Colloidal mercury (Hg) distribution in soil samples by sedimentation field-flow fractionation coupled to mercury cold vapour generation atomic absorption spectroscopy
A.
Santoro
a,
R.
Terzano
*a,
L.
Medici
b,
M.
Beciani
c,
A.
Pagnoni
c and
G.
Blo
c
aDipartimento di Biologia e Chimica Agroforestale ed Ambientale, University of Bari, Via Amendola 165/A, 70126, Bari, Italy. E-mail: r.terzano@agr.uniba.it
bConsiglio Nazionale delle Ricerche (CNR), Istituto di Metodologie per l'Analisi Ambientale (IMAA), Contrada S. Loja, I-85050, Tito Scalo, Potenza, Italy
cDipartimento di Chimica, University of Ferrara, Via L. Borsari 46, 44100, Ferrara, Italy
First published on 16th November 2011
Abstract
Diverse analytical techniques are available to determine the particle size distribution of potentially toxic elements in matrices of environmental interest such as soil, sediments, freshwater and groundwater. However, a single technique is often not exhaustive enough to determine both particle size distribution and element concentration. In the present work, the investigation of mercury in soil samples collected from a polluted industrial site was performed by using a new analytical approach which makes use of sedimentation field-flow fractionation (SdFFF) coupled to cold vapour generation electrothermal atomic absorption spectroscopy (CV-ETAAS). The Hg concentration in the SdFFF fractions revealed a broad distribution from about 0.1 to 1 μm, roughly following the particle size distributions, presenting a maximum at about 400–700 nm in diameter. A correlation between the concentration of Hg in the colloidal fraction and organic matter (O.M.) content in the soil samples was also found. However, this correlation is less likely to be related to Hg sorption to soil O.M. but rather to the presence of colloidal mercuric sulfide particles whose size is probably controlled by the occurrence of dissolved O.M. The presence of O.M. could have prevented the aggregation of smaller particles, leading to an accumulation of mercuric sulfides in the colloidal fraction. In this respect, particle size distribution of soil samples can help to understand the role played by colloidal particles in mobilising mercury (also as insoluble compounds) and provide a significant contribution in determining the environmental impact of this toxic element.
Environmental impactSoil colloidal particles, if mobilised, can spread potentially toxic elements throughout the environment, thus endangering the health of living organisms and human beings. This issue is particularly relevant for elements in insoluble forms which anyway can migrate in water fluxes as submicronic particles. In this research an analytical method to correlate Hg concentration with particle size for the analysis of the colloidal fraction of soil samples has been developed and applied to soil samples collected in an area polluted by the activity of a chlor-alkali plant. Hg was found mostly concentrated in the fraction between 400 and 700 nm. Despite Hg being mostly speciated as insoluble mercuric sulfides, the small size of the particles could still create serious environmental risks. |
1. Introduction
In recent years, interest in the nature of fine particles in soils has increased, mostly arising from the understanding that contaminants move through soils at a much faster rate than hydrological models predict.1 This discrepancy has been attributed to colloid transport, which can carry the contaminants strongly sorbed to colloidal particles.2,3 These particles have a size range from 0.001 to 1.0 μm and are often found in low concentrations and in the form of diverse organo-mineral complexes. Since significant concentrations of pollutants are usually found in smaller soil particles,4 it is reasonable to think that soil colloidal particles (either organic or inorganic) if mobilised can spread potentially toxic compounds throughout the environment, thus endangering the health of living organisms and human beings.As a recognised global environmental pollutant,5–7mercury attracts special interest in soil science, also because it is often found concentrated in the clay (<2 μm) fraction.8
However, for many decades, determination of particle size distributions involving Hg, in natural heterogeneous samples, has been limited not only by the techniques available but also by modifications in the original sample (e.g. aggregation and disaggregation processes) likely to occur during sampling and sample preparation that can affect the measurements.9 More recently, together with traditional filtration and centrifugation, a wide range of methods (electron microscopy, laser diffraction, field-flow fractionation—FFF—techniques, etc.) for the characterisation of particle size distribution has been developed.9,10 In a detailed overview on fractionation methods, Leadet al.11 have pointed out that conventional techniques such as filtration, ultrafiltration and ultracentrifugation are all affected by numerous factors which can lead to a poor resolution of size distribution; whereas different FFF sub-techniques allow a higher resolution.12,13
The main advantage of FFF techniques attracting many scientists is the relatively low interaction between the sample and the separation system as well as the reduced alteration of the sample (e.g.coagulation) during fractionation steps. Another important advantage is the possibility of off-line or on-line coupling of a FFF technique with specific detection systems for a subsequent chemical particle characterisation.14–19
Among the FFF techniques, sedimentation field-flow fractionation (SdFFF) exploits a centrifugal field to separate the particles according to their buoyant mass, which is in turn proportional to the equivalent spherical diameter provided the particle density is constant.20 In a typical SdFFF instrument, the particles are first injected into the separation channel without the field or flows applied, and are distributed by diffusion across the thickness of the channel (injection point). When the field is applied, the solute zone is compressed by the applied centrifugal field into a narrow layer against one wall (relaxation at the accumulation wall). Then, the liquid flow is started and the solute zone is carried downstream at a rate depending on the particle layer thickness, which is related to particle size and density (separation). In the normal (or Brownian) mode of elution, this results in an increase in elution time for larger particles.
When SdFFF is coupled on-line or off-line to an atomic absorption spectrometer (AAS), the elemental characterisation of the separated particles can be determined.21 The UV-Vis signal versus the particle diameter fractogram obtained from SdFFF separation can be transformed into a graph showing the concentration of the element in particles (determined by AAS) versus diameters, giving the size distribution of the element of interest.22
The technique has already been used for the fractionation of colloids in natural water samples;12,16 however only a few studies, involving colloidal distribution of Mn23 or Cd,24 have been made on soil and sediment samples. In this respect, mercury determination in soils has rarely been considered due to the usual difficulties encountered in its analytical determination.
In this study, soil samples contaminated with Hg have been examined using SdFFF separation coupled to cold vapour generation electrothermal atomic absorption spectrometry (CV-ETAAS) to characterise the colloidal Hg-bearing fractions.
2. Materials and methods
2.1 Soil samples: collection and preparation
Soil samples were collected from an industrial area located in the south of Italy near the village of Ferrandina in the district area of Matera.25 The site has been recognised as contaminated by diffuse pollution because of the large number of industrial plants that have been installed one after the other during the past fifty years.26,27 In particular, a decommissioned chlor-alkali plant seems to have acted as a significant source of mercury contamination to the environment.28–30In the frame of a monitoring project aimed at evaluating the state of pollution of the area, numerous samples were collected all around the industrial site at different soil depths and by following different sampling schemes.25 Taking into account the dramatic dilution imposed by the SdFFF technique, only three samples (A1, A2, A3) containing the highest amounts of Hg were used in this study. These samples can be considered representative of the investigated area. A1 and A2 were collected at a depth of 0–10 cm, while A3 at 40–50 cm.
After collection, soil samples were homogenised, air dried, sieved at 2 mm, and analysed for the total Hg content. The soil clay fraction (<2 μm) was separated by using an in-house developed procedure, based on repeated sonication–centrifugation steps. This procedure facilitated the separation of the soil aggregates without modifying Hg speciation.28
An amount of 3.5 g of soil was mixed with 50 mL of ultrapure deionised water and stirred for 10 min. Then, the solution was sonicated for 10 min at 100 W by means of a Sonics Vibra-Cell (Sonics and Materials Inc., USA) to break the soil aggregates. After sonication, the suspension was centrifuged for 2.07 minutes at 600 rpm (according to Stokes' equation and assuming a particle density of 2.65 g cm−3 as these conditions yield the 2 μm diameter of the silt/clay boundary31) by using a Mod. CR15 (B. Braun Biotech International) centrifuge. Then the supernatant containing the clay fraction was recovered. The optimal number of repeated centrifugations, performed in order to quantitatively extract the clay fraction, was experimentally established as five repetitions. The pellets obtained by centrifugation of the supernatant for 30 minutes at 5500 rpm from each repetition were finally combined, weighed, lyophilised by using a Hetosicc lyophiliser (Hetolab equipment, Denmark) and stored at 4 °C for further analysis.
In order to verify the reliability of this procedure, two other methods for clay fraction extraction, i.e. repeated sedimentation–syphonation cycles32 and wet sieving,33 were used. The gravity sedimentation was performed by adding 40 g of soil to 400 mL of water in a 500 mL beaker.34 The solution was stirred overnight and then allowed to settle for 3 hours and 30 minutes. The material in the top 5 cm (<2 μm fraction) was recovered by syphonation, and the entire process was repeated 5 times. The five fractions were then collected together and the clay sized material was recovered by centrifugation at 5500 rpm for 30 minutes.
For the wet sieving, 60 g of air-dried 2 mm sieved soil were shaken in 400 mL of deionised water for 1 h at 50 rpm in an end-over-end stirrer, followed by wet sieving using ultrapure water through 1.18, 0.42 and 0.02 mm sieves; then the last fraction was filtered at 2 μm by using polycarbonate Nuclepore® (Whatman) filters.
The Hg concentration was determined in the <2 μm fraction for one of the samples (A3) extracted with all the three methods and the results were compared by using Student's t-test at 95% confidence interval.
2.2 Soil samples: physico-chemical and mineralogical analyses
Soil properties such as pH, total organic carbon (TOC) and texture were determined for the three soil samples, according to standard methodologies of soil analysis. In addition, the mineralogical composition was assessed both for the fraction <2 mm and <2 μm.XRD patterns were obtained using a Rigaku D-max Rapid micro-diffractometer operating at 40 kV and 30 mA with CuKα radiation and a flat graphite monochromator. Different mounts with corundum NIST 676 as internal standard were prepared and analysed: air-dried, glycolated at 60 °C, and heated at 375 °C. The XRD data were collected as two-dimensional images and converted into 2θ–I profiles. The quantification of the mineralogical phases was obtained by Rietveld refinement using EXPGUI software.35
2.3 Particle distribution analysis and Hg determination
The total Hg content in the samples sieved at 2 mm and 2 μm was determined for solid samples by using an advanced mercury analyser (AMA 254, FKV, Altec), equipped for thermal decomposition of the sample, amalgam formation and atomic absorption.Dimensional characterisation of soil colloidal sub-samples was performed by using an SdFFF Model S101 instrument (Postnova Analytics, Salt Lake City, UT, USA). The fractionation channel was made of two Hallestoy C inox bars and a Mylar spacer of nominal thickness 0.0254 cm, 2.0 cm nominal width and ca. 90 cm tip-to-tip in length. The channel void volume was 4.8 mL (w = 0.0282) and the axis-to-channel distance was ca. 15 cm. The flow stream, delivered by a Mod. 422 Master HPLC Pump (Kontron Instruments, Italy) was 1.5 mL min−1. The carrier solution used was 0.001% FL70 (Fisher Scientific, France), commonly used in the FFF experiments, to avoid the accumulation of impurities in the FFF channel.36,37
The characterisation of colloidal soil samples by SdFFF was performed using the normal mode, avoiding interference due to the steric mode (co-elution of smaller and larger particles).38 The soil <1 μm fraction was extracted by following the same procedure adopted for the fraction <2 μm with the exception of using a centrifugation time of 4.13 minutes.31 The soil sub-samples for injection in the SdFFF system were prepared by suspending 50 mg of <1 μm soil fraction in 50 mL of 0.001% FL70 surfactant. The suspended soil sub-samples were vigorously stirred for 1 min by using a vortex vibrator at a maximum frequency of 60 Hz before being injected into the SdFFF channel by using a 10 μL chromatographic syringe.
The outlet tube of the SdFFF system was connected to a UV detector operated at 254 nm (Uvidec 100, Jasco Ltf., Japan) and the signal was fed to a Linseis Mod. L6512 X-Y recorder (Linsel GmbH Selb, Germany). UV absorbance data were collected by an in-house data acquisition program and handled by using the FFFractionation Inc. software.
To achieve a good balance between resolution of the sample profile, speed of the analysis and also in order to collect sufficiently concentrated SdFFF eluted fractions to be used in further analytical determinations, some factors affecting SdFFF fractionation were checked and optimised.20 In general, better resolution is often achieved by increasing the field strength (with potential sample loss and longer time of analysis), while increasing the flow rate can speed up the analysis but at the expense of peak resolution.9 By carefully taking into account these factors, the optimal instrumental elution parameters for the particle separation in the analysed soil samples were determined to be the following: initial field G0 = 400 rpm, hold field = 20 rpm, initial delay time tl = 5 min, waiting time = 5 s and flow rate = 1.5 mL min−1.
Eluted sample fractions were collected every 3.5 min by a Mod. 2110 collector (Bio-Rad Laboratories, Italy). Since SdFFF dilutes the injected sample by a factor of 50, in order to have a detectable amount of Hg in the collected fractions, five FFF runs were repeated and the corresponding five fractions obtained were combined and then concentrated in an oven at 30 °C for 24 hours, producing a final volume of 25 mL.
The determination of mercury in the resulting suspension (10 fractions for each soil sample) was performed by cold vapour electrothermal atomic absorption spectroscopy (CV-ETAAS), by using a Mod. A Analyst™ 800 instrument (Perkin Elmer, USA), equipped with a Transversely Heated Graphite Furnace (THGA) with longitudinal Zeeman-effect background corrector. The reducing agent used for cold vapour generation was NaBH4 (0.2% in NaOH 0.05%), while HCl 3% was used as a carrier.
Samples were introduced into the system by an autosampler Mod. AS800 (Perkin Elmer, USA), equipped with a flow injection device (FIAS 100, Perkin Elmer, USA).
Samples were acidified with concentrated HNO3 to promote Hg solubilization and a known amount of sample (500 μl) was introduced into the FIAS loop and then transported into the system by a flow of 3% HCl. After reduction by NaBH4, Hg cold vapours were produced and transported by an inert gas (Ar) to the graphite tube, previously coated with an iridium pure standard solution (PerkinElmer, 1000 mg L−1) to improve the sensitivity of the instrument in determining Hg.39 The detection was operated by using a mercury electrodeless discharge lamp (EDL) at a wavelength of 253.7 nm.
The graphite furnace heating program is summarised in Table 1.
| Step | Temperature/°C | Ramp/s | Hold/s | Ar flow/mL min−1 |
|---|---|---|---|---|
| Dry | 30 | 1 | 50 | 250 |
| Pyrolysis | 50 | 1 | 20 | 250 |
| Atomisation | 1200 | 0 | 4 | 0 |
| Cleaning | 1400 | 1 | 3 | 250 |
2.4 Microscopic analyses
To check the size of the particles in the collected fractions with the equivalent spherical diameter calculated using the SdFFF theory, microscopic analyses using scanning electron microscopy (SEM) were performed. The apparatus used for this study was a FESEM (field emission scanning electron microscope) SUPRA™40 (Carl Zeiss AG), equipped with a 3rd generation Gemini® column, controlled by the software SmartSEM™.Energy dispersive spectra were collected on a liquid nitrogen cooled Si(Li) INCAx-sight EDS detector (Oxford Instruments, England).
A few drops of the SdFFF fractions were distributed evenly over double-sided polycarbonate adhesive tape, and allowed to evaporate to dryness at room temperature. The samples were then placed on aluminium stubs for microscopic analysis.
In order to collect proper images and to avoid the accumulation of charges on the surface of the sample when bombarded with the electron beam, all the samples were sputter coated with a thin layer of conductive carbon.
2.5 Method quality control
No certified soil reference material (CRM) with a similar particle size to the one of interest (≤1 μm) was available, therefore in order to examine the possibility of soil matrices causing major sources of errors, the certified soil reference material NIST SRM 2710 (Montana soil, 75 μm average particle size) was used. The SdFFF fractionation could not be performed as the CRM particle size was >1 μm. However, it could help in highlighting the major instrumental problems (e.g. anomalous elution profile) related to the soil matrix. Three aliquots of 50 mg of untreated standard material were diluted with 50 mL of FL 70 0.001%, sonicated and injected into the SdFFF channel. No specific instrumental problems, such as blockage of the syringe, unusual or absent UV signal or irregular elution of the fractions were observed in this preliminary test.Before analysing the soil samples (A1, A2, A3) with the CV-ETAAS system, two suitable calibration experiments (external calibration and standard additions) were also performed and the slopes of the two different calibration curves were evaluated for statistically significant difference at 95% confidence level (Student's t-test). The slopes of the two different calibration curves were not found to be significantly different, suggesting a reduced or inexistent matrix effect on Hg determinations by CV-ETAAS analysis.40
Instrumental Limit of Detection (LoD) and Quantification (LoQ) were evaluated using two approaches, according to Massart et al.41 by using linear regression analyses (95% confidence interval, α = 0.05), and as 3 and 10 times the standard deviation (σ) of 10 repeated measurements of the blank solution.42 According to these procedures, LoD and LoQ were evaluated as 0.44 μg L−1 and 1.5 μg L−1 or resulted 0.08 μg L−1 (3σ) and 0.27 μg L−1 (10σ). The highest values found were considered as the LoD and LoQ of the method: 0.44 μg L−1 and 1.5 μg L−1, respectively.
3. Results and discussion
3.1 Mercury concentration in soil samples
The total Hg concentration in the considered soil <2 mm and <2 μm sub-samples is reported in Table 2, together with some characterising physico-chemical parameters. In samples A1 and A3, Hg was more concentrated in the clay fraction (<2 μm) as already observed by Terzano et al.,29 for samples collected in the same site.| Sample | pH | TOC (%) | Sand (%) | Silt (%) | Clay (%) | Total Hg/μg g−1 | |
|---|---|---|---|---|---|---|---|
| <2 mm | <2 μm | ||||||
| A1 | 8.0 | 3.0 | 53 | 30 | 16 | 50 ± 5 | 130 ± 10 |
| A2 | 8.5 | 1.4 | 48 | 39 | 13 | 230 ± 40 | 200 ± 10 |
| A3 | 8.5 | 0.7 | 40 | 39 | 21 | 12 ± 2 | 30 ± 2 |
The extraction of the fraction <2 μm with the sonication–centrifugation method developed in-house was compared to two other methods, a traditional wet sieving and sedimentation–syphonation, for sample A3. The amount of Hg determined by using the latter two methods was 28 ± 3 μg g−1 and 31.5 ± 0.5 μg g−1 respectively, which is not significantly different from that obtained by using the in-house developed method (Table 2). Nevertheless, the adopted sonication–centrifugation method proved to be faster than the two other methods and the soil aggregates were almost completely broken up. It must be pointed out that this type of sample preparation on one side could alter the real size of soil particles (i.e. by destroying natural aggregates), but on the other side allows one to obtain better separated materials. This aspect is relevant to study small primary soil particles which are of high environmental concern, as they can more actively sorb pollutants and easily migrate.
3.2 Colloidal particle size and Hg distribution
Under the optimised fractionation conditions (Section 2.3), characteristic SdFFF fractograms for A1, A2 and A3 sub-samples (<1 μm) were obtained. All the elution profiles, reported in Fig. 1, showed a main peak at an elution time between 15 and 25 minutes. The plots of Fig. 2, derived by conversion of the fractograms reporting the UV response profile vs. elution time into UV response profile vs. equivalent spherical diameter (μm),40 show a maximum peak of the particles in the size range between 0.4 and 0.7 μm for all the three samples.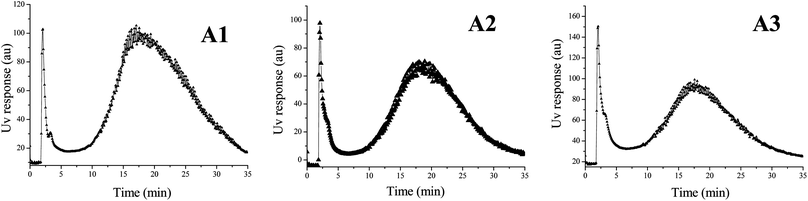 | ||
| Fig. 1 Fractograms obtained for the collected soil samples A1 (left), A2 (middle), A3 (right). | ||
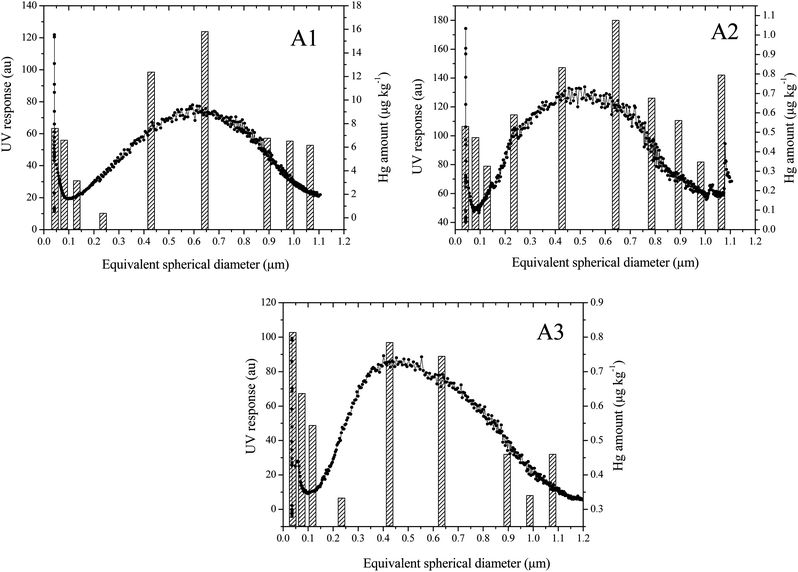 | ||
| Fig. 2 SdFFF fractograms for A1, A2 and A3 samples, showing the particle distribution (μm) vs. the UV detector response (a.u.) (black line); and distribution of Hg concentrations (μg kg−1) detected by CV-ETAAS (dashed bars). In A1 and A3 graphs, the seventh fraction is not reported as it was below the limit of quantification (LoQ). | ||
To check the correspondence of the particle size in the eluted fractions to the particle size calculated by the SdFFF instrumentation, three fractions were collected, concentrated and submitted to SEM-EDS investigation. Fig. 3 shows that colloidal particles were eluted in the expected order of increasing size, confirming the expected theoretical fractionation from ca. 0.05 to 1.1 μm. The particles belonging to the three eluted fractions (Fig. 3A–C) show different characteristics (as for shape and size) suggesting a correct separation of the colloids. However, particle aggregates were observed, especially in fractions B and C, presumably due to self-assembly of the particles after fractionation and sample preparation for SEM analyses. In proximity to the void volume, the concurrent elution of particles with different sizes might have occurred, also due to the presence of non-spherical particles (especially disc shaped particles16,43).
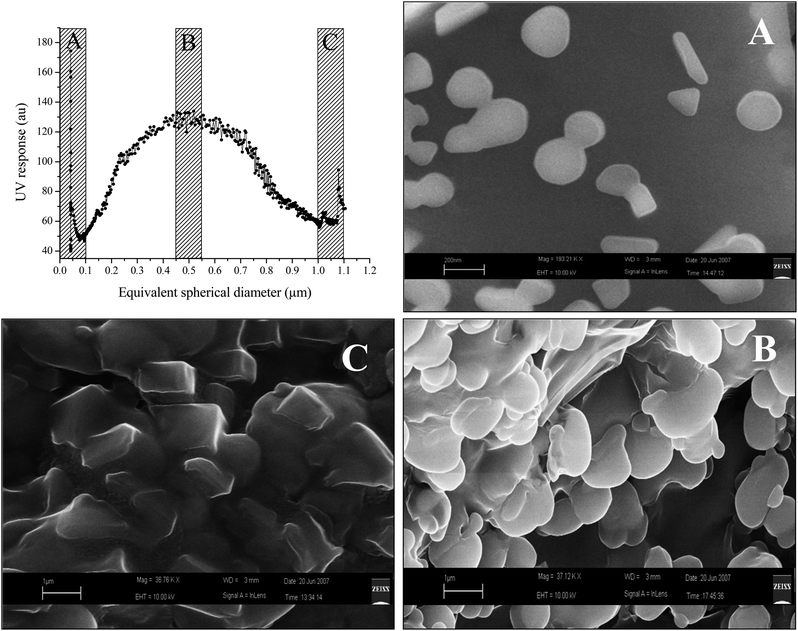 | ||
| Fig. 3 SdFFF fractogram for A2 sample and related SEM-EDS micrographs corresponding to the fractions ≤100 nm (A), 500–700 nm (B) and ≥1 μm (C). | ||
The combined corresponding fractions collected from five repeated SdFFF runs for A1, A2 and A3 were analysed for total mercury content by using CV-ETAAS (Fig. 2, vertical bars).
Regarding sample A1, it was found that the mercury level was higher for the fraction around 0.65 μm, where the maximum of the particle distribution was also observed. Sample A2 presented a similar trend in Hg amount, with a maximum in concentration around 0.65 μm. Interestingly, the Hg distribution levels obtained for sample A2 are ca. one order of magnitude lower than the ones determined for A1, despite the fact that the total amount of Hg for the A2 soil is higher (230 μg g−1) than for A1 (50 μg g−1). As reported in Table 2, Hg concentration in A1 seems to increase going down with particle size from 2 mm to 2 μm, while the opposite happens for A2 (Table 2). Thus, it is likely that Hg concentration increases further in the colloidal fraction (<1 μm) for A1, while for A2 Hg is more concentrated between 1 μm and 2 μm. In the case of A3, the maximum of Hg distribution was observed in particles with a size of around 0.4 μm, where the maximum of particle distribution was also registered. In A3, the overall concentrations of Hg in the different colloidal fractions were found to be lower than for A1 and similar to those recorded for A2.
3.3 Environmental implications
It is known that metal transport in the colloidal fraction is often mediated by organic matter or by reactive minerals such as iron oxides or clays. By looking at the mineralogical composition of the fraction <2 μm (Table 3), no crystalline iron mineral was detected and no relevant difference can be observed in the clay composition of the three samples. The most obvious difference is in the organic carbon content (Table 2). In particular, sample A1, which shows the highest concentration of Hg in the colloidal fraction, has a content of organic matter which is more than twice that of A2 and four times that of A3. However, a previous study by Terzano et al.29 performed at the same site showed that Hg is not directly bound to organic matter, iron oxides (either crystalline or amorphous) or other soil components but is mainly present as individual mercuric sulfides or amorphous mixed sulfides/chlorides. Nonetheless, the observed higher amount of Hg in the colloidal fraction of the soil could be related to the higher organic carbon content. As observed by Ravichandran et al.44 and Horzempa and Helz,45 the presence of dissolved organic matter can prevent the aggregation of HgS colloids through electrostatic repulsion. Therefore, considering that the growth of colloidal particles into larger crystals can be suppressed by organic matter adsorption,46 it is feasible that a higher concentration of Hg can be found in the colloidal fractions of sample A1 when compared to A2 and A3, where the amount of organic carbon is much lower. In conclusion, O.M., by influencing the size of HgS colloids, can increase the risks of Hg mobilisation in soil since the smaller the particle size, the higher can be the transportation rates of Hg in hydrologic fluxes or as airborne particles.| Qtz | Cal | Pla | Fel | Ill | Ver | I/S | Kao | Dol | Chl | Oth | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a Qtz: quartz; Cal: calcite; Pla: plagioclase; Fel: feldspate; Ill: illite; Ver: vermiculite; I/S: interstratified illite/smectite; Kao: kaolinite; Dol: dolomite; Chl: chlorite; Oth: others. | |||||||||||
| <2 mm | |||||||||||
| A1 | 49 | 21 | 11 | 8 | 4 | n.d | n.d. | 3 | 2 | 1 | 1 |
| A2 | 27 | 19 | 11 | 4 | 34 | n.d. | n.d. | 4 | n.d. | 1 | |
| A3 | 30 | 22 | 13 | 8 | 11 | n.d. | n.d. | 7 | 4 | 4 | 1 |
| <2 μm | |||||||||||
| A1 | 9 | 1 | 5 | 2 | 32 | n.d. | 13 | 18 | 10 | 10 | |
| A2 | 20 | 13 | 3 | n.d. | 24 | n.d. | 25 | 10 | n.d. | 5 | |
| A3 | 11 | 9 | 17 | 2 | 18 | 3 | 10 | 13 | 2 | 14 | 1 |
As a practical consideration, although in the soils investigated here Hg seems to be present in sparingly soluble forms (e.g. HgS), the levels of mercury found in the colloidal fractions need to be taken into account when assessing the environmental risk in the area, since pollutant mobility through soil may be higher than expected because of colloidal transport.
In this respect, a more extensive investigation, on a larger scale, taking into account a broader range of influential factors as well as the element speciation, needs to be carried out.
4. Conclusion
In this study, SdFFF was successfully employed to carry out particle size distribution analysis of soil samples contaminated with high levels of mercury. Furthermore, by coupling SdFFF to a cold vapour ETAAS for mercury content determination, useful information on Hg content along the profile of particle size distribution could be derived.In the collected samples, a maximum in Hg concentration was also found to correspond to the maximum in the particle size distribution, which was in a size range of a few hundred nanometres (400–700 nm). An apparent relation between Hg concentration in the colloidal fraction and organic matter (O.M.) content in the soil samples was observed but it seems to be determined not by Hg sorption to O.M., but rather to colloidal mercuric sulfide particles whose size is likely to be influenced by dissolved O.M.
Several authors3,24,47 have already demonstrated that small colloidal particles can diffuse more easily through the environmental compartments via transportation with hydrologic fluxes, and turn into a serious threat to the environment. Mobilisation of colloidal particles such as small airborne particles can also be a source of potential risk for humans if inhaled.
Therefore, even if Hg is found in the environment as scarcely soluble forms (e.g. HgS), the colloidal size of the Hg-bearing particles should raise the level of attention of regulators, as in this form it can more easily migrate, dissolve, be chemically modified or ingested.
Acknowledgements
This research was partially financed by the MIUR (COFIN 2005) project “Innovative chemical, physical, and biological methods to characterize and remediate soils polluted by heavy metals (MICROS)”. The authors thank Prof. Ruggiero for coordinating the project and for scientific support. Sincere thanks also to the Research Centre ENEA-Trisaia of Rotondella (Italy), and in particular to Massimo Morgana and Assunta Romanelli, for providing the automated mercury analyser.References
- D. J. Chittleborough, S. Tadjiki, J. F. Ranville, F. Shanks and R. Beckett, Supersoil 2004: 3rd Australian New Zealand Soils Conference, Australia, 2004 Search PubMed.
- J. M. Thomas, Pure Appl. Chem., 1988, 60, 1517–1528 CrossRef CAS.
- T. Baumann, P. Fruhstorfer, T. Klein and R. Niessner, Water Res., 2006, 40, 2776–2786 CrossRef CAS.
- J. J. D'Amore, S. R. Al-Abed, K. G. Scheckel and J. A. Rya, J. Environ. Qual., 2005, 34, 1707–1745 CrossRef CAS.
- Mercury substitution priority working list. An input to global considerations on mercury management, Nordic Council of Ministers, 2007, http://www.basel.int/techmatters/mercury/comments/240707hsweden-2.pdf, accessed 26th May 2011.
- UNEP, Decisions adopted by the Governing Council/Global Ministerial Environment Forum at its twenty-fourth session, 2007, http://www.unep.org/gc/gc24/docs/GC24_decisions.pdf, accessed 26th May 2011.
- S. Lindberg, R. Bullock, R. Ebinghaus, D. Engstrom, X. Feng, W. Fitzgerald, N. Pirrone, E. Prestbo and C. Seigneur, Ambio, 2007, 36, 62–66 CrossRef.
- P. R. G. Barrocas and J. C. Wasserman, Environmental Geochemistry in the Tropics, 1998, vol. 72, pp. 171–184 Search PubMed.
- M. Hassellov, B. Lyvén, H. Bengtsson, R. Jansen, D. R. Turner and R. Beckett, Aquat. Geochem., 2001, 7, 155–171 CrossRef CAS.
- J. Slowey, J. J. Rytuba and G. Brown, Jr, Environ. Sci. Technol., 2005, 39, 1547–1554 CrossRef.
- J. R. Lead, W. Davison, J. Hamilton-Taylor and J. Buffle, Aquat. Geochem., 1997, 3, 213–232 CrossRef CAS.
- Y. Ran, J. M. Fu, G. Y. Sheng, R. Beckett and B. T. Hart, Chemosphere, 2000, 41, 33–43 CrossRef CAS.
- D. Schmitt, H. E. Taylor, G. R. Aiken, D. A. Roth and F. H. Frimmel, Environ. Sci. Technol., 2002, 36, 2932–2938 CrossRef CAS.
- R. Chantiwas, R. Beckett, J. Jakmunee, I. D. McKelvie and K. Grudpan, Talanta, 2002, 58, 1375–1383 CrossRef CAS.
- L. J. Gimbert, K. N. Andrew, P. M. Haygarth and P. J. Worsfold, TrAC, Trends Anal. Chem., 2003, 22, 615–633 CrossRef CAS.
- C. Contado, G. Blo, F. Fagioli, F. Dondi and R. Beckett, Colloids Surf., A, 1997, 120, 47–49 CrossRef CAS.
- F. V. D. Kammer, M. Babolowski and K. Friese, J. Chromatogr., A, 2005, 81–89 Search PubMed.
- M. Baalousha, F. V. D. Kammer, M. Motelica-Heino, M. Baborowsky, C. Hofmeister and P. Le Costumer, Environ. Sci. Technol., 2006, 40, 2156–2162 CrossRef CAS.
- E. Bolea, M. P. Gorriz, M. Bouby, F. Laborda, J. R. Castillo and H. Geckeis, J. Chromatogr., A, 2006, 1129, 236–246 CrossRef CAS.
- J. F. Ranville, D. J. Chittleborough, F. Shanks, R. J. S. Morrison, T. Harris, F. Doss and R. Beckett, Anal. Chim. Acta, 1999, 381, 315–329 CrossRef CAS.
- B. Chen and R. Beckett, Analyst, 2001, 126, 1588–1593 RSC.
- G. Blo, C. Contado, F. Fagioli, M. H. Bollain Rodriguez and F. Dondi, Chromatographia, 1995, 41, 715–721 CAS.
- G. Nádaská, K. Polčová and J. Lesný, Nova Biotechnologica, 2009, 9, 295–301 Search PubMed.
- L. Zhaoli and L. Zhou, J. Environ. Sci., 2010, 22, 106–115 CrossRef.
- A. Santoro, R. Terzano, M. Spagnuolo, S. Fiore, M. Morgana and P. Ruggiero, Int. J. Environ. Waste Manage., 2010, 5, 79–92 CrossRef CAS.
- D. M. 308/06-Decreto Ministeriale 28 Novembre 2006, n. 308, Italy, 2006, in Italian.
- D. Chianese, M. D'Emilio, M. Bavusi, V. Lapenna and M. Macchiato, Environ. Geol., 2006, 49, 389–404 CrossRef CAS.
- A. Santoro, R. Terzano, G. Blo, S. Fiore, S. Mangold and P. Ruggiero, J. Synchrotron Radiat., 2010, 17, 187–192 CrossRef CAS.
- R. Terzano, A. Santoro, M. Spagnuolo, B. Vekemans, L. Medici, K. Janssens, J. Gottligher, M. Denecke, S. Mangold and P. Ruggiero, Environ. Pollut., 2010, 158, 2702–2709 CrossRef CAS.
- P. Ruggiero, R. Terzano, M. Spagnuolo, L. Cavalca, M. Colombo, V. Andreoni, M. Rao, P. Perucci and E. Monaci, J. Environ. Monit., 2011, 13, 145–156 RSC.
- T. E. Faber, Fluid Dynamics for Physicists, Cambridge University Press, New York, 1995 Search PubMed.
- F. Caravaca, A. Lax and J. Albaladejo, Geoderma, 1999, 93, 161–176 CrossRef CAS.
- P. K. Mutuo, K. D. Shepherd, A. Albrecht and G. Cadish, Soil Biol. Biochem., 2006, 38, 1658–1664 CrossRef CAS.
- M. L. Jackson, Soil Chemical Analysis—Advanced Course, Dept. of Soil Science, University of Wisconsin, Madison, WI, 2nd edn, 1973, 8th printing Search PubMed.
- B. H. Toby, EXPGUI, a graphical user interface for GSAS, J. Appl. Crystallogr., 2001, 34, 210–213 CrossRef CAS.
- G. Johannes, S. K. Wiedmer, M. Jussila and M. L. Riekkola, Chromatographia, 2005, 61, 359–364 Search PubMed.
- J. R. Kassab, P. J. P. Cardot, R. A. Zahoransky and S. Battu, J. Chromatogr., B: Anal. Technol. Biomed. Life Sci., 2005, 826, 8–16 CrossRef CAS.
- R. Beckett, The Composition and Surface Properties of Suspended Particulate Matter, in The Role of Particulate Matter in the Transport and Fate of Pollutants, ed. B. T. Hart, Water Studies Centre, Chisholm Institute of Technology, 1986, pp. 113–142 Search PubMed.
- E. Vassileva, H. Baeten and M. Honig, Fresenius' J. Anal. Chem, 2001, 369, 491–495 CrossRef CAS.
- G. Blo, A. Ceccarini, C. Conato, C. Contado, F. Fagioli, F. Roger, A. Pagnoni and F. Dondi, Anal. Bioanal. Chem., 2006, 384, 922–930 CrossRef CAS.
- D. L. Massart, B. G. M. Vandegiste, L. M. Buydens, S. De Jong, P. J. Lewi and J. Smeyers-Verbeke, Handbook of Chemometrics and Qualimetrix: Part A, Elsevier, 1997 Search PubMed.
- Eurachem Guide: The Fitness for Purpose of Analytical Methods, A Laboratory Guide to Method Validation and Related Topics, LGC, Teddington, UK, 1998. Available at: http://www.eurachem.org/guides/pdf/valid.pdf Search PubMed.
- J. C. Giddings, Science, 1993, 260, 1456–1465 CAS.
- M. Ravichandran, G. R. Aiken, J. R. Ryan and M. M. Reddy, Environ. Sci. Technol., 1999, 33, 1418–1423 CrossRef CAS.
- L. M. Horzempa and G. R. Helz, Geochim. Cosmochim. Acta, 1979, 43, 1645–1650 CrossRef CAS.
- H. Kodama and M. Schnitzer, Geoderma, 1978, 19, 279–291 CrossRef.
- E. F. Covelo, F. A. Vega and M. L. Andrade, J. Hazard. Mater., 2007, 140, 308–315 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |