship between the WSOC and EC (see Fig. 4), both the WSOCHPI and WSOCHPO were poorly correlated with the EC, with R2 values of 0.06 and 0.13, respectively (not shown). This implies that primary combustion emissions were not important production routes of either the WSOCHPI or WSOCHPO fractions during summer at the site. Also, the OC/EC was more highly correlated with the WSOCHPO (R2 = 0.55) than WSOCHPI (R2 = 0.37) (see Fig. 5). This indicated that an increase in the OC/EC at the site resulted in the increased mass of the WSOCHPO and was accompanied by the addition of the secondary organic aerosol mass.
Conclusion
Daily measurements of the total and chemically isolated water-soluble organic carbon (WSOC) in the PM2.5 were made at an urban site in Gwangju, Korea, for the period between July 28 and August 28, 2009. Quantifying the mass of the broad chemical groupings of WSOC aerosols could provide unique insights into the sources of organic aerosols. XAD7HP resin, with total organic carbon (TOC) detection, was employed to chemically quantify the hydrophilic and hydrophobic WSOC fractions (WSOCHPI and WSOCHPO).The poor correlation between the WSOC and EC (R2 = 0.13) suggests that the WSOC observed at the site was likely due to atmospheric processing of primary organic species during the transport of air masses. Also the good relationship between the WSOC/OC and SO42−/SOx (R2 = 0.64) would suggest that a significant fraction of the observed WSOC is formed in a process similar to the SO42− aerosols, probably through in-cloud processing, or the oxidation processes of SO2 using OH radicals. On average, the WSOC concentration accounted for 61% of the total OC at the sampling site during the study period, of which WSOCHPO accounted for 75.3% of the total WSOC. It follows that the sources of the WSOC fraction of OC, especially the WSOCHPO fraction, were a large contributor to the OC during the summer period at the site. Based on the air mass types, the average WSOC/OC was highest for the type III period when the air masses originated from the northeastern polluted areas of China and passed over the metropolitan areas of Korea. This result could be explained by further aging of the air masses, as demonstrated by the high SO42−/SOx observed during the type III air mass period. Finally, our results indicated that the photochemical production of hydrophobic WSOC was a significant contributor to the total OC.
Acknowledgements
This work was supported by the Mid-career Researcher Program through a NRF grant funded by the Korea government (MEST) (no. R01-2008-000-20255-0). This work also was partially supported by the General Researcher Program through a NRF grant funded by the Korea government (MEST) (no. 2011-0007222).References
- P. Saxena and L. M. Hildemann, J. Atmos. Chem., 1996, 24, 57–109 CrossRef CAS.
- S. Decesari, M. C. Facchini, E. Matta, S. Fuzzi and E. Tagliavini, J. Geophys. Res., 2000, 105, 1481–1489 CrossRef CAS.
- S. Zappoli, A. Andracchio, S. Fuzzi, M. C. Facchini, A. Gelencsér, G. Kiss, Z. Krivácsy, A. Molnár, E. Mészáros, H. C. Hansson, K. Rosman and Y. Zebühr, Atmos. Environ., 1999, 33, 2733–2743 CrossRef CAS.
- A. P. Sullivan, R. J. Weber, A. L. Clements, J. R. Turner, M.-S. Bae and J. J. Schauer, Geophys. Res. Lett., 2004, 31, L13105 CrossRef.
- J.-L. Jaffrezo, G. Aymoz, C. Delaval and J. Cozic, Atmos. Chem. Phys., 2005, 5, 2809–2821 CrossRef CAS.
- H. Yang, J. Z. Yu, S. S. H. Ho, J. Xu, W.-S. Wu, C. H. Wan, X. Wang and L. Wang, Atmos. Environ., 2005, 39, 3735–3749 CrossRef CAS.
- B. T. Madar, J. Z. Yu, J. H. Xu, Q. F. Li, W. S. Wu, R. C. Flagan and J. H. Seinfeld, J. Geophys. Res., 2004, 109, D06206, DOI:10.1029/2003jd004105.
- Y. Miyazaki, Y. Kondo, N. Takegawa, Y. Komazaki, M. Fukuda, K. Kawamura, M. Mochida, K. Okuzawa and R. J. Weber, J. Geophys. Res., 2006, 111, D23206, DOI:10.1029/2006jd007125.
- S. S. Park, J. Y. Hur, S. Y. Cho, S. J. Kim and Y. J. Kim, J. Korean Soc. Atmos. Environ., 2007, 23, 675–688 CrossRef , in Korean with English abstract.
- S. S. Park and S. Y. Cho, Atmos. Environ., 2011, 45, 60–72 CrossRef CAS.
- C. N. Cruz and S. N. Pandis, Environ. Sci. Technol., 2000, 35, 4313–4319 CrossRef.
- S. Decesari, M. C. Facchini, S. Fuzzi, G. B. McFiggans, H. Coe and K. N. Bower, Atmos. Environ., 2005, 39, 211–222 CrossRef CAS.
- R. Sorjamaa, B. Svenningsson, T. Raatikainen, S. Henning, M. Bilde and A. Laaksonen, Atmos. Chem. Phys., 2004, 4, 2107–2117 CrossRef CAS.
- P. Saxena and L. M. Hildemann, J. Atmos. Chem., 1996, 24, 57–109 CrossRef CAS.
- Y. Kondo, Y. Miyazaki, N. Takegawa, T. Miyakawa, R. J. Weber, J. L. Jimenez, Q. Zhang and D. R. Worsnop, J. Geophys. Res., 2007, 112, D01203, DOI:10.1029/2006jd007056.
- R. J. Weber, et al. , J. Geophys. Res., 2007, 112, D13302 CrossRef.
- S. Decesari, M. C. Facchini, E. Matta, F. Lettini, M. Mircea, S. Fuzzi, E. Tagliavini and J.-P. Putaud, Atmos. Environ., 2001, 35, 3691–3699 CrossRef CAS.
- Z. Krivácsy, A. Gelencsér, G. Kiss, E. Mészáros, A. Molnár, A. Hoffer, T. Mészáros, Z. Sárvári, D. Temesi, B. Varga, U. Baltensperger, S. Nyeki and E. Weingartner, J. Atmos. Chem., 2001, 39, 235–259 CrossRef.
- G. Kiss, B. Varga, I. Galambos and I. Ganszky, J. Geophys. Res., 2002, 107(D21), 8339 CrossRef.
- Y. Miyazaki, Y. Kondo, M. Shiraiwa, N. Takegawa, T. Miyakawa, S. Han, K. Kita, M. Hu, Z. Q. Deng, Y. Zhao, N. Sugimoto, D. R. Blake and R. J. Weber, J. Geophys. Res., 2009, 114, D14208 CrossRef.
- P. Sannigrahi, A. P. Sullivan, R. J. Weber and E. D. Ingall, Environ. Sci. Technol., 2006, 40, 666–672 CrossRef CAS.
- A. P. Sullivan and R. J. Weber, J. Geophys. Res., 2006, 111, D05314 CrossRef.
- A. P. Sullivan and R. J. Weber, J. Geophys. Res., 2006, 111, D05315 CrossRef.
- R. M. B. Duarte and A. C. Duarte, J. Atmos. Chem., 2005, 51, 79–93 CrossRef CAS.
- J. H. Jeong, J. H. Kim, S. S. Park, K. J. Moon and S. J. Lee, J. Korean Soc. Atmos. Environ., 2011, 27, 337–346 CrossRef , (in Korean with English abstract).
- N. Havers, P. Burba, J. Lambert and D. Klockow, J. Atmos. Chem., 1998, 29, 45–54 CrossRef CAS.
- M. C. Facchini, S. Decesari, M. Mircea, S. Fuzzi and G. Loglio, Atmos. Environ., 2000, 34, 4853–4857 CrossRef CAS.
- G. Kiss, E. Tombacz and H. C. Hansson, J. Atmos. Chem., 2005, 50, 279–294 CrossRef CAS.
- H. L. Lee, S. S. Park, K. W. Kim and Y. J. Kim, Atmos. Res., 2008, 88, 199–211 CrossRef CAS.
- S. S. Park, Y. J. Kim, S. Y. Cho and S. J. Kim, J. Air Waste Manage. Assoc., 2007, 57, 434–443 CAS.
- R. R. Draxler and G. D. Rolph, HYSPLIT (Hybrid Single-Particle Lagrangian Integrated Trajectory) Model, NOAA ARL, Silver Spring, MD, 2003, http://www.arl.noaa.gov/ready/hysplit4.html. Search PubMed.
- National Institute of Occupational Safety and Health (NIOSH), Method 5040 Issue 1: Elemental Carbon (Diesel Exhaust), NIOSH Manual of Analytical Methods, Cincinnati, 4th edn, 1996 Search PubMed.
- S. S. Park, J.-M. Ko and S. Y. Cho, Atmos. Environ., 2011, 45, 3257–3266 CrossRef CAS.
- B. Varga, G. Kiss, I. Ganszky, A. Gelencsér and Z. Krivácsy, Talanta, 2001, 55, 561–572 CrossRef CAS.
- S. S. Park, J.-H. Kim, J. U. Jeong and S. Y. Cho, Environ. Eng. Sci., submitted Search PubMed.
- B. J. Turpin and H.-J. Lim, Aerosol Sci. Technol., 2001, 35, 602–610 CAS.
- L. K. Sahu, Y. Kondo, Y. Miyazaki, M. Kuwata, M. Koike, N. Takegawa, H. Tanimoto, H. Matsueda, S. C. Yoon and Y. J. Kim, J. Geophys. Res., 2009, 114, D03301 CrossRef.
- B. R. T. Simoneit, M. Kobayashi, M. Mochida, K. Kawamura, M. Lee, H.-J. Lim, B. J. Turpin and Y. Komazaki, J. Geophys. Res., 2004, 109, D19S10 CrossRef.
- X.-F. Huang, J. Z. Yu, L.-Y. He and Z. Yuan, J. Geophys. Res., 2006, 111, D22212 CrossRef.
- Y. Miyazaki, Y. Kondo, S. Han, M. Koike, D. Kodama, Y. Komazaki, H. Tanimoto and H. Matsueda, J. Geophys. Res., 2007, 112, D22S30 CrossRef.
- S. S. Park, Y. J. Kim and K. Fung, Atmos. Environ., 2002, 36, 1287–1297 CrossRef CAS.
- M. Jang, N. M. Czoschke, S. Lee and R. M. Kamens, Science, 2002, 298, 814–817 CrossRef CAS.
- S.-H. Chu, Atmos. Environ., 2004, 38, 5237–5246 CrossRef CAS.
- S. Takahama, C. I. Davidson and S. N. Pandis, Environ. Sci. Technol., 2006, 40, 2191–2199 CrossRef CAS.
- Q. Zhang, J. L. Jimenez, D. R. Worsnop and M. Canagaratna, Environ. Sci. Technol., 2007, 41, 3213–3219 CrossRef CAS.
- R. L. Tanner, K. J. Olszna, E. S. Edgerton, E. Knipping and S. L. Shaw, Atmos. Environ., 2009, 43, 3440–3444 CrossRef CAS.
- R. K. Pathak, T. Wang, K. F. Ho and S. C. Lee, Atmos. Environ., 2011, 45, 318–325 CrossRef CAS.
- S. L. Clegg, P. Brimblecombe and A. S. Wexler, J. Phys. Chem., 1998, 102A, 2155–2171 Search PubMed.
- R. K. Pathak, W. S. Wu and T. Wang, Atmos. Chem. Phys., 2009, 9, 1711–1722 CrossRef CAS.
- N. Kaneyasu, S. Ohta and N. Murao, Atmos. Environ., 1995, 29, 1559–1568 CrossRef CAS.
- E. V. Fischer, L. D. Ziemba, R. W. Talbot, J. E. Dibb, R. J. Griffin, L. Husain and A. N. Grant, J. Geophys. Res., 2007, 112, D02303, DOI:10.1029/2006jd007253.
- H. Yang, J. Z. Yu, S. S. H. Ho, J. Xu, W.-S. Wu, C. H. Wan, X. Wang and L. Wang, Atmos. Environ., 2005, 39, 3735–3749 CrossRef CAS.
- J. Z. Yu, H. Yang, H. Zhang and A. K. H. Lau, Atmos. Environ., 2004, 38, 1061–1071 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |