Graphene quantum dots: an emerging material for energy-related applications and beyond
Zhipan
Zhang
*,
Jing
Zhang
,
Nan
Chen
and
Liangti
Qu
*
Key Laboratory of Cluster Science, Ministry of Education of China, School of Chemistry, Beijing Institute of Technology, Beijing 100081, P. R. China. E-mail: lqu@bit.edu.cn; zhipan@bit.edu.cn
First published on 22nd August 2012
Abstract
In this perspective, we focus on a new type of quantum dots, graphene quantum dots (GQDs). Due to quantum confinement and edge effects, GQDs have presented extraordinary properties, attracting extensive attention from scientists in the fields of chemistry, physics, materials, biology, and other interdisciplinary sciences. Herein, we summarize the significant advances achieved by us and other groups in the past few years on both the experimental and theoretical fronts. Synthetic strategies, unique optical and electronic properties, and the promise of GQDs in energy-related devices, such as photovoltaic devices, fuel cells, and light-emitting diodes, are systematically discussed.
 Zhipan Zhang | Zhipan Zhang received his doctorate degree from École Polytechnique Fédérale de Lausanne in 2008. After working at Dyesol Ltd and staying in Monash University and the University of Calgary, he is now an Associate Professor at Beijing Institute of Technology. His research focuses on energy-related applications of new materials, particularly in photovoltaics and solar fuels. |
 Liangti Qu | Liangti Qu received a PhD in Chemistry from Tsinghua University (Beijing, China) in 2004. He is now a Professor of Chemistry at Beijing Institute of Technology and leads the nanocarbon research group. His research interest in materials chemistry mainly focuses on the synthesis, functionalization and application of nanomaterials with carbon–carbon conjugated structures, including carbon nanotubes, graphenes and conducting polymers. |
Broader contextBoth graphene and quantum dots (QDs) possess interesting and useful optical and electronic properties associated with their nanoscale structures. In the case of graphene, the two-dimensional (2D) single-atom carbon sheet has attracted tremendous research interest due to its large surface area, high carrier transport mobility, superior mechanical flexibility and excellent thermal/chemical stability. On the other hand, 0D QDs are interesting materials and the development of new types of QDs will allow control of the fundamental properties of materials through size/shape effects, which will further allow new devices to be developed with distinctive properties and functions for numerous applications. Graphene quantum dots (GQDs), single- or few-layer graphene with a tiny size of only several nanometers, stand for a new type of QDs with the unique properties associated with both graphene and QDs. Therefore, many interesting phenomena that are different from those in normal QDs are expected in view of the remarkable quantum-confinement and edge effects of 0D GQDs. Indeed, the extraordinary optoelectronic properties of GQDs have been recently revealed and their important potential in solar cells, fuel cells and others have emerged. In this perspective, the exciting developments and existing challenges regarding the new quantum-confined system of GQDs are discussed. |
1. Introduction
Carbonaceous materials have been an old companion of mankind for thousands of years, first supplying the much needed energy for basic living and then providing varieties of important substances for the development of modern science and technology. With the advent of nanotechnology, advanced carbonaceous materials of nanoscale sizes and unique properties were discovered in succession, with buckminsterfullerene C60 in 1985,1 carbon nanotubes in 1991,2 and most recently graphene in 2004.3 As a two-dimensional (2D) single-atom carbon sheet, graphene has ignited tremendous research interest due to its large surface area, high intrinsic carrier mobility,4–6 strong mechanical strength,7 and superior flexibility.8 In particular, its high carrier mobility qualifies it as a promising alternative material to silicon in electronic circuitry of nanometer sizes.9–12 While the application of 2D graphene sheets (GSs) is unfortunately limited by their nature of easy aggregation and poor dispersion in common solvents, the etching of graphene into one-dimensional (1D) stripes, known as graphene nanoribbons (GNRs), mitigates these problems and demonstrates the intriguing properties of confined transport gaps and quantum dot behaviors associated with the geometry of the ribbons.13–17 In the past few years, there has been an ongoing enthusiasm to further convert graphene to 0D graphene quantum dots (GQDs) and study the new phenomena from GQDs associated with quantum confinement and edge effects.18–20 These dots are usually biocompatible, strongly luminescent and well dispersed in various solvents, showing bright promise for integration into devices of bioimaging, photovoltaic and light emitting applications. In this perspective, we aim to review recent advances in the field of GQDs, focusing on the synthetic methodology, modulation of intrinsic properties and energy-oriented applications of GQDs.Another type of 0D carbon nanomaterial is known as carbon nanodots (C-dots).21 Common C-dots comprise discrete, quasi-spherical carbon nanoparticles with sizes below 10 nm, which were first found from arc-discharge soot in 200422 and subsequently prepared by other benign methods.23–40 Despite the fact that some C-dots demonstrate the existence of sp2 (graphitic) π bonds and signs of nanocrystalline graphite composition,30,33,36,41 many lack explicit structural information and only a handful show signs of graphitic features.34 In comparison, GQDs are generally either produced from graphene-based starting materials or synthesized with an unambiguous structure by solution wet chemistry (see Synthetic strategies), thus they clearly possess graphene lattices inside the dots regardless of the dot size. Particularly, we discuss GQDs in the range of a few nm to ∼100 nm prepared by different strategies, as quantum confinement has been clearly observed in a graphene dot of 110 nm in diameter.42 In addition, only single-, double- and few- (3 to <10) layer graphene is considered here, with the rest being thin graphitic particles or films.9 Therefore, we define GQDs as graphene dots of smaller than 100 nm in size and less than 10 layers in thickness, for clarification purposes in this perspective, and specifically draw GQDs out from conventional C-dots and other graphene nanostructures.21,43,44
2. Synthetic strategies
The strategies for synthesizing GQDs with tunable properties can be divided into two methods. One is the cutting approach that GS assemblies are directly cut into 0D GQDs via physical, chemical or electrochemical techniques. As GQDs can be regarded as quantum sized graphene fragments, intuitively, direct cutting or exfoliation of large GSs will naturally yield GQDs. The other idea lies in chemical synthesis, that graphene moieties are controllably synthesized by stepwise reactions of molecular precursors. As listed in Table 1, these strategies provide considerable room to tune the properties of as-synthesized GQDs for various specific applications.| Synthetic strategies | Subclassification | Size of GQDs (nm) | Ref. |
|---|---|---|---|
| Cutting approaches | Nanolithography technique | 30, 40, 80, 110, 250 | 42 |
| 50 | 46 | ||
| 90 | 47 | ||
| Hydrothermal cutting of GSs | 5–13 | 48 | |
| 1.5–5 | 49 | ||
| Solvothermal cutting of GSs | 5.3 (in average) | 50 and 51 | |
| Electrochemical scissoring of GSs | 3–5 | 52 and 53 | |
| 5–10 | 54 | ||
| 60 | 55 | ||
| 5–19 | 56 | ||
| Chemical exfoliation | 1–4, 4–8, 7–11 | 58 | |
| 15, 18 | 59 | ||
| 2–7 | 60 | ||
| Nanotomy assisted exfoliation | 10–50, according to the demand | 61 | |
| Ultrasonic shearing of GSs | 3–5 | 63 | |
| Chemical synthesis | Stepwise organic synthesis of GQDs | ∼5 | 68 and 69 |
| Precursor pyrolysis | 15 | 71 | |
| 1.65–21, depending on reaction time | 72 | ||
| Surface-catalyzed decomposition of fullerene | 2.7–10 | 73 |
2.1 Cutting approaches
 | ||
| Fig. 1 (a) A graphene-based single-electron transistor. A 30 nm GQD is connected to contact regions through narrow constrictions of 20 nm wide graphene. (Reprinted with permission from ref. 42. Copyright 2008 the American Association for the Advancement of Science.) (b) A scanning force microscope image of an etched GQD device with source (S) and drain (D) leads and a plunger gate (PG) for electrostatic tenability. (Reprinted with permission from ref. 46. Copyright 2009 Wiley-VCH.) (c) A scanning electron microscope image of the photo-etched sample structure. The upper small GQD as the main device has a diameter of 90 nm while the bottom single electron transistor as a charge sensor has a diameter of 180 nm. (Reprinted with permission from ref. 47. Copyright 2010 American Institute of Physics.) | ||
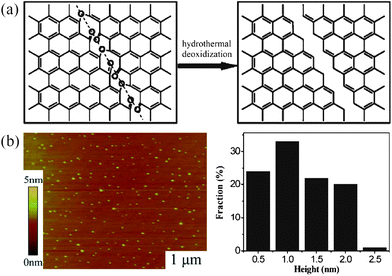
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O–COOH, OH and C–O–C) at the edges and on the basal planes so that the sheets became soluble in water. A following hydrothermal treatment was carried out under weakly alkaline (pH = 8) conditions at 200 °C for 10 h before the product was dialyzed in a dialysis bag overnight to give GQDs with diameters in the range of 5–13 nm (averaged at 9.6 nm). AFM results indicated that their topographic heights were mostly between 1 and 2 nm, implying the formation of GQDs with 1–3 graphene layers. In a later report, the same group modified this approach and selected monolayer GO sheets with a height of 1.1 nm as the precursor to prepare well-crystallized GSs through high temperature thermal reduction.49 After the oxidation in concentrated H2SO4 and HNO3, the ordered GO sheets were further cut hydrothermally under strongly alkaline (pH > 12) conditions to produce GQDs with a lateral size ranging from 1.5–5 nm (3 nm in average) and a narrow height distribution from 1.5–1.9 nm, indicating that the GQDs typically consisted of 2–3 graphene layers. In the mechanism, epoxy groups appear linearly on a carbon lattice upon acid oxidization and the cooperative alignment induces a rupture of the underlying C–C bonds to form an epoxy chain, where epoxy pairs can be further oxidized to energetically favorable carbonyl pairs at room temperature, as illustrated in Fig. 2a (left). During the hydrothermal deoxidization process, ultrafine pieces in the vicinity of the mixed epoxy lines and/or edges were vulnerable to further attack and can break up by removing the bridging O atoms in the epoxy lines to finally form the GQDs. Since there is no essential change in the lateral structure of GSs, the crystallinity of the final GQDs is primarily dictated by the starting material, i.e. highly ordered monolayer GSs will produce well-crystallized GQDs in the end.49
O–COOH, OH and C–O–C) at the edges and on the basal planes so that the sheets became soluble in water. A following hydrothermal treatment was carried out under weakly alkaline (pH = 8) conditions at 200 °C for 10 h before the product was dialyzed in a dialysis bag overnight to give GQDs with diameters in the range of 5–13 nm (averaged at 9.6 nm). AFM results indicated that their topographic heights were mostly between 1 and 2 nm, implying the formation of GQDs with 1–3 graphene layers. In a later report, the same group modified this approach and selected monolayer GO sheets with a height of 1.1 nm as the precursor to prepare well-crystallized GSs through high temperature thermal reduction.49 After the oxidation in concentrated H2SO4 and HNO3, the ordered GO sheets were further cut hydrothermally under strongly alkaline (pH > 12) conditions to produce GQDs with a lateral size ranging from 1.5–5 nm (3 nm in average) and a narrow height distribution from 1.5–1.9 nm, indicating that the GQDs typically consisted of 2–3 graphene layers. In the mechanism, epoxy groups appear linearly on a carbon lattice upon acid oxidization and the cooperative alignment induces a rupture of the underlying C–C bonds to form an epoxy chain, where epoxy pairs can be further oxidized to energetically favorable carbonyl pairs at room temperature, as illustrated in Fig. 2a (left). During the hydrothermal deoxidization process, ultrafine pieces in the vicinity of the mixed epoxy lines and/or edges were vulnerable to further attack and can break up by removing the bridging O atoms in the epoxy lines to finally form the GQDs. Since there is no essential change in the lateral structure of GSs, the crystallinity of the final GQDs is primarily dictated by the starting material, i.e. highly ordered monolayer GSs will produce well-crystallized GQDs in the end.49
 | ||
| Fig. 2 (a) Mechanism of the hydrothermal cutting of oxidized GSs into GQDs: a mixed epoxy chain consisting of epoxy and carbonyl pair groups (left) ruptured under the hydrothermal treatment and led to a complete cut (right). (Reprinted with permission from ref. 48. Copyright 2010 Wiley-VCH.) (b) An AFM image of the GQDs synthesized by the solvothermal route and their height distributions. (Reprinted with permission from ref. 50. Copyright 2011 Royal Society of Chemistry.) | ||
In another approach, Zhu et al. prepared GQDs on a large scale by implementing a one-step solvothermal route from GO.50,51 Briefly, the DMF solutions of GO were first sonicated and then heated in an autoclave at 200 °C for 5 h. The brown transparent suspension was separated from black precipitates and evaporation of the solvent gave GQDs with an average diameter of 5.3 nm and an average height of 1.2 nm, suggesting most of the GQDs were single-layered or bi-layered (Fig. 2b).
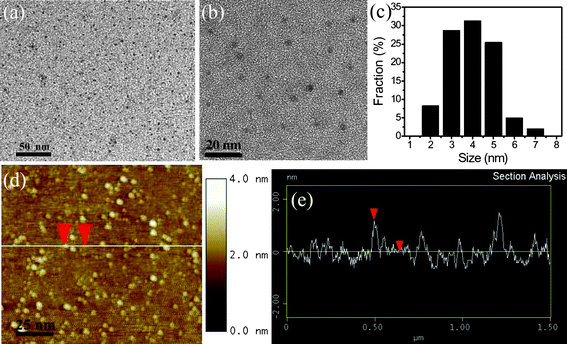 | ||
| Fig. 3 (a and b) TEM images of as-prepared GQDs with different magnifications, (c) the size distribution of GQDs, (d) an AFM image of the GQDs on Si substrate, and (e) the height profile along the line in (d). (Reprinted with permission from ref. 52. Copyright 2011 Wiley-VCH.) | ||
The mechanism of electrochemical cutting of GSs is postulated and thus depicted in Fig. 4a and b. Akin to the oxidation of other carbon materials by a strong oxidant with a high oxidizing potential (e.g., KMnO4), the applied potential of 3 V was high enough to drive the electrolyte ions into the graphene layers and oxidize the C–C bonds of the graphene sheets. The physical and/or chemical defects along the GS film provide a myriad of active sites for preferential electrochemical oxidation at the defect sites, leading to breakage of the graphene film into tiny graphene dots. As shown in Fig. 4c, the surface of the graphene film was compact and smooth before the electrochemical scan. With potential scans of a certain period, nanopores and nanograins (<10 nm) uniformly appeared on the whole surface of graphene film (Fig. 4d), indicating that the electrochemical process had led to the production of nanodots from a graphene film. The surface uniformity of the electrochemically treated graphene film also suggested that the electrochemical approach was a mild and well-controlled process. Accordingly, on judicious selection of electrolytes, one may deliberately adjust the incorporated dopant ions and accordingly tailor the property of the doped GQDs (vide infra),53 greatly diversifying the types of QDs that could be offered by this electrochemical approach.
 | ||
| Fig. 4 A scheme of the stacking structure of graphene layer(s) in a filtration-formed graphene film (a) and electrochemically produced GQDs (b), and the surface SEM images of the original graphene film (c) and the one after CV scan for 2000 cycles (d). (Reprinted with permission from ref. 53. Copyright 2012 American Chemical Society.) | ||
In addition, Zhang et al. synthesized water soluble GQDs in high yield by electrolyzing a graphite rod in 0.1 M NaOH aqueous solution and subsequently reducing the resulting nanoscale GQDs with hydrazine at room temperature.54 A homogeneous black solution formed during the electrolysis of graphite, while black material precipitated at the bottom after reduction with hydrazine. The condition of room temperature reduction was found to be critical. Centrifuging the solution above the precipitate gave a clear solution of yellow luminescent GQDs with an average diameter of 5–10 nm. The AFM image confirmed that all of the GQDs have a thickness <0.5 nm, mostly consisting of a single graphene layer. It is believed that the O and OH radicals produced by anodic oxidation of water can serve as electrochemical “scissors” to cut carbon nanocrystals and form oxygenated groups in the electrochemical oxidative cleavage of the graphite anode. After the subsequent reduction and functionalization with hydrazine, large graphene sheets and graphite nanocrystals tend to agglomerate and precipitate, therefore leaving a homogeneous colloidal solution of GQDs.
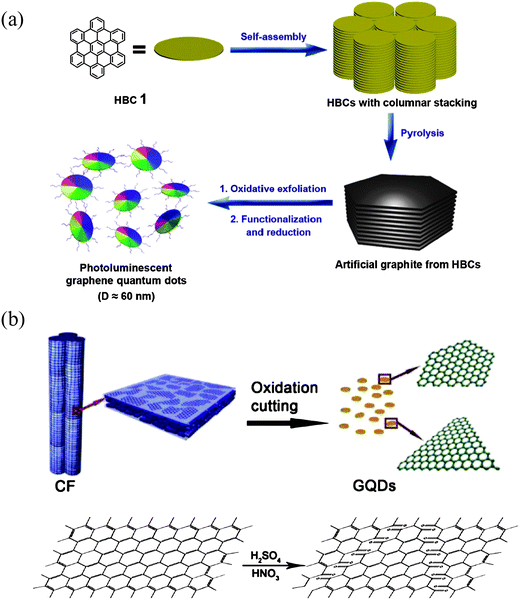 | ||
| Fig. 5 (a) The processing diagram for the preparation of GQDs by using HBC as carbon source. (Reprinted with permission from ref. 55. Copyright 2011 American Chemical Society.) (b) Representation scheme of oxidation cutting of CF into GQDs and proposed mechanism. (Reprinted with permission from ref. 58. Copyright 2012 American Chemical Society.) | ||
As GO nanocolloids can be made from graphite nanofibers,57 it is natural to infer that GQDs may also be synthesized from similar inexpensive materials. Recently, Peng et al. reported a convenient synthesis of one-step wet chemically derived GQDs by exfoliating micrometer-sized carbon fibers (CFs) with a resin-rich surface.58 It is deemed that if the starting material already has small domains of sp2 carbons, they can be easily extracted and controlled in size during the exfoliation. Indeed, the dimension of these CF derived GQDs can be tuned by simply choosing different reaction temperatures. Stirring pitch CFs in a mixture of concentrated H2SO4 and HNO3 for 24 hours at different temperatures of 80 °C, 100 °C and 120 °C gave GQDs of 1–4 nm, 4–8 nm and 7–11 nm in diameter, respectively. The AFM image demonstrated that the topographic heights of GQDs were between 0.4 and 2 nm, corresponding to 1–3 graphene layers. Fig. 5b shows a possible process by which the fiber structure breaks up. The planar graphitic domains are chemically initiated by the lining up of chemical functionalities (such as epoxy or carbonyl groups), making the graphitic domains prone to fracture, preferably along the zigzag direction. As was also found in the experiment, a large number of GQDs made by this method preferred zigzag edges to armchair ones. More recently, GQDs were prepared by chemically oxidizing a commonly used CX-72 carbon black, aggregates of spherical graphite particles with an approximate diameter of 30 nm.59 The chemical oxidation was used to break down the aggregates, solubilize the carbon nanoparticles, and further produced GQDs with a high yield. Interestingly, single-layer GQDs of about 15 nm and few-layered (2–6) GQDs of 18 nm in size can be obtained at the same time, which could be separated by simply centrifuging in nitric acid solution. The prominent advantages of using a low-cost common carbon source, high yield and one-step facile process make this method very attractive in the large scale synthesis of GQDs.
Microwave-assisted techniques have been widely applied to materials synthesis. Microwave irradiation can offer rapid and uniform heating for the reaction medium, and thus reaction time is dramatically shortened, with product yields and purities greatly improved. Li et al. reported a facile one-pot microwave-assisted chemical oxidation approach for the preparation of stabilizer-free greenish yellow-luminescent GQDs from GO nanosheets under acid conditions with 3 h.60 The diameters of GQDs were mainly distributed in the range of 2–7 nm with an average diameter of 4.5 nm and the topographic heights of GQDs were mostly between 0.5–2 nm with an average height of 1.2 nm, suggesting that most of GQDs were single-layered or bi-layered. A moderate reduction of these GQDs by NaBH4 gave brightly blue-luminescent GQDs with no perceptible change in the dimensions and height of the dots. The percentages of oxygen/nitrogen-bonded carbon in GQDs decreased progressively in comparison to the starting GO, verifying that reduction occurred during the microwave treatment.
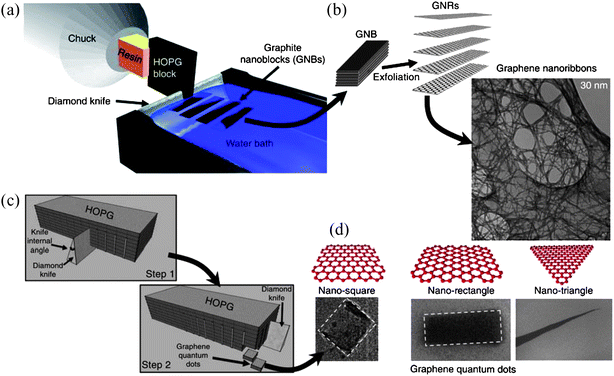 | ||
| Fig. 6 A schematic of the nanotomy process showing (a) the production and (b) subsequent exfoliation of graphene nanoblocks cut by the diamond-knife-based mechanical cleaving of a HOPG block. (c) A sketch of the two-step nanotomy process to produce graphene nanoblocks for GQD production. (d) Micrographs of GQDs of different shapes (square, rectangle and triangle). (Reprinted with permission from ref. 61. Copyright 2012 Nature Publishing Group.) | ||
2.2 Chemical synthesis
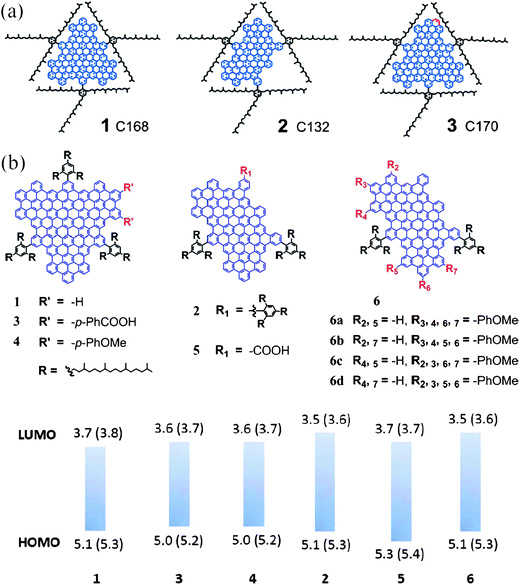 | ||
| Fig. 7 (a) Structures of three colloidal GQDs. (Reprinted with permission from ref. 68. Copyright 2010 American Chemical Society.) (b) Molecular structures of GQDs with different sizes and functional groups, together with their HOMO and LUMO levels obtained experimentally. (Reprinted with permission from ref. 69. Copyright 2011 American Chemical Society.) | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1, might be used as the carbon source to prepare GQDs, provided that H and O exist in the forms of hydroxyl, carboxyl, or carbonyl groups that may dehydrate under hydrothermal conditions.
:2:1, might be used as the carbon source to prepare GQDs, provided that H and O exist in the forms of hydroxyl, carboxyl, or carbonyl groups that may dehydrate under hydrothermal conditions.
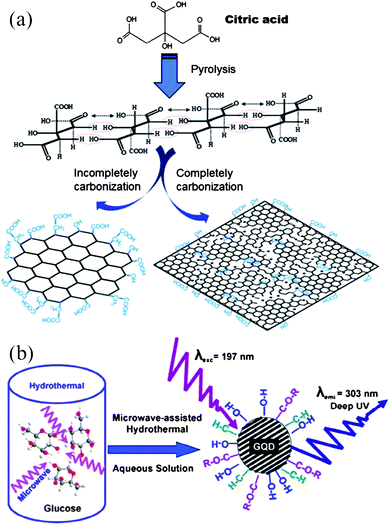 | ||
| Fig. 8 (a) A diagram for the synthesis of GQDs and GO by pyrolyzing citric acid (reprinted with permission from ref. 71. Copyright 2012 Elsevier B.V.) and (b) preparation of GQDs by pyrolyzing glucose via MAH method. (Reprinted with permission from ref. 72. Copyright 2012 American Chemical Society.) | ||
2.3 Surface-catalyzed decomposition of fullerene
Lu et al. presented an aesthetically beautiful work on the synthesis of regularly sized GQDs on a Ru(0001) substrate using C60.73 With the aid of a scanning tunneling microscope (STM), direct observation of the Ru-catalyzed cage-opening of C60 and the assembly of its fragments into surface-stabilized carbon clusters were achieved. Thermal hopping of the C60 molecules and dissociation of C60 clusters on the terrace initiated in an adatom-vacancy mechanism when the sample was annealed to 500–550 K (Fig. 9a and b), and at the higher annealing temperature of 650 K, embedded C60 molecule decomposed to form carbon clusters, as shown in Fig. 9c. Compared to carbon adatoms derived from other precursors, such as C2H4, the restricted mobility of these C60-derived clusters (diffusion coefficient in the range 10−15–10−16 cm2 s−1) enabled them to coalesce and form a series of atomically defined GQDs. Triangular, trapezoid-shaped and parallelogram-shaped GQDs were found after annealing at 725 K for 2 min (Fig. 9d and e) and the fragmentation processes followed the trapezoid → parallelogram → triangle sequence, with triangles being the equilibrium shape. Further annealing at 825 K for another 2 min produced hexagon-shaped GQDs 5 nm and 10 nm in size, indicating that instead of triangles, hexagons were the equilibrium shape at the elevated temperature (Fig. 9f).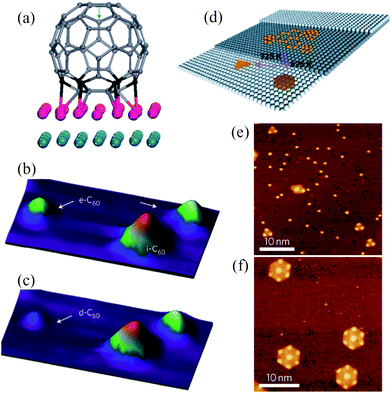 | ||
| Fig. 9 (a) The simulated ‘on-top_vac’ configuration of a C60 molecule on Ru(0001), and (b and c) three-dimensional STM images of a carbon cluster derived from the decomposition of embedded C60 molecules on Ru(0001). (b) A constant current image at 600 K. e-C60: embedded C60; i-C60: intact C60. (c) A constant current image at 650 K. d-C60: decomposition of embedded C60. (d) Temperature-dependent growth of GQDs with different equilibrium shape from the aggregation of the surface diffused carbon clusters, and (e and f) corresponding STM images for the well-dispersed triangular and hexagonal equilibrium shaped GQDs produced from C60-derived carbon clusters. (Reprinted with permission from ref. 73. Copyright 2011 Nature Publishing Group.) | ||
3. Physical properties of GQDs
3.1 Electronic properties
Electronically, graphene has unique properties due to its 2D nature, with monolayer graphene being the most perfect 2D electronic material possible in nature, also described as “ultimate flatland”.74 Owing to the confinement of carriers in this strict 2D layer, electronic band dispersion of 2D graphene follows a gapless and linear function of the wavevector k, near the charge neutrality point (Dirac point), i.e. E = ħνFk, where νF is the effective speed of light.75,76 Charge carriers, so-called massless Dirac fermions, in 2D graphene mimic relativistic particles with zero mass, and the Dirac spectrum in graphene leads to a number of exotic phenomena. For instance, graphene's conductivity never falls below a minimum value even when carrier concentrations approach zero, setting an obstacle for fabricating useful transistors due to the lack of an “off” state.76 Additionally, confining electrons in graphene is also challenging, as the presence of Klein tunneling matches wavefunctions of electron and positron across the barrier, making the graphene p–n junctions essentially transparent.77–79 Fortunately, quantum mechanically confining carriers in a low-dimensional structures, such as 1D GNRs and 0D GQDs, has rendered an effective way to circumvent these problems, with finite bandgaps and quantized conductance directly observed in GNRs.13,80–82As for GQDs, theoretical investigations by the tight-binding model revealed that state energies and energy spacings in a GQD vary significantly with not only the electric field strength it perceives, but also the field direction, a characteristic resulting from the spatial anisotropy of the dot.83 Libisch et al. simulated quantum confinement effects in GQDs with linear dimensions of 10–40 nm and demonstrated marked deviations from a simple Dirac billiard for massless fermions as a result of disorders originated from edge roughness, charge impurities, or short-ranged scatterers.84 Experimentally, Güttinger studied energy levels of GQDs in magnetic fields applied perpendicular to the graphene plane and observed indication of the formation of Landau levels, where the zeroth Landau level can be used to locate the electron–hole crossover in GQDs.46 As illustrated in Fig. 10a, Ponomarenko et al. showed that Coulomb blockade peaks became strongly nonperiodic in GQDs, with random peak spacing and its statistics well described by the theory of chaotic Dirac (neutrino) billiards.42 In some cases, when GQDs were further narrowed down to a few nm, they exhibited Coulomb blockade effects even at room temperature, indicating that electron interactions build up as the dimensions of graphene sheets are reduced. The pronounced carrier–carrier Columbic interactions also contributed significantly to an exceptionally slow carrier cooling process found in defect-free, colloidal GQDs, where lifetimes of hot carriers about 2 orders of magnitude longer than those in bulk graphene materials were observed.85 In this system, hot carriers survived up to hundreds of picoseconds, enabling intersystem crossing to efficiently compete with internal conversion.86 This has important implications for spin quantum computation, as intersystem crossing is generally much slower than internal conversion and the increased access to triplet states would offer a wider time window for spin manipulation. Nanostructured carbon has long been a promising candidate for spin-based quantum computation, due to its weak spin-orbital coupling and hyperfine interaction with nuclear spins, both of which are the dominant sources of spin decoherence.87–91 One of the most counterintuitive but fascinating phenomena ever observed in GQDs is the formation of spin qubits that are coupled in a long distance by means of Klein tunneling.92 As depicted in Fig. 10b, two distant qubits can be strongly coupled without touching the states of intermediate qubits that might be located between the two, an indispensible feature in fault-tolerant quantum computing.93 Lately and as a complement, Recher et al. demonstrated that the perpendicularly applied magnetic field can efficiently and controllably break valley degeneracy, opening up another feasible route to create well-defined and well-controlled spin-qubits in GQDs.94
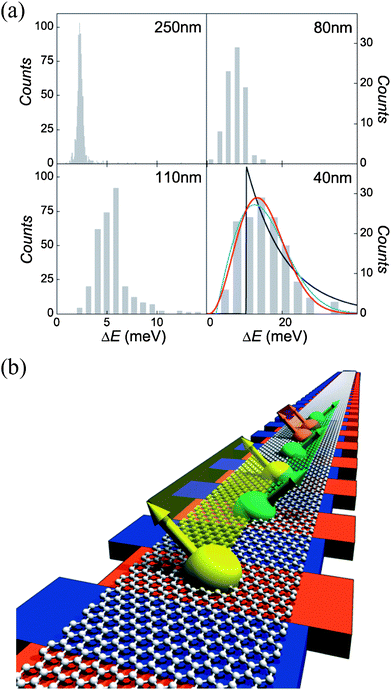 | ||
| Fig. 10 (a) Level statistics in Dirac billiards. Histograms of the nearest-neighbor level spacing in GQDs of different diameters. The level statistics becomes increasingly non-Poissonian for smaller GQDs, where the red, black, and blue curves are the best fits for the Gaussian unitary, Poisson, and Gaussian orthogonal ensembles, respectively. (Reprinted with permission from ref. 42. Copyright 2008 the American Association for the Advancement of Science.) (b) Illustration of many spin qubits. Red bars and blue bars represent QDs and barrier regions, respectively. The same color band denotes different spin qubits that are strongly coupled to each other via Klein tunneling. (Reprinted with permission from ref. 93. Copyright 2007 Nature Publishing Group.) | ||
3.2 Optical properties: absorption, photoluminescence and electroluminescence
GQDs usually show well-established absorption bands in the UV region, with an absorption peak around 230 nm due to the π → π* transition of aromatic sp2 domains,3,48,72 and a long tail extending into the visible range (a typical absorption spectrum of GQDs is presented in Fig. 11a). We note that many GQDs also have another absorption (shoulder) peak centered at a wavelength between 270 and 390 nm, which has been associated with the n → π* transition of CO,44 however, we deem that this assignment might not reflect the whole story. The peak position was shown to be more dependent on the preparation method than the size of the dots. For example, GQDs (9.6 nm average diameter) prepared by the hydrothermal route showed an absorption band at 320 nm,48 the same position as those of GQDs (average diameter of 5.3 nm) from a solvothermal synthesis50,51 and GQDs (3–5 nm) obtained from an electrochemical method,52 while a further decrease in GQD size down to 1.5–5 nm (3 nm on average) by a hydrothermal approach red-shifted the shoulder band to ca. 360 nm,49 close to those of GQDs (diameter of 5–10 nm) from electrolysis of graphite,54 GQDs (∼15 nm in size) prepared by pyrolysis,71 and GQDs (around 15 nm) derived from carbon black.59Large disk-like GQDs of around 60 nm diameter exhibited a weak absorption shoulder at 280 nm,55 which approximates to N-doped GQDs (2– 5 nm in size) synthesized electrochemically with a band at 270 nm (ref. 53) and GQDs (3–5 nm) collected in an ultrasonic preparation with an absorption band at ca. 300 nm.63 Although the absorption spectra of GQDs derived from CFs implied that this band position can be size-related, where the absorption peak changed from 330 nm to 270 nm as the size of the dots decreased from 7–11 nm to 1–4 nm,58 self-passivated GQDs prepared by a microwave-assisted hydrothermal route clearly showed zero-dependence of absorption peak position on the size of the dots, with an obvious peak centered at 282 nm regardless that the size of the GQDs changed from 1.65 nm to 21 nm.72 These somewhat confusing but intriguing properties are quite different from most of inorganic semiconductor QDs, such as CdS,95 InP,96 PbS,97 and even some of the C-dots.34 While the exact cause of these phenomena is in debate, they appear to be closely associated with dopants, edge structure and defects/surface states of the GQDs and thus highly sensitive to the way GQDs were prepared. For instance, various functional groups (C–OH, CO, O–CO, CN, etc.) introduced during the growth of GQDs could form surface states with their energy levels between π and π* states of CC,53,72 and induce absorption bands due to the electron transitions within one or several of these groups. The peak of the band became less pronounced in the presence of any chemical reducing process during the synthesis of GQDs, a scenario found in GQDs mildly reduced at room temperature by hydrazine.54 When strong reducing conditions were involved, as is the case for GQDs (average 13.3 nm) produced by high-temperature reduction with hydrazine56 and GQDs (average diameter of 4.5 nm) reduced by NaBH4,60 the band could completely disappear, confirming the correlation between this absorption band and surface functional groups. Nevertheless, exceptions to this mechanism have also been reported. Chen et al. found that the absorption band at ca. 370 nm was attributed to the self-assembled J-type aggregation in high concentration GQDs solutions,98 while on the other hand, this band was still present in the defect-free, colloidal GQDs synthesized by well-controlled carbon chemistry.68 As illustrated in Fig. 11c, a shoulder peak is still seen at 380 nm for GQDs 1 and 3, and at ca. 390 nm for GQD 2 (refer to Fig. 7a for the structures of GQDs 1–3), implying that another mechanism is functioning here. In contrast to other GQDs mentioned above, the absorption spectra of 1–3 additionally show a strong peak in the visible range and a stretched tail down to the near infrared region, presumably due to the large dimension of conjugation in these compounds that is rarely seen in other GQDs with defects.99
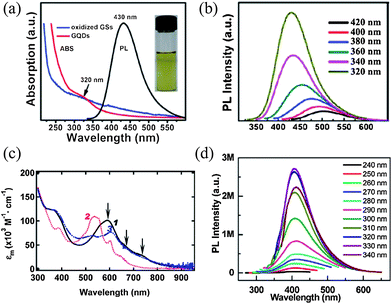 | ||
| Fig. 11 (a) UV-vis absorption (ABS, red) and PL (at 320 nm excitation) spectra of the GQDs synthesized hydrothermally. (b) PL spectra of the GQDs synthesized hydrothermally at different excitation wavelengths. (Reprinted with permission from ref. 48. Copyright 2010 Wiley-VCH.) (c) UV-vis absorption spectra of GQDs 1–3 in solution. The structures of 1–3 are shown in Fig. 7a. (Reprinted with permission from ref. 68. Copyright 2010 American Chemical Society.) (d) PL spectra of the GQDs prepared in an ultrasonic reaction at different excitation wavelengths. (Reprinted with permission from ref. 63. Copyright 2012 American Chemical Society.) | ||
Similar to C-dots, most GQDs reported so far in the literature are luminescent, despite the fact that the exact origins of their photoluminescence (PL) remain elusive. As shown in Fig. 12, GQDs prepared in different ways can emit deep UV,72 blue,43,48,53,55,63,100,101 green,49–52,101 yellow,58 red86 and even white PL,101 with the quantum yield (QY) ranging from 2–22.4%.50,86 It is reported that the QYs of C-dots are strongly dependent on surface passivation,25,27 and C-dots with only COOH groups at the surface exhibit lower QYs than those passivated by organic ligands,21 rendering QY values of 0.8–1.9%,23 0.43%,30 and 1.2%.33 Therefore, the QYs of GQDs seem to be higher than those of pure C-dots without organic passivating ligands, possibly due to the fact that GQDs are generally self-passivated in the exfoliating or growing process.72 The lowest reported QY of 2% comes from colloidal GQDs where edges are merely passivated by hydrogen atoms, highlighting the ineffectiveness of hydrogen atoms as passivation groups.86 This is also reflected in the photostability study, where PL of colloidal GQDs diminished within minutes under ambient light but GQDs prepared hydrothermally (thus passivated with groups of carbonyl, epoxy, etc.) showed stable emission in the presence of O2 for up to hours.49,86
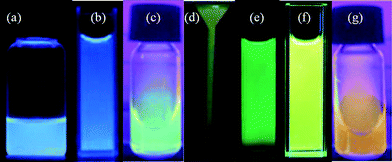 | ||
| Fig. 12 Colorful photographs taken for solutions of different GQD under 365 nm UV light irradiation (except for (e) where 375 nm was used). (a) Reprinted with permission from ref. 56. Copyright 2011 Royal Society of Chemistry. (b) Reprinted with permission from ref. 71. Copyright 2012 Elsevier B.V. (c) and (g) Reprinted with permission from ref. 59. Copyright 2012 Royal Society of Chemistry. (d) Reprinted with permission from ref. 60. Copyright 2012 Wiley-VCH. (e) Reprinted with permission from ref. 50. Copyright 2011 Royal Society of Chemistry. (f) Reprinted with permission from ref. 54. Copyright 2012 Royal Society of Chemistry. | ||
Analogous to what has been observed in absorption spectra of GQDs, PL color in certain systems showed dependence on the size of GQDs, changing from blue and green to yellow as dots got larger,58 while PL emission of the GQDs by the MAH method was still size-independent.72 However, one of the common phenomena found in GQDs is the excitation-dependent PL emission, which is shown in Fig. 11b. When the excitation wavelengths changed from 320 to 420 nm, the PL peak of hydrothermally cut GQDs shifted to longer wavelengths and the PL intensity decreased remarkably,48 as was also observed in GQDs synthesized from other methods.49–56,59,60,72 This is generally interpreted to be related to emissive traps,27 electronic conjugate structures,53 and free zigzag sites having a carbene-like triplet ground state,48 with some researchers claiming that the surface state also have an important role.72 It is possible that a combination of these pictures might be at play, as both the edge structure and the defects/surface states can significantly change the electronic properties of GQDs.19 Exceptionally, a handful GQDs have been reported to emit PL independent of excitation wavelength. As shown in Fig. 11d, when the excitation wavelength changed from 240 to 340 nm, the strong PL peak of GQDs synthesized by an ultrasonic reaction remained at ca. 407 nm, a phenomenon attributed to the formation of ultra-narrow GNRs with ca. 0.4 nm in width during the ultrasonic preparation.63 The PL of GQDs prepared by pyrolysis was also excitation-independent, which was understood as a result of high uniformity both in the size and the surface state of those sp2 clusters contained in GQDs.71 Similarly, Zhang et al. found that the PL peak of their GQDs remained constant at 540 nm when excited by light with wavelengths from 340–410 nm.54 Theoretical calculations showed the strong yellow luminescence of the GQDs was from a high concentration of modified phthalhydrazide-like and hydrazide groups chemically linked to the GQDs after absorption through the graphene π–π* and n–π* transitions, leading to the excellent PL stability of the GQDs.
Meanwhile, some of the GQDs are reported to possess upconversion PL properties and as illustrated in Fig. 13a, Zhu et al. found their GQDs prepared by a solvothermal method showed upconverted PL spectra when excited from 600–900 nm.51 Interestingly, apart from the excitation-independent downconversion PL, the ultrasonically produced GQDs also showed excitation-independent upconversion PL, with its peak almost unchanged at ca. 407 nm as the excitation wavelength red-shifted from 500 to 700 nm (Fig. 13b),63 and the upconversion PL was attributed to the multiphoton active process, similar to previous reported C-dots.26,34 Alternatively, Shen et al. observed upconverted PL emissions located at about 525 nm when GQDs passivated with PEG1500N were excited by a 980 nm laser (Fig. 13c).56 As the excitation wavelength changed from 600 to 800 nm, the PL peaks of GQDs shifted accordingly from 390 to 468 nm, with the energy difference between upconverted emission light (Em) and excitation light (Ex) remaining almost unchanged at about 1.1 eV (Fig. 13d). Instead of explaining the upconverted PL property by the popular multiphoton active process,26,34 the authors speculated that the upconversion luminescence in their GQDs was due to the anti-Stokes photoluminescence (ASPL). The π orbital electrons first got excited to a high-energy state and then returned to the σ orbital, accounting for why the upconversion excitation and emission light was in a constant energy difference.
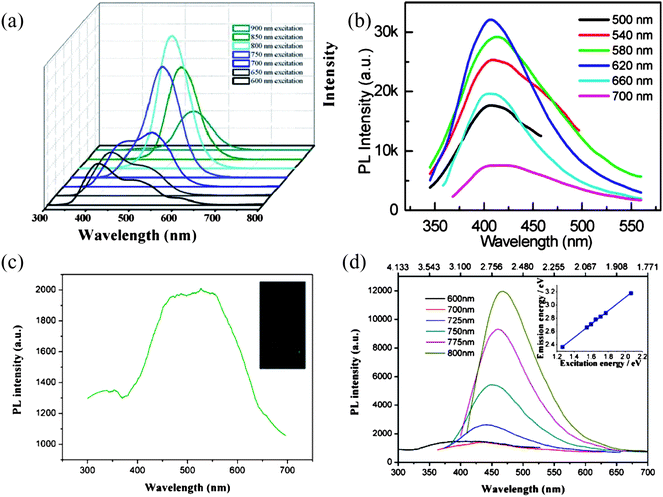 | ||
| Fig. 13 (a) The upconversion PL properties of GQDs by a solvothermal method. (Reprinted with permission from ref. 51. Copyright 2012 Royal Society of Chemistry.) (b) Upconverted PL spectra of the ultrasonically produced GQDs at different excitation wavelengths. (Reprinted with permission from ref. 63. Copyright 2012 American Chemical Society.) (c) PL spectrum of GQDs passivated with PEG1500N excited at 980 nm laser. Inset: photograph of the GQD aqueous solution taken under a 980 nm laser. (d) Upconverted PL properties of GQDs, inset is the energy of the excitation light as a function of the emission, and the function of the fit line is Em = 1.00Ex + dE (R2 = 0.9983) with dE = 1.1 eV. (Reprinted with permission from ref. 56. Copyright 2011 Royal Society of Chemistry.) | ||
The PL of some GQDs is pH-dependent and solvent-dependent. For GQDs prepared by the hydrothermal exfoliation method, the GQDs emitted strong PL under alkaline conditions, whereas under acidic conditions, the PL was nearly completely quenched. If pH was switched repeatedly between 13 and 1, the PL intensity varied reversibly.48,49 These reversible phenomena were understood as the protonation and deprotonation of free zigzag sites under acid and alkaline conditions. In another case, GQDs with their surface passivated by PEG1500N exhibited bright PL in a water solution of neutral pH, with the intensity of PL peaks decreased by about 25% under both acidic and alkaline conditions.56 Similarly, Zhu et al. also found that PL intensities of GQDs decreased in a solution of high or low pH, but remained constant in a solution of pH 4–8. Moreover, in a solution of pH over 12, the PL peak blue-shifted and the FWHM becomes narrow.50,51 Interestingly, the PL of their GQDs was also sensitive to the species of solvent and the peak shifted from 475 to 515 nm in THF, acetone, DMF, water, respectively. The incoherence in pH-dependent PL behaviors and the distinctive solvent-dependent PL clearly indicate dissimilar PL mechanisms in different GQDs, a puzzle needs to be solved with more experimental and theoretical results in the near future.
Electrochemical luminescence (ECL) behaviors of GQDs have only been identified recently. Li et al. found that when K2S2O8 served as coreactant, their green emitting GQDs showed an intense ECL emission about 9 times higher than background signal at −1.45 V, with an onset potential at about −0.9 V, while the same concentration of GO had no obvious ECL signal, ruling out the interference from the starting materials in the ECL process of GQDs.60 One of the advantages of GQDs in ECL study is the well-defined ECL peak at relatively positive potential in comparison with previously reported C-dots,31,32 which might benefit from the high content of sp2 carbon domains in GQDs inherited from graphene accelerating electron transport during the ECL process. The ECL of GQDs was shown to possess excellent stability, where constant signals with relative standard deviation (RSD) of 1.3% were measured upon continuous cyclic scans in the buffer solution.
3.3 Crystalline structure and Raman spectrum
Bilayer or few-layer GQDs feature an interlayer spacing that manifests itself as a (002) diffraction peak around 2θ = 25° in X-ray diffraction (XRD) profiles, and the peak is often broad due to the small size of GQDs. Depending on the preparation methods, the exact interlayer spacing can vary significantly. GQDs prepared by the hydrothermal, electrochemical exfoliation and precursor pyrolysis methods showed an interlayer spacing of ∼0.34 nm, a similar value to bulk graphite,48,52–54,60,71 while GQDs derived from CFs had a much larger interlayer spacing of 0.403 nm.58 The increase in interlayer distance was attributed to the oxygen-containing groups introduced in the exfoliation and oxidation of CF. Similarly, GQDs synthesized by the MAH method possessed interlayer spacing ranging from 0.343–0.481 nm, which was deemed to be associated with the presence of O–H, C–H, and C–O–R at the edges of the GQDs that enlarged the spacing of graphene layers.72Raman spectra of GQDs generally show characteristic peaks of the “disordered” D-band and the crystalline G-band, with the 2D band visible at ca. 2700 cm−1. The D and G bands originate from the disorder band caused by the graphite edges and the in-phase vibration of the graphite lattice, respectively,102 and the relative intensity ratio of D-band and the G-band (ID/IG) is an indicator of GQD edge-quality.103 Values between 0.5 and 1.26 have been reported,48,49,52–54,58 among which the electrochemical method produced GQDs with the smallest ID/IG of 0.5, highlighting its uniqueness in preparing high quality GQDs.52 We note that ref. 61 reported a very low ID/IG of 0.22–0.28 for the as-synthesized GNRs, but unfortunately, ID/IG information on their GQDs was missing, though similar values can be reasonably expected. Notably, the doubly degenerate G peak can split into two sub-bands, namely G+ and G−, in the ZnO–graphene hybrid QDs, which was attributed to strain induced symmetry breaking caused by bending of the graphene surrounding the ZnO quantum dots.100
4. Functionalization and assembly of GQDs
The functional modification and assembly of GQDs are important in the application of GQDs, as the former dictates the properties of individual GQDs and the latter opens up opportunities to control over the optical and electronic coupling between the separate GQD units, therefore accessing the full potential of the ensembles by virtue of their collective properties. For example, the aforementioned variance in QY of PL can be the direct consequence of functionalization of GQDs by different surface passivating ligands, while the assemblies of GQDs, even in the form of disordered aggregation, have produced some unusual properties, such as emission transformation.98 With recent progress in both directions, GQDs with new interesting properties have emerged.Doping GQDs with heteroatoms would effectively tune their intrinsic properties, including electronic characteristics, surface and local chemical features, which thus could offer more active sites and induce new properties of GQDs for new device applications. In this regard, our group has prepared N-doped GQDs (N-GQDs) by following the electrochemical approach we reported previously52 but changing the PBS electrolyte to the N-containing tetrabutylammonium perchlorate (TBAP) in acetonitrile.53 The N-GQDs were continuously produced by CV scanning over a potential window of ±3.0 V, and the as-prepared N-doped GQD solution was found to exhibit a long-term homogeneous phase without any noticeable precipitation at room temperature. Compared to N-free GQDs prepared by the same electrochemical method,52 the N-doped GQDs were about the same in size but presented a blue luminescence different from that of N-free counterparts with green luminescence (Fig. 14, left). X-ray photoelectron spectroscopy (XPS) measurements confirmed the incorporation of N atoms into the GQDs and the high-resolution N 1s spectrum of the GQDs revealed the presence of N atoms in both pyridine-like (398.5 eV) and pyrrolic (401 eV) forms, despite this, the former form was predominant. O-rich groups were also found and a possible structure of an O-rich N-doped GQD is depicted in Fig. 14 (top right). It would be interesting to compare N-doped GQDs with N-doped TiO2, a well developed photocatalyst with a better response in the visible light region than that of pure TiO2.104 N-doping in TiO2 is believed to generate localized states in the band-gap and induce a new absorption band centered at 430–450 nm, thus red-shifting the absorption band edge of TiO2 and increasing its vis-photocatalytic activities.105 Alternatively, visible light response could be indirectly induced by doping, i.e. oxygen vacancies that act as color centers are efficiently stabilized by the presence of nitrogen as a result of charge compensation.106–108 Additionally, high temperature calcinations are generally required to incorporate N atoms into the rigid lattice of TiO2. In contrast, the blue-shift in the absorption spectrum associated with N-doped GQDs implies a different mechanism functioning in the N-doping of GQDs. Despite the need for more structural characterization and information, the heat-free electrochemical doping of GQDs has well been established as an effective way to tailor the properties of GQDs.
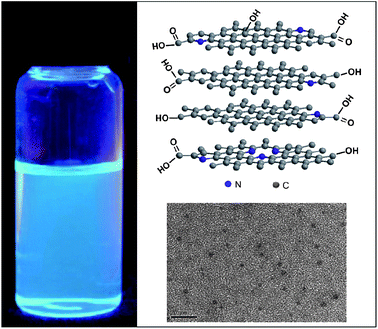 | ||
| Fig. 14 A photograph of the N-GQD solution in water under 365 nm UV irradiation (left), the possible structure of an O-rich N-GQD (not drawn to scale), and TEM image of the as-prepared N-GQDs (bottom right). (Reprinted with permission from ref. 53. Copyright 2012 American Chemical Society.) | ||
Lately, Son et al. reported a synthesis of ZnO–graphene hybrid quasi QD assemblies.100 In a mixture of zinc acetate dihydrate and graphene oxide in dimethyl formamide (DMF), embryo ZnO QDs preferably formed first, followed by the grafting of graphene layers onto ZnO QDs by the formation of Zn–O bonding. During this process, parts of graphene detached from the graphene oxide layers via a chemical exfoliation process and partially encapsulated the ZnO QDs to form quasi-core–shell nanoparticles, as shown in Fig. 15. The coupling between graphene and ZnO induced a splitting of the pristine LUMO level to LUMO, LUMO + 1 and LUMO + 2 in the graphene layer, with electron transitions to the ZnO quantum-dot ground state only allowed from LUMO and LUMO + 2, and resulted in new features in the PL spectrum.
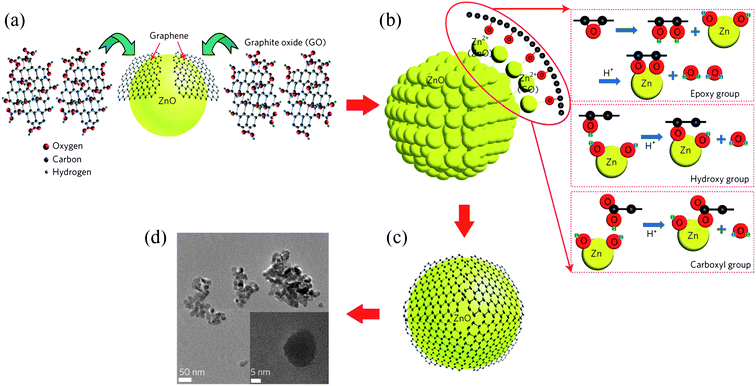 | ||
| Fig. 15 Chemical synthesis process for the ZnO–graphene quasi-QDs. A schematic of (a) chemical exfoliation of graphene sheets from graphene oxide, (b) synthesis of ZnO–graphene QDs from graphene oxide and zinc acetate dehydrate and (c) graphene-covered ZnO QDs. (d) A TEM image of the ZnO–graphene QDs. (Reprinted with permission from ref. 100. Copyright 2012 Nature Publishing Group.) | ||
Hamilton et al. reported the assemblies of colloidal GQDs, a new type of disk-shaped nanostructures, on polar surfaces with controlled orientations.109 Following the principle that the molecular orientation on a water surface is determined by the interplay between cofacial intermolecular attraction (e.g., π-stacking) and molecule–water interactions, they employed Langmuir techniques and AFM to illustrate the design principles to control the assemblies of the GQDs on water and mica surfaces. Typically, the disk-like molecules can either align parallel to the surface (i.e., “face-on”) if the cofacial molecule–water interaction is stronger than the cofacial intermolecular attraction, or align out of plane from the surface (i.e., “edge-on”) if the cofacial molecule–water interaction is weaker. The proper modification of GQDs with a desirable group of –COOH successfully manipulated the orientation of GQDs either in- or out-of-plane with the substrate (Fig. 16), and the orientation control of the GQDs could have practical significance in determining their performance in devices. For example, to use them as sensitizers in dye-sensitized solar cells (DSCs), the orientation of the GQDs adsorbed on semiconducting metal oxides not only determines the efficient coverage of the light absorbers on the oxides and thus the photocurrent but also dictates the photoinduced charge injection.
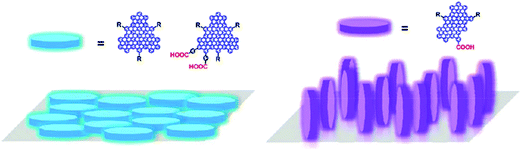 | ||
| Fig. 16 A scheme of the orientations of GQDs either in- or out-of-plane with the substrates. (Reprinted with permission from ref. 109. Copyright 2011 American Chemical Society.) | ||
In a templated manner, we have presented the well-organized assembly of 0D functional GQDs into 1D nanotube (NT) arrays by direct electrophoresis deposition of GQDs within a nanoporous anodized aluminum oxide (AAO) template (Fig. 17a). The used GQDs with a size of 3–5 nm were rich in negatively charged carboxyl groups and could be well dispersed in water, thus facilitating the electrophoresis deposition. The as-formed GQD-NTs had a diameter of ca. 200–300 nm, remaining a free-standing array structure (Fig. 17b and c), and TEM investigation revealed the quasi-orientation of GQDs within the assembled tubes (Fig. 17d). On the basis of the unique porous nanotube architecture of GQDs, the GQD-NTs ensured a more efficient charge transfer between the target molecules and the GQDs, and thus produced a much stronger surface-enhanced Raman scattering (SERS) effect exceeding that on flat graphene sheets, demonstrating their remarkable potential as a new metal-free platform for efficient SERS applications. Meanwhile, microspheres of GQDs (MGQDs) have also been prepared by assembly of GQDs via a water-in-oil (W/O) emulsion technique.111 Although made of quantum-sized graphene dots, the as-formed MGQDs were solid and remained intact after slight ultrasonication. The versatile W/O emulsion method allows the in situ intercalation of functional nanocomponents into the MGQDs for specific applications. As exemplified by the Fe3O4-containing MGQDs, they exhibited a large magnetic response, and the embedded Fe3O4 nanoparticles can further act as the catalysts for the growth of carbon nanotubes (CNTs) towards the hierarchical carbon nanostructures.
 | ||
| Fig. 17 (a) A scheme of the fabrication of GQD-NTs and their SERS function, (b and c) SEM images of assembled nanotube arrays of GQDs with different magnifications after release from the AAO template, and (d) a TEM image of an individual GQD nanotube. (Reprinted with permission from ref. 110. Copyright 2012 American Chemical Society.) | ||
5. Application of GQDs: energy-related, environment-oriented, and more beyond
5.1 Photovoltaics
QDs have played a leading role in revolution of the photovoltaic devices owing to their size-tuned optical response, efficient multiple carrier generation and potential in exceeding the Shockley–Queissar limit.112,113 With their high specific surface areas, high mobilities and tunable band gaps, GQDs should also hold great promise for efficient photovoltaic devices.Our group fabricated a bulk heterojunction (BHJ) polymer solar cell based on an active layer of poly(3-hexylthiophene) (P3HT) and GQDs (Fig. 18a).52 As depicted in Fig. 18b, the LUMO level of the electrochemically synthesized GQDs is estimated to be in the range 4.2–4.4 eV, roughly lying between the LUMO of P3HT and the work function of Al and thus forming an electron transport cascade. Generally, effective exciton dissociation and fast carrier transport are key factors for efficient BHJ solar cells, and GQDs are ideal candidates for these functions. The large surface areas of GQDs can conveniently afford ample interfaces for exciton dissociation, while the high electron mobility of GQDs is expected to increase the conductivity of the active layer and greatly facilitate the charge transport through the active layer. Indeed, the addition of GQDs to P3HT significantly improved the photovoltaic performance of the device, with a decent open-circuit voltage of 0.67 V and a photo-to-electric conversion efficiency of 1.28% (Fig. 18c). The low fill factor of the device is merely a result from the lack of device optimization and suggests there is ample room to further increase the performance of this type device. Nonetheless, the validation of the concept clearly demonstrates that GQDs could be a promising alternative to the widely used C60/C70 as the electron acceptor in BHJ solar cells.
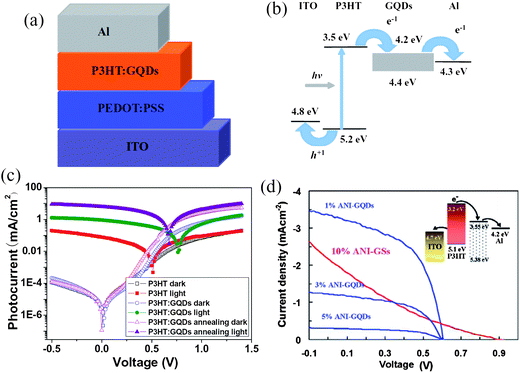 | ||
| Fig. 18 Schematic (a) and energy band (b) diagrams of the ITO/PEDOT:PSS/P3HT:GQDs/Al device. (c) J–V characteristic curves of different devices. Annealing was carried out at 140 °C for 10 min. (Reprinted with permission from ref. 52. Copyright 2011 Wiley-VCH.) (d) J–V characteristics of the photovoltaic devices based on ANI-GQDs with different GQDs contents and ANI-GS (under optimized condition). All devices were annealed at 160 °C for 10 min and measured under simulated AM1.5G illumination at 100 mW cm−2. (Reprinted with permission from ref. 101. Copyright 2011 American Chemical Society.) | ||
In a later work, Gupta et al. employed their aniline-functionalized GQDs (ANI-GQDs) and aniline-functionalized graphene sheets (ANI-GSs) as the electron acceptor and prepared BHJ solar cells based on an optimized cell structure.101 As shown in Fig. 18d, the effect of GQDs on the photovoltaic performance of the solar cells was examined by adding different concentrations of ANI-GQDs to P3HT in the active layer. The device with 1 wt% ANI-GQD in P3HT had the highest conversion efficiency and was much more efficient than the device with 10% ANI-GSs that showed signs of shunting. AFM images displayed that the P3HT/ANI-GSs film contained phase separation domains (about 100–200 nm diameter) much larger than the diffusion length of excitons (10 nm), whereas P3HT/ANI-GQD films showed uniform and nanoscale phase separation. Therefore, the improved morphology of the P3HT/ANI-GQD film was responsible for the improved performance.
In another device configuration, colloidal GQD 1 was selected as the sensitizer in the DSCs.99 The structure of GQD 1 is shown in Fig. 7a, with its absorption spectrum plotted in Fig. 11c. GQD 1 showed an absorbance maximum at 591 nm with a molar extinction coefficient of 1.0 × 105 M−1 cm−1, about an order of magnitude larger than those of the widely used metal complexes in DSCs,114 and the absorption edge extended up to 900 nm, which is highly sought in the DSC field. However, the fabricated DSC only demonstrated a limited photovoltaic performance, with a short-circuit current density in the domain of µA cm−2. This was attributed to low affinity of GQD 1 for the TiO2 film due to the pure physical absorption, resulting in a slightly colored film even after long time sensitization. Additionally, the large size of GQD 1 (diameter ca. 5 nm from molecular modeling) may prevent an effective packing of GQDs on the surface of TiO2 nanoparticles, a prerequisite for high photocurrent and high conversion efficiency in DSCs.
5.2 Organic light-emitting diodes (OLEDs)
OLEDs can also benefit from the high electron mobility and the strong luminescence of GQDs. Mixtures of poly(2-methoxy-5-(2-ethylhexyloxy)-1,4-phenylenevinylene) (MEH-PPV) and methylene blue functionalized GQDs (MB-GQDs) with different concentrations were employed as the light emitting layer and tested in OLEDs under the optimized conditions.101 As shown in Fig. 19a, the turn-on voltage of the device decreased from ∼6 V for the pure MEHPPV sample (trace i) to ∼4 V for MEH-PPV with 1% MB-GQDs (trace ii). At a higher concentration of 3% MB-GQDs, charge trapping as well as a shortening effect occurred, possibly due to agglomeration (trace iv). The maximum light-emission intensity increased with the addition of MB-GQDs to MEH-PPV, directly reflecting the enhanced internal quantum efficiency. In mechanism, the MB-GQDs dispersed in the MEH-PPV afforded additional electrical transport paths that improved charge injection and consequently the carrier density, therefore requiring a lower turn-on voltage.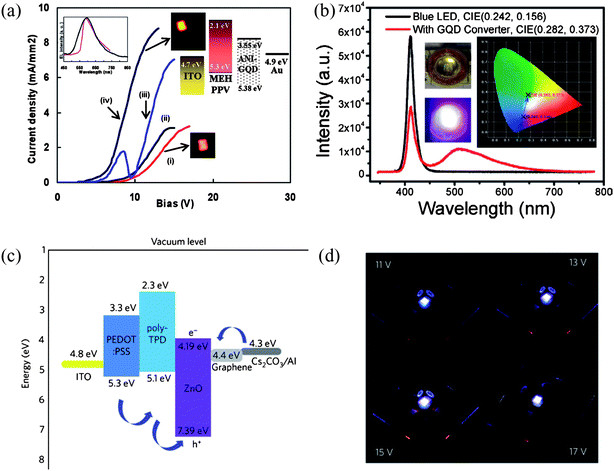 | ||
| Fig. 19 (a) Measured current density of MEH-PPV with MB-GQDs (a) 0%, (b) 0.5%, (c) 1%, and (d) 3% as a function of the applied voltage. The inset plots the electroluminescence spectrum of the MEH-PPV (red) and MEH-PPV/MB-GQDs (1%) (black). The inset also shows the band diagram of the MEH-PPV/MB-GQDs and the recorded brightness of MEH-PPV LED and MEH-PPV/MB-GQDs (1%) LED. (Reprinted with permission from ref. 101. Copyright 2011 American Chemical Society.) (b) Luminescence spectra of the blue LED with and without GQDs coating. Left inset: photographs of the GQD coated LED without (top) and with applied voltage (bottom). Right inset: the CIE chromaticity coordinates for the illuminating blue LED with and without GQD layer. (Reprinted with permission from ref. 72. Copyright 2012 American Chemical Society.) (c) Band diagram of the ZnO–graphene QD LED device. The pathways of holes and electrons are indicated by arrows. (d) Photograph of light emission at 11, 13, 15 and 17 V applied bias. (Reprinted with permission from ref. 100. Copyright 2012 Nature Publishing Group.) | ||
Tang et al. showed another interesting application of GQDs as an excellent light converter by coating a layer of their GQDs onto a commercially available light-emitting diode (LED) emitting blue light centered at 410 nm.72 Upon the coating of the GQDs, the intensity of the blue light weakened and a broad band peaking at ∼510 nm arose, as is illustrated in Fig. 19b. Concomitantly, the Commission International d'Eclairage (CIE) chromaticity coordinates of the LED shifted from (0.242, 0.156) to (0.282, 0.373), confirming the ability of GQDs to convert blue light into white light. This color conversion behavior was understood as the mixing of the 410 nm emission from the original LED and a broad emission at 510 nm given by GQDs under the 410 nm excitation.
Additionally, Son and coworkers explored the potential of hybrid GQDs in OLEDs and the ZnO–graphene QD LED showed a white electroluminescence with a rather high turn-on voltage of 11–12 V.100 Cs2CO3 was selected and used as both an electron-injection layer (EIL) and a hole-blocking layer (HBL), and as depicted in Fig. 19c, electrons are injected from the Cs2CO3 to the graphene layer rather than to the conduction band of ZnO, as the energy level of the former is lower than that of the latter. The wavelength of excitonic emission of ZnO QD was found to be red-shifted by the conjugation with graphene, opening up a new way to tune the centre of the electroluminescence of a metal-oxide semiconductor. Fig. 19d further shows a photograph of the light emission from the device at applied biases of 11, 13, 15 and 17 V, where pixels seemed uniformly luminescent and appeared bluish-white to the naked eye due to the combination of a series of blue and yellow emissions. At 15 V applied bias and with optimal CIE coordinates (0.23, 0.20), the maximum brightness reached ∼798.1 cd m−2, with an external quantum efficiency of 0.04%.
5.3 Fuel cells
N-doped carbon nanomaterials, such as carbon nanotubes115 and graphene sheets116 have been successfully demonstrated as metal-free electrocatalysts to replace the commercially available Pt-based catalysts for the oxygen reduction reaction (ORR) in fuel cell. It is curious to see if N-doped GQDs possess any catalytic effects superior to the previous reported materials. We studied N-doped GQDs supported by a graphene film (N-GQD/graphene) (Fig. 20a) and found that, similar to the commercial Pt/C, a well-defined cathodic peak clearly occurred in the O2-saturated but not N2-saturated KOH solution for N-doped GQDs/graphene (Fig. 20b).53 The ORR onset potential was at ca. −0.16 V with a reduction peak at ca. −0.27 V, which was close to those of the commercial Pt/C catalyst. Unlike the commercial Pt/C electrode where the cathodic peaks for oxygen reduction disappeared in the O2-saturated electrolyte containing 3 M methanol (a typical fuel molecule) coupled with one pair of peaks characteristic of methanol reduction/oxidation, the N-GQD/graphene electrode exhibited a stable ORR without any electroactivity specific to methanol in the methanol-containing electrolyte, suggesting a remarkable tolerance to possible crossover effects. No obvious ORR electrocatalytic activity was found in ORR tests on pure graphene and N-free GQD/graphene performed in O2-saturated KOH solution. Therefore, the observed electrocatalytic activity for N-doped GQDs can be exclusively attributed to the N-doping effect. Electrochemical kinetics studies showed the ORR on N-GQD/graphene was first-order with respect to dissolved oxygen and the electron transfer number involved in the reaction was derived to be 3.6–4.4 (Fig. 20c), well corresponding to the theoretical four-electron process for the ORR. The N-GQD/graphene also demonstrated an excellent stability, with no obvious decrease in current observed after 2 days of continuous cycling in O2-saturated 0.1 M KOH (Fig. 20d), qualifying it as a promising alternative to Pt as the ORR catalyst in fuel cells.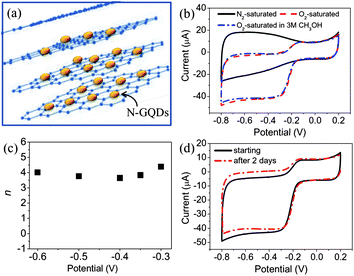 | ||
| Fig. 20 (a) Scheme of graphene supported N-GQDs, (b) CVs of N-GQD/graphene electrode in N2-saturated 0.1 M KOH, O2-saturated 0.1 M KOH, and O2-saturated 3 M CH3OH solutions, (c) the dependence of electron transfer number on the potential. (d) Electrochemical stability of N-GQD/graphene as determined by continuous cyclic voltammetry in O2-saturated 0.1 M KOH. (Reprinted with permission from ref. 53. Copyright 2012 American Chemical Society.) | ||
5.4 Environment-oriented applications
With the increase of world population, environmental problems are aggravating particularly in water resources, as human activity has led to a huge production of wastewaters containing organic compounds that are difficult to remove. TiO2 is regarded as the most promising photocatalyst in decontaminating wastewaters in lieu of the Fenton process,117 but its narrow spectrum has limited its activity under visible light. The upconversion properties of GQDs thus render a solution. Zhuo et al. prepared rutile TiO2/GQD and anatase TiO2/GQD nanocomposites and studied their kinetics in degrading methylene blue (MB) under Xe lamp with a 420 nm cutoff filter.63 As shown in Fig. 21a, the photodegradation efficiency is up to 97% for rutile TiO2/GQDs and 31% for anatase TiO2/GQDs in 60 min, respectively, both of which are significantly higher than those of pure rutile and anatase TiO2. The superior photocatalytic activity of the rutile TiO2/GQDs is rationalized by the different bandgap values of rutile and anatase TiO2 as depicted in Fig. 21b. Under visible light illumination, the upconverted PL peak of GQDs was located at ca. 407 nm (3.05 eV), with the energy lying between the bandgap energy of rutile TiO2 3.0 eV (414 nm) and that of anatase TiO2 3.2 eV (388 nm). Therefore, the upconversion PL emission of GQDs was only able to excited rutile TiO2 to form reactive holes to degrade MB.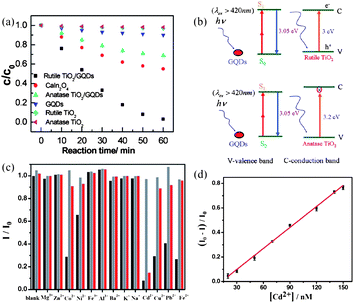 | ||
| Fig. 21 (a) The relationship between MB concentration and reaction time for different catalysts: rutile TiO2/GQDs, CaIn2O4, anatase TiO2/GQDs, GQDs, rutile TiO2 nanoparticles, and anatase TiO2 nanoparticles. (b) A schematic of the photocatalytic process for rutile TiO2/GQDs and anatase TiO2/GQDs under visible light (λ > 420 nm). (Reprinted with permission from ref. 63. Copyright 2012 American Chemical Society.) (c) Relative ECL intensity (I/I0) of GQDs (20 ppm) in the presence of various metal ions (20 µM) without any chelator (black) or with 40 µM EDTA (grey) and 1 mM Cys (red). (d) A linear calibration plot for Cd2+ detection in the presence of 1 mM Cys. (Reprinted with permission from ref. 60. Copyright 2012 Wiley-VCH.) | ||
Cd2+ is a highly toxic metal ion of disastrous potential to the environment, thus an accurate determination of Cd2+ is very important. Li et al. developed a new method of determining Cd2+ on the basis of the intense ECL of their GQDs.60 As shown in Fig. 21c, six ions of Ni2+, Pb2+, Cu2+, Co2+, Fe2+ and Cd2+ could quench the ECL due to the interaction between metal ions and GQDs, with Cd2+ being the most effective. It was found that the addition of cysteine (Cys) could recover the quenched ECL induced by all the metal ions except Cd2+, which still quenched the GQDs ECL by 85% even in the presence of 1 mM Cys, making Cys an effective masking agent for the determination of Cd2+ with acceptable selectivity. In the presence of 1 mM Cys, the ECL intensity of GQDs gradually decreased with increasing Cd2+ concentration, exhibiting a linear dependence over the range from 20–150 nM (R = 0.998) with a detection limit of 13 nM at a signal-to-noise ratio of 3 (Fig. 21d).
5.5 Bioimaging, biosensors and beyond
Besides the potentials in energy- and environment-related fields, GQDs are also promising for biological applications, such as bioimaging, biosensors and cell isolations, due to their low cytotoxicity as confirmed by different groups. Peng et al. evaluated the cytotoxicity of green or blue GQDs by using two different human breast cancer cell lines MDA-MB-231 and T47D, and concluded that low doses of GQDs (up to 50 µg mL−1) did not impose considerable toxicity to these cells compared to the untreated control cells,58 where 1 µM of doxorubicin was used as a positive control, toxicity at 24 and 48 h is 50% and 35%, respectively. Cell proliferation at low dosage as the exposure time increases was not affected either. In another study, average cell viability of two stem cells, neurospheres cells (NSCs) and cardiac progenitor cells (CPCs), was above 80% after 3 days culturing with GQDs at a concentration of 100 µg mL−1, and that of pancreas progenitor cells (PPCs) was about 65%,54 whereas these stem cells could be adequately observed after incubation in a 25 µg mL−1 solution of GQDs. Similarly, Dong et al. also reported that single-layer GQDs derived from CX-72 carbon black did not pose a considerable toxicity to human breast cancer MCF-7 cells at the same concentration of 100 µg mL−1.59 More impressively, Zhu et al. found that very high concentrations of GQDs (up to 400 µg in 150 µL of culture medium, 104 cells) did not weaken the MG-63 cell activity significantly, as shown by the MTT viability assay (Fig. 22c).50 All these studies suggest the low cytotoxicity of GQDs is comparable to that of C-dots and shows promise for in vitro and in vivo imaging,118 despite more toxicity studies, such as LD50 (median lethal dose) measurements, are further required before applying GQDs in clinical medicine.21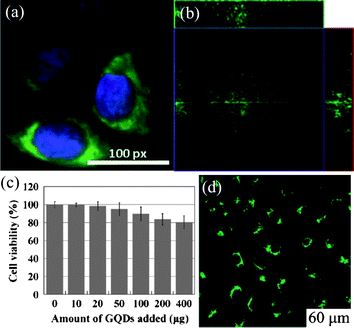 | ||
| Fig. 22 (a) The overlay high contrast image of a nucleolus stained with blue DAPI and GQDs (green) staining. (Reprinted with permission from ref. 58. Copyright 2012 American Chemical Society.) (b) Section analysis of human breast cancer MCF-7 cells labeled with GQDs derived from carbon black. (Reprinted with permission from ref. 59. Copyright 2012 Royal Society of Chemistry.) (c) Effect of solvothermally produced GQDs on MG-63 cells viability. (d) Cellular imaging of GQDs under bright field of 405 nm. (Reprinted with permission from ref. 50. Copyright 2011 Royal Chemical Society.) | ||
Green GQDs was selected to incubate with T47D human breast cancer cells with the nucleus stained by 4′,6-diamidino-2-phenylindole (DAPI, blue color). As shown in Fig. 22a, the obtained fluorescent image clearly shows the phase contrast of labeled T47D cells, with the nucleus stained in blue by DAPI and green GQDs agglomerated around each nucleus.58 In another experiment, Dong et al. found that the GQDs-stained MCF-7 cells exhibited a bright green color when imaged with excitation at 488 nm on the confocal laser scanning microscope and the section analysis of one MCF-7 cell indicated that GQDs could label the cell membrane, the cytoplasm and the nucleus simultaneously, the first time for luminescent carbon nanomaterials to label the cell nucleus (Fig. 22b).59 Additionally, Zhu et al. showed a case of two-color imaging with GQDs. After the uptake of hydrothermally cut GQDs, bright green areas inside the MG-63 cells were observed when excited at 405 nm (Fig. 22d) and cellular imaging changed to a green-yellow color when the excitation shifted to 488 nm, both indicating translocation of GQDs through the cell's membrane.50 Furthermore, Pan et al. cultured HeLa cells with their green GQDs and demonstrated that unlike dye-labeled cells, the GQD-labeled HeLa cells did not experience any obvious reduction in PL brightness during continuous excitation for 10 min.49 All the above results evidently confirm the viability of these photostable GQDs as a feasible substitute for fluorescent dyes in high contrast bioimaging and other biomedical applications.
It has also been demonstrated that GQD modified pyrolytic graphite (PG) electrode coupled with probe single-stranded DNA (ssDNA) can be used as a platform to develop different kinds of electrochemical biosensors for the sensitive and selective detection of various target molecules.119 Generally, the ssDNA effectively inhibits the electron transfer between the electrochemical active species [Fe(CN)6]3−/4− and the electrode after the probe molecules are strongly bound to the surface of the modified PG electrode via their interaction with graphene. However, in the case where the target molecules, such as target ssDNA or target protein, coexist in the test solution, the probe ssDNA prefers to bind with the target instead of graphene, if the sequence of the probe ssDNA is designed as complementary to the target DNA or as the aptamer of the target protein, resulting in the observed peak currents of [Fe(CN)6]3−/4− that increases with the target molecules. As more and more aptamers are discovered, study of such kinds of label-free, electrochemical biosensors can be extended for the detection of more molecules, not necessarily limited to complementary DNA or proteins with known aptamers.
6. Summary and outlooks
As presented in this perspective, GQDs are new materials that have exhibited varieties of intriguing electronic and optical properties promising for applications in energy-, environment-, and biology-related fields. The availability of various physical, chemical or electrochemical methods for preparation of GQDs provides immense room to tune the intrinsic and surface nature of GQDs as desired for specific purposes. With a well adjustable band gap, GQDs possess unique absorption, photoluminescence and electroluminescence. Functional assemblies of GQDs generate new properties beyond that of individual GQDs. As a result, applications in photovoltaic cells, fuel cells, LEDs, catalysis, bioimaging and biosensors are emerging for GQDs.On the other hand, the research on GQDs is still at its early stage in line with the onrush of graphene research, with 20% of our references coming from last 6 months and more than half from the last two years or so. Some important issues are waiting for solution to further development of GQDs. For example, an explicit and comprehensive understanding of the optical properties of GQDs is still absent, including the effect of factors such as size, surface chemistry, doping atoms, and so on. Large scale production of high quality GQDs with structural uniformity via a straightforward approach is also a challenge for most of current synthetic techniques, where a multistep procedure with a rigorous separation process is generally necessary for preparing GQDs. Moreover, the research on GQD assembly, hybrid structure or composition with other functional components is even less developed thus far.
Besides the further enhancement of device performance through device optimization, GQDs with appropriate functionalization, assembly and/or hybrid structures can be involved in different energy-related devices beyond those presented above. As illustrated in Fig. 23, the tunability of GQDs' spectrum conveniently renders a variety of sensitizers for DSCs,69,99 the catalytic effect of doped GQDs demonstrates well their potential as an effective replacement for the commonly used Pt counter electrode in DSCs, and the high electron mobility of GQDs could improve the conductivity of the gel/solid electrolyte or possibly enable GQDs themselves as a suitable hole conductor in DSCs. In BHJ solar cells, apart from what we have seen as the electron acceptor,52,101 modified GQDs with an improved energy alignment could theoretically work as an electron donor as well, showing the possibility that charge transfer may take place even between two types of GQDs and therefore form an interesting junction completely based on graphene. Meanwhile, the capability of GQDs to upconvert incident beams is of particular interest to both DSCs and BHJ solar cells, where a better use of the infrared spectrum is always welcome. It is also natural but important to back check the performance of GQDs in fields where conventional carbonaceous materials have already shown promise. As revealed in Fig. 17, GQDs can be readily organized into a 1D nanotube array with good conductivity and high surface areas and thus will be favorable as an electrode material for lithium ion batteries and supercapacitors. With the development of new assembling methods and techniques, the formed multidimensional structure120 and the hybrid/composition of GQDs with other high-surface-area substances will further benefit the related devices. Beyond the energy-oriented applications of GQDs discussed above, GQDs should find applications in various chemical and biological sensors. For example, high-performance detection towards small toxic gas molecules, biomolecules, and bioactivities of living cells has been achieved with graphene-based field-effect transistors (GFETs),121 electrochemical sensors, and fluorescence sensors.11 Additionally, the outstanding photoluminescent properties also qualify GQDs as a viable material for applications in cell imaging and even in clinical medicine, provided that more cytotoxicological data and in vivo experiments test and validate the biological safety of GQDs. In summary, we envisage that GQDs will soon shine as a gem in the crown of carbon materials with the advance of new synthetic strategies, the discovery of novel physical/chemical properties and the unprecedented functions in devices in various fields.
 | ||
| Fig. 23 Some potential applications of GQDs in different energy-related devices. | ||
Acknowledgements
We thank the financial support from the 973 program (2011CB013000) of China and NSFC (21004006, 21174019 and 51161120361), Fok Ying Tong Education Foundation (no. 131043), the research foundation for the doctoral program of higher education of China (20101101120036), the 111 Project 807012, and the Program for the New Century Excellent Talents in University (NCET-10-0047).Notes and references
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef CAS.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, S. V. Dubonos, I. V. Grigorieva and A. A. Firsov, Science, 2004, 306, 666–669 CrossRef CAS PubMed.
- K. I. Bolotin, K. J. Sikes, Z. Jiang, M. Klima, G. Fudenberg, J. Hone, P. Kim and H. L. Stormer, Solid State Commun., 2008, 146, 351–355 CrossRef CAS.
- S. V. Morozov, K. S. Novoselov, M. I. Katsnelson, F. Schedin, D. C. Elias, J. A. Jaszczak and A. K. Geim, Phys. Rev. Lett., 2008, 100, 016602 CrossRef CAS PubMed.
- X. Du, I. Skachko, A. Barker and E. Y. Andrei, Nat. Nanotechnol., 2008, 3, 491–495 CrossRef CAS PubMed.
- T. J. Booth, P. Blake, R. R. Nair, D. Jiang, E. W. Hill, U. Bangert, A. Bleloch, M. Gass, K. Novoselov, M. I. Katsnelson and A. K. Geim, Nano Lett., 2008, 8, 2442–2446 CrossRef CAS PubMed.
- Z. Dong, C. Jiang, H. Cheng, Y. Zhao, G. Shi, L. Jiang and L. T. Qu, Adv. Mater., 2012, 24, 1856–1861 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183–191 CrossRef CAS PubMed.
- A. H. Castro Neto, F. Guinea, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS.
- X. Huang, Z. Y. Yin, S. X. Wu, X. Y. Qi, Q. Y. He, Q. C. Zhang, Q. Y. Yan, F. Boey and H. Zhang, Small, 2011, 7, 1876–1902 CrossRef CAS PubMed.
- X. Huang, X. Y. Qi, F. Boey and H. Zhang, Chem. Soc. Rev., 2012, 41, 666–686 RSC.
- M. Y. Han, B. Ozyilmaz, Y. Zhang and P. Kim, Phys. Rev. Lett., 2007, 98, 206805 CrossRef PubMed.
- K. Todd, H. Chou, S. Amasha and D. Goldhaber-Gordon, Nano Lett., 2009, 9, 416–421 CrossRef CAS PubMed.
- C. Stampfer, J. Güttinger, S. Hellmüller, F. Molitor, K. Ensslin and T. Ihn, Phys. Rev. Lett., 2009, 102, 056403 CrossRef CAS PubMed.
- F. Molitor, A. Jacobsen, C. Stampfer, J. Güttinger, T. Ihn and K. Ensslin, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 075426 CrossRef.
- M. Wang, E. B. Song, S. Lee, J. Tang, M. Lang, C. Zeng, G. Xu, Y. Zhou and K. L. Wang, ACS Nano, 2011, 5, 8769–8773 CrossRef CAS PubMed.
- C. O. Girit, J. C. Meyer, R. Erni, M. D. Rossell, C. Kisielowski, L. Yang, C. H. Park, M. F. Crommie, M. L. Cohen, S. G. Louie and A. Zettl, Science, 2009, 323, 1705–1708 CrossRef CAS PubMed.
- K. A. Ritter and J. W. Lyding, Nat. Mater., 2009, 8, 235–242 CrossRef CAS PubMed.
- J. Shen, Y. Zhu, X. Yang and C. Li, Chem. Commun., 2012, 48, 3686–3699 RSC.
- S. N. Baker and G. A. Baker, Angew. Chem., Int. Ed., 2010, 49, 6726–6744 CrossRef CAS PubMed.
- X. Y. Xu, R. Ray, Y. L. Gu, H. J. Ploehn, L. Gearheart, K. Raker and W. A. Scrivens, J. Am. Chem. Soc., 2004, 126, 12736–12737 CrossRef CAS PubMed.
- H. Liu, T. Ye and C. Mao, Angew. Chem., Int. Ed., 2007, 46, 6473–6475 CrossRef CAS PubMed.
- J. G. Zhou, C. Booker, R. Y. Li, X. T. Zhou, T. K. Sham, X. L. Sun and Z. F. Ding, J. Am. Chem. Soc., 2007, 129, 744–745 CrossRef CAS PubMed.
- Y. P. Sun, B. Zhou, Y. Lin, W. Wang, K. A. S. Fernando, P. Pathak, M. J. Meziani, B. A. Harruff, X. Wang, H. F. Wang, P. G. Luo, H. Yang, M. E. Kose, B. L. Chen, L. M. Veca and S. Y. Xie, J. Am. Chem. Soc., 2006, 128, 7756–7757 CrossRef CAS PubMed.
- L. Cao, X. Wang, M. J. Meziani, F. Lu, H. Wang, P. G. Luo, Y. Lin, B. A. Harruff, L. M. Veca, D. Murray, S. Y. Xie and Y. P. Sun, J. Am. Chem. Soc., 2007, 129, 11318–11319 CrossRef CAS PubMed.
- Y. P. Sun, X. Wang, F. Lu, L. Cao, M. J. Meziani, P. G. Luo, L. R. Gu and L. M. Veca, J. Phys. Chem. C, 2008, 112, 18295–18298 CAS.
- X. Wang, L. Cao, S. T. Yang, F. Lu, M. J. Meziani, L. Tian, K. W. Sun, M. A. Bloodgood and Y. P. Sun, Angew. Chem., Int. Ed., 2010, 49, 5310–5314 CrossRef CAS PubMed.
- J. G. Zhou, J. Cheiftz, R. Y. Li, F. P. Wang, X. T. Zhou, T. K. Sham, X. L. Sun and Z. F. Ding, Carbon, 2009, 47, 829–838 CrossRef CAS.
- L. Tian, D. Ghosh, W. Chen, S. Pradhan, X. Chang and S. Chen, Chem. Mater., 2009, 21, 2803–2809 CrossRef CAS.
- L. Y. Zheng, Y. W. Chi, Y. Q. Dong, J. P. Lin and B. B. Wang, J. Am. Chem. Soc., 2009, 131, 4564–4565 CrossRef CAS PubMed.
- Y. Dong, N. Zhou, X. Lin, J. Lin, Y. Chi and G. Chen, Chem. Mater., 2010, 22, 5895–5899 CrossRef CAS.
- Q. Zhao, Z. Zhang, B. Huang, J. Peng, M. Zhang and D. Pang, Chem. Commun., 2008, 5116–5118 RSC.
- H. Li, X. He, Z. Kang, H. Huang, Y. Liu, J. Liu, S. Lian, C. H. A. Tsang, X. Yang and S. Lee, Angew. Chem., Int. Ed., 2010, 49, 4430–4434 CrossRef CAS PubMed.
- J. Lu, J. Yang, J. Wang, A. Lim, S. Wang and K. P. Loh, ACS Nano, 2009, 3, 2367–2375 CrossRef CAS PubMed.
- S. C. Ray, A. Saha, N. R. Jana and R. Sarkar, J. Phys. Chem. C, 2009, 113, 18546–18551 CAS.
- H. Zhu, X. L. Wang, Y. L. Li, Z. J. Wang, F. Yang and X. R. Yang, Chem. Commun., 2009, 5118–5120 RSC.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, V. Georgakilas and E. P. Giannelis, Chem. Mater., 2008, 20, 4539–4541 CrossRef CAS.
- A. B. Bourlinos, A. Stassinopoulos, D. Anglos, R. Zboril, M. Karakassides and E. P. Giannelis, Small, 2008, 4, 455–458 CrossRef CAS PubMed.
- L. Yuan, J. Dai, X. Fan, T. Song, Y. T. Tao, K. Wang, Z. Xu, J. Zhang, X. Bai, P. Lu, J. Chen, J. Zhou and Z. L. Wang, ACS Nano, 2011, 5, 4007–4013 CrossRef CAS PubMed.
- J. G. Zhou, X. T. Zhou, R. Y. Li, X. L. Sun, Z. F. Ding, J. Cutler and T. K. Sham, Chem. Phys. Lett., 2009, 474, 320–324 CrossRef CAS.
- L. A. Ponomarenko, F. Schedin, M. I. Katsnelson, R. Yang, E. W. Hill, K. S. Novoselov and A. K. Geim, Science, 2008, 320, 356–358 CrossRef CAS PubMed.
- J. Shen, Y. Zhu, X. Yang and C. Li, Chem. Commun., 2012, 48, 3686–3699 RSC.
- S. J. Zhu, S. J. Tang, J. H. Zhang and B. Yang, Chem. Commun., 2012, 48, 4527–4539 RSC.
- K. S. Novoselov, D. Jiang, F. Schedin, T. J. Booth, V. V. Khotkevich, S. V. Morozov and A. K. Geim, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 10451–10453 CrossRef CAS PubMed.
- J. Güttinger, C. Stampfer, T. Frey, T. Ihn and K. Ensslin, Phys. Status Solidi B, 2009, 246, 2553–2557 CrossRef.
- L. J. Wang, G. Cao, T. Tu, H. O. Li, C. Zhou, X. J. Hao, Z. Su, G. C. Guo, H. W. Jiang and G. P. Guo, Appl. Phys. Lett., 2010, 97, 262113 CrossRef.
- D. Pan, J. Zhang, Z. Li and M. Wu, Adv. Mater., 2010, 22, 734–738 CrossRef CAS PubMed.
- D. Pan, L. Guo, J. Zhang, C. Xi, Q. Xue, H. Huang, J. Li, Z. Zhang, W. Yu, Z. Chen, Z. Li and M. Wu, J. Mater. Chem., 2012, 22, 3314–3318 RSC.
- S. J. Zhu, J. H. Zhang, C. Y. Qiao, S. J. Tang, Y. F. Li, W. J. Yuan, B. Li, L. Tian, F. Liu, R. Hu, H. N. Gao, H. T. Wei, H. Zhang, H. C. Sun and B. Yang, Chem. Commun., 2011, 47, 6858–6860 RSC.
- S. J. Zhu, J. H. Zhang, X. Liu, B. Li, X. F. Wang, S. J. Tang, Q. N. Meng, Y. F. Li, C. S. Shi, R. Hu and B. Yang, RSC Adv., 2012, 2, 2717–2720 RSC.
- Y. Li, Y. Hu, Y. Zhao, G. Q. Shi, L. E. Deng, Y. B. Hou and L. T. Qu, Adv. Mater., 2011, 23, 776–780 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. H. Cheng, Y. Hu, G. Q. Shi, L. M. Dai and L. T. Qu, J. Am. Chem. Soc., 2012, 134, 15–18 CrossRef CAS PubMed.
- M. Zhang, L. L. Bai, W. H. Shang, W. J. Xie, H. Ma, Y. Y. Fu, D. C. Fang, H. Sun, L. Z. Fan, M. Han, C. M. Liu and S. H. Yang, J. Mater. Chem., 2012, 22, 7461–7467 RSC.
- R. Liu, D. Wu, X. Feng and K. Müllen, J. Am. Chem. Soc., 2011, 133, 15221–15223 CrossRef CAS PubMed.
- J. H. Shen, Y. H. Zhu, C. Chen, X. L. Yang and C. Z. Li, Chem. Commun., 2011, 47, 2580–2582 RSC.
- J. Y. Luo, L. J. Cote, V. C. Tung, A. T. L. Tan, P. E. Goins, J. S. Wu and J. X. Huang, J. Am. Chem. Soc., 2010, 132, 17667–17669 CrossRef CAS PubMed.
- J. Peng, W. Gao, B. K. Gupta, Z. Liu, R. Romero-Aburto, L. Ge, L. Song, L. B. Alemany, X. Zhan, G. Gao, S. A. Vithayathil, B. A. Kaipparettu, A. A. Marti, T. Hayashi, J. J. Zhu and P. M. Ajayan, Nano Lett., 2012, 12, 844–849 CrossRef CAS PubMed.
- Y. Q. Dong, C. Q. Chen, X. T. Zheng, L. L. Gao, Z. M. Cui, H. B. Yang, C. X. Guo, Y. W. Chi and C. M. Li, J. Mater. Chem., 2012, 22, 8764–8766 RSC.
- L. L. Li, J. Ji, R. Fei, C. Z. Wang, Q. Lu, J. R. Zhang, L. P. Jiang and J. J. Zhu, Adv. Funct. Mater., 2012, 22, 2971–2979 CrossRef CAS.
- N. Mohanty, D. Moore, Z. Xu, T. S. Sreeprasad, A. Nagaraja, A. A. Rodriguez and V. Berry, Nat. Commun., 2012, 3, 844 CrossRef PubMed.
- H. T. Li, X. D. He, Y. Liu, H. Huang, S. Y. Lian, S. T. Lee and Z. H. Kang, Carbon, 2011, 49, 605–609 CrossRef CAS.
- S. Zhuo, M. Shao and S. T. Lee, ACS Nano, 2012, 6, 1059–1064 CrossRef CAS PubMed.
- X. Yang, X. Dou, A. Rouhanipour, L. Zhi, H. J. Räder and K. Müllen, J. Am. Chem. Soc., 2008, 130, 4216–4217 CrossRef CAS PubMed.
- X. Yan and L. S. Li, J. Mater. Chem., 2011, 21, 3295–3300 RSC.
- C. D. Simpson, J. D. Brand, A. J. Berresheim, L. Przybilla, H. J. Räder and K. Müllen, Chem.–Eur. J., 2002, 8, 1424–1429 CrossRef CAS.
- J. Sakamoto, J. Van Heijst, O. Lukin and A. Schlüter, Angew. Chem., Int. Ed., 2009, 48, 1030–1069 CrossRef CAS PubMed.
- X. Yan, X. Cui and L. S. Li, J. Am. Chem. Soc., 2010, 132, 5944–5945 CrossRef CAS PubMed.
- X. Yan, B. Li, X. Cui, Q. Wei, K. Tajima and L. S. Li, J. Phys. Chem. Lett., 2011, 2, 1119–1124 CrossRef CAS PubMed.
- L. S. Li and X. Yan, J. Phys. Chem. Lett., 2010, 1, 2572–2576 CrossRef CAS.
- Y. Q. Dong, J. W. Shao, C. Q. Chen, H. Li, R. X. Wang, Y. W. Chi, X. M. Lin and G. N. Chen, Carbon, 2012, 50, 4738–4743 CrossRef CAS.
- L. Tang, R. Ji, X. Cao, J. Lin, H. Jiang, X. Li, K. S. Teng, C. M. Luk, S. Zeng, J. Hao and S. P. Lau, ACS Nano, 2012, 6, 5102–5110 CrossRef CAS PubMed.
- J. Lu, P. S. E. Yeo, C. K. Gan, P. Wu and K. P. Loh, Nat. Nanotechnol., 2011, 6, 247–252 CrossRef CAS PubMed.
- A. K. Geim and A. H. MacDonald, Phys. Today, 2007, 60, 35–41 CrossRef CAS.
- S. Das Sarma, S. Adam, E. H. Hwang and E. Rossi, Rev. Mod. Phys., 2011, 83, 407–470 CrossRef CAS.
- K. S. Novoselov, A. K. Geim, S. V. Morozov, D. Jiang, Y. Zhang, M. I. Katsnelson, I. V. Grigorieva, S. V. Dubonos and A. A. Firsov, Nature, 2005, 438, 197–200 CrossRef CAS PubMed.
- O. Klein, Z. Phys., 1929, 53, 157–165 CrossRef CAS.
- M. I. Katsnelson, K. S. Novoselov and A. K. Geim, Nat. Phys., 2006, 2, 620–625 CrossRef CAS.
- F. Miao, S. Wijeratne, Y. Zhang, U. Coskun, W. Bao and C. Lau, Science, 2007, 317, 1530–1533 CrossRef CAS PubMed.
- Z. Chen, Y. Lin, M. Rooks and P. Avouris, Phys. E., 2007, 40, 228–232 CrossRef CAS.
- F. Sols, F. Guinea and A. H. CastroNeto, Phys. Rev. Lett., 2007, 99, 166803 CrossRef CAS PubMed.
- Y. M. Lin, V. Perebeinos, Z. Chen and P. Avouris, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 78, 161409(R) CrossRef.
- R. B. Chen, C. P. Chang and M. F. Lin, Phys. E., 2010, 42, 2812–2815 CrossRef CAS.
- F. Libisch, C. Stampfer and J. Burgdörfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 115423 CrossRef.
- M. L. Mueller, X. Yan, B. Dragnea and L. S. Li, Nano Lett., 2011, 11, 56–60 CrossRef CAS PubMed.
- M. L. Mueller, X. Yan, J. A. McGuire and L. S. Li, Nano Lett., 2010, 10, 2679–2682 CrossRef CAS PubMed.
- A. V. Khaetskii, D. Loss and L. Glazman, Phys. Rev. Lett., 2002, 88, 186802 CrossRef PubMed.
- J. R. Petta, A. C. Johnson, J. M. Taylor, E. A. Laird, A. Yacoby, M. D. Lukin, C. M. Marcus, M. P. Hanson and A. C. Gossard, Science, 2005, 309, 2180–2184 CrossRef CAS PubMed.
- F. H. L. Koppens, C. Buizert, K. J. Tielrooij, I. T. Vink, K. C. Nowack, T. Meunier, L. P. Kouwenhoven and L. M. K. Vandersypen, Nature, 2006, 442, 766–771 CrossRef CAS PubMed.
- P. Recher and B. Trauzettel, Nanotechnology, 2010, 21, 302001 CrossRef PubMed.
- M. T. Allen, J. Martin and A. Yacoby, Nat. Commun., 2012, 3, 934 CrossRef CAS PubMed.
- B. Trauzettel, D. V. Bulaev, D. Loss and G. Burkard, Nat. Phys., 2007, 3, 192–196 CrossRef CAS.
- V. Fal'ko, Nat. Phys., 2007, 3, 151–152 CrossRef.
- P. Recher, J. Nilsson, G. Burkard and B. Trauzettel, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 085407 CrossRef.
- A. P. Alivisatos, Science, 1996, 271, 933–937 CAS.
- O. Mićić, H. Cheong, H. Fu, A. Zunger, J. Sprague, A. Mascarenhas and A. Nozik, J. Phys. Chem. B, 1997, 101, 4904–4912 CrossRef.
- L. Cademartiri, E. Montanari, G. Calestani, A. Migliori, A. Guagliardi and G. A. Ozin, J. Am. Chem. Soc., 2006, 128, 10337–10346 CrossRef CAS PubMed.
- S. Chen, J. W. Liu, M. L. Chen, X. W. Chen and J. H. Wang, Chem. Commun., 2012, 48, 7637–7639 RSC.
- X. Yan, X. Cui, B. Li and L. S. Li, Nano Lett., 2010, 10, 1869–1873 CrossRef CAS PubMed.
- D. I. Son, B. W. Kwon, D. H. Park, W. S. Seo, Y. Yi, B. Angadi, C. L. Lee and W. K. Choi, Nat. Nanotechnol., 2012, 7, 465–471 CrossRef CAS PubMed.
- V. Gupta, N. Chaudhary, R. Srivastava, G. D. Sharma, R. Bhardwaj and S. Chand, J. Am. Chem. Soc., 2011, 133, 9960–9963 CrossRef CAS PubMed.
- K. N. Kudin, B. Ozbas, H. C. Schniepp, R. K. Prud'homme, I. A. Aksay and R. Car, Nano Lett., 2008, 8, 36–41 CrossRef CAS PubMed.
- C. Casiraghi, A. Hartschuh, H. Qian, S. Piscanec, C. Georgi, A. Fasoli, K. S. Novoselov, D. M. Basko and A. C. Ferrari, Nano Lett., 2009, 9, 1433–1441 CrossRef CAS PubMed.
- J. L. Zhang, Y. M. Wu, M. Y. Xing, S. A. K. Leghari and S. Sajjad, Energy Environ. Sci., 2010, 3, 715–726 CAS.
- C. Di Valentin, E. Finazzi, G. Pacchioni, A. Selloni, S. Livraghi, M. C. Paganini and E. Giamello, Chem. Phys., 2007, 339, 44–56 CrossRef CAS.
- F. Spadavecchia, G. Cappelletti, S. Ardizzone, M. Ceotto and L. Falciola, J. Phys. Chem. C, 2011, 115, 6381–6391 CAS.
- T. Ihara, M. Miyoshi, Y. Iriyama, O. Matsumoto and S. Sugihara, Appl. Catal., B, 2003, 42, 403–409 CrossRef CAS.
- A. V. Emeline, V. N. Kuznetsov, V. K. Rybchuk and N. Serpone, Int. J. Photoenergy, 2007, 2008, 1–19 CrossRef.
- I. P. Hamilton, B. S. Li, X. Yan and L. S. Li, Nano Lett., 2011, 11, 1524–1529 CrossRef CAS PubMed.
- H. H. Cheng, Y. Zhao, Y. Q. Fan, X. J. Xie, L. T. Qu and G. Q. Shi, ACS Nano, 2012, 6, 2237–2244 CrossRef CAS PubMed.
- Y. Ding, H. H. Cheng, C. Zhou, Y. Q. Fan, J. Zhu, H. B. Shao and L. Q. Qu, Nanotechnology, 2012, 23, 255605 CrossRef PubMed.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 CAS.
- O. E. Semonin, J. M. Luther, S. Choi, H. Y. Chen, J. Gao, A. J. Nozik and M. C. Beard, Science, 2011, 334, 1530–1533 CrossRef CAS PubMed.
- M. K. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- K. P. Gong, F. Du, Z. H. Xia, M. Durstock and L. M. Dai, Science, 2009, 323, 760–764 CrossRef CAS PubMed.
- L. T. Qu, Y. Liu, J. B. Baek and L. M. Dai, ACS Nano, 2010, 4, 1321–1326 CrossRef CAS PubMed.
- I. T. Peternel, N. Koprivanac, A. M. Lončarić Božić and H. M. Kušić, J. Hazard. Mater., 2007, 148, 477–484 CrossRef CAS PubMed.
- S. T. Yang, L. Cao, P. G. Luo, F. S. Lu, X. Wang, H. F. Wang, M. Meziani, Y. F. Liu, G. Qi and Y. P. Sun, J. Am. Chem. Soc., 2009, 131, 11308–11309 CrossRef CAS PubMed.
- J. Zhao, G. F. Chen, L. Zhu and G. X. Li, Electrochem. Commun., 2011, 13, 31–33 CrossRef CAS.
- Y. Q. Fan, H. Cheng, C. Zhou, X. J. Xie, Y. Liu, L. M. Dai, J. Zhang and L. T. Qu, Nanoscale, 2012, 4, 1776–1781 RSC.
- Q. Y. He, S. X. Wu, Z. Y. Yin and H. Zhang, Chem. Sci., 2012, 3, 1764–1772 RSC.
| This journal is © The Royal Society of Chemistry 2012 |