Liquid-phase chemical hydrogen storage materials
Mahendra
Yadav
and
Qiang
Xu
*
National Institute of Advanced Industrial Science and Technology (AIST), Ikeda, Osaka 563-8577, Japan. E-mail: q.xu@aist.go.jp; Fax: +81 72 751 9629; Tel: +81 72 751 9562
First published on 25th September 2012
Abstract
In the search for future energy supplies, the application of hydrogen as an energy carrier is seen as a prospective issue. However, the implementation of a hydrogen economy is suffering from several unsolved problems. Particularly challenging is the storage of appropriate amounts of hydrogen. In this context one of the promising hydrogen storage techniques relies on liquid-phase chemical hydrogen storage materials, in particular, aqueous sodium borohydride, ammonia borane, hydrazine, hydrazine borane and formic acid. The use of these materials in hydrogen storage provides high gravimetric and volumetric hydrogen densities, low potential risk, and low capital investment because it is largely compatible with the current transport infrastructure. In this review, we survey the research progresses in hydrogen generation from these liquid-phase chemical hydrogen storage materials and their regeneration.
 Mahendra Yadav | Mahendra Yadav was born in Kushinagar, U. P., India in 1984. He received his MSc in Chemistry from Deen Dayal Upadhyay Gorakhpur University, India in 2006 and PhD degree in Inorganic Chemistry under the supervision of Prof. D. S. Pandey from Banaras Hindu University, Varanasi, India in 2010. He then joined Prof. Qiang Xu's group at AIST as a JSPS (Japan Society for the Promotion of Science) postdoctoral fellow in 2010. He is currently interested in development of homo-/heterogeneous catalysts for the activation of small molecules for chemical hydrogen storage. |
 Qiang Xu | Qiang Xu received his PhD degree in Physical Chemistry in 1994 at Osaka University, Japan. After one year working as a postdoctoral fellow at Osaka University, he started his career as a Research Scientist in Osaka National Research Institute in 1995. Currently, he is a Senior Research Scientist at the National Institute of Advanced Industrial Science and Technology (AIST, Japan) and adjunct professor at Kobe University. He received the Thomson Reuters Research Front Award in 2012. His research interests include porous and nanostructured materials and related functional applications, especially for clean energy. He has published more than 250 papers in refereed journals. |
Broader contextHydrogen is foreseen to become a major energy carrier in the relatively close future. The shift towards a so-called hydrogen economy is driven by both a shortage in fossil fuels (while it seems possible to gain “green” hydrogen from renewable sources) and tremendous progresses in the production of fuel cells (making them suitable for transportation applications). A major problem to complete this shift is to find suitable ways to store hydrogen. Indeed, to be implemented at an industrial scale, storage devices must store as much hydrogen as possible within the smallest possible volume. Moreover, the storage as well as the release temperatures must be as close as possible to ambient conditions. Achieving such goals is proving to be a rather difficult task. Among the many possible systems, one of the promising hydrogen storage techniques relies on liquid-phase chemical hydrogen storage materials, in particular, aqueous sodium borohydride, ammonia borane, hydrazine, hydrazine borane and formic acid. In this review, we present a survey on the research progresses in catalytic hydrogen generation from these liquid-phase chemical hydrogen storage materials and their regeneration. |
1. Introduction
The ever-increasing demands of society put more-and-more stringent conditions on the efficiency of production, distribution and use of energy.1 Society awaits a major technological breakthrough before we can reduce our overuse and dependence on fossil fuels that are rapidly depleting and causing serious damage to the planet. Thus, the development of clean alternative fuel sources based on renewable energy is the subject of recent attention. Hydrogen is one of the most promising candidates to replace nonrenewable fuel sources used nowadays because it is a renewable, environmentally friendly energy carrier, and in addition hydrogen has the greatest specific energy of any fuel directly fed into a PEM fuel cell; in fact, H2 has twice as much specific energy as its closest competitor, methane.2 There is no doubt that a great part of the world energy demands of this century will be fulfilled by hydrogen-based fuel cell technology.3The establishment of a sustainable hydrogen-based energy future forces us to develop clean/renewable hydrogen production, efficient hydrogen storage and convenient distribution. The storage of large quantities of hydrogen at a low pressure is one of the key factors in establishing such a hydrogen-based economy.4 In particular, for on-board energy storage, vehicles need compact, light, safe, and affordable hydrogen containment. Although liquid hydrogen has high gravimetric and volumetric hydrogen densities, it has several disadvantages, including its continuous boil-off, in particular for on-board storage. Compressed hydrogen gas is an alternative; high-pressure hydrogen-storage tanks up to 700 bar have been developed, which enables a vehicle driving range of ∼300 miles.4 Systems under higher pressures that could hold higher hydrogen densities are complicated by safety concerns and logistical obstacles. Other storage materials and methods,5 including metal hydrides,3,6 metal–organic frameworks,7,8 on-board reforming of hydrocarbon into hydrogen,9 and organic materials,10,11 have also been investigated extensively. However, many of the candidate materials are still not able to meet the practical requirements such as volumetric (>82 gH2 L−1) and gravimetric hydrogen capacities (>90 gH2 kg−1), handling pressure and temperature, recycling of by-products, cost, and so on, for mobile applications.4
Chemical hydrogen storage which involves storing of hydrogen in the form of chemical bonds is one of the safe alternatives to physical hydrogen storage.12 Over the past decades, solid-state hydrogen storage materials have received considerable attention as promising chemical hydrogen storage materials due to their attractive features, such as high hydrogen density, relative stability and safe storability.12−14 There are many solid-state materials with high hydrogen storage capacities, which, however, display drawbacks, like the high temperature required to desorb hydrogen, slow hydrogen release kinetics or deterioration with successive cycling, different loading/unloading logistics and heat dissipation issues.15 Thus, the search for safe and efficient liquid-phase hydrogen storage materials to conveniently release hydrogen under mild conditions is desired urgently. In this respect, hydrolysis of aqueous boron-based compounds, such as, NaBH4, NH3BH3 and N2H4BH3 has received much attention.16–25 Recently, selective decomposition of hydrous hydrazine (H2NNH2·H2O) to hydrogen and nitrogen at room temperature makes hydrazine a promising hydrogen carrier for storage and transportation.26,27 In addition, formic acid, one of the major products formed in biomass processing, is non-toxic and a liquid at room temperature (density, 1.22 g cm−3) containing 4.4% (w/w) of hydrogen, has also shown to be a potential hydrogen storage material.28–30 This review discusses the state-of-the-art of these representative material systems for hydrogen storage, aiming at providing an outline of the forefront of catalytic hydrogen release and regeneration of the liquid-phase chemical hydrogen storage materials.
2. Sodium borohydride
2.1. Catalytic hydrogen generation from sodium borohydrides in liquid phase
Sodium borohydride (NaBH4, SB) is one of the most studied chemical hydrides owing to its combined advantages of high hydrogen capacity (with a theoretical value of 10.8 wt%),31 easy control of the hydrogen generation rate, friendly operation (low reaction-initiation temperature, stability in air under no pressure, and the NaBH4 solution is nonflammable) and the environmentally benign hydrolysis product (NaBO2 sodium metaborate).31–33 Hydrogen stored in NaBH4 can be released either by thermolysis34,35 or by hydrolysis.36–40 However, in a practical hydrogen generation system only the hydrolysis approach has been used. This is because in hydrolysis one-half of the hydrogen produced derives from water, resulting in a high hydrogen storage capacity. In addition, the generated H2 has high purity (no CO, S) and is humidified (heat generates some water vapour), which facilitates its use in fuel cells. Hydrolysis is typically conducted in the aqueous phase at lower temperatures.NaBH4 undergoes hydrolysis at room temperature and liberates a theoretical hydrogen content of 10.8 wt% via the following reaction [eqn (1)].33,36 Ideally, one mole of sodium borohydride reacts with 2 moles of water to liberate 4 moles of hydrogen.
| NaBH4 + 2H2O → NaBO2 + 4H2 | (1) |
However, in real conditions hydrolysis reaction needs more water to release 4 moles of hydrogen because of the low solubility of both NaBH4 and the borate byproducts in water. The use of excess of water lowers the theoretical gravimetric hydrogen storage capacity of NaBH4; for example, the use of 4 moles of water results in a hydrogen content of 7.3 wt% in comparison to the use of 2 moles of water [eqn (1)], which corresponds to a hydrogen content of 10.8 wt%.25,38
Hydrolysis is thermodynamically favored and is a very exothermic process. NaBH4 undergoes self-hydrolysis (hydrolysis without catalyst) in aqueous solution. However, aqueous phase reactions with pure water give very slow hydrogen generation, because after some initial hydrolysis the reaction mixture becomes basic and the reaction intermediates are stabilized at elevated pH. This allows the use of bases as stabilizers to prevent premature reaction and acids as catalysts to improve the kinetics of sodium borohydride hydrolysis.31,33 In addition, metal catalysts have also been found to catalyze the hydrolysis reaction.33,36,37,41–43
These two catalytic approaches (use of acid additives or the use of metal catalysts) have been widely investigated to improve the hydrolysis rate of the sodium borohydride reaction.31,33,36–44 Early in the 1950s, Schlesinger et al. reported acid-catalyzed hydrolysis of NaBH4 at ambient conditions.33 It was observed that in the presence of acid, NaBH4 solution released 90% of the stoichiometric amount of hydrogen from the hydrolysis reaction. However, this technique requires large amounts of acid, making this method unsafe, heavy and bulky. The difficulty of controlling the reaction is a further drawback. Because of this, the use of acid as a catalyst for NaBH4 hydrolysis has not been considered as a potential accelerator for hydrogen release. However, the effect of acid accelerators on hydrogen generation from solid sodium borohydride for small-scale portable applications has been studied. Prosini and Gilson designed a hydrogen generator for a fuel cell powered cellular phone based on the “hydrogen on demand” concept and taking advantage of the hydrolysis of solid NaBH4 with HCl–water solution.44 They also evaluated the optimum HCl–hydride (= 1), HCl–H2O (= 1.6), and acid solution–boride (= 3.6 cm3 g−1) ratios and hydrogen mass flow rates upon operation. Murugesan and Subramanian also studied hydrogen generation using acidified water and solid NaBH4 for small-scale portable applications.45 They investigated various acids (mineral acids: HCl, H2SO4, HNO3, H3PO4; organic benign acids: HCOOH, CH3COOH) on hydrogen yield from solid NaBH4. With all the mineral acids studied, they observed an increase in the hydrogen production rate, which increased with increasing acid concentration. In the case of benign organic acids, higher concentrations, in general, are required to provide hydrogen production rates similar to those with mineral acids.
Since Schlesinger et al.33 found the NaBH4 hydrolysis by acids, few investigations on homogeneous catalysts have been carried out due to the difficulty of the control of the reaction process. Recently, Keçeli and Özkar suggested using ruthenium(III) acetylacetonate as the homogeneous catalyst for the NaBH4 hydrolysis reaction.46 A number of heterogeneous catalysts based on metals or metal compounds or metal alloys have been reported to be active for catalytic hydrolysis of sodium borohydride in alkaline solutions under ambient conditions, including Co, Ni, Fe, Ru, Pt, Pd, Ru–Cu, Ru–Pd, Ru–Ag, Ru–Pt, Pt–Ag etc.33,47–62 The efficiency of the catalyst has been investigated in terms of the type and form of the catalyst.54,55
Initially, noble metal catalyzed hydrolysis of NaBH4 was investigated by using metal salts e.g. RuCl3, RhCl3, H2PtCl6, IrCl4, PdCl2etc. Among them RuCl3, RhCl3, H2PtCl6 were found most active, and during the hydrolysis reaction these metals are reduced to the elementary state.33,41 After that Amendola et al. reported Ru catalyst supported on ion exchange resin beads which show high activity for hydrolysis of NaBH4.36,37 They investigated the effects of NaBH4, NaOH concentrations and temperature on the rate of the hydrolysis reaction. Ru metal supported on anionic resins showed better activities than cationic resins. The A-26 and IRA-400 anion exchange resins gave the highest hydrogen generation rates. They claimed that the slow rate at the beginning with high concentrations of NaBH4 was due to the high viscosity of the solution. Further, it was observed that high concentrations of hydroxyl ions from NaOH decreased water molecules available for hydrolysis reaction. Demirci and Garin investigated the activity of different alloy based catalysts and the activity decreased in the order Ru, Ru2Pt1 > RuPt, Ru1Pt2 > RuPd > RuAg, Pt > RuCu > PtAg.52 Kojima et al. reported the first study dealing with transition metals supported on various oxides (Co3O4, TiO2, SiO2, NiO, LiMn2O4, TiO, CoO, Ti2O3, LiNiO3 and LiCoO2).43 The 1.5 wt% Pt/LiCoO2 catalyst showed the best performance. A hydrogen generation rate higher than 200 L(H2) min−1 g−1 (Pt) was observed and it was claimed that this rate was ten times faster than the Ru catalyst reported by Amendola et al.36,37 Krishnan et al. alloyed Pt to Ru and compared 10 wt% Pt–Ru/LiCoO2 to both 10 wt% Ru/LiCoO2 and 10 wt% Pt/LiCoO2. The efficiency of Pt–Ru/LiCoO2 was almost double of that of the Ru- and Pt-based catalysts.67 Later, Yang and coworkers tested various supports for dispersing Pt–Ru: e.g. Co3O4, NiO, LiCoO2, LiNiO2, LiMnO2, TiO2 and ZrO2. The best catalytic systems were Pt–Ru/Co3O4 followed by Pt–Ru/LiCoO2.64 Zeolite-confined ruthenium(0) nanoclusters catalyst with turnover frequency (TOF) up to 33![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 have been reported by Zahmakiran and Özkar.65 In general, noble metals show high activity in comparison with non-noble metals.
000 h−1 have been reported by Zahmakiran and Özkar.65 In general, noble metals show high activity in comparison with non-noble metals.
Non-noble metal catalysts, especially nickel and cobalt, are attractive because of their low-cost and comparable catalytic activity. As a result, improvement of the hydrogen generation activity of the low-cost catalyst is becoming imperative and necessary. In the beginning, a number of non-noble metal salts (manganese, iron, cobalt, nickel and copper chlorides) were tested and results show that Co and Ni metal based catalysts were the most active.33,41 Typically it was observed that the salts reacted rapidly with NaBH4 solutions and gave finely divided black precipitates of metal borides. Accordingly, metal borides like cobalt or nickel borides have been prepared by the chemical reduction method using NaBH4 as a reduction chemical and their catalytic activity has been investigated by many groups.54,60,66,67 In addition, Wu et al. heat-treated the obtained CoB at various temperatures.67 It was observed that the catalyst treated at 500 °C exhibited the best catalytic activity because it had the best crystallisation (the more crystallised, the more active). Liu et al. compared the catalytic activity of Co2B to those of cobalt powder, cobalt chloride and RANEY® cobalt. Co2B [468 mL(H2) min−1 g−1 (Co2B)] was better than the powder [126 mL(H2) min−1 g−1 (Co)] and the RANEY® [268 mL(H2) min−1 g−1 (Co)] but was less active than the cobalt chloride [570 mL(H2) min−1 g−1 (Co2B)].59 Patel et al. used CoB-based thin film catalyst synthesised by a pulsed laser deposition technique which permitted the formation of CoB nanoparticles (NPs).68,69 It was observed that cobalt was efficient only when alloyed with boron, which partially prevents metal oxidation. Hanxi and coworkers claimed the preparation of a highly stable and active nickel boride catalyst (NixB), even if its performance was four times lower than that of a Ru-based catalyst at room temperature.70 Furthermore, it has been reported that the structure and catalytic activity of the produced catalyst are generally sensitive to its preparation conditions. The conditions include the type of reducing agent,62 the pH of the reduction medium during the preparation of the catalyst,36,59 the type of precursor and phase of the precursor,71,72 the ratio of reducing ion–metal ion,73 and heat treatment.73,74 Performances of some selected catalysts are summarized in Table 1.
| Catalyst | HGR (L(H2) min−1 g−1) | T (°C) | Ref. |
|---|---|---|---|
| a HGR = hydrogen generation rate, CF = carbon fibres. | |||
| CoCl2 | 11.4 | 20 | 89 |
| CoB | 2.8 | 30 | 60 |
| CoB–Ni | 11.0 | 30 | 75 |
| CoWB–Ni | 15.0 | 30 | 76 |
| CoP–Cu | 1.0 | 30 | 77 |
| Ru | 4.0 | 25 | 37 |
| Ru60Co40 | 17.5 | 25 | 78 |
| Ru80Fe20 | 18.3 | 25 | 78 |
| Ru60Co20Fe20 | 26.8 | 25 | 78 |
| 10 wt% Ru/LiCoO2 | 428 | 25 | 63 |
| 1.5 wt% Pt/LiCoO2 | 203 | 22 | 43 |
| 10 wt% Pt/LiCoO2 | 300 | 25 | 63 |
| 10 wt% PtRu/LiCoO2 | 560 | 25 | 63 |
| 10 wt% Ru/CF | 14.2 | 20 | 78 |
| 20 wt% Pt/C | 115 | 20 | 79 |
| 9 wt% Co/γAl2O3 | 1 | 30 | 80 |
| 2 wt% Ru/γAl2O3 | 4.8 | 30 | 81 |
| 16.6 wt% Ru60Co20Fe20/CF | 41.7 | 20 | 78 |
| 13.3 wt% Ru75Co25/CF | 37.1 | 20 | 78 |
In addition to hydrolysis, the hydrogen generation from NaBH4 in other solvents, like alcohols, was also assessed.82–84 NaBH4 is known to be reactive to e.g. methanol and ethanol and methanol has the highest reactivity towards NaBH4.82,83 The overall reaction between methanol and NaBH4 can be described as [eqn (2)]:
| NaBH4 + 4CH3OH → NaB(OCH3)4 + 4H2 | (2) |
Karan and coworkers assessed the H2 generation in methanol and mixtures of methanol and water (H2O/NaBH4 molar ratio of either 2 or 10).82 The best system was the mixture with a molar ratio of 10. Demirci and coworkers investigated metal catalyzed methanolysis by using Co/TiO2 and Ru/TiO2 catalysts. Co/TiO2 shows high catalytic performances, higher than that of Ru–TiO2. Hydrogen generation rates of 144 to 644 L(H2) min−1 g−1 (Co) were measured as the Co loading on TiO2 was decreased from 20 to 1 wt%.85
2.2 Regeneration of sodium borohydride
For NaBH4 to be used as a viable hydrogen fuel, an economical and environmentally sound process for recycling of spent fuel to NaBH4 is needed. In the hydrolysis of NaBH4, it has been reported that BH4− transforms to B(OH)4−,86,87 and at pH < 9, B(OH)3 is the predominant byproduct in aqueous solutions, whereas at pH > 9, mainly B(OH)4− forms.31 However, upon drying at <110 °C, the species present are primarily NaBO2·2H2O and NaBO2·4H2O.88 The formation of Na2B4O7·5H2O has also been reported.89 At higher drying temperatures, the hydrated borates dehydrate to form, for example, NaBO2·1/2H2O and then NaBO2.88 Considerable efforts have been devoted to find a suitable route to prepare NaBH4 from NaBO2. The borate species are thermodynamically stable,31 thus requiring a large quantity of energy to regenerate the parent material. This is one major demerit which limits the on-board vehicular applications of NaBH4.90,91 In general, the recycling process of byproducts formed by hydrolysis of NaBH4 involves the separation of borates from unreacted NaBH4 of the reaction slurry and then drying at temperatures of approximately 300 °C to obtain anhydrous NaBO2.92,93 The as-obtained NaBO2 can then be transformed back into NaBH4 by several reported processes.94,95 Some notable NaBH4 preparation processes are briefly discussed here.The Bayer process has been employed for commercial scale synthesis of NaBH4. In this process NaBH4 was prepared by the reaction of borax (Na2B4O7), metallic sodium (Na), hydrogen (H2) and silica (SiO2) at high temaparture.96 This process was further modified by employing less expensive reducing metal magnesium instead of sodium. The modified Bayer process is a one-pot synthesis of sodium borohydride by reduction of NaBO2 using magnesium (Mg)97,98 or magnesium hydride (MgH2)99,100 as a reducing agent [eqn (3)]. Although this employed the less expensive reducing metal, high yields and fast reaction rates could not be achieved. Theoretically, the reaction could occur at room temperature in accordance with the standard Gibbs free energy (ΔG0 = −269.7 kJ mol−1). However, the investigators found that the process should be carried out at high temperature because of poor mass transfer between the solid particles of the reactants, as well as high hydrogen pressure providing MgH2 decomposition at high temperatures.
| NaBO2 + 2MgH2 → NaBH4 + 2MgO | (3) |
It was reported that NaBH4 produced from the modified Bayer process is in molten form and does not decompose at 550 °C and 7 MPa (under hydrogen atmosphere) for 2 h.98 Because the modified Bayer process needs to be conducted under severe reaction conditions with hazardous materials, it is a high-risk process, especially if one contemplates a large-scale industrial production. Furthermore, an effective method to separate the reaction products is required to obtain a reasonable yield of NaBH4.94
A one-step synthetic approach should be desirable e.g., using the direct thermal reduction of sodium borate by a reducing agent such as, methane (or natural gas), H2 or carbon. However, investigations of free energies of the reactions indicated that they are not thermodynamically feasible under reasonable conditions.94,101
While developing an all-thermal synthetic process for NaBH4, Millenium Cell Inc. proposed a family of processes102–105 which can be modified in various ways to optimize the tradeoffs among energy efficiency, cost and greenhouse gas emissions. One example is based on the use of disproportionation, a classic reaction in the chemistry of boron compounds.106 Another proposed carbon-based reduction method is based on formaldehyde as a reducing agent.107
An electrolytic process, which was first proposed by Cooper,108 was thought of as the most attractive process for NaBO2 recycling. The electrolytic reaction [eqn (4)] was carried out in the NaBO2 caustic solution.
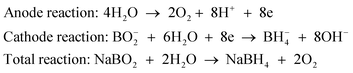 | (4) |
The plenty of OH− in caustic solution could be electrolyzed prior to BO2−, and thus, the electrolysis of BO2− to BH4− in aqueous media proved to be difficult, if not impossible. Traces of NaBH4 were detected in the products resulting from the electrosynthesis in aqueous media and it is suggested that this embryonic study should be pursued.109 Calabretta and Davis and Calabretta110 demonstrated an anhydrous molten Na–B–O–H system, which could be used as a potential medium in the electrolysis of BO2− to BH4−. This process is currently under investigation.110
At present, none of these processes is sufficiently simple to lower the cost of borohydride manufacture below its current level. Based on the current scenario of NaBH4 synthesis, the most attractive route to achieving the low-cost production of sodium borohydride lies with the electrochemical methods, which, however, need to be realized.
2.3 Conclusions
Apart from its established use for hydrogenation and reduction processes in the chemical industry, sodium borohydride (NaBH4) has been demonstrated as an effective hydrogen storage material. Its hydrolysis generates high purity humidified hydrogen suitable for use in PEM fuel cells. Although the technology can be scaled and tailored for many different applications from very small portable devices to fuel cell vehicles, the current high cost of NaBH4 is limiting early adoption of the technology to premium power applications. The utilization of NaBH4 in high demand, continuous power generation and fuel cell vehicles will not be economically feasible until the production cost of NaBH4 can be significantly reduced. A substantial decrease in the NaBH4 cost might be achieved by finding an effective method of recycling NaBO2 back to NaBH4, if possible, in electrochemical way.3. Ammonia borane
3.1 Catalytic hydrogen generation from ammonia borane in liquid phase
Ammonia borane (NH3BH3, AB) has a hydrogen capacity of 19.6 wt%, exceeding that of gasoline and making it an attractive candidate for chemical hydrogen-storage applications.16–23,111–116 It possesses a volumetric density of 146 gH2 L−1 and a gravimetric density of 196 gH2 kg−1. These values are well above the US Department of Energy targets (2015) of a volumetric density greater than 82 gH2 L−1 and a gravimetric density greater than 90 gH2 kg−1.117 AB is a colorless molecular crystal at room temperature118 with a density of 0.74 g cm−3.119 It is stable in air, and soluble in water and other relatively polar solvents.21,120 Several methods have been developed for laboratory-scale preparation of AB, including reaction of ammonium salts with lithium or sodium borohydride,119,121–123 and direct reaction of ammonia with diborane (B2H6),124 BH3·THF125 or BH3·SMe2.126 According to the literature reports, the most efficient one involves the reaction of NaBH4 and ammonium formate (HCO2NH4) in dioxane, which gives high purity (≥98%) AB in high yield (≥95%).123Ammonia borane is hydrogen-dense and thus mainly attractive for portable and automotive applications, and the primary challenge is to recover as much H2 as possible and, where possible, under mild conditions (<85 °C). Release of hydrogen from AB can be accomplished either by thermolysis (neat or metal catalyzed) in the solid state and nonaqueous medium (ethereal solvents) or metal catalyzed reactions in protic solvents (water and methanol).16–23,111–116
To date, considerable works involving the release of hydrogen from the thermal dehydrogenation of AB have been reported.112,113,127–130 Thermolysis is a three-step process [eqn (5)–(7)]. The first step commences at approximately 100 °C and releases 1 equiv. of H2 (6.5 wt%). The second step occurs at a broad temperature range centered at approximately 150 °C. The final step, which requires high temperatures (>1200 °C), provides the third equivalent of H2. Unwanted gaseous byproducts, such as borazine (B3N3H6), are also liberated.131 To reduce the threshold temperature and volatile byproducts, various approaches have been considered, including nano-scaffolding,132 catalysis,133,134 dispersion in an ionic liquid,127 and the synthesis of derivatives (e.g., amidoboranes).129
| NH3BH3 → 1/n[NH2–BH2] + H2 | (5) |
| 1/n[NH2–BH2] → 1/n[NH–BH]n + H2 | (6) |
| 1/n[NH–BH]n → BN + H2 | (7) |
Hydrogen release from AB is exothermic while the complete release of hydrogen needs high temperature. A conceptually different approach was conceived for the solvolysis of AB, mainly hydrolysis16,17,135–138 and methanolysis,123,139,140 where H2 is released at ambient temperatures in the presence of suitable catalysts. The Hδ− bound to the B atom reacts with the Hδ+ provided by the solvent. AB is highly soluble and quite stable in these media. The 11B NMR spectrum remains unchanged in water for more than 80 days under an argon atmosphere, indicating the high stability of AB in water.16,111 The aqueous medium should either be neutral or weakly basic for AB to be stable,141 in a strongly basic medium AB is unstable.142 The hydrolysis of AB occurs at an appreciable rate only in the presence of a suitable catalyst at ambient temperature, thus one of the major obstacles of the practical application of this system is to develop efficient, economical and easily recyclable catalysts for improving the kinetic properties under moderate conditions. The hydrolysis reaction can be briefly expressed as follows [eqn (8)]:16
| NH3BH3 + 2H2O → NH4+ + BO2− + 3H2 | (8) |
A variety of Lewis and Brønsted acids,21 solid and gaseous acids,135 zeolites,135 and even CO2,135 are efficient catalysts for the hydrolysis. The performances of several metal-based hydrolysis catalysts are summarized in Table 2. Noble metal-based catalysts were firstly found by us to have considerable activities toward hydrolytic dehydrogenation of AB.16 The Pt-based catalysts were reported to have high activity toward this reaction with the released H2 to AB ratio up to 3.0 (Fig. 1) and the catalytic activities are in the order of 20 wt% Pt/C > 40 wt% Pt/C > PtO2 > Pt black > K2PtCl4. The 20 wt% Pt/C catalyst shows the highest activity and the reaction is completed in less than 2 min (Pt/AB = 0.018).16 In contrast, [Rh(1,5-COD)(μ-Cl)]2 and Pd black have lower activity and some noble metal oxides (RuO2, Ag2O, Au2O3, IrO2) are almost inactive.16 The Ru, Rh, Pd, Pt and Au NPs supported on different supports (γ-Al2O3, VULCAN® carbon and SiO2) were also investigated for hydrolysis of AB by our group.143 It has been found that the Ru, Rh and Pt catalysts exhibit high activities to generate a stoichiometric amount of hydrogen with fast kinetics, whereas the Pd and Au catalysts are less active. In order to achieve well dispersed noble metal clusters/NPs with small sizes and study their kinetics, Özkar and co-workers have recently used different polymers (laurate or poly(4-styrenesulfonic acid-co-maleic acid)(PSSA-co-MA)) as stabilizers for metal NPs (Rh, Ru, Pd).138,144,145 The resulting catalysts show significant enhancement in activity. Furthermore, they also investigated the catalytic activities of zeolite-Y confined Rh and Pd NPs,146,147 which show high catalytic activities (TOF = 92 min−1 for Rh NPs) and long lifetime (47200 turnovers for Rh NPs and 15600 turnovers for Pd NPs) in the hydrolysis of AB at 25 °C.
| Catalyst | Catalyst/AB molar ratio (mol mol−1) | Maximum H2/NH3BH3 ratio (mol mol−1) | Time for reaction completion (min) | Ref. |
|---|---|---|---|---|
| a No reaction. | ||||
| 2 wt% Ru/γ-Al2O3 | 0.018 | 3.0 | 3 | 143 |
| 2 wt% Rh/γ-Al2O3 | 0.018 | 3.0 | 1.3 | 143 |
| [Rh(1,5-COD)(μ-Cl)]2 | 0.018 | 2.6 | 15 | 16 |
| 2 wt% Pd/γ-Al2O3 | 0.018 | 2.9 | 120 | 143 |
| Pd black | 0.018 | 2.6 | 250 | 16 |
| 2 wt% Pt/γ-Al2O3 | 0.018 | 3.0 | 0.8 | 143 |
| 2 wt% Pt/C | 0.018 | 3.0 | 1.5 | 16 |
| 2 wt% Pt/SiO2 | 0.018 | 3.0 | 3 | 143 |
| PtO2 | 0.018 | 3.0 | 8 | 16 |
| Pt black | 0.018 | 3.0 | 12 | 16 |
| 2 wt%Au/γ-Al2O3 | 0.018 | 1.9 | 610 | 143 |
| Laurate stabilized Rh(0) | 0.0025 | 3.0 | 6 | 138 |
| Laurate stabilized Ru(0) | 0.00125 | 3.0 | 22 | 144 |
| Zeolite stabilized Rh(0) | 0.004 | 3.0 | 67 | 146 |
| 10 wt% Co/γ-Al2O3 | 0.018 | 2.9 | 70 | 17 |
| 10 wt% Co/SiO2 | 0.018 | 2.9 | 70 | 17 |
| 10 wt% Co/C | 0.018 | 2.9 | 55 | 17 |
| 10 wt% Ni/γ-Al2O3 | 0.018 | 2.9 | 65 | 17 |
| 10 wt% Cu/γ-Al2O3 | 0.018 | 2.9 | 590 | 17 |
| 10 wt% Fe/γ-Al2O3 | 0.018 | —a | —a | 17 |
| In situ synthesized Fe particle | 0.12 | 3.0 | 8 | 148 |
| Ni in starch | 0.1 | 3.0 | 6 | 149 |
| Bare Ni NPs | 0.1 | 2.8 | 11 | 150 |
| PVP–Ni NPs | 0.1 | 2.7 | 9 | 150 |
| Fe0.5Ni0.5 | 0.12 | 3.0 | 2.2 | 151 |
| In situ synthesized Co | 0.04 | 3.0 | 1.7 | 155 |
 | ||
| Fig. 1 Hydrogen release from aqueous NH3BH3 (0.33 wt%) solution in the presence of various metal catalysts (metal/NH3BH3 = 0.018). Reprinted with permission from ref. 16. Copyright 2006 Elsevier. | ||
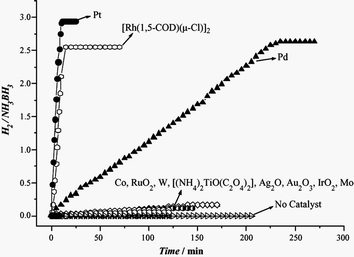
Although the noble metal-based catalysts show very high activities for hydrolysis of AB as mentioned above, from the viewpoint of practical application, the development of efficient, low-cost, and stable catalysts to improve further the kinetic properties under moderate conditions is very important. As shown in Table 2, supported non-noble metal (Ni or Co) catalysts exhibit high activities, with which hydrogen is released in an almost stoichiometric amount from aqueous NH3BH3, and the reaction can be completed in 55–70 min (metal/NH3BH3 = 0.018), whereas supported Fe is catalytically inactive.17 Afterwards, we systematically investigated the catalytic activities of unsupported first-row transition metal NPs, which were pre-reduced by NaBH4 or in situ reduced in the presence of AB and NaBH4 during the hydrolysis reaction. It is found that the catalytic activities are highly dependent on their particle sizes, crystallinities, compositions, etc.148–153 Most importantly, amorphous Fe NPs prepared by in situ reduction with AB/NaBH4 were found to have an excellent catalytic activity for the hydrolysis of aqueous NH3BH3 under argon and even in air at room temperature, releasing hydrogen of H2/NH3BH3 = 3.0 in ca. 8 min (Fe/AB = 0.12) (Fig. 2).148 In addition, the amorphous Co and Ni and Fe–Ni alloy nanoparticles also exhibit much better catalytic activity than their crystalline ones.149,151,154
 | ||
| Fig. 2 Hydrogen generation by hydrolysis of aqueous AB (0.16 M, 10 mL) in the presence of (a) in situ synthesized Fe catalysts and (b) the pre-synthesized (Fe/AB = 0.12) at room temperature under argon. The volume of released gas includes 20 mL H2 from the reducing agent NaBH4, the volume of 1 equiv. H2 = ∼59 mL, 2 equiv. H2 = ∼97 mL, 3 equiv. H2 = ∼136 mL. Inset: TEM micrographs and the corresponding SAED patterns of the Fe NPs. Scale bar: 20 nm. Reprinted with permission from ref. 148. Copyright 2007 Wiley-VCH. | ||
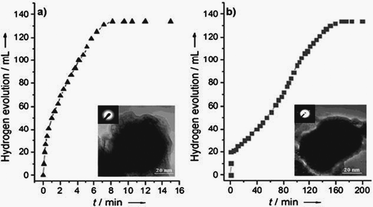
The M@SiO2 nanospheres (M = Co, Ni) prepared with the reversed micelle method were found to have higher catalytic activity for hydrolysis of AB to generate a stoichiometric amount of H2 compared to the M/SiO2 nanosphere and Ni/commercial SiO2 catalysts, which were synthesized by depositing Ni or Co NPs on SiO2 nanospheres and commercial SiO2, respectively, with the impregnation method.152,155 Monodisperse nickel NPs supported on the Ketjen carbon support, synthesized by the reduction of Ni(acac)2 with borane tributylamine (BTB) in the presence of oleylamine (OAm) and oleic acid (OA), were reported to exhibit high catalytic activity in hydrogen generation from the hydrolysis of the AB even at low catalyst and substrate concentrations at room temperature.156 In a recent report by Song, Cai and coworkers, nanoporous Ni spheres showed a high activity with a TOF value 19.6, which was the highest among all the Ni-based catalysts and was about one fourth of the activity of the Pt catalyst (Fig. 3). Furthermore, the stability of these catalysts was improved by silica coating of the Ni spheres.157
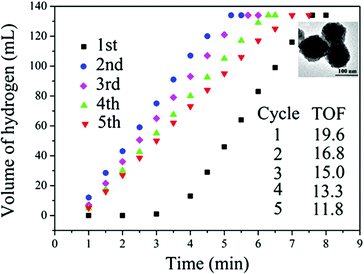 | ||
| Fig. 3 Time versus volume of hydrogen generated from hydrolysis of AB catalyzed by nanoporous Ni spheres and corresponding TOF values up to five cycles. Volume of 1 equiv. H2 = ∼48 mL, 2 equiv. H2 = ∼95 mL, 3 equiv. H2 = ∼142 mL. Insect: TEM image of nanoporous Ni sphere catalyst. Reprinted with permission from ref. 157. Copyright 2010 Wiley-VCH. | ||
Li and co-workers employed a Ni based metal–organic framework (MOF) as precursor for preparing a mixture of Ni NPs with undegradated Ni–MOF reduced by AB in methanol. The mixture exhibits a high catalytic activity for complete hydrogen generation from aqueous AB solution at 25 °C in air.153 This group also reported a highly active Co(0) catalyst, synthesized by the NaBH4 reduction of a metal–organic framework (MOF) precursor Co2(bdc)2(dabco) (bdc = 1,4-benzenedicarboxylate; dabco = 1,4-diazabicyclo[2.2.2]octane), for the hydrogen generation from AB.158 In the presence of the Co-MOF based catalyst, the hydrogen generation from aqueous AB solution (0.32 M) was completed within 1.4 min (MOF/NaBH4/AB = 0.057/0.08/1) at room temperature.
Recently our group reported highly dispersed and small sized Ni NPs (∼2.7 nm) immobilized by the frameworks of ZIF-8, which exhibit highly catalytic activity and durability for hydrolysis of ammonia borane (AB).159 Among the samples, the sample prepared by using the chemical vapor deposition (CVD) approach (CVD–Ni@ZIF-8) showed the highest activity, with which the reaction can be completed (H2/AB = 3.0) in 13 min (Ni/AB = 0.016), giving a TOF value of 14.2 min−1 in comparison to the sample prepared by using a chemical liquid deposition (CLD) approach (CLD–Ni@ZIF-8), with which the reaction can be completed in 19 min (Ni/AB = 0.019), giving a TOF value of 8.4 min−1.
The copper nanoparticles synthesized using the Solvated metal atom dispersion (SMAD) method has been employed for the hydrolysis of ammonia borane. Controlled oxidation of the as-prepared nanoparticles resulted in the formation of Cu@Cu2O core–shell nanoparticles. The Cu@Cu2O core shell and Cu2O NPs show better activities than pure Cu NPs for the generation of hydrogen in the AB hydrolysis reaction.139 Zeolite confined Cu NPs were prepared by the ion-exchange of Cu2+ ions with the extra framework Na+ ions in zeolite-Y followed by reduction of the Cu2+ ions within the cavities of zeolite with NaBH4 in aqueous solution. Zeolite confined Cu NPs are active in the hydrolysis of AB with an average TOF value of 46.5 h−1 and provide 1300 turnovers in the catalytic hydrolytic dehydrogenation of AB.160
Bimetallic catalysts usually show enhanced catalytic performance in comparison to their monometallic counterparts. Various bimetallic (Ni–Fe, Ni–Au, Ni–Ag, Ni–Pt, Ni–Pd) catalysts have also been employed in the AB hydrolysis reaction.151,161 These catalysts show superior catalytic activity due to a synergistic effect in comparison to their monometallic counterparts. Recently, we have prepared Au–Ni and Au–Co NPs with small diameters (3–4 nm) within silica nanospheres (around 15 nm) by using Au(en)2Cl3, Ni(NH3)6Cl2 and Co(NH3)6Cl2 as precursors in a NP-5/cyclohexane reversed-micelle system, followed by in situ reduction in an aqueous solution of NaBH4/AB. Compared with monometallic Au@SiO2, Ni@SiO2 and Co@SiO2 counterparts, the Au–Ni@SiO2 and Au–Co@SiO2 catalysts exhibit superior performances in the hydrolytic dehydrogenation of AB.162,163 The synergistic effect between Au and Ni or Au and Co inside the silica nanospheres plays an important role in the catalytic hydrolysis of NH3BH3. Furthermore, we have also investigated the effect of heat treatment of core–shell structured Au–Co@SiO2 nanospheres. During heat treatment in vacuum, it has been observed that multiple Au–Co NPs embedded in SiO2 nanospheres (Au–Co@SiO2–RT) merged into single Au–Co NPs in SiO2 (Au–Co@SiO2–HT), resulting in a size increase of the Au–Co NPs. The Au–Co@SiO2–HT nanospheres showed better catalytic activity than the Au–Co@SiO2–RT. The higher catalytic activity of Au–Co@SiO2–HT could be attributed to the decrease in the content of basic ammine by the decomposition of metal ammine complexes during the heat treatment.
Similar to alloy NPs, core–shell structured bimetallic NPs are also very important for catalytic applications. Recently, our group prepared magnetically recyclable Au@Co core–shell NPs by a rational and general strategy through the one-step seeding-growth route with AB as the reducing agent under ambient conditions.164 Au NPs can be formed first, and serve as the in situ seeds for successive catalytic reduction, resulting in the growth of outer Co NPs as a shell in the AB aqueous solution, although the Au3+ and Co2+ precursors were added simultaneously. Unexpectedly, compared to the monometallic and alloy counterparts, the resultant magnetically recyclable Au@Co NPs displayed excellent catalytic activity and long-term stability towards hydrolytic dehydrogenation of aqueous AB under ambient conditions.165 A similar approach was adopted to prepare triple layered Au/Co/Fe core–shell nanoparticles. Transmission electron microscope, energy dispersive X-ray spectroscopic, and electron energy-loss spectroscopic measurements revealed that the trimetallic Au/Co/Fe NPs have a triple-layered core–shell structure composed of an Au core, a Co-rich inter-layer, and an Fe-rich shell. The Au/Co/Fe core–shell NPs exhibit much higher catalytic activities for hydrolytic dehydrogenation of ammonia borane than the monometallic (Au, Co, Fe) or bimetallic (AuCo, AuFe, CoFe) counterparts.165
Very recently, we reported a facile one-step and general route for in situ synthesis of a series of Cu@M (M = Co, Fe, Ni) core–shell NPs under ambient conditions (Fig. 4).166 In a typical synthesis of Cu@M (M = Co, Fe, Ni) core–shell NPs, an aqueous solution of copper(II) chloride and cobalt(II) or nickel(II) chloride or iron(II) sulfate, and polyvinylpyrrolidone K 30 (PVP) as a capping agent was introduced to a round-bottom flask containing ammonia borane. The Cu2+ and M2+ were reduced in sequence to produce core–shell structured NPs during the reduction process, in which Cu2+ with high reduction potentials (EoCu(II)/Cu(I) = +0.159 eV vs. SHE; EoCu(I)/Cu = +0.520 eV vs. SHE) was first reduced by NH3BH3. The generated Cu–H or/and subsequent M–H species with a strong reducing ability can further reduce M2+, although it is difficult to reduce them by NH3BH3 due to their lower reduction potentials (EoCo(II)/Co = −0.28 eV vs. SHE; EoFe(II)/Fe = −0.44 eV vs. SHE; EoNi(II)/Ni = −0.25 eV vs. SHE). In this process, the preformed Cu0 NPs, serving as in situ seeds/core NPs, could induce the successive growth of M0 as a shell to thus yield Cu@M core–shell NPs. Such in situ reduction and one-pot synthetic protocol for core–shell NPs is to take advantage of the differences in the reduction potentials of core and shell metal salts, where the key point is to employ a suitable reducing agent. NH3BH3, a moderate reducing agent, is suitable for this process. In contrast to the monometallic and alloy counterparts, the in situ generated bimetallic core–shell NPs have exhibited synergistic and superior catalytic activity for hydrolytic dehydrogenation of ammonia borane.
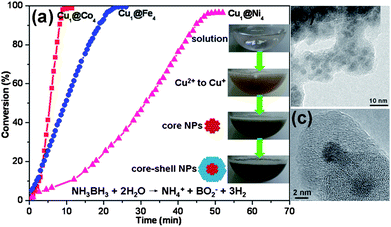 | ||
| Fig. 4 (a) Hydrogen generation from NH3BH3 aqueous solution over Cu@Co, Cu@Fe Cu@Ni core–shell nanocatalysts under ambient conditions ((Cu2+ + M2+)/NH3BH3 = 0.02). (b) Representative TEM image of Cu@Co core–shell nanocatalyst. (c) Representative HRTEM image of Cu@Co core–shell nanocatalyst. Reprinted with permission from ref. 166. Copyright 2011 Royal Society of Chemistry. | ||
Notably, the convenience and reliability of the AB hydrolysis reaction make suitable for applications, and it has already been extensively used as a test (model) reaction for examining the catalytic activity of new nanomaterials.148,157,166–168
In addition to the hydrolysis of AB, methanolysis of AB has also been developed to generate hydrogen at room temperature over various catalysts [eqn (9)], such as RuCl3, RhCl3, CoCl2, NiCl2, Pd/C, RANEY® Ni, PVP-stabilized Pd NPs, zeolite stabilized Rh NPs, Co–Co2B, Ni–Ni3B, and Co–Ni–B, etc.123,139,169,170 Hydrogen capacity from this system is about 3.9 wt%, which is lower than that from the hydrolytic system (about 7.8 wt%).
| NH3BH3 + 4MeOH → NH4B(OMe)4 + 3H2 | (9) |
In order to explore the liquid phase dehydrogenation of ammonia borane, a number of studies have been carried out in organic liquids and ionic liquids.171–175 Heinekey, Goldberg and coworkers used an Ir Pincer catalyst and achieved about one equivalent of H2 from the solution of ammonia borane in tetrahydrofuran at room temperature.171 Baker and coworkers demonstrated that a homogeneous nickel catalyst with N-heterocyclic carbene (NHC) ligands releases >2.5 equiv. of H2 gas based on ammonia borane from the solution of ammonia borane in diglyme at 60 °C.172 Burrell and coworkers investigated dehydrogenation of AB assisted with heterogeneous Pt, Pd, Ru-catalysts in non-aqueous solution (2-methoxyethyl ether) at room temperature to 70 °C.173 The best catalytic activity was observed for Pt. Half an equiv. of a possible three equiv. H2 was extracted within 30 min at 70 °C, and overall close to two equiv. of H2 was extracted from AB. Very recently, Kang and coworkers used Pd NPs as catalyst and tetraglyme as solvent for the dehydrogenation of ammonia borane (AB) at 85 °C. Remarkably an enhanced catalytic performance was achieved to release 2.3 equiv. of H2 in 1 h (Pd/AB = 0.019).174 Baker, Sneddon and coworkers investigated a range of transition metal complexes as active catalysts for dehydrogenation of ammonia borane in different ionic liquids.175 All of the complexes screened were found to give enhanced H2 release relative to the background reaction (in the absence of catalyst under similar reaction conditions), demonstrating the viability of metal-catalyzed dehydrogenation in this ionic liquid medium. While the most active catalysts contained the precious metals Rh, Ru and Pd, the base metal precursor Ni(COD)2 showed comparably high initial rates and NiCl2 gave a high total H2 release.175 Although the hydrogen productivity from ammonia borane in organic liquid or ionic liquid is lower than hydrolysis, this process produces the BNH byproduct, which can easily be recycled (vide infra).
Recently, two methods involving water but no catalysts have been developed for releasing hydrogen from AB.176–178 One is based on combustion of AB mixtures with aluminum powder and gelled water. It was experimentally shown that these mixtures, upon ignition, exhibit self-sustained combustion with hydrogen release from both AB and water, 7.7 wt% H2 in total. The other method involves external heating of aqueous AB solutions to ∼120 °C or higher under argon pressure to avoid water boiling. Experiments show that heating aqueous AB solutions to temperatures 117–170 °C releases 3 equiv. of hydrogen per mole AB, where 2.0–2.1 equiv. originate from AB and 0.9–1.0 equiv. from water.
In order to search for new BN-based liquid-phase hydrogen storage materials, recently, Liu and coworkers developed BN-methylcyclopentane (1), which is an air- and moisture-stable liquid at room temperature.179 It is capable of releasing 2 equiv. of H2 per molecule (4.7 wt%) both thermally, at temperatures above 150 °C, and catalytically, using a variety of cheap and abundant metal halides at temperatures below 80 °C (Fig. 5). The exclusive product of dehydrogenation is the trimer (2), which is also a liquid at room temperature. They also demonstrated that the conversion of the spent fuel (2) back to the charged fuel (1) can be accomplished in high yield, making this system a viable candidate for liquid-phase hydrogen storage in mobile applications.
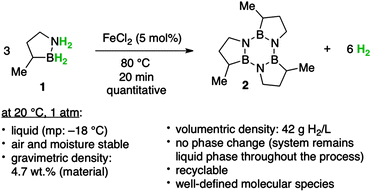 | ||
| Fig. 5 A single-component liquid-phase hydrogen storage material. Reprinted with permission from ref. 179. Copyright 2011 American Chemical Society. | ||
3.2 Regeneration of ammonia borane
The hydrolysis and dehydrogenation processes can effectively release hydrogen from ammonia borane under mild conditions. However, the viability of any chemical hydrogen storage system is critically dependent on efficient recyclability, but reports on the latter subject are sparse.21,123,180–184 The 11B NMR spectroscopy analyses of spent fuels indicated that the byproducts in the hydrolysis of AB are mainly borate species.16,87,135,185–188 Generally speaking, a significant drawback of solvolysis, as in the case of NaBH4, is that B–H bonds are converted to much stronger B–O bonds, resulting in a more exothermic reaction than dehydrogenation. Therefore, regeneration of spent fuel requires strong reducing agents.It has been reported that ammonia borane can be synthesized in high yield by the combination of sodium borohydride and ammonium sulfate ((NH4)2SO4) or ammonium chloride (NH4Cl) (eqn (10)).123,180
 | (10) |
Further, it has also been reported that hydrolysis of sodium borohydride and ammonia borane both generate borates as spent fuel.189 Therefore, the processes developed to recycle the spent fuel of sodium borohydride (Section 2.2) could be used for recycling the spent fuel of ammonia borane. Chen and coworkers suggested a total life cycle of hydrogen release and regeneration of NH3BH3 and NaBH4 (Fig. 6).190 In brief, since boric acid is the main hydrolysis product of ammonia borane hydrolysis, it is possible at first to produce trimethyl borate, B(OCH3)3, from esterification of boric acid with methanol.95,191 The Brown–Schlesinger process192 would further convert as-obtained trimethyl borate to sodium borohydride by reacting it with sodium hydride (NaH). Lastly, ammonia borane is synthesized from sodium borohydride reacting with ammonia sulfate in tetrahydrofuran (THF) at 40 °C.123
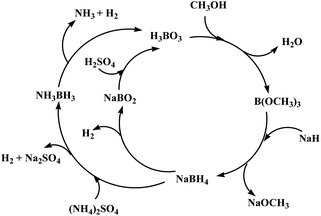 | ||
| Fig. 6 Proposed total life cycle of ammonia borane and sodium borohydride for hydrogen generation and regeneration. Reprinted with permission from ref. 190. Copyright 2011 Elsevier. | ||
Ramachandran and Gagare demonstrated a system based on transition metal catalyzed solvolysis of AB to yield [NH4][B(OMe)4] [eqn (9)], which could be converted back to AB by treatment with NH4Cl and lithium aluminium hydride [eqn (11)].123 It is likely to be energetically costly to convert the oxidation product, Al(OMe)3, back to the complex hydride.
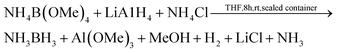 | (11) |
Studies have been revealed that the spent fuels obtained after dehydrogenation of ammonia borane are generally BNH materials. The composition of spent fuel depends on the dehydrogenation method.21,22 A homogeneous nickel catalyst with N-heterocyclic carbene (NHC) ligands, developed by Baker and coworkers, is capable of greater release than two equivalents of H2 from the solution of ammonia borane in diglyme at 60 °C and gives polyborazylene (PB), which is a BNH material (containing a B–N linkage), as a single spent fuel component.172 The strategies presented to date for regeneration of BNH-spent fuel involve two important steps, digestion and reduction. Addition of an acid (HX) can protonate these linkages, releasing amine and making B–X bonds (digestion). The B–X bonds can then be reduced by chemical reductants to form B–H bonds.181,193,194 Sutton, Gordon, Power and coworkers described an energy efficient regeneration process for polyborazylene (PB) spent fuel.182 In this scheme, effective digestion of polyborazylene with 1,2-benzenedithiol was demonstrated to form ammonia adducts of dithioboron compounds. These compounds contain relatively weak B–S bonds that are readily reduced by Sn–H. Very recently, this group demonstrated that the spent fuel derived from the removal of greater than two equivalents of H2 per molecule of AB (i.e., polyborazylene, PB) can be converted back to AB nearly quantitatively by treatment with hydrazine (N2H4) in liquid ammonia (NH3) at 40 °C in a sealed pressure vessel (Scheme 1).184 For practical application this regeneration process of ammonia borane must be as energetically efficient as possible, so no reaction step can be too exo- or endothermic.
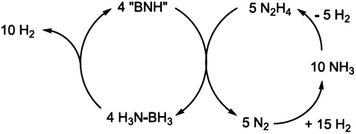 | ||
| Scheme 1 Ideal overall reaction scheme for AB (NH3BH3) regeneration from PB (“BNH”) with hydrazine (N2H4). Reprinted with permission from ref. 184. Copyright 2011. The American Association for the Advancement of Science. | ||
3.3 Conclusions
The 19.6 wt% H2 in AB has made it an attractive molecule for chemical hydrogen storage. Besides dehydrogenation of AB, hydrolysis of AB presents a high hydrogen capacity up to 7.8 wt% of the starting materials. In comparison with NaBH4, which undergoes self-hydrolysis in water without catalysts, AB has the advantage that it is stable in aqueous solution, which makes AB a promising hydrogen storage system. A portable hydrogen generation system is expected to be established on the basis of the metal-catalyzed dissociation and hydrolysis of AB. The AB hydrolysis reaction proceeds under ambient conditions with rapid kinetics in the presence of suitable catalysts; not only noble metal but also many highly active non-noble metal-based catalysts have been developed. For practical applications of AB hydrolytic dehydrogenation in portable electric devices, further experimental and theoretical studies for pending limitations, such as (i) how to break the strong B–O bonds formed during the H2 generation to recycle the end products and (ii) how to reduce the catalyst deactivation, etc., are highly desired to overcome the high material cost issue.4. Hydrazine
4.1 Catalytic decomposition of hydrous hydrazine
Anhydrous hydrazine (H2NNH2, HZ), a liquid at room temperature, has a hydrogen content as high as 12.5 wt%. It is a colorless oily liquid at room temperature and used as a monopropellant in satellite propulsion.195,196 This compound is hypergolic; it explosively reacts upon exposure to a metal surface. Studies, mostly on the reactions of hydrazine highly diluted in inert gases such as argon, have shown that hydrazine can be decomposed on supported metals,197 metal nitrides,198,199 or metal carbides200,201 in two ways: incomplete decomposition [eqn (12)],| 3H2NNH2 → 4NH3 + N2(g) | (12) |
| H2NNH2 → N2(g) + 2H2(g) | (13) |
The decomposition pathway depends on the catalyst used and also on applied reaction condtions.197,199,201–206 Most of the reported catalysts show a high activity for reaction [eqn (12)] at temperatures below 300 °C.197,199–206 Hydrogen generation from hydrazine over these catalysts was observed at high temperature due to the decomposition of NH3. Interestingly, the SiO2-supported Ni, Pd, and Pt catalysts were active even at around room temperature and the Ni/SiO2 catalyst shows 90% selectivity for hydrogen at 50 °C.197 The hydrogen selectivity increases with the reaction temperature in the range of 30–80 °C, and then quickly decreases when the reaction temperature is further increased. It is suggested that two competing reactions (12) and (13) are occurring during hydrazine decomposition over these supported catalysts. At higher temperature (>400 °C), the hydrogen selectivity tends to increase again (∼100%) due to the decomposition of NH3.
The explosive nature of anhydrous hydrazine (>98%) upon exposure to metal catalysts surface limits its application from safety point of view. Hydrous hydrazine, such as hydrazine monohydrate, H2NNH2·H2O, still contains a large amount of hydrogen, 8.0 wt%, which is available for hydrogen generation and is much safer,196 while efforts need to be made from the engineering side to minimize the influence of toxicity. Notably, generation of only nitrogen as byproduct in addition to hydrogen, which does not need on-board collection for recycling, and easy recharging using the current infrastructure of liquid fuels are distinct advantages of hydrous hydrazine. These advantages of hydrous hydrazine make it a promising hydrogen carrier for storage and transportation. The key to exploit effectively the hydrogen-storage properties of hydrazine is to develop efficient and selective catalysts for H2 generation from hydrous hydrazine.26,27,207–214 It has been reported that an Ir/Al2O3 catalyst is active for hydrazine monohydrate decomposition, while no quantitative results were reported in this work.207 In the beginning of our study toward catalytic decomposition of hydrous hydrazine, we investigated the catalytic activity of various metal (Fe, Co, Ni, Cu, Ru, Rh, Ir, Pt and Pd) nanoparticles at room temperature (Fig. 7A and Table 3).26 Among the various NPs examined, the Rh NPs were found to be the most selective (∼44%) for hydrogen release from hydrous hydrazine decomposition. Other metal NPs, such as Co, Ru and Ir, exhibited only 7% selectivity for hydrogen, and Fe, Cu, Ni, Pt and Pd are totally inactive under the described reaction condition.26
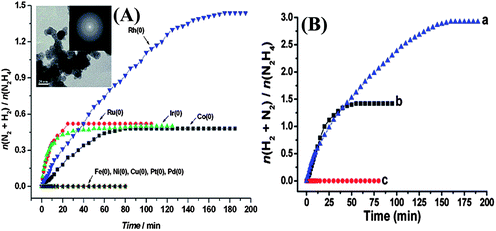 | ||
| Fig. 7 (A) Time-course plots for the decomposition of hydrazine in aqueous solutions in the presence of different metal NPs (metal/N2H4 = 1:10) at 298 K. The inset shows a TEM image of Rh(0) NPs and the corresponding SAED pattern. (B) Time course plots for decomposition of hydrous hydrazine (0.5 M) catalyzed by (a) Rh4Ni, (b) Rh, and (c) Ni nanocatalysts (M/N2H4 = 1:10) at 25 °C. Reprinted with permission from ref. 26 and 27. Copyright 2011 American Chemical Society. | ||
| Catalysts | Temp. (°C) | H2-selectivity (%) | Ref. |
|---|---|---|---|
| Ni | 25 | 0 | 26 |
| 50 | 33 | 211 | |
| Co | 25 | 7 | 26 |
| Fe | 25 | 0 | 26 |
| Cu | 25 | 0 | 26 |
| Ru | 25 | 7 | 26 |
| Rh | 25 | 43.8 | 26 |
| Pd | 25 | 0 | 26 |
| Ir | 25 | 7 | 26 |
| Pt | 25 | 0 | 26 |
| Rh4Ni | 25 | 100 | 27 |
| Ni–Pt | 25 | 100 | 208 |
| Ni–Ir | 25 | 100 | 209 |
| Ni–Pd | 50 | 80 | 210 |
| Ni–Fe | 25 | 0 | 212 |
| 70 | 100 | 212 | |
| Rh–Ni/graphene | 25 | 100 | 213 |
| Ni–Al hydrotalcite | 30 | 93 | 214 |
Bimetallic NPs generally show composition-dependent surface structures and therefore potential for improving catalytic performances.215,216 Usually, co-reduction with a relatively strong reducing agent can be used for the preparation of bimetallic alloy NPs. We also adopted this method and investigated the catalytic activities of a series of bimetallic alloy NPs towards catalytic decomposition of hydrous hydrazine. We investigated the catalytic activities of RhxNiy (Rh/Ni = 1:16–64:1) nanocatalysts to hydrous hydrazine decomposition reaction with the Rh/N2H4 molar ratio (1:10) kept unchanged and found that the selectivity to hydrogen strongly depends on the Rh/Ni ratio.27 Surprisingly, despite Ni NPs being inactive and the low H2 selectivity of Rh NPs to this reaction, the alloying of Ni and Rh drastically enhances the hydrogen selectivity. The H2 selectivity was found to be strongly dependent on the Rh–Ni ratio, and a maximum of H2 selectivity at 100% was reached at Rh–Ni = 4:1 (Fig. 7B and Table 3).27 Moreover, alloying Rh with other first-row transition metals, such as Fe, Co, and Cu, exhibits a loss in hydrogen selectivity, under the analogous synthetic and reaction conditions.27 Generally speaking, alloy materials have distinct binding properties with reactants, in contrast to those for monometallic metal catalysts. It is reasonable to assert that the alloying of Rh and Ni leads to a modification of the catalyst surface and tunes the interactions of Rh with the N–N and N–H bonds as well as the stability of reaction intermediates on the catalyst surface.201,206 Consequently, the reaction prefers pathway (13) to pathway (12), resulting in a complete conversion of hydrazine to hydrogen and nitrogen.
In another report, we demonstrated that the alloying of nickel and platinum makes it possible to achieve 100% selectivity for the decomposition of hydrazine in aqueous solution to hydrogen at room temperature, whereas the monometallic nickel and platinum counterparts are inactive for this reaction.208 The bimetallic Ni–Pt nanocatalyst with the platinum content as low as 7 mol% (Ni0.93Pt0.07) presents a step toward a high-performance catalyst system.208 In order to search for new catalysts, we found a surfactant-stabilized highly active bimetallic Ni0.95Ir0.05 alloy nanocatalyst prepared by alloying Ir (5 mol%) with Ni (95 mol%), which exhibits 100% H2 selectivity for complete decomposition of hydrous hydrazine at room temperature.209 Notably, the corresponding monometallic counterpart has poor H2 selectivity (7% H2 selectivity, Ir NPs) or is inactive (Ni NPs).
Further, we reported Ni–Pd bimetallic nanoparticle catalysts (Ni1−xPdx), synthesized by alloying Ni and Pd with varying Pd contents, which exhibit appreciably high H2 selectivity (>80% at x = 0.40) from the decomposition of hydrous hydrazine at 50 °C, whereas the corresponding monometallic counterparts are either inactive (Pd NPs) or poorly active (Ni NPs exhibit 33% H2 selectivity).210 Unlike the high activity of Ni–Pd nanocatalysts, Pd–M (M = Fe, Co and Cu) bimetallic nanocatalysts exhibit poor catalytic activity.
A high-performance catalyst system with low noble metal content might facilitate the application of hydrous hydrazine as a highly promising practical material for hydrogen storage. Therefore, emphasis has been placed on the development of suitable reaction conditions for hydrazine decomposition to hydrogen by using low-cost nanocatalysts. We have observed that Ni NPs, which are inactive for the decomposition of hydrous hydrazine at room temperature, can show drastically enhanced catalytic activity with an H2 selectivity of 33% when the reaction temperature is raised to 50 °C (Table 3).211 This significant temperature effect can be suitably exploited to achieve 100% H2 selectivity at 50 °C by alloying Ni and Pt with a Pt content as low as 1 mol%. These results indicate that a suitable reaction temperature may make it possible to achieve high catalytic performance for hydrogen generation by decomposition of hydrous hydrazine with Ni-based nanocatalysts with low content of noble metals.211
After having the high catalytic performance of the Ni and noble metal (Rh, Pt, Ir) based bimetallic nanoparticle catalysts (nanocatalysts), we extended our studies toward the development of a completely noble-metal-free catalyst, which is crucial for promoting the potential application of hydrous hydrazine as a hydrogen storage material. In this regard, we found high-performance noble-metal-free bimetallic Ni–Fe NP catalysts for selective decomposition of hydrous hydrazine to hydrogen under moderate conditions.212 Bimetallic Ni–Fe nanocatalysts were prepared using a surfactant-aided coreduction process (Scheme 2). To an aqueous solution of nickel(II) chloride and ferrous(II) sulfate was added an aqueous solution of sodium borohydride in the presence of hexadecyltrimethylammonium bromide (CTAB) (Scheme 2).212 Ni3Fe, NiFe, and NiFe3 represent the Ni–Fe nanocatalysts prepared with varying compositions of Ni and Fe, with Ni–Fe molar ratios of 3:1, 1:1 and 1:3, respectively.
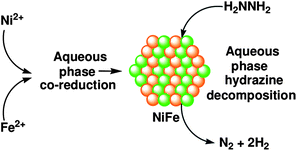 | ||
| Scheme 2 NiFe nanocatalyst preparation and hydrazine decomposition. Reprinted with permission from ref. 212. Copyright 2011 American Chemical Society. | ||
Investigations of the catalytic performance of Ni–Fe nanocatalysts with various Ni–Fe molar ratios for the decomposition of hydrous hydrazine at 70 °C suggested a significant dependence of the hydrogen selectivity on the composition of the nanocatalyst (Fig. 8). Although the NiFe nanocatalyst exhibited the highest catalytic performance among the examined Ni–Fe nanocatalysts, only 81% hydrogen selectivity was achieved.212 Surprisingly, it was found that the addition of NaOH significantly enhanced the H2 selectivity. The NiFe nanocatalyst released gases in a stoichiometric amount (3.0 equiv.) from the decomposition of hydrous hydrazine in 190 min (catalyst/H2NNH2 = 1:10) with NaOH (0.5 M) at 70 °C (Fig. 8), corresponding to decomposition of hydrous hydrazine to hydrogen with 100% selectivity via pathway (13).212 In addition, the NiFe catalyst exhibited high stability. The possible reason for the effects of the alkaline additive might be understood as follows: pathway (12) gives the basic product NH3; the addition of NaOH may make the catalyst surface highly basic, which may be unfavorable for the formation of basic NH3 and therefore for pathway (12). In contrast to the NiFe nanocatalyst, 89% hydrogen selectivity was observed for the Ni3Fe nanocatalyst, whereas the NiFe3 nanocatalyst exhibited 71% hydrogen selectivity for the decomposition of hydrous hydrazine at 70 °C (Fig. 8A). Notably, addition of weaker bases (e.g., NH3, CH3COONa) had no effect on the catalytic performance of the NiFe catalysts. Improvements in the catalytic performance due to addition of NaOH were also observed for Ni45Pt55 and Ni50Ir50 catalysts; the addition of NaOH (0.5 M) resulted in increases in the H2 selectivity from 61 to 86% for Ni45Pt55 and 7 to 95% for Ni50Ir50 at 25 °C, indicating that the presence of alkaline additives is commonly beneficial in promoting pathway (13).212
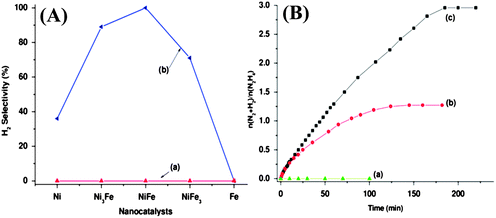 | ||
| Fig. 8 (A) Comparison of H2 selectivities in the decomposition of hydrous hydrazine (0.5 M) to hydrogen in the presence of Ni, Ni3Fe, NiFe, NiFe3, and Fe nanocatalysts (catalyst/H2NNH2 = 1:10) with NaOH (0.5 M) at (a) 25 and (b) 70 °C. (B) Time-course plots for the decomposition of hydrous hydrazine (0.5 M) to hydrogen in the presence of (a) Fe, (b) Ni, and (c) NiFe nanocatalysts (catalyst/H2NNH2 = 1:10) with NaOH (0.5 M) at 70 °C. Reprinted with permission from ref. 212. Copyright 2011 American Chemical Society. | ||
None of the Ni–Fe NPs catalysts was found to be active for this reaction when examined at 25 °C with or without NaOH, indicating the crucial role of temperature in the decomposition reaction (Fig. 8A). Fe NPs prepared under analogous conditions exhibit no activity for this reaction at 25 °C or at elevated temperatures (thermal decomposition of hydrazine in absence of catalyst is ∼250 °C).196 Ni NPs, which are also inactive at room temperature for the decomposition of hydrous hydrazine, can show an enhancement in the activity with an increase in the reaction temperature.211,212 An investigation of the role of temperature on the NiFe nanocatalyst showed enhanced activity and H2 selectivity for decomposition of hydrazine to hydrogen at elevated temperatures. Although the NiFe catalyst was inactive at 25 °C, the activity was enhanced along with the enhancement in H2 selectivity to 80% at 50 °C and 100% at 70 °C in the presence of NaOH (0.5 M).
Recently, Zhang and coworkers prepared graphene-supported RhNi catalyst by a facile co-reduction route, wherein the graphene plays a key role as a dispersion agent and distinct support for the RhNi NPs.213 The RhNi catalyst prepared in the presence of GO and NaOH exhibits 100% H2 selectivity [n(N2 + H2)/nN2H4 = 3.0] and high activity to complete the decomposition reaction of hydrous hydrazine within only 49 min (Rh/N2H4 = 1/10) at room temperature, which is faster (as high as 327%) than that of the most active catalysts previously reported for this reaction.26,27,208,209
Very recently, Zhang and coworkers found that the hydrotalcite supported Ni catalyst (78 wt% Ni–Al2O3–HT) exhibits 100% conversion and 93% H2 selectivity for the decomposition of hydrous hydrazine. In the presence of 78 wt% Ni–Al2O3–HT (50 mg) catalyst, the reaction can be completed in 13 min (Ni/N2H4 = 1:2.4) at 50 °C and in 70 min at 30 °C.214 This unique catalysis is due to the cooperation of Ni NPs and the strong basic sites of the hydrotalcite host.
4.2 Synthesis of hydrazine
Decomposition of hydrazine generates nitrogen gas as a byproduct in addition to hydrogen (H2). The recycling of nitrogen (N2) to hydrazine is very crucial for the development of hydrazine-based hydrogen storage cycling. There are several methods reported in the literature for synthesis of hydrazine, but most of the synthetic processes involve different nitrogen containing materials other than nitrogen gas.196,217 The commercial-scale production of hydrazine is based on the Raschig process [eqn (14)–(16)], the Schestakov synthesis [eqn (17)], the Bayer process [eqn (18) and (19)], and the H2O2 process [eqn (20) and (21)] etc.196,217 These processes mainly involve NH3 or urea as the starting material.Raschig process:
| 2NaOH + Cl2 → NaOCl + NaCl + H2O | (14) |
| NaOCl + NH3 → NH2Cl + NaOH | (15) |
| 2NH3 + NH2Cl → N2H4 + NH4Cl | (16) |
Schestakov synthesis:
(H2N)2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O + NaOCl + 2NaOH → N2H4 + H2O + NaCl + Na2CO3 O + NaOCl + 2NaOH → N2H4 + H2O + NaCl + Na2CO3 | (17) |
Bayer process:
| NaOCl + 2NH3 + 2CH3COCH3 → (CH3)2CN–NC(CH3)2 + NaCl + 3H2O | (18) |
| (CH3)2CN–NC(CH3)2 + 2H2O → 2CH3COCH3 + N2H4 | (19) |
H2O2 process:
| H2O2 + 2NH3 + 2C2H5COCH3 → (C2H5)(CH3)CN–NC(CH3)(C2H5) + 4H2O | (20) |
| (C2H5)(CH3)CN–NC(CH3)(C2H5) + 2H2O → 2C2H5COCH3 + N2H4 | (21) |
The search for selective reduction of nitrogen has not yet found a practical or economic solution. Ammonia remains a valuable nitrogen-containing starting material for the production of hydrazine.196,217,218 The approach from nitrogen and hydrogen to hydrazine might be the transformation of nitrogen to ammonia (the Haber–Bosch process, homogeneous catalytic processes, or an electrolytic process) and then to hydrazine on a large scale [eqn (22)] or perhaps, preferably, transformed directly to hydrazine by means of an electrolytic process similar to that for ammonia synthesis.217–223
| N2 + 3H2 → 2NH3 → N2H4 + H2 | (22) |
4.3 Conclusions
Hydrazine seems to be a promising hydrogen storage system due to its uncomplicated byproduct and easy storage. For the decomposition, many efficient catalysts have been developed and studied in detail. Further developments of low-cost, high-performance catalysts are needed for the practical use of hydrous hydrazine as an effective hydrogen storage material. The effective practical application of hydrazine will be economically feasible if low-cost production of hydrazine can be achieved by finding an effective method of conversion of nitrogen to hydrazine.5. Hydrazine borane
5.1 Catalytic hydrogen generation from aqueous hydrazine borane
Hydrazine borane (N2H4BH3, HB) has a gravimetric hydrogen storage capacity of 15.4 wt%.117 Like AB, the aqueous solution of HB (solubility of 6 g per 100 g H2O at 25 °C)224,225 is quite stable against spontaneous hydrolysis; for instance, over 3 weeks of storage under inert atmosphere at room temperature. Hydrazine borane can release hydrogen through chemical pathways such as thermolysis or hydrolysis.226 HB starts to decompose slowly at around 60 °C. Better dehydrogenation kinetics and extents are achieved at elevated temperatures, higher than 100 °C. The release of hydrogen from HB in the presence of LiH has been reported upon heating at a temperature range of 100–150 °C (for example, 4.4 equiv. H2 per HB + LiH at 100 °C upon 45 h heating).227 Its dehydrogenation by catalytic hydrolysis has been also reported by Özkar and coworkers.224 They reported that the hydrogen generation can readily be achieved from the hydrolysis of hydrazine borane by using RhCl3 precatalyst, in which bulk Rh(0) was found to be an active catalyst providing a TOF value of 12000 h−1 even at room temperature. They used rhodium(0) nanoparticles supported on hydroxyapatite (Ca10(OH)2(PO4)6, HAP) as catalyst and observed a TOF value of 6700 h−1.228 However, only 3/7 of its hydrogen was recovered by hydrolysis of the BH3 group and the hydrazine group was not decomposed [eqn (23)].224,228 Karahan et al. reported the metal catalyzed methanolysis of hydrazine borane using a nickel(II) chloride precatalyst at room temperature.229 The methanolic solution of HB (HB/Ni ≥ 200) can release 3 equiv. of H2, corresponding to methanolysis of the BH3 unit, with a rate of 24 mol H2 (mol Ni min)−1 at room temperature.229| N2H4BH3 + 3H2O → N2H4 + B(OH)3 + 3H2 | (23) |
Hydrolysis of BH3 unit of HB leads to occurrence of free N2H4.224,228 It is therefore reasonable to consider that the presence of a selective catalyst is crucial to achieve the complete HB dehydrogenation. Unlike the NH3 group of AB, the N2H4 group of HB can also be dehydrogenated in the presence of a selective catalyst [eqn (24)], although this reaction is in competition with NH3 release [eqn (25)].
| αN2H4 → αN2 + 2αH2 | (24) |
| (1 − α)N2H4 → 4(1 − α)/3NH3 + (1 − α)/3N2 | (25) |
| N2H4BH3 + 3H2O → B(OH)3 + (3 + 2α)H2 + (2α + 1)/3N2 + 4(1 − α)/3NH3 | (26) |
Being inspired by studies on hydrous hydrazine decomposition,27,208,209 bimetallic metallic nanocatalysts that are active for decomposition of hydrous hydrazine, which are also active for hydrolysis of –BH3 group, have been used for complete dehydrogenation of HB in aqueous solution.24 Use of a Pt nanocatalyst at 50 °C only permits release of ca. 2.98 ± 0.05 equiv. H2 per HB, which originates from hydrolysis of the BH3 group [eqn (23)]. On the other hand, in the presence of the Ni nanocatalyst, some H2 and N2 from the N2H4 group evolve, which corresponds to 3.22 ± 0.05 equiv. H2 + N2 (Fig. 9). The presence of a small amount of Pt in a Ni–Pt NP has a significant beneficial effect on N2H4 decomposition (Fig. 9). Ni0.97–Pt0.03 releases 5.07 ± 0.5 equiv. H2 + N2 per HB. By increasing the Pt content up to 0.11 mol% (Ni0.89–Pt0.11), there is an increase up to 5.79 ± 0.05 equiv. H2 + N2 per HB whereas the hydrogen selectivity reaches the value 93 ± 1% [eqn (26)]. A further increase of the Pt content (0.17 and 0.23 mol%) has a detrimental effect since the H2 mol number slightly decreases to 5.29 ± 0.05 equiv. H2 + N2 per HB. In fact, the variation of the mol number of [H2 + N2] as a function of the Pt content has a volcano shape (Fig. 9b), and the maximum peak is observed for Pt = 0.11 mol% (Ni0.89Pt0.11). Electronic and/or geometric effects could rationalize such reactivity changes.230,231 It can be seen that the observed kinetics of the dehydrogenation of the N2H4 group is similar to that of the decomposition of neat hydrazine with the same NiPt catalysts.208
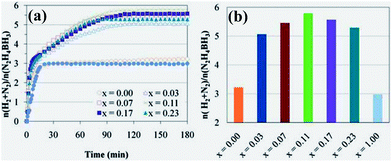 | ||
| Fig. 9 (a) Time course profiles and (b) Pt-content dependence of n(H2 + N2)/n(N2H4BH3) for dehydrogenation of N2H4BH3 in the presence of the Ni1−xPtx nanocatalysts at 50 °C. Reprinted with permission from ref. 24. Copyright 2011 Royal Society of Chemistry. | ||
Very recently, a series of Ni-based bimetallic systems have been investigated with Pt, Ru, Rh or Ir as the second metal.232 The results show that most of the Ni1−xMx nanocatalysts outperform the monometallic Ni, Pt, Ru, Rh and Ir catalysts. The best performance achieved is 5.1 ± 0.05 mol (N2 + H2) per mol(HB) with Ni0.89Rh0.11 and Ni0.89Ir0.11, suggesting the occurrence of the following reaction [eqn (27)]:
| N2H4BH3 + 3H2O → B(OH)3 + 4.3H2 + 0.8N2 + 0.6NH3 | (27) |
5.2 Synthesis of hydrazine borane
Several attempts by Goubeau and Ricker using various reagents (B2H6, LiBH4, NaBH4, N2H4, and (N2H5)2SO4) showed that the most efficient HB synthesis process involved NaBH4 and (N2H5)2SO4 in dioxane [eqn (28)].233| 2NaBH4 + (N2H5)2SO4 → 2N2H4BH3 + 2H2 + Na2SO4 | (28) |
Recently, Demirci and coworkers reported a modified procedure of this synthetic method by using a 2-step synthesis (salt metathesis and solvent extraction–drying) through which N2H4BH3 is successfully obtained in 3 days, with a yield of about 80% and a purity of 99.6%.226 In addition, several alternative processes for preparing HB have also been reported,234–236 which typically consisted of a BH3 source, mainly NaBH4, and a N2H4 source, either N2H4·HCl or MgCl2·4N2H4, in an organic solvent (benzene, dioxane, hexane, THF) under ambient condition (yields 33–98%). Another approach was to use directly N2H4 as solvent at 0 °C. For a 2 days process, the yield was low (34%) with a purity of 98% whereas for a longer time (1 week) yields up to 79% were achieved.
Like sodium borohydride and ammonia borane, hydrazine borane also produces borates as byproducts in the dehydrogenation reaction in water. Therefore, the processes developed to recycle the spent fuel of sodium borohydride (Section 2.2) could be used for recycling the spent fuel of hydrazine borane. The as-sythesized sodium borohydride upon reaction with (N2H5)2SO4 can generate hydrazine borane. More recently, Sutton, Gordon and coworkers interestingly obtained HB as a major product (70–100%) while they were working on reacting spent fuel polyborazylene (PB) with N2H4 (12 h) to obtain AB.184,237 Future study in this direction could be to find a low cost and efficient regeneration of hydrazine borane from the byproduct obtained after hydrogen release.
5.3 Conclusions
Hydrazine borane is emerging as one of the most promising solid hydrogen carriers due to its high gravimetric hydrogen storage capacity (15.4 wt%). The current catalyst systems are able to generate hydrogen from hydrolysis of the BH3 part, or both hydrolysis of BH3 and decomposition of the hydrazine part. However, like sodium borohydride and AB, HB will certainly face issues related to the use of water in excess (because of low solubility and of hydration of spent fuel, borate by-products), catalyst durability, by-products recyclability, and cost. Sodium borohydride, AB and HB should then be judged as equal for these specific problems. Nevertheless, the present challenges are now to find a more selective catalyst enabling one to reach 100% selectivity to hydrogen – the catalyst activity should be also stable – and to better understand the fundamentals of the reactions occurring.6. Formic acid
6.1 Catalytic decomposition of formic acid
Formic acid, the simplest carboxylic acid, is a non-toxic liquid (although neat formic acid is corrosive and its vapour is harmful) at room temperature with a density of 1.22 g cm−3, which is suitable for easy transportation, refueling, and handling.28 Formic acid and its conjugated base, formate, have been used widely as a hydrogen source in transfer hydrogenation reactions.238,239 Formic acid has a hydrogen content of 43 g kg−1, corresponding to 4.4 wt% hydrogen, and produces only gaseous products (H2–CO2) by decomposition. The H2–CO2 mixture can be easily separated under certain conditions.28| HCOOH → H2 + CO2 ΔG = −48.4 kJ mol−1 | (29) |
| HCOOH → H2O + CO ΔG = −28.5 kJ mol−1 | (30) |
The decomposition of formic acid follows two principal pathways, in which the process producing CO2 and H2 [eqn (29)] is the desired reaction and that producing CO and H2O [eqn (30)] is the undesired side reaction.240 The selective decomposition of formic acid to CO2 and H2, which is the reversible reaction of CO2 hydrogenation, is crucial for formic acid based hydrogen storage.241 The combination of carbon dioxide and formic acid as a hydrogen storage system might act as an elegant and simple concept wherein selective decomposition of formic acid to H2 and CO2 and recycling of CO2 by H2 reduction to formic acid can be achieved (Scheme 3).242,243 Meanwhile, CO2 is abundant on the Earth; it is cheap, and readily available. In this case, a reduction of CO2 emissions is the use of CO2 itself as a hydrogen carrier.
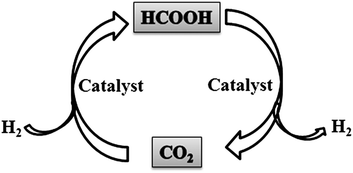 | ||
| Scheme 3 The hydrogen storage system based on carbon dioxide–formic acid (CO2–HCOOH). | ||
| Catalyst (precursor) and ligands | TOF (h−1) | T (°C) | Ref. |
|---|---|---|---|
| a Abbreviations used: dppm = 1,1-bis(diphenylphosphino)methane, dppe = 1,2-bis(diphenylphosphino)ethane tppts = trisodium-3,3′,3′′-phosphinidynetris(benzenesulfonate), tppds = ddiphenylphosphine disulfonate, bpy = 2,2′-bipyrdine, bpm = 2,2′-bipyrimidine, PP3 = tris[(2-diphenylphosphino)ethyl] phosphine. | |||
| [Ru2(m-CO)(CO)4(m-dppm)2] | 500 | RT | 250 |
| RuBr3·xH2O, 3 equiv. (PPh)3 | 3630 | 40 | 242 |
| RuCl2(PPh3)3 | 2688 | 40 | 254 |
| [RuCl2(benzene)]2, 6 equiv. dppe | 900 | 40 | 257 |
| RuCl3·xH2O, 2 equiv. TPPTS | 460 | 120 | 30 |
| RuCl3, 2 equiv. mTPPDS | 476 | 90 | 265 |
| [RhIII(Cp*)(H2O)(bpy)]2+ | 28 | RT | 251 |
| [IrIII(Cp*)(H2O)(bpm)RuII(bpy)2]4+ | 426 | RT | 253 |
| [Ir(Cp*)-4,4′-hydroxy-2,2′-bipyridine] | 3100 | 60 | 252 |
| 1.4 × 104 | 90 | 252 | |
| [Ru2(HCO2)2(CO)4] | 1.8 × 104 | 120 | 266 |
| [Ru4(CO)12H4] | 1470 | 107 | 267 |
| [{RuCl2(p-cymene)}2] | 1540 | 80 | 268 |
| RuCl3 | 280 | 80 | 262 |
| [Fe(BF)4)2]·6H2O, 2 equiv. PP3 | 1942 | 40 | 264 |
| 5390 | 80 | 264 | |
Fukuzumi et al. reported that HCOOH selectively decomposed to afford H2 and CO2 in aqueous solution at room temperature in the presence of a catalytic amount of a water-soluble Rh catalyst, [RhIII(Cp*)(bpy)(H2O)](SO4) (Cp* = pentamethylcyclopentadienyl, bpy = 2,2′-bipyridine).251 A similar non-phosphine catalyst [IrIII(Cp*)(dhbpy)(H2O)](SO4) (Cp* = pentamethylcyclopentadienyl, dhbpy = 4,4′-dihydroxy-2,2′-bipyridine) was reported by Himeda for generation of CO-free hydrogen from the decomposition of formic acid.252 The highest catalytic activity (turnover frequency (TOF) of up to 14000 h−1 at 90 °C) and an almost complete consumption of formic acid were obtained for the catalytic system. No deterioration of the catalyst was observed during the catalytic decomposition of HCOOH in the continuous runs of reaction. More recently, Fukuzumi et al. demonstrated that heteronuclear iridium–ruthenium complexes are highly active catalysts for hydrogen generation in an aqueous solution under ambient conditions giving a TOF of about 426 h−1.253
In 2008, important investigations were made by two independent groups, Beller and Laurenczy et al.30,242,254–258 Beller and coworkers investigated the decomposition of formic acid with different homogeneous catalysts at 40 °C, including RhCl3·xH2O, RuBr3·xH2O, [{RuCl2(p-cymene)}2], [RuCl2(PPh3)3], [{RuCl2(benzene)2}2], etc. in the presence of amine adducts.242,254,255 They investigated the influence of different amines on catalytic activity by using 1000 ppm [{RuCl2(p-cymene)}2] as the standard catalyst precursor, and a formic acid to amine ratio of 5:2. Generally, for the reaction catalyzed by [{RuCl2(p-cymene)}2], various alkyl dimethylamines with longer alkyl chains showed a higher degree of reaction rate promotion. High decomposition rates (TOF = 21 h−1) were also observed by using triethylamine.242,255 Interestingly, using the ruthenium phosphine complex [RuCl2(PPh3)3] they observed much higher turnover frequencies: up to 417 and 302 h−1 after 2 and 3 hours, respectively, and a formic acid conversion of 90% after 3 hours.254 A similar activity is obtained with an in situ catalyst prepared from ruthenium trichloride hydrate and triphenylphosphine in DMF. Among all examined catalysts under different reaction conditions, the decomposition of formic acid in a 5HCOOH–2NEt3 mixture over the catalyst in situ prepared from RuBr3·xH2O and three equivalents of PPh3 after pretreatment in DMF at 80 °C for 2 h yielded an initial TOF of 3630 h−1 at 40 °C.242 Recently, they developed a highly active and stable catalyst system (in situ generated (benzene)ruthenium dichloride dimer [RuCl2(benzene)]2/6 equiv. 1,2-bis(diphenylphosphino)ethane (dppe)) for both batch and continuous experiments. In the presence of this in situ generated catalyst and N,N-dimethyl-n-hexylamine a total TON of approximately 260000, with an average TOF of more than 900 h−1, at room temperature has been achieved. In the produced gas mixture only hydrogen and CO2 are detected, which allows direct use in fuel cells after simple cleaning by a charcoal column.257 In another report they demonstrated for the first time the light-accelerated hydrogen generation reaction from formic acid with a catalyst system based on a ruthenium precursor (i.e. [RuCl2(benzene)]2, RuCl3·xH2O, [Ru(cod)(methylallyl)2]) and aryl phosphines (PPh3, dppe etc.) (Fig. 10).258 The best productivity is observed with a [RuCl2(benzene)]2/dppe catalyst, where gas evolution increased from 407 to 2804 turnovers, which is an almost 7-fold increase.
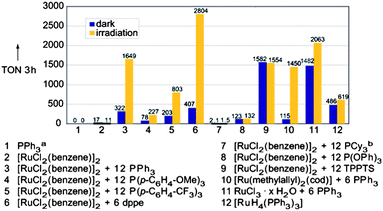 | ||
| Fig. 10 The influence of light on the decomposition of 5 mL 5HCOOH–2NEt3 with ruthenium catalyst systems (320 ppm Ru, 40 °C). (a) No hydrogen detected by GC; (b) no dark experiment, performed under lab conditions, environmental light. Reprinted with permission from ref. 258. Copyright 2009 Royal Society of Chemistry. | ||
In 2008, Laurenczy and coworkers developed a homogenous catalytic system which can efficiently and selectively decompose formic acid into hydrogen and carbon dioxide.30,256 [Ru(H2O)6]2+, [Ru(H2O)6]3+ and RuCl3·xH2O were found to be excellent precatalysts in the presence of TPPTS (TPPTS = meta-trisulfonated triphenylphosphine) for the formic acid decomposition in the aqueous phase under mild conditions and over a large range of pressures. With increased temperatures, the reaction rate became much higher (TOF = 670 h−1 at 120 °C) and at all reaction temperatures, the conversion was higher than 90%. The performance of the catalytic system for continuous hydrogen generation is also investigated. A detailed mechanism study was also carried out by this group using multinuclear NMR spectroscopy.256 They proposed a tentative reaction mechanism consisting of two competitive catalytic cycles involving a monohydride ruthenium complex [RuH(tppts)2(H2O)3]+ as a common intermediate. Furthermore, they developed heterogeneous catalysts based on ruthenium(II)–TPPTS catalyst using different immobilization/solidification methods, such as ion exchange, polymer immobilization and physical absorption.259 Both ion exchange and coordination to the phosphine-containing polymers gave stable supported heterogeneous catalysts for selective decomposition of formic acid. The leaching of the metal was negligible and the catalysts could be easily separated from the reactant/solution, and could be reused directly.
The finding by Beller and co-workers showed that the high catalytic activity for formic acid decomposition was obtained with a high ratio of organic amine to formic acid. In the formic acid decomposition process developed by Laurenczy and co-workers, sodium formate (HCOONa) was used, which is non-volatile but only active at high temperature. Shi, Deng and co-workers realized that amine-functionalized ionic liquids (ILs), defined as organic salts with melting points below 100 °C, can be substituted for volatile organic amines.260,261 They prepared and used a series of amine-functionalized ionic liquids for hydrogen generation by the selective catalytic decomposition of formic acid in the presence of [{RuCl2(p-cymene)}2] catalyst (Table 5).261 Among the ILs investigated, the 1-(2-diisopropylaminoethyl)-3-methylimidazolium chloride–sodium formate (iPr2NEMimCl–HCOONa) system exhibited high activity (TOF > 600 h−1) at 60 °C. However, the authors stated that they were not able to recycle the catalyst solution. In a separate study Wasserscheid and coworkers investigated an outstanding simple, active and recyclable ionic liquid-based system for the catalytic decomposition of formic acid. The most efficient system, RuCl3 dissolved in [EMMIM][OAc], was shown to produce hydrogen and carbon dioxide as the only products and was recyclable for at least nine cycles.262 During these cycles no deactivation or change in selectivity was observed. It is worth noting that this simple catalytic system exhibits turnover frequencies of 150 h−1 at 80 °C and 850 h−1 at 120 °C.
| Catalysts | V 1h/V2h (mL)a | TON1h/TON2h | Conv. (x%, 2 h) |
|---|---|---|---|
|
a Measured by gas burette (H2:CO2 = 1:1).
b
i-Pr2NEMImCl:i-Pr2NEMImHCOO = 88.2:11.8 (mol).
c 5 mmol Et2NEMImCl + 5 mmol HCOONa.
d 5 mmol i-Pr2NEMImCl + 5 mmol HCOONa.
e 5 mmol i-Pr2NEPyCl + 5 mmol HCOONa.
f 10 mmol i-Pr2NEMImCl + 5 mmol HCOONa, 7.84 μmol [RuCl2(p-cymene)]2.261,262
|
|||
| H2NEMImBr | 9/17 | 3/6 | 1.1 |
| Me2NEMImCl | 33/58 | 11/19 | 3.8 |
| Me2NPMImCl | 28/51 | 9/17 | 1.4 |
| Et2NEMImCl | 41/71 | 14/24 | 4.7 |
| i-Pr2NEMImCl | 108/206 | 36/68 | 13.6 |
| i-Pr2NEMMImCl | 156/311 | 52/103 | 20.6 |
| i-Pr2NEPyCl | 113/210 | 37/70 | 13.9 |
| i-Pr2NEMImBF4 | 52/87 | 17/29 | 5.8 |
| i-Pr2NEMImOTf | 74/122 | 25/40 | 8.1 |
| i-Pr2NEMImNTf2 | 35/55 | 12/18 | 3.6 |
| i-Pr2NEMImCl/HCOOb | 207/445 | 69/147 | 29.5 |
| Et2NEMImCl–HCOONac | 172/331 | 57/110 | 21.9 |
| i-Pr2NEMImCl–HCOONad | 382/862 | 127/286 | 57.1 |
| i-Pr2NEPyCl–HCOONae | 252/455 | 84/151 | 30.1 |
| i-Pr2NEMImCl–HCOONaf | 481/971 | 627/1267 | 64.3 |
Recently, Beller, Ludwig and coworkers reported the first non-noble metal-based homogeneous catalyst system for hydrogen generation from formic acid. By application of a catalyst formed in situ from inexpensive Fe3(CO)12, 2,2′:6′2′′-terpyridine or 1,10-phenanthroline, and triphenylphosphine, hydrogen generation is possible under visible light irradiation and ambient temperature.263 The best catalyst system identified was triirondodecacarbonyl (Fe3(CO)12) in the presence of triphenylphosphine (PPh3), 2,2′:6′2′′-terpyridine (tpy), and dimethylformamide (DMF). This system was capable of generating hydrogen from formic acid–amine adducts at temperatures above 100 °C, whereas in the presence of in situ generated catalyst system under visible light irradiation, hydrogen generation even occurred at room temperature. Depending on the kind of N-ligands significant catalyst turnover numbers (>100) and turnover frequencies (up to 200 h−1) were observed, which are the highest known to date for nonprecious metal catalyzed hydrogen generation from formic acid. NMR, IR and DFT studies of the iron complexes formed under reaction conditions confirm that PPh3 plays an active role in the catalytic cycle and that N-ligands enhance the stability of the system. It was shown that the reaction mechanism includes iron hydride species which are generated exclusively under irradiation with visible light.
In another report in 2011, Beller, Ludwig, Laurenczy and coworkers demonstrated a highly active catalyst system based on iron complexes for the selective decomposition of formic acid (Table 6).264 This catalyst consists of an iron cation that is permanently coordinated by four phosphorus centers of a tetradentate phosphine ligand, namely tris[(2-diphenylphosphino)ethyl] phosphine (PP3). The remaining two coordination sites of the FeII center are occupied by the HCOOH substrate and/or product-derived species during the catalytic cycle. The catalyst can be formed in situ from [Fe(BF4)2] and the PP3 ligand under the reaction conditions or can be added to the reaction mixture in a presynthesized form as [FeH(PP3)]+. This catalyst functions in a common organic solvent (propylene carbonate) without any further additives or light. It achieves a high TON of up to 100000 and a high TOF of nearly 10000 h−1. The catalyst shows high selectivity for H2 formation, whereas the rate of the competing HCOOH decomposition pathway to CO and H2O is negligible. Spectroscopic studies and density functional theory calculations suggest two viable pathways (I and II, Fig. 11) for H2 release from HCOOH, both of which go through a common Fe-hydride species, [FeH(PP3)]+(1). In the first candidate of catalytic cycle, the Fe-hydride combines with a proton from HCOOH, and H2 is formed and released. The HCO2− counter anion remains coordinated to the Fe center and undergoes β-hydride elimination (transferring the hydride from HCO2− to Fe) in the rate-determining step; subsequent CO2 release reforms [FeH(PP3)]+. In the second candidate of catalytic cycle, formate coordinates to [FeH(PP3)]+ to form a neutral [FeH(HCO2)(PP3)] (2) species. β-Hydride elimination and protonation releases CO2 and closes the catalytic cycle after H2 release. The Fe center remains exclusively in the formal +2 oxidation state during both catalytic cycles.
| Catalysts | V 2h (mL) | V 3h (mL) | TON2h | TON3h |
|---|---|---|---|---|
| [Fe(BF4)2]·6H2O/PP3 | 146 | 215 | 562 | 825 |
| [Fe(BF4)2]·6H2O/2PP3 | 333 | 505 | 71279 |
1942 |
| [FeH(PP3)]BF4 | 194 | 295 | 745 | 1135 |
| [FeH(PP3)]BF4/PP3 | 319 | 500 | 1227 | 1923 |
| [FeH(H2)(PP3)]BF4 | 189 | 294 | 727 | 1129 |
| [FeH(H2)(PP3)]BPh4 | 174 | 264 | 670 | 1015 |
| [FeCl(PP3)]BF4 | 0.4 | 0.4 | — | — |
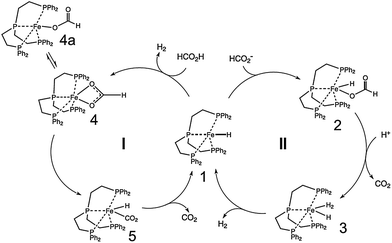 | ||
| Fig. 11 Proposed mechanisms for the selective iron-catalyzed hydrogen generation from formic acid. Reprinted with permission from ref. 264. Copyright 2011 The American Association for the Advancement of Science. | ||
| Catalyst | TOF (h−1) | T (°C) | Ref. |
|---|---|---|---|
| 20 wt% PdAu/C | 27 | 92 | 232 |
| 20 wt% PdAu/C–CeO2 | 227 | 92 | 232 |
| 20 wt% Pd–Au–Dy/C | 269 | 92 | 281 |
| 20 wt% Pd–Au–Eu/C | 387 | 92 | 281 |
| 20 wt% Pd–Au–Ho/C | 224 | 92 | 281 |
| 30 wt% PtRuBiOx | 312 | 80 | 283 |
| 20.4 wt% PdAu/MIL-101 | — | 90 | 286 |
| Pd–S–SiO2 | 803 | 85 | 300 |
| Au@Pd/C(core–shell) | 125 | 20 | 284 |
| 626 | 90 | 284 | |
| 2 wt% Au@SiO2 | — | 90 | 287 |
| 0.8 wt% Au/ZrO2 | 1590 | 50 | 301 |
Recently, Xing and coworkers developed carbon supported Pd–Cu, Pd–Ag and Pd–Au alloy catalysts and overcame the poisoning by CO byproduct from the decomposition of formic acid at low temperatures.240 The best decomposition of formic acid/sodium formate was demonstrated by Pd–Au/C. They also found that the activities of Pd–Au/C and Pd–Ag/C can be markedly enhanced by co-deposition with CeO2. The TOFs of Pd–Au/C–CeO2 and Pd–Ag/C–CeO2 are 227 and 76 h−1, respectively, at 92 °C. Inspired by the promotion effect of Ce on Pd–Au/C and Pd–Ag/C catalysts, they extended their investigations to other rare earth elements (REs) (Dy, Eu, and Ho) and obtained TOFs 269 ± 202, 387 ± 292, 224 ± 73, and 45 ± 11 h−1 for Pd–Au–Dy/C, Pd–Au–Eu/C, Pd–Au–Ho/C and Pd–Au/C catalysts, respectively, at 92 °C.293 In addition, these catalysts were even active at room temperature temporarily, and above 52 °C steadily. All the REs-promoted Pd–Au/C catalysts showed lower activation energies for decomposition of formic acid than Pd–Au/C and Pd–Au–Eu/C had the lowest value of 84.2 ± 7.4 kJ mol−1. These authors have also developed a PdAu bimetallic catalyst with a PdAu@Au core–shell nanostructure supported on carbon, which shows higher activity than the monometallic counterparts.294 In a report by Chan and coworkers, the metal–metal oxide catalyst composed of platinum ruthenium and bismuth, denoted as PtRuBiOx, was found to catalyze selective dehydrogenation of formic acid in water at nearly ambient temperature.295 The observed activation energy was 37.3 kJ mol−1 and the turnover frequency was estimated to be 312 h−1 in the first hour of the decomposition.
In an effort, Tsang and coworkers prepared various core–shell nanoparticles having an inner core of a metal element and an external shell of palladium (Table 8).296,297 Among all metals tested, the highest activity toward formic acid decomposition at room temperature was found for Ag@Pd NPs (diameter 8 nm) with the thinnest continuous Pd shell (1–2 atomic layers) and corresponding Ag/Pd alloy and pure Pd catalysts show very low activity (Fig. 12).296,297 Turnover frequencies per surface Pd site were comparable to homogeneous catalysts: 125 h−1 at 20 °C and 252 h−1 at 50 °C. At 20 °C, an equimolar mixture of hydrogen and CO2 was continuously produced without any trace of CO; on the other hand, CO was detected at temperatures higher than 50 °C. In contrast to previous reports on Pd catalysts, no deactivation was observed during the experiments, and the rate closely followed the first order kinetics of formic acid decomposition by reaction (29). Furthermore, theoretical calculations showing a strong correlation between the catalytic activity and the work function of the metal core: the largest net difference with the work function of the Pd shell will lead to the highest adsorption energy by charge transfer from the core to the shell, hence to the best possible activity of the resulting bimetallic structure for formic acid decomposition (Fig. 12b). The very short range of this so-called “ligand” electronic effect between the two metals explains why the highest performance is achieved for the thinnest Pd layer. Nanomaterials interface definitely plays a key role in catalysis.
| Catalysts | Temp (°C) | H2/CO2 | CO (ppm) | TOF (h−1) |
|---|---|---|---|---|
| Ag@Pd (1:1) |
20 | 0.96 | — | 125 |
| Ag@Pd (1:1) |
35 | 0.98 | — | 156 |
| Ag@Pd (1:1) |
50 | 1.02 | — | 252 |
| Ag@Pd (1:1) |
70 | 1.03 | 74 | 500 |
| Ag@Pd (1:1) |
90 | 1.02 | 84 | 626 |
| Ag/Pd alloy (1:1) |
20 | 1.03 | — | 144 |
| Pd (2.3 nm) | 20 | 0.98 | — | 24 |
| Pd (3.2 nm) | 20 | 1.01 | — | 25 |
| Ag@Pd/C (1:1) |
20 | 1.09 | — | 192 |
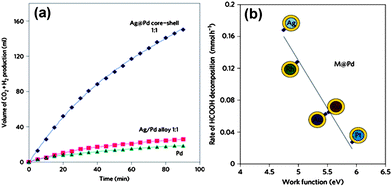 | ||
| Fig. 12 (a) Time course plots of hydrogen generation from aqueous formic acid (10 mL of 1 M) in the presence of Ag@Pd, Ag–Pd alloy and pure Pd catalysts (2.0 × 10−4 mol, 0.021 g). (b) Correlation with the work function of the M core, where M = fcc (111) Ag, Rh, Au, Ru and Pt or hexagonal close-packed (hcp) (0001) Ru. Ag, with the largest difference in work function in relation to Pd, gives the strongest electron promotion to the Pd shell. Reprinted with permission from ref. 296. Copyright 2011 Nature Publishing Group. | ||
Recently, we reported bimetallic Au–Pd NPs immobilized in mesoporous MOFs as efficient catalysts for decomposition of formic acid.298 MIL-101 was chosen as a support because of its large pore sizes (2.9–3.4 nm) and window sizes (1.2–1.4 nm) and its hybrid pore surface, which facilitates the encapsulation of metal NPs and the adsorption of the catalytic substrate formic acid inside the pores. In order to improve the interactions between the metal precursors and the MIL-101 support, we grafted the electron-rich functional group ethylenediamine (ED) into MIL-101, which contains coordinatively unsaturated Cr3+ centers, to form ED–MIL-101, which exhibited improved immobilization of small metal NPs. The resulting bimetallic Au–Pd NPs immobilized in the MOF, MIL-101, and ethylenediamine (ED)–grafted MIL-101 (ED–MIL-101), Au–Pd/MIL-101 and Au–Pd/ED–MIL-101, represent the first highly active MOF-immobilized metal catalysts for the complete conversion of formic acid to hydrogen at a convenient temperature.
Very recently, we have developed a high-performance nanoreactor composed of silica nanospheres encapsulating amine-functionalized gold nanoparticles.299 A microemulsion-templating approach was employed to fabricate gold nanoparticles encapsulated inside the hollow silica nanospheres using the procedure shown in Fig. 13. The gold@silica nanospheres synthesized under different conditions show distinct catalytic activities in the decomposition of formic acid in aqueous media. Similar to the monometallic gold nanoparticles supported on MOF or carbon, the gold nanoparticles encapsulated in SiO2 nanospheres without amine functionalization (Au@SiO2, prepared using TEOS and HAuCl4; Fig. 13a) are inactive for the decomposition of aqueous formic acid. However, the gold nanoparticles encapsulated in amine-functionalized silica nanospheres, surprisingly, exhibit remarkable catalytic activities and 100% H2 selectivity. At 90 °C, 139 mg of formic acid (3.0 mmol) can be completely converted to hydrogen (H2) and carbon dioxide (CO2), which were identified by mass spectrometry and gas chromatography, in 360 and 240 min in the presence of 60 mg of Au@SiO2_EN (2 wt% Au; prepared using TEOS and Au(en)2Cl3; Fig. 13b) and Au@SiO2_AP (2 wt% Au; prepared using TEOS/APTS and HAuCl4; Fig. 13c), respectively (Fig. 14).
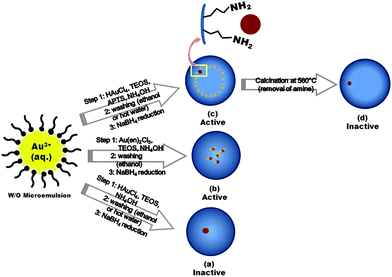 | ||
| Fig. 13 Microemulsion-based syntheses of gold nanocatalysts encapsulated within hollow silica nanospheres of (a) Au@SiO2, (b) Au@SiO2_EN, (c) Au@SiO2_AP, and (d) Au@SiO2_AP_C. Reprinted with permission from ref. 299. Copyright 2012 Royal Society of Chemistry. | ||
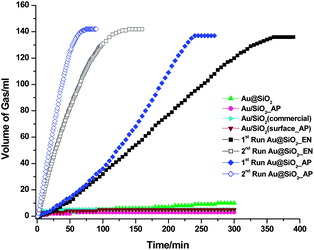 | ||
| Fig. 14 Time-course plots for hydrogen generation from the aqueous solution (1.0 mL) of formic acid (3.0 M) and sodium formate (1.0 M) in the presence of different Au NP catalysts (60 mg, 2 wt% Au) at 90 °C. Reprinted with permission from ref. 299. Copyright 2012 Royal Society of Chemistry. | ||
Remarkably, it is found that all the Au NPs supported on the outer surface of SiO2 are inactive no matter whether the outer surface is functionalized with amine, indicating that the encapsulation of Au NPs in the amine-functionalized silica nanosphere is important for obtaining a suitable environment around the Au NPs for effective catalysis of formic acid decomposition. It should be noted that the amine-functionalized Au@SiO2 nanosphere catalyst (Au@SiO2_AP) keeps its catalytic activity for the four runs, showing a high stability/durability that is important for practical application of catalysts.
Very recently, Cao and coworkers demonstrated the selective dehydrogenation of FA–amine mixtures using ultradispersed subnanometric (∼1.8 nm) gold as catalysts. The reaction catalyzed by Au subnanoclusters dispersed on acid-tolerant ZrO2, proceeds efficiently and selectively under ambient conditions, without the generation of any unwanted byproduct, such as CO, They achieved a TOF of 1590 h−1 and TON of more than 118400 at 50 °C.301
6.2. Synthesis of formic acid
Formic acid decomposition generates a major byproduct, carbon dioxide, that is a major green-house gas. For better hydrogen storage systems, efficient recycling of the byproduct has to be achieved. It has been proposed that the carbon dioxide can be reduced by molecular hydrogen to formic acid or methanol. Even if methanol has a higher storage capacity of two hydrogen molecules compared to one in formic acid, the use of carbon dioxide to synthesize formic acid should be preferred.302 The need for three equivalents of hydrogen to produce one equivalent of methanol results in a loss of one equivalent of hydrogen, since water is formed. In contrast, a transfer rate of 100% for formic acid formation is found. Therefore, the reversible storage of hydrogen in formic acid by reduction of carbon dioxide is an efficient process.291| H2 + CO2 → HCOOH ΔG = +33 kJ mol−1 | (31) |
The development of this reaction and the state-of-the art catalyst systems are well described in the literature.303–305 Therefore, we will discuss some selective catalysts.
6.2.1.1 Homogeneous catalytic formation of formic acid. Extensive studies have been undertaken to develop the homogenous catalysis of hydrogenation of carbon dioxide to yield formic acid. Numerous transition metal complexes based on e.g. Ru, Rh, Ir, Pd, Ni, Fe, Ti, and Mo have demonstrated excellent activities.303–312Table 9 summarizes some of the most active homogenous catalytic systems for hydrogenation of carbon dioxide. The activity of catalysts significantly depends on the pH of the reaction solution.311–315 An excellent activity is feasible in the presence of a base, because the abstraction of protons forces the reaction, which is in contrast to the thermodynamically unfavorable “base-free” reduction. In organic media, amines are preferred, whereas in water, NaOH or carbonates are used.
| Catalysts | TOF (h−1) | T (°C) | Ref. |
|---|---|---|---|
| a Abbreviation used: tppms = 3-sulfonatophenyldiphenylphosphine, tppts = trisodium-3,3′,3′′-phosphinidynetris(benzenesulfonate), hfacac = hexafluoroacetylacetonate, dcpb = 1,4-di(dicyclohexylphosphino) butane, PNP = PNP-type pincer ligand, dhbipy = 4,4′-dihydroxy-2,2′-bipyrdine, dhpt = 4,7-dihydroxy-1,10-phenantroline, thbpym = 4,4′,6,6′-tetrahydroxy-2,2′-bipyrimidine. | |||
| [RuCl(OAc)(PMe3)4] | 95000 |
50 | 306 |
| [RuCl2(tppms)2]2 | 9600 | 80 | 307 |
| [RhCl(tppts)3] | 7260 | 81 | 305 |
| [RuH2(PMe3)4] | 1400 | 50 | 308 |
| [Rh(hfacac)(dcpb)] | 1335 | 25 | 309 |
| [(PNP)IrH3] | 73000 |
120 | 310 |
| [Cp*Ir(dhbipy)Cl] | 42000 |
120 | 311 |
| [Cp*Ir(dhpt)Cl] | 33000 |
120 | 311 |
| [{Cp*Ir(Cl)}2(thbpym)] | 53800 |
80 | 312 |
Interestingly, the removal of the base is not essential, since the adduct of formic acid and base allows the straightforward decomposition to hydrogen. Besides this, the reaction outcome can be improved by applying supercritical conditions, due to the high availability of carbon dioxide, while in the solution approach the solubility of carbon dioxide is the limiting factor. One of the most active systems has been invented by Jessop and co-workers.306 A turnover frequency (TOF) of 95000 h−1 was achieved by [RuCl(OAc)(PMe3)4] under supercritical conditions (CO2-pressure: 190 bar, temperature: 50 °C). The group of Joó reported a [RuCl2(tppms)2]2 (tppms = meta-monosulfonated triphenylphosphine) catalyst with a TOF value of 9600 h−1,307 with which the reaction was carried out under non-supercritical conditions (CO2-pressure: 95 bar, temperature: 80 °C) in water. Leitner and co-workers reported that the complex Rh(hfacac)(dcpb) (hfacac = hexafluoroacetylacetonate; dcpb = 1,4-di(dicyclohexylphosphino)butane) demonstrated an outstanding activity of 1335 h−1 (TOF) at room temperature and a low CO2–H2 pressure (40 atm).309
An approach for reversible hydrogen storage using CO2 and H2 has been demonstrated by Hull, Himeda, Fujita and coworkers (Fig. 15).312 Herein, a water-soluble, pH-modulated catalyst drives the hydrogenation of CO2 to formate under basic conditions, and hydrogen release is easily triggered by acidifying the solution to protonate the catalyst. Progress has been reported in this regard by Himeda and coworker using [Cp*IrCl] complexes with 4,4′-dihydroxy-2,2′-bipyrdine (dhbipy) and 4,7-dihydroxy-1,10-phenanthroline (dhpt) ligands.311 They observed significantly higher activities of these complexes with TOFs of 42000, and 33000 h−1. It has been demonstrated that the hydroxy groups on the aromatic bipyridine and phenanthroline ligands have an enormous effect on the Lewis basicity of the ligand and, hence, on the metal center and its catalytic activity – when the ligand is protonated, the catalyst is inactive. The deprotonated hydroxypyridine ligand, for example, forms a pyridinolate, which is a much stronger σ-donor than the protonated pyridinol, which can be tuned by changing the pH value of the reaction solution.313 The pH-dependent activation and deactivation of the catalyst also has an effect on the reusability of the catalyst: in acidic media, the catalyst precipitates and can thus be separated from the reaction mixture. Very recently, Hull, Himeda, Fujita and coworkers developed a binuclear iridium catalyst containing [Cp*IrCl] and 4,4′,6,6′-tetrahydroxy-2,2′-bipyrimidine (thbpym) ligand, the first catalyst capable of reversible H2 storage using CO2 in aqueous media under mild temperatures and pressures.312 A recyclable pressurization sequence demonstrates that low-pressure (atmospheric) H2 gas can be stored as liquid formate, and then used to generate high-pressure H2 and CO2 for possible fuel applications. The –OH moieties on the thbpym ligand are pH-responsive, and H2 storage can be turned on or off by adjusting the pH of the solution. The rate of CO2 hydrogenation was as high as 70 h−1 (25 °C and 0.1 MPa) and 53800 h−1 (80 °C and 5 MPa). This catalyst was also very active for the decomposition of formic acid or formate to give CO-free H2 and CO2 at low pH with a TOF of 158000 at 80 °C and 228000 h−1 at 90 °C.
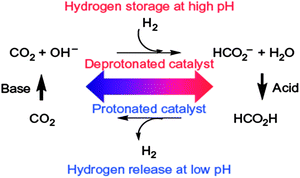 | ||
| Fig. 15 Reversible H2 storage is achieved by switching the pH to protonate or deprotonate the catalyst. Reprinted with permission from ref. 312. Copyright 2012 Nature Publishing Group. | ||
Very recently, Fukuzumi and cowrkers reported a organoiridium complex, [IrIII(Cp*)(4-(1H-pyrazol-1-yl-κN2)benzoic acid-κC3)(H2O)]2SO4, as an efficient catalyst for interconversion between H2 and HCOOH depending on pH. Hydrogenation of carbon dioxide by hydrogen occurs in the presence of a catalyst in weakly basic water (pH 7.5) under an atmospheric pressure of H2 and CO2 at room temperature, whereas formic acid efficiently decomposes to afford H2 and CO2 in the presence of the same catalyst in acidic water (pH 2.8).316
6.2.1.2 Heterogeneous catalytic formation of formic acid. There are only a few reports on the direct conversion of carbon dioxide to form formic acid with heterogeneous catalysts. Possible reasons are the unfavorable reaction conditions (high temperature, high pressure) compared to homogeneous catalysts and low chemoselectivity of heterogeneous processes, as formic acid can act as an intermediate to form methanol or methane.317–321 The first attempt for synthesis of formic acid over heterogeneous catalyst was reported as early as 1935.322 The reactions were carried out with RANEY® nickel as catalyst under 200–400 bar hydrogen pressure and 80–150 °C. However, one equivalent of amine had to be added in order to shift the thermodynamic equilibrium towards the formation of the product formic acid.
Recently, Fachinetti and coworkers developed a heterogeneous gold catalyst for synthesis of formic acid by hydrogenation of carbon dioxide.323 They found gold black catalyst promotes the CO2 hydrogenation in the presence of neat NEt3 to form HCOOH/NEt3 adducts. A continuous production process for catalyst- and solvent-free adducts has been demonstrated using a robust Au/TiO2 (AUROlite) catalyst and production of 1.326 kg (7.365 mol) HCOOH–NEt3 adduct with an acid/amine ratio of 1.7 has been achieved from CO2–H2 (1:1) and NEt3 in the presence of 13 g AUROlite (Au = 0.7 mmol) catalyst in a steel net cage in 37 days. The adducts have been split into pure HCOOH (anhydrous) and neat NEt3 with the help of high-boiling tri-n-hexylamine (n-C6H13)3N.
The reduction of carbonates to formic acid under mild reaction conditions was demonstrated by Wiener et al. applying Pd/C as catalyst.324 However, due to the chemical equilibrium between carbonate and formate the reaction could not be run to completeness. Further problems are the decrease of the solubility of carbonate with increasing formate concentration and the separation of the product from the reaction mixture.
Inspired by high activity of homogeneous catalysts, some immobilized catalysts have also been investigated. Ruthenium complexes immobilized over amine-functionalized silica have been developed with an in situ synthetic approach for CO2 hydrogenation to formic acid.325 The catalyst not only exhibits high activity (TOF = 1384 h−1) and 100% selectivity, but also offers practical advantages such as easy separation and recycling.
Ionic liquids have some unique properties, such as excellent thermal stability, wide liquid regions, and favorable solvation properties for various substances. Han and coworkers reported that the combination of a basic ionic liquid [mammim][OTf] (1-(N,N-dimethylaminoethyl)-2,3-dimethylimidazolium trifluoromethane-sulfonate) and a silica-supported ruthenium complex (“Si”–(CH2)3–NH(CSCH3)–RuCl3–PPh3) promoted CO2 hydrogenation to formic acid with satisfactory activity and selectivity.326 At a total pressure of 18 bar (H2:CO2 = 1:1) a maximum TOF of 103 h−1 was achieved. The activity has been proven to be constant over 5 cycles.
In addition to thermal catalytic reactions, electrochemical reduction of carbon dioxide on an amalgamated nickel cathode has been performed for production of formic acid by Williams and co-workers. While the maximal energetic efficiency is 60% and the concentration of formic acid is limited to 4 mol L−1, further improvement is expected.291
Willner and coworkers reported the enzymatic formic acid synthesis from HCO3− with FDH and photoreduction of various 4,4′- or 2,2′-bipyridinium salts by a system containing [Ru(bpy)3]2+ (bpy = 2,2′-bipyridine) as a photosensitizer and mercaptoethanol (RSH) as an electron donor (Fig. 16a).332–335 The efficiency of formic acid synthesis depends on the nature of the bipyridinium salts, and the quantum yield for formic acid from HCO3− is in the range of 0.5–1.6%.
![Photochemical and enzymatic CO2 conversion to formic acid (a) with a system consisting of an electron donor (RSH or TEOA), [Ru(bpy)3]2+, bipyridinium salt (EC) and FDH, and (b) with a system consisting of TEOA, ZnTMPyP4+, bipyridinium salt (MV2+), and FDH. Reprinted with permission from ref. 331. Copyright 2011 Wiley-VCH.](/image/article/2012/EE/c2ee22937d/c2ee22937d-f16.gif) | ||
| Fig. 16 Photochemical and enzymatic CO2 conversion to formic acid (a) with a system consisting of an electron donor (RSH or TEOA), [Ru(bpy)3]2+, bipyridinium salt (EC) and FDH, and (b) with a system consisting of TEOA, ZnTMPyP4+, bipyridinium salt (MV2+), and FDH. Reprinted with permission from ref. 331. Copyright 2011 Wiley-VCH. | ||
In a report from Kodaka et al., direct formic acid synthesis from CO2 gas with the system consisting of triethanolamine (TEOA), [Ru(bpy)3]2+, various bipyridinium salts (1,1′-dialkyl-4,4′-bipyridinium salts or 1,1′-dialkyl-2,2′-bipyridinium salts), and FDH has also been described (Fig. 16a).336 They investigated the effects of the structure and redox potentials of various bipyridinium salts on the synthesis of formic acid from CO2. After 7 h irradiation, the formation of formic acid in the concentration range of 0.2–0.95 mM was achieved from a sample solution containing 0.5 M TEOA, 0.5 mM [Ru(bpy)3]2+, 3.0 mM bipyridinium salt, and 8 mg FDH. The most effective formic acid synthesis from CO2 was observed by using N,N′-trimethylene-2,2′-bipyridinium salt and the yield depends on the relative magnitude of the redox potentials of the bipyridinium salts.
To develop a methodology for the conversion of CO2 to formic acid with FDH by visible light sensitization, the dye molecule should possess a high sensitization activity. In this regard, the synthesis of formic acid from HCO3− with FDH and the photoreduction of N,N′-dimethyl-4,4′-bipyridinium (MV2+) with systems using water soluble zinc porphyrins (zinc tetrakis(4-methylpyridyl)porphyrin, ZnTMPyP) have been introduced by Amao and Miyatani.337–339 ZnTMPyP4+ has been used as a photosensitizer, as shown in Fig. 16b. After 3 h irradiation, the formation of 60 μM of formic acid was achieved from the sample solution containing 0.3 M TEOA, 9.0 μM ZnTMPyP4+, 15 mM MV2+, 20 units FDH and 1.0 mM NaHCO3. The conversion yield of HCO3− to formic acid was estimated to be 6%. Later, they used Mg chlorophyll-a (MgChl-a) as visible light photosensitizer.340 The sample solution in this case consisted of NADPH, MgChl-a, MV2+, FDH, and CO2 gas. When the sample solution containing NADPH (3.0 mM), MgChl-a (9.0 μM), MV2+ (0.1 mM), FDH (5 units), and CO2 gas (saturated) was irradiated at 30 °C, 56 μm of formic acid was produced after 4 h of irradiation. Further, it has been observed that formic acid was not produced in the absence of any one of the components.
Sato and coworkers reported immobilization of a molecular CO2 reduction catalyst onto a semiconductor surface for engineering hybrid photocatalytic materials.341–345 The ruthenium catalysts were grafted onto different p-type semiconductors such as N-doped Ta2O5, Z-doped InP, etc. via covalent linkages or via a polymerization process. It was reported that the robustness of a hybrid system depends strongly on the choice of grafting linkages. The [Ru(dpbpy)(bpy)(CO)2]/N–Ta2O5 (dpbpy = 4,4′-diphosphonate-2,2′-bipyridine, bpy = 2,2′-bipyridine) system with phosphate linker displayed a 5 times higher rate of CO2 reduction to HCOOH (in acetonitrile solution using visible light) compared with the [Ru(dcbpy)(bpy)(CO)2]/N–Ta2O5 (dcbpy: 4,4′-dicarboxy-2,2′-bipyridine, bpy = 2,2′-bipyridine) system with carbonate linker (Fig. 17).341,345 By using a Deronzier's-type ruthenium bipyridine-based polymeric film catalyst, a robust system adapted for working in aqueous solution was achieved.344
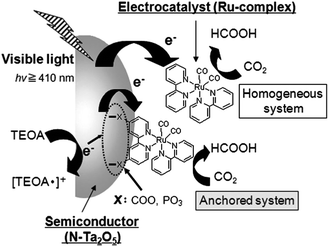 | ||
| Fig. 17 Mechanism for the reduction of CO2 by photocatalysis under visible-light with an Ru-complex and an N–Ta2O5 hybrid catalyst. Reprinted with permission from ref. 345. Copyright 2011 Royal Society of Chemistry. | ||
Recently Sato, Arai and coworkers reported that the photoelectrochemical reduction of CO2 to HCOO− (formate) over a p-type InP/Ru complex polymer hybrid photocatalyst was highly enhanced by introducing an anchoring complex into the polymer (Fig. 18).343 By functionally combining the hybrid photocatalyst with TiO2 for water oxidation, selective photoreduction of CO2 to HCOO− was achieved in aqueous media, in which H2O was used as both an electron donor and a proton source. The so-called Z-scheme (or two-step photoexcitation) system operated with no external electrical bias. The selectivity for HCOO− production was >70%, and the conversion efficiency of solar energy to chemical energy was 0.03–0.04%.
 | ||
| Fig. 18 Total reaction of the Z-scheme system for CO2 reduction. Reprinted with permission from ref. 343. Copyright 2011 American Chemical Society. | ||
6.3 Conclusions
Formic acid, having 4.4 wt% hydrogen content, represents a convenient hydrogen carrier in fuel cells designed for portable use. A power supply system could be feasibly based on the catalytic processes of formation and decomposition of formic acid. The present scenario of catalytic decomposition and formation of formic acid shows the presence of many effective homogeneous and heterogeneous catalysts. To meet practical applications, further work is needed for the development of low-cost, more efficient catalysts, especially heterogeneous catalysts. Moreover, development of photocatalytic systems for both the processes (fixation and the evolution of hydrogen to/from formic acid), which employs sustainable/renewable energy resources, is highly desirable.7. Other liquid organic hydogen storage materials
Hydrogen storage in liquid organic hydrogen carriers (LOHCs) has attracted much attention in the past few decades because of its simple, safe, and feasible handling of hydrogen.346–360 The concept of hydrogen storage in LOHCs is based on reversible catalytic hydrogenation–dehydrogenation reactions. First, the hydrogen is chemically stored in an organic carrier through a catalytic hydrogenation reaction. When the demand for energy exists, the hydrogen is extracted from the organic carrier by a catalytic dehydrogenation reaction and fed into fuel cells to generate electricity. The practical barrier for LOHCs is appreciably lower than for other storage media because they are chemically similar to the current distribution medium, gasoline. The tanks, piping and refinery systems used to make and deliver gasoline are appropriate for LOHCs. Therefore, a number of research efforts have been resulted in the development of various liquid organic carriers for hydrogen storage.346–360 Initial efforts to develop liquid organic hydrogen carriers were primarily focused on cycloalkanes.346–351 The dehydrogenation of these cheap, abundant materials to the corresponding aromatic compounds releases approximately 7 wt% hydrogen. However, the large enthalpy of dehydrogenation of cycloalkanes (∼60 kJ mol−1 H2) is a major drawback to their utilization in practical systems. Pez et al. calculated the enthalpy of formation of hydrogenation for a variety of aromatic heterocycles and their saturated analogs.352,353 Their calculations show that the ΔH of dehydrogenation is significantly lowered upon introduction of a hetero-atom into the ring system as it significantly reduces the aromaticity of the dehydrogenated molecule. This led several groups to explore the reversible dehydrogenation of heterocyclic aromatic compounds as N-ethyl perhydrocarbazole to ethylcarbazole.352–360 Although promising hydrogen cycling performances have been demonstrated, these investigators utilized very high loadings of heterogeneous, precious metal catalysts in order to achieve acceptable dehydrogenation kinetics at temperatures (≤150 °C) near the operating temperatures of PEM fuel cells. The high cost of the massive quantities of precious metals precludes the commercialization of these systems.8. Conclusions and perspective
Safe, efficient storage and delivery of hydrogen is essential for the development of a hydrogen-based energy infrastructure. All the liquid-phase chemical hydrogen storage materials reviewed above have relatively high hydrogen content and have the potential to be used as hydrogen sources suitable for portable fuel cells. However, each of them has its own merits and drawbacks. The materials and systems presented herein can effectively work under mild conditions (even at room temperature) with suitable catalysts compared to the hydrogen generation from organic compounds (for example, various cyclohexane derivatives and alcohols as feedstock) and thermolysis systems of solid-state chemical hydrogen storage materials, which usually need high temperature reforming or decomposition processes. Easy recharging using liquid filling pumps and the availability of the current infrastructure of liquid fuels for recharging as well as the production of only nitrogen/carbon dioxide, which does not need on-board collection for recycling, besides hydrogen, of hydrous hydrazine and formic acid as hydrogen storage materials, are extraordinary advantages over solid-state hydrogen storage materials. Furthermore, the development of chemical hydrogen storage materials containing abundant elements (C, H and O for formic acid, and H and N for hydrazine) is encouraging in comparison to much less abundant boron-containing hydrogen storage materials. From these points of view, hydrous hydrazine and formic acid, which contain only abundant elements, do not need on-board collection of spent fuel for recycling, and can be easily recharged, are suitable for large-scale automotive application, while boron-containing materials, which contain less abundant elements and need for on-site collection of spent fuel for recycling, are suitable for small-scale, mobile single-use devices where reusability is not important. Recent research efforts have greatly improved the hydrogen generation temperature and the reaction kinetics in these systems and, especially, intensive efforts have been made to develop low-cost non-noble metal catalysts, which is important for implementation of hydrogen storage as a global energy solution. However, for practical applications in mobile electric devices, limitations are still pending, such as, cost, catalyst deactivation, regeneration of byproducts, and control of the reaction kinetics. Further developments of these systems will certainly contribute to the establishment of future sustainable energy infrastuctures.We have tried to present an up-to-date overview in such a rapidly growing field, while the subject is very active and many papers are contributed each year (even during the writing of this article) from chemists, physical and materials scientists, etc. Therefore, it is hard to take into account all publications to this field in our limited number of pages. We apologize here if some significant contributions were left out.
Acknowledgements
The authors gratefully acknowledge the reviewers for valuable comments and constructive suggestions. The authors are pleased to acknowledge the fine work of the talented and dedicated graduate students, postdoctoral fellows, and colleagues who have worked with us in this area and whose names can be found in the references. The authors would like to thank National Institute of Advanced Industrial Science and Technology (AIST) and Japan Society for the Promotion of Science (JSPS) for financial support. M. Y. thanks JSPS for postdoctoral fellowship.References
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 668–681 RSC.
- Hydrogen as a Future Energy Carrier, ed. A. Züttel, A. Borgschulte and L. Schlapbach, Wiley-VCH, Weinheim, 2008 Search PubMed.
- L. Schlapbach and A. Zuttel, Nature, 2001, 414, 353–358 CrossRef.
- Fuel Cell Technologies Program, http://www1.eere.energy.gov/hydrogenandfuelcells/mypp/.
- G. Ferey, Nature, 2005, 436, 187–188 CrossRef CAS.
- S. Orimo, Y. Nakamori, J. R. Eliseo, A. Züttel and C. M. Jensen, Chem. Rev., 2007, 107, 4111–4132 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS.
- A. M. Seayad and D. M. Antonelli, Adv. Mater., 2004, 16, 765–777 CrossRef CAS.
- G. A. Deluga, J. R. Salge, L. D. Schmidt and X. E. Verykios, Science, 2004, 303, 993–997 CrossRef CAS.
- P. Makowski, A. Thomas, P. Kuhn and F. Goettmann, Energy Environ. Sci., 2009, 2, 480–490 RSC.
- Y. G. Wang, N. Shah and G. P. Huffman, Catal. Today, 2005, 99, 359–364 Search PubMed.
- U. Eberle, M. Felderhoff and F. Schüth, Angew. Chem., Int. Ed., 2009, 48, 6608–6630 CrossRef CAS.
- P. Chen, Z. T. Xiong, J. Z. Luo, J. Y. Lin and K. L. Tan, Nature, 2002, 420, 302–304 CrossRef CAS.
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 253–254, 1–9 CrossRef CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283–1316 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 156, 190–194 CrossRef CAS.
- Q. Xu and M. Chandra, J. Power Sources, 2006, 163, 364–370 CrossRef CAS.
- H. L. Jiang, S. K. Singh, J. M. Yan, X. B. Zhang and Q. Xu, ChemSusChem, 2010, 3, 541–549 CrossRef CAS.
- P. Wang and X. D. Kang, Dalton Trans., 2008, 5400–5413 RSC.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 2303–2311 CrossRef CAS.
- F. H. Stephens, V. Pons and R. T. Baker, Dalton Trans., 2007, 2613–2626 RSC.
- C. W. Hamilton, R. T. Baker, A. Staubitz and I. Manners, Chem. Soc. Rev., 2009, 38, 279–293 RSC.
- H. W. Langmi and G. S. McGrady, Coord. Chem. Rev., 2007, 251, 925–935 CrossRef CAS.
- J. Hannauer, O. Akdim, U. B. Demirci, C. Geantet, J. M. Herrmann, P. Miele and Q. Xu, Energy Environ. Sci., 2011, 4, 3355–3358 RSC.
- U. B. Demirci and P. Miele, Energy Environ. Sci., 2011, 4, 3334–3341 RSC.
- S. K. Singh, X. B. Zhang and Q. Xu, J. Am. Chem. Soc., 2009, 131, 9894–9895 CrossRef CAS.
- S. K. Singh and Q. Xu, J. Am. Chem. Soc., 2009, 131, 18032–18033 CrossRef CAS.
- F. Joó, ChemSusChem, 2008, 1, 805–808 CrossRef CAS.
- (a) B. Loges, A. Boddien, F. Gartner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS; (b) M. Grasemann and G. Laurenczy, Energy Environ. Sci., 2012, 5, 8171–8181 RSC; (c) A. Boddien, C. Federsel, P. Sponholz, D. Mellmann, R. Jackstell, H. Junge, G. Laurenczy and M. Beller, Energy Environ. Sci., 2012, 5, 8907–8911 RSC.
- C. Fellay, P. J. Dyson and G. Laurenczy, Angew. Chem., Int. Ed., 2008, 47, 3966–3968 CrossRef CAS.
- E. Y. Marrero-Alfonso, A. M. Beaird, T. A. Davis and M. A. Matthews, Ind. Eng. Chem. Res., 2009, 48, 3703–3712 Search PubMed.
- B. H. Liu and Z. P. Li, J. Power Sources, 2009, 187, 527–534 CrossRef CAS.
- H. I. Schlesinger, H. C. Brown, A. B. Finholt, J. R. Gilbreath, H. R. Hockstra and E. K. Hydo, J. Am. Chem. Soc., 1953, 75, 215–219 CrossRef CAS.
- U. B. Demirci and P. Miele, C. R. Chim., 2009, 12, 943–950 CrossRef CAS.
- Y. Nakamori, H. W. Li, M. Matsuo, K. Miwa, S. Towata and S. Orimo, J. Phys. Chem. Solids, 2008, 69, 2292–2296 CrossRef CAS.
- S. C. Amendola, S. L. Sharp-Goldman, M. S. Janjua, N. C. Spencer, M. T. Kelly, P. J. Petillo and M. Binder, Int. J. Hydrogen Energy, 2000, 25, 969–975 CrossRef CAS.
- S. C. Amendola, S. L. Sharp-Goldman, M. S. Janjua, M. T. Kelly, P. J. Petillo and M. Binder, J. Power Sources, 2000, 85, 186–189 CrossRef CAS.
- Y. Shang and R. Chen, Energy Fuels, 2006, 20, 2142–2148 CrossRef CAS.
- J. H. Wee, K. Y. Lee and S. H. Kim, Fuel Process. Technol., 2006, 87, 811–819 CrossRef CAS.
- E. Fakioglu, Y. Yurum and N. T. Veziroglu, Int. J. Hydrogen Energy, 2004, 29, 1371–1376 CrossRef CAS.
- H. C. Brown and C. A. Brown, J. Am. Chem. Soc., 1962, 84, 1493–1494 CrossRef CAS.
- S. Suda, Y. M. Sun, B. H. Liu, Y. Zhou, S. Morimitsu, K. Arai, N. Tsukamoto, M. Uchida, Y. Candra and Z. P. Li, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 209–212 CAS.
- Y. Kojima, K. Suzuki, K. Fukumoto, M. Sasaki, T. Yamamoto and Y. Kawai, Int. J. Hydrogen Energy, 2002, 27, 1029–1034 CrossRef CAS.
- P. P. Prosini and P. Gislon, J. Power Sources, 2006, 161, 290–293 Search PubMed.
- S. Murugesan and V. R. Subramanian, J. Power Sources, 2009, 187, 216–223 Search PubMed.
- E. Keçeli and S. Özkar, J. Mol. Catal. A: Chem., 2008, 286, 87–91 Search PubMed.
- C. M. Kaufman and B. Sen, J. Chem. Soc., Dalton Trans., 1985, 307–313 RSC.
- A. Levy, J. B. Brown and C. J. Lyons, Ind. Eng. Chem., 1960, 52, 211–214 CrossRef CAS.
- K. A. Holbrook and P. J. Twist, J. Chem. Soc. A, 1971, 890–894 RSC.
- R. Paul, P. Buisson and N. Joseph, Ind. Eng. Chem., 1952, 44, 1006–1010 CrossRef CAS.
- G. Guella, B. Patton and A. Miotello, J. Phys. Chem. C, 2007, 111, 18744–18750 CrossRef CAS.
- U. B. Demirci and F. Garin, J. Alloys Compd., 2008, 463, 107–111 CrossRef CAS.
- T. F. Hung, H. C. Kuo, C. W. Tsai, H. M. Chen, R. S. Liu, B. J. Weng and J. F. Lee, J. Mater. Chem., 2011, 21, 11754–11759 RSC.
- U. B. Demirci, O. Akdim, J. Andrieux, J. Hannauer, R. Chamoun and P. Miele, Fuel Cells, 2010, 10, 335–350 CrossRef CAS.
- C. W. Tsai, H. M. Chen, R. S. Liu, J. F. Lee, S. M. Chang and B. J. Weng, Int. J. Hydrogen Energy, 2012, 37, 3338–3343 CrossRef CAS.
- M. Zahmakiran and S. Özkar, J. Mol. Catal. A: Chem., 2006, 258, 95–103 CrossRef CAS.
- V. I. Simagina, P. A. Storozhenko, O. V. Netskina, O. V. Komova, G. V. Odegova, T. Yu. Samoilenko and A. G. Gentsler, Kinet. Catal., 2007, 48, 168–175 CrossRef CAS.
- U. B. Demirci and F. Garin, Catal. Commun., 2008, 9, 1167–1172 Search PubMed.
- B. H. Liu, Z. P. Li and S. Suda, J. Alloys Compd., 2006, 415, 288–293 CrossRef CAS.
- S. U. Jeong, R. K. Kim, E. A. Cho, H. J. Kim, S. W. Nam, I. H. Oh, S. A. Hong and S. H. Kim, J. Power Sources, 2005, 144, 129–134 CrossRef CAS.
- Ö. Metin and S. Özkar, Int. J. Hydrogen Energy, 2007, 32, 1707–1715 CrossRef CAS.
- J. H. Park, P. Shakkthivel, H. J. Kim, M. K. Han, J. H. Jang, Y. R. Kim, H. S. Kim and Y. G. Shu, Int. J. Hydrogen Energy, 2008, 33, 1845–1852 CrossRef CAS.
- P. Krishnan, T. H. Yang, W. Y. Lee and C. S. Kim, J. Power Sources, 2005, 143, 17–23 CrossRef.
- P. Krishnan, K. L. Hsueh and S. D. Yim, Appl. Catal., B, 2007, 77, 206–214 CrossRef CAS.
- M. Zahmakiran and S. Özkar, Langmuir, 2009, 25, 2667–2678 CrossRef CAS.
- C. Wu, F. Wu, Y. Bai, B. Yi and H. Zhang, Mater. Lett., 2005, 59, 1748–1751 CrossRef CAS.
- J. C. Walter, A. Zurawski, D. Montgomery, M. Thornburg and S. Revankar, J. Power Sources, 2008, 179, 335–339 CrossRef CAS.
- N. Patel, G. Guella, A. Kale, A. Miotello, B. Patton, C. Zanchetta, L. Mirenghi and P. Rotolo, Appl. Catal., A, 2007, 323, 18–24 CrossRef CAS.
- N. Patel, R. Fernandes, G. Guella, A. Kale, A. Miotello, B. Patton and C. Zanchetta, J. Phys. Chem. C, 2008, 112, 6968–6976 CrossRef CAS.
- D. Hua, Y. Hanxi, A. Xinping and C. Chuansin, Int. J. Hydrogen Energy, 2003, 28, 1095–1100 CrossRef CAS.
- O. Akdim, U. B. Demirci, D. Muller and P. Miele, Int. J. Hydrogen Energy, 2009, 34, 2631–2637 CrossRef CAS.
- S. U. Jeong, E. A. Cho, S. W. Nam, I. H. Oh, U. H. Jung and S. H. Kim, Int. J. Hydrogen Energy, 2007, 32, 1749–1754 CrossRef CAS.
- X. Yuan, C. Jia, X. L. Ding and Z. F. Ma, Int. J. Hydrogen Energy, 2012, 37, 995–1001 Search PubMed.
- D. Xu, P. Dai, X. Liu, C. Cao and Q. Guo, J. Power Sources, 2008, 182, 616–620 CrossRef CAS.
- H. B. Dai, Y. Liang, P. Wang and H. M. Cheng, J. Power Sources, 2008, 177, 17–23 CrossRef CAS.
- H. B. Dai, Y. Liang, P. Wang, X. D. Yao, T. Rufford, M. Lu and H. M. Cheng, Int. J. Hydrogen Energy, 2008, 33, 4405–4412 Search PubMed.
- K. W. Cho and H. S. Kwon, Catal. Today, 2007, 120, 298–304 CrossRef CAS.
- J. H. Park, P. Shakkthivel, H. J. Kim, M. K. Han, J. H. Jang, Y. R. Kim, H. S. Kim and Y. G. Shu, Int. J. Hydrogen Energy, 2008, 33, 1845–1852 CrossRef CAS.
- C. Wu, H. Zhang and B. Yi, Catal. Today, 2004, 93–95, 477–483 CrossRef CAS.
- W. Ye, H. Zhang, D. Xu, L. Ma and B. Yi, J. Power Sources, 2007, 164, 544–548 CrossRef CAS.
- A. J. Hung, S. F. Tsai, Y. Y. Hsu, J. R. Ku, Y. H. Chen and C. C. Yu, Int. J. Hydrogen Energy, 2008, 33, 6205–6215 CrossRef CAS.
- C. T. F. Lo, K. Karan and B. R. Davis, Ind. Eng. Chem. Res., 2007, 46, 5478–5484 CrossRef CAS.
- H. C. Brown, E. J. Mead and B. C. S. Rao, J. Am. Chem. Soc., 1955, 77, 6209–6213 CrossRef CAS.
- R. E. Davis and J. A. Gottbrath, J. Am. Chem. Soc., 1962, 84, 895–898 CrossRef CAS.
- J. Hannauer, U. B. Demirci, G. Pastor, C. Geantet, J. M. Herrmann and P. Miele, Energy Environ. Sci., 2010, 3, 1796–1803 RSC.
- J. Andrieux, U. B. Demirci, J. Hannauer, C. Gervais, C. Goutaudier and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 224–233 CrossRef CAS.
- J. Hannauer, U. B. Demirci, C. Geantet, J. M. Herrmann and P. Miele, Phys. Chem. Chem. Phys., 2011, 13, 3809–3818 RSC.
- E. Y. Marrero-Alfonso, J. R. Gray, T. A. Davis and M. A. Matthews, Int. J. Hydrogen Energy, 2007, 32, 4723–4730 CrossRef CAS.
- B. H. Liu, Z. P. Li and S. Suda, J. Alloys Compd., 2009, 468, 493–498 CrossRef CAS.
- US Department of Energy, Office of Energy Efficiency and Renewable Energy and the Freedom CAR and Fuel Partnership, “Targets for Onboard Hydrogen Storage Systems for Light-Duty Vehicles”, September 2009, http://www1.eere.energy.gov Search PubMed.
- J. Yang, A. Sudik, C. Wolverton and D. J. Siegel, Chem. Soc. Rev., 2010, 39, 656–675 RSC.
- H. K. Atiyeh and B. R. Davis, Int. J. Hydrogen Energy, 2007, 32, 229–236 Search PubMed.
- Ç. Çakanyildirim and M. Gürü, Int. J. Hydrogen Energy, 2008, 33, 4634–4639 CrossRef CAS.
- Y. Wu, M. T. Kelly and J. V. Ortega, Review of Chemical Processes for the Synthesis of Sodium Borohydride, 2007, http://205.254.148.40/hydrogenandfuelcells/pdfs/review_chemical_processes.pdf Search PubMed.
- C. H. Liu, B. H. Chen, D. J. Lee, J. R. Ku and F. Tsau, Ind. Eng. Chem. Res., 2010, 49, 9864–9869 Search PubMed.
- G. Broja and W. Schlabacher, DE Pat., 1108670, 1959.
- T. Haga and Y. Kojima, JP Pat., 2002–241109, 2002.
- Y. Kojima and T. Haga, Int. J. Hydrogen Energy, 2003, 28, 989–993 CrossRef CAS.
- Z. P. Li, B. H. Liu, N. Morigasaki and S. Suda, J. Alloys Compd., 2003, 354, 243–247 CrossRef CAS.
- Z. P. Li, N. Morigazaki, B. H. Liu and S. Suda, J. Alloys Compd., 2003, 349, 232–236 CrossRef CAS.
- A. Roine, HSC Chemistry: V5.0, Outokompu Research Oy, Pori, 2004 Search PubMed.
- S. C. Amendola and M. T. Kelly, US Pat., 6433129, 2002.
- S. C. Amendola, M. T. Kelly, J. V. Ortega and Y. Wu, US Pat., 6670444, 2003.
- S. C. Amendola, M. T. Kelly and Y. Wu, US Pat., 6524542, 2003.
- J. V. Ortega, Y. Wu, S. C. Amendola and M. T. Kelly, US Pat., 6586563, 2003.
- Production of the Boranes and Related Research, ed. R. T. Holzmann, Academic Press, New York, 1967 Search PubMed.
- O. Glemser, DE Pat., 949943, 1956.
- H. B. H. Cooper, US Pat., 3734842, 1973.
- D. M. F. Santos and C. A. C. Sequeira, Int. J. Hydrogen Energy, 2010, 35, 9851–9861 Search PubMed.
- D. L. Calabretta and B. R. Davis, J. Power Sources, 2007, 164, 782–791 CrossRef CAS.
- Q. Xu and M. Chandra, J. Alloys Compd., 2007, 446–447, 729–732 CrossRef CAS.
- A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS.
- Z. Li, G. Zhu, G. Lu, S. Qiu and X. Yao, J. Am. Chem. Soc., 2010, 132, 1490–1491 CrossRef CAS.
- F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746–749 CrossRef CAS.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS.
- U. B. Demirci and P. Miele, Energy Environ. Sci., 2009, 2, 627–637 RSC.
- http://www.eere.energy.gov/hydrogenandfuelcells/pdfs/freedomcar_targets_explanations.pdf .
- R. Custelcean and Z. A. Dreger, J. Phys. Chem. B, 2003, 107, 9231–9235 CrossRef CAS.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8–12 CrossRef CAS.
- A. T. Raissi, Hydrogen From Ammonia and Ammonia–Borane Complex for Fuel Cell Applications, Proceedings of the 2002 US DOE Hydrogen Program Review, 2002, http://www1.eere.energy.gov/hydrogenandfuelcells/pdfs/32405b15.pdf Search PubMed.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1955, 77, 6084–6085 CrossRef CAS.
- M. G. Hu, J. M. Van Paasschen and R. A. Geanangel, J. Inorg. Nucl. Chem., 1977, 39, 2147–2150 CrossRef CAS.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 46, 7810–7817 CrossRef CAS.
- E. Mayer, Inorg. Chem., 1972, 11, 866–869 CrossRef CAS.
- S. G. Shore and K. W. Boeddeker, Inorg. Chem., 1964, 3, 914–915 CrossRef CAS.
- R. A. Adams, J. Beres, A. Dodds and A. J. Morabito, Inorg. Chem., 1971, 10, 2072–2074 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. Butterick III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748–7749 CrossRef CAS.
- L. Li, X. Yao, C. H. Sun, A. J. Du, L. N. Cheng, Z. H. Zhu, C. Z. Yu, J. Zou, S. C. Smith, P. Wang, H. M. Cheng, R. L. Frost and G. Q. M. Lu, Adv. Funct. Mater., 2009, 19, 265–271 CrossRef.
- Z. T. Xiong, C. K. Yong, G. T. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2007, 7, 138–141.
- H. V. K. Diyabalanage, R. P. Shrestha, T. A. Semelsberger, B. L. Scott, M. E. Bowden, B. L. Davis and A. K. Burrell, Angew. Chem., Int. Ed., 2007, 46, 8995–8997 CrossRef.
- V. Sit, R. A. Geanangel and W. W. Wendlandt, Thermochim. Acta, 1987, 113, 379–382 CrossRef CAS.
- P. E. de Jongh and P. Adelhelm, ChemSusChem, 2010, 3, 1332–1348 CrossRef CAS.
- C. A. Jaska, K. Temple, A. J. Lough and I. Manners, J. Am. Chem. Soc., 2003, 125, 9424–9434 CrossRef CAS.
- S. K. Kim, W. S. Han, T. J. Kim, T. Y. Kim, S. W. Nam, M. Mitoraj, L. Piekoś, A. Michalak, S. J. Hwang and S. O. Kang, J. Am. Chem. Soc., 2010, 132, 9954–9955 CrossRef.
- M. Chandra and Q. Xu, J. Power Sources, 2006, 159, 855–860 CrossRef CAS.
- T. J. Clark, G. R. Whittell and I. Manners, Inorg. Chem., 2007, 46, 7522–7527 CrossRef CAS.
- S. B. Kalidindi, M. Indirani and B. R. Jagirdar, Inorg. Chem., 2008, 47, 7424–7429 CrossRef CAS.
- F. Durap, M. Zahmakıran and S. Özkar, Appl. Catal., A, 2009, 369, 53–59 CrossRef CAS.
- S. B. Kalidindi, A. A. Vernekar and B. R. Jagirdar, Phys. Chem. Chem. Phys., 2009, 11, 770–775 RSC.
- M. Couturier, J. L. Tucker, B. M. Andresen, P. Dubé and J. T. Negri, Org. Lett., 2001, 3, 465–467 CrossRef CAS.
- P. A. Storozhenko, R. A. Svitsyn, V. A. Ketsko, A. K. Buryak and A. V. Ul'yanov, Russ. J. Inorg. Chem., 2005, 50, 980–985.
- J. S. Wang and R. A. Geanangel, Inorg. Chim. Acta, 1988, 148, 185–190 CrossRef CAS.
- M. Chandra and Q. Xu, J. Power Sources, 2007, 168, 135–142 CrossRef CAS.
- F. Durap, M. Zahmakıran and S. Özkar, Int. J. Hydrogen Energy, 2009, 34, 7223–7230 CrossRef CAS.
- O. Metin, S. Sxahin and S. Özkar, Int. J. Hydrogen Energy, 2009, 34, 6304–6313 CrossRef CAS.
- M. Zahmakıran and S. Özkar, Appl. Catal., B, 2009, 89, 104–110 CrossRef.
- M. Rakap and S. Özkar, Int. J. Hydrogen Energy, 2010, 35, 1305–1312 Search PubMed.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, Angew. Chem., Int. Ed., 2008, 47, 2287–2289 CrossRef CAS.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, Inorg. Chem., 2009, 48, 7389–7393 CrossRef CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, Int. J. Hydrogen Energy, 2009, 34, 3816–3822 CrossRef CAS.
- J. M. Yan, X. B. Zhang, S. Han, H. Shioyama and Q. Xu, J. Power Sources, 2009, 194, 478–481 CrossRef CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, J. Power Sources, 2009, 191, 209–216 CrossRef CAS.
- Y. Li, L. Xie, Y. Li, J. Zheng and X. Li, Chem.–Eur. J., 2009, 15, 8951–8954 CrossRef CAS.
- J. M. Yan, X. B. Zhang, H. Shioyama and Q. Xu, J. Power Sources, 2010, 195, 1091–1094 CrossRef CAS.
- T. Umegaki, J. M. Yan, X. B. Zhang, H. Shioyama, N. Kuriyama and Q. Xu, J. Power Sources, 2010, 195, 8209–8214 CrossRef CAS.
- Ö. Metin, V. Mazumder, S. Özkar and S. Sun, J. Am. Chem. Soc., 2010, 132, 1468–1469 CrossRef CAS.
- C. Y. Cao, C. Q. Chen, W. Li, W. G. Song and W. Cai, ChemSusChem, 2010, 3, 1241–1244 Search PubMed.
- P. Song, Y. Li, W. Li, B. He, J. Yang and X. Li, Int. J. Hydrogen Energy, 2011, 36, 10468–10473 Search PubMed.
- P. Z. Li, K. Aranishi and Q. Xu, Chem. Commun., 2012, 48, 3173–3175 RSC.
- M. Zahmakıran, F. Durap and S. Özkar, Int. J. Hydrogen Energy, 2010, 35, 187–197 CrossRef CAS.
- R. Yi, R. Shi, G. Gao, N. Zhang, X. Cui, Y. He and X. Liu, J. Phys. Chem. C, 2009, 113, 1222–1226 CrossRef CAS.
- H. L. Jiang, T. Umegaki, T. Akita, X. B. Zhang, M. Haruta and Q. Xu, Chem.–Eur. J., 2010, 16, 3132–3137 CrossRef CAS.
- Z. H. Lu, H. L. Jiang, M. Yadav, K. Aranishia and Q. Xu, J. Mater. Chem., 2012, 22, 5065–5071 RSC.
- J. M. Yan, X. B. Zhang, T. Akita, M. Haruta and Q. Xu, J. Am. Chem. Soc., 2010, 132, 5326–5327 CrossRef CAS.
- K. Aranishi, H. L. Jiang, T. Akita, M. Haruta and Q. Xu, Nano Res., 2011, 4, 1233–1241 Search PubMed.
- H. L. Jiang, T. Akita and Q. Xu, Chem. Commun., 2011, 47, 10999–11001 RSC.
- Y. Yamada, K. Yano, Q. Xu and S. Fukuzumi, J. Phys. Chem. C, 2010, 114, 16456–16462 CrossRef CAS.
- Y. Yamada, K. Yano and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 5356–5363 RSC.
- H. Erdoğan, Ö. Metin and S. Özkar, Phys. Chem. Chem. Phys., 2009, 11, 10519–10525 RSC.
- S. Çalışkan, M. Zahmakıran and S. Özkar, Appl. Catal., B, 2010, 93, 387–394 CrossRef.
- M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048–12049 CrossRef CAS.
- R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844–1845 CrossRef CAS.
- R. P. Shrestha, H. V. K. Diyabalanage, T. A. Semelsberger, K. C. Ott and A. K. Burrell, Int. J. Hydrogen Energy, 2009, 34, 2616–2621 CrossRef CAS.
- S. Kim, T. Kim, T. Kim, G. Lee, J. T. Park, S. W. Nam and S. O. Kang, Chem. Commun., 2012, 48, 2021–2023 RSC.
- W. R. H. Wright, E. R. Berkeley, L. R. Alden, R. T. Baker and L. G. Sneddon, Chem. Commun., 2011, 47, 3177–3179 RSC.
- M. Diwan, V. Diakov, E. Shafirovich and A. Varma, Int. J. Hydrogen Energy, 2008, 33, 1135–1141 CrossRef CAS.
- M. Diwan, D. Hanna and A. Varma, Int. J. Hydrogen Energy, 2010, 35, 577–584 Search PubMed.
- S. Basu, M. Diwan, M. G. Abiad, Y. Zheng, O. H. Campanella and A. Varma, Int. J. Hydrogen Energy, 2010, 35, 2063–2072 Search PubMed.
- W. Luo, P. G. Campbell, L. N. Zakharov and S. Y. Liu, J. Am. Chem. Soc., 2011, 133, 19326–19329 CrossRef CAS.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8–12 CrossRef CAS.
- S. Hausdorf, F. Baitalow, G. Wolf and F. O. R. L. Mertens, Int. J. Hydrogen Energy, 2008, 33, 608–614 CrossRef CAS.
- B. L. Davis, D. A. Dixon, E. B. Garner, J. C. Gordon, M. H. Matus, B. Scott and F. H. Stephens, Angew. Chem., Int. Ed., 2009, 48, 6812–6816 CrossRef CAS.
- A. D. Sutton, B. L. Davis, K. X. Bhattacharyya, B. D. Ellis, J. C. Gordon and P. P. Power, Chem. Commun., 2010, 46, 148–149 RSC.
- A. D. Sutton, A. K. Burrell, D. A. Dixon, E. B. Garner III, J. C. Gordon, T. Nakagawa, K. C. Ott, J. P. Robinson and M. Vasiliu, Science, 2011, 331, 1426–1429 CrossRef CAS.
- N. Mohajeri, A. T. Raissi and O. Adebiyi, J. Power Sources, 2007, 167, 482–485 CrossRef CAS.
- G. P. Rachiero, U. B. Demirci and P. Miele, Catal. Today, 2011, 170, 85–92 Search PubMed.
- G. P. Rachiero, U. B. Demirci and P. Miele, Int. J. Hydrogen Energy, 2011, 36, 7051–7065 CrossRef CAS.
- C. G. Salentine, Inorg. Chem., 1983, 22, 3920–3924 CrossRef.
- U. Sanyal, U. B. Demirci, B. R. Jagirdar and P. Miele, ChemSusChem, 2011, 4, 1731–1739 Search PubMed.
- C. H. Liu, Y. C. Wu, C. C. Chou, B. H. Chen, C. L. Hsueh, J. R. Ku and F. Tsau, Int. J. Hydrogen Energy, 2009, 48, 2950–2959 Search PubMed.
- H. I. Schlesinger, H. C. Brown, D. L. Mayfield and J. R. Gilbreath, J. Am. Chem. Soc., 1953, 75, 213–215 CrossRef CAS.
- H. I. Schlesinger, H. C. Brown and A. E. Finholt, J. Am. Chem. Soc., 1953, 75, 205–209 CrossRef CAS.
- L. G. Sneddon, Amineborane Based Chemical Hydrogen Storage, DoE Hydrogen Annual Merit Review, 2007, http://www.hydrogen.energy.gov/pdfs/review07/st_27_sneddon.pdf Search PubMed.
- F. M. Taylor and J. Dewing, US Pat., 3103417, 1963.
- Primer on Spontaneous Heating and Pyrophoricity, DOE Handbook, 1994, vol. 1081–1094, p. 16, http://www.hss.energy. gov/nuclearsafety/techstds/standard/hdbk1081/hdbk, 1081.pdf Search PubMed.
- E. W. Schmidt, Hydrazine and Its Derivatives: Preparation, Properties, Applications, John Wiley & Sons, New York, 2nd edn, 1984 Search PubMed.
- M. Zheng, R. Cheng, X. Chen, N. Li, L. Li, X. Wang and T. Zhang, Int. J. Hydrogen Energy, 2005, 30, 1081–1089 CrossRef CAS.
- X. Chen, T. Zhang, L. Xia, T. Li, M. Zheng, Z. Wu, X. Wang, Z. Wei, Q. Xin and C. Li, Catal. Lett., 2002, 79, 21–25 CrossRef CAS.
- M. Zheng, X. Chen, R. Cheng, N. Li, J. Sun, X. Wang and T. Zhang, Catal. Commun., 2006, 7, 187–191 CrossRef CAS.
- X. Chen, T. Zhang, P. Ying, M. Zheng, W. Wu, L. Xia, T. Li, X. Wang and C. Li, Chem. Commun., 2002, 288–289 RSC.
- J. B. O. Santos, G. P. Valença and J. A. J. Rodrigues, J. Catal., 2002, 210, 1–6 CrossRef CAS.
- W. E. Armstrong, L. B. Ryland and H. H. Voge, US Pat., 4 124 538, 1978.
- X. Chen, T. Zhang, M. Zheng, Z. Wu, W. Wu and C. Li, J. Catal., 2004, 224, 473–478 CrossRef CAS.
- Y. K. Al-Haydari, J. M. Saleh and M. H. Matloob, J. Phys. Chem., 1985, 89, 3286–3290 CrossRef CAS.
- D. J. Alberas, J. Kiss, Z. M. Liu and J. M. White, Surf. Sci., 1992, 278, 51–61 Search PubMed.
- J. Prasad and J. L. Gland, Langmuir, 1991, 7, 722–726 CrossRef CAS.
- S. J. Cho, J. Lee, Y. S. Lee and D. P. Kim, Catal. Lett., 2006, 109, 181–187 CrossRef CAS.
- S. K. Singh and Q. Xu, Inorg. Chem., 2010, 49, 6148–6152 CrossRef CAS.
- S. K. Singh and Q. Xu, Chem. Commun., 2010, 46, 6545–6547 RSC.
- S. K. Singh, Y. Iizuka and Q. Xu, Int. J. Hydrogen Energy, 2011, 36, 11794–11801 Search PubMed.
- S. K. Singh, Z. H. Lu and Q. Xu, Eur. J. Inorg. Chem., 2011, 2232–2237 Search PubMed.
- S. K. Singh, A. K. Singh, K. Aranishi and Q. Xu, J. Am. Chem. Soc., 2011, 133, 19638–19641 CrossRef CAS.
- J. Wang, X. B. Zhang, Z. L. Wang, L. M. Wang and Y. Zhang, Energy Environ. Sci., 2012, 5, 6885–6888 RSC.
- L. He, Y. Huang, A. Wang, X. Wang, X. Chen, J. J. Delgado and T. Zhang, Angew. Chem., Int. Ed., 2012, 48, 4800–4803 Search PubMed.
- A. M. Cao and G. Veser, Nat. Mater., 2009, 9, 75–81.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- H. Hayashi, Res. Chem. Intermed., 1998, 24, 183–196 Search PubMed.
- N. Hazari, Chem. Soc. Rev., 2010, 39, 4044–4056 RSC.
- J. M. Chin, R. R. Schrock and P. Müller, Inorg. Chem., 2010, 49, 7904–7916 CrossRef CAS.
- V. Smil, in Enriching the Earth: Fritz Haber, Carl Bosch and the Transformation of World Food Production, MIT Press, Cambridge, MA, 2004 Search PubMed.
- T. Murakami, T. Nishikiori, T. Nohira and Y. Ito, J. Am. Chem. Soc., 2003, 125, 334–335 CrossRef CAS.
- S. Sridhar, T. Srinivasan, U. Virendra and A. A. Khan, Chem. Eng. J., 2003, 94, 51–56 Search PubMed.
- H. Hayashi, Catal. Rev. Sci. Eng., 1990, 32, 229–277 CAS.
- S. Karahan, M. Zahmakiran and S. Özkar, Int. J. Hydrogen Energy, 2011, 36, 4958–4966 CrossRef CAS.
- V. S. Nguyen, S. Swinnen, J. Leszczynski and M. T. Nguyen, Phys. Chem. Chem. Phys., 2011, 13, 6649–6656 RSC.
- R. Moury, G. Moussa, U. B. Demirci, J. Hannauer, S. Bernard, E. Petit, A. Lee and P. Miele, Phys. Chem. Chem. Phys., 2012, 14, 1768–1777 RSC.
- T. Hügle, M. F. Kühnel and D. Lentz, J. Am. Chem. Soc., 2009, 131, 7444–7446 CrossRef.
- D. Çelik, S. Karahan, M. Zahmakıran and S. Özkar, Int. J. Hydrogen Energy, 2012, 37, 5143–5151 Search PubMed.
- S. Karahan, M. Zahmakıran and S. Özkar, Dalton Trans., 2012, 41, 4912–4918 RSC.
- J. H. Sinfelt, Acc. Chem. Res., 1977, 10, 15–20 CrossRef CAS.
- J. H. Sinfelt, Acc. Chem. Res., 1987, 20, 134–139 CrossRef CAS.
- Ç. Çakanyıldırım, U. B. Demirci, T. Şener, Q. Xu and P. Miele, Int. J. Hydrogen Energy, 2012, 37, 9722–9729 Search PubMed.
- V. J. Goubeau and E. Ricker, Z. Anorg. Allg. Chem., 1961, 310, 123–142 CrossRef.
- F. C. Gunderloy Jr, Inorg. Chem., 1963, 2, 221–222 CrossRef.
- F. C. Gunderloy Jr, Inorg. Synth., 1967, 9, 13–16.
- F. C. Gunderloy Jr and M. Park, US Pat., 3375087, 1968.
- A. Sutton, J. C. Gordon, K. C. Ott and A. K. Burrell, US Pat., 2010/0272622, 2010.
- J. R. Hyde and M. Poliakoff, Chem. Commun., 2004, 1482–1483 RSC.
- J. Zhang, P. G. Blazecka, M. M. Bruendl and Y. Huang, J. Org. Chem., 2009, 74, 1411–1414 CrossRef CAS.
- X. Zhou, Y. Huang, W. Xing, C. Liu, J. Liao and T. Lu, Chem. Commun., 2008, 3540–3542 RSC.
- T. C. Johnson, D. J. Morris and M. Wills, Chem. Soc. Rev., 2010, 39, 81–88 RSC.
- A. Boddien, B. Loges, H. Junge and M. Beller, ChemSusChem, 2008, 1, 751–758 CrossRef CAS.
- B. Loges, A. Boddien, F. Gartner, H. Junge and M. Beller, Top. Catal., 2010, 53, 902–914 CrossRef CAS.
- R. S. Coffey, Chem. Commun., 1967, 923–924 Search PubMed.
- S. H. Strauss, K. H. Whitmire and D. F. Shriver, J. Organomet. Chem., 1979, 174, C59–C62 CrossRef CAS.
- R. S. Paonessa and W. C. Trogler, J. Am. Chem. Soc., 1982, 104, 3529–3530 CrossRef CAS.
- R. B. King and N. K. Battacharyya, Inorg. Chim. Acta, 1995, 237, 65–69 CrossRef CAS.
- J. H. Shin, D. G. Churchill and G. Parkin, J. Organomet. Chem., 2002, 642, 9–15 CrossRef CAS.
- Y. Gao, J. Kuncheria, G. P. A. Yap and R. J. Puddephatt, Chem. Commun., 1998, 2365–2366 RSC.
- Y. Gao, J. K. Kuncheria, H. A. Jenkins, R. J. Puddephatt and G. P. A. Yap, J. Chem. Soc., Dalton Trans., 2000, 3212–3217 RSC.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, ChemSusChem, 2008, 1, 827–834 CrossRef CAS.
- Y. Himeda, Green Chem., 2009, 11, 2018–2022 RSC.
- S. Fukuzumi, T. Kobayashi and T. Suenobu, J. Am. Chem. Soc., 2010, 132, 1496–1497 CrossRef CAS.
- B. Loges, A. Boddien, H. Junge and M. Beller, Angew. Chem., Int. Ed., 2008, 47, 3962–3965 CrossRef CAS.
- H. Junge, A. Boddien, F. Capitta, B. Loges, J. R. Noyes, S. Gladiali and M. Beller, Tetrahedron Lett., 2009, 50, 1603–1606 CrossRef CAS.
- C. Fellay, N. Yan, P. J. Dyson and G. Laurenczy, Chem.–Eur. J., 2009, 15, 3752–3760 CrossRef CAS.
- A. Boddien, B. Loges, H. Junge, F. Gartner, J. R. Noyes and M. Beller, Adv. Synth. Catal., 2009, 351, 2517–2520 CrossRef CAS.
- B. Loges, A. Boddien, H. Junge, J. R. Noyes, W. Baumann and M. Beller, Chem. Commun., 2009, 4185–4187 RSC.
- W. Gan, P. J. Dyson and G. Laurenczy, React. Kinet. Catal. Lett., 2009, 98, 205–213 CrossRef CAS.
- X. Li, X. Ma, F. Shi and Y. Deng, ChemSusChem, 2010, 3, 71–74 CrossRef CAS.
- X. Li, F. Shi, X. Ma, L. Lu and Y. Deng, J. Fuel Chem. Technol., 2010, 38, 544–553 Search PubMed.
- M. E. M. Berger, D. Assenbaum, N. Taccardi, E. Spiecker and P. Wasserscheid, Green Chem., 2011, 13, 1411–1415 RSC.
- A. Boddien, B. Loges, F. Gärtner, C. Torborg, K. Fumino, H. Junge, R. Ludwig and M. Beller, J. Am. Chem. Soc., 2010, 132, 8924–8934 CrossRef CAS.
- A. Boddien, D. Mellmann, F. Gärtner, R. Jackstell, H. Junge, P. J. Dyson, G. Laurenczy, R. Ludwig and M. Beller, Science, 2011, 333, 1733–1736 CrossRef CAS.
- W. Gan, C. Fellay, P. J. Dyson and G. Laurenczy, J. Coord. Chem., 2010, 63, 2685–2694 CrossRef CAS.
- D. J. Morris, G. J. Clarkson and M. Wills, Organometallics, 2009, 28, 4133–4140 CrossRef CAS.
- M. Czaun, A. Goeppert, R. May, R. Haiges, G. K. S. Prakash and G. A. Olah, ChemSusChem, 2011, 4, 1241–1248 CrossRef CAS.
- J. D. Scholten, M. H. G. Prechtl and J. Dupont, ChemCatChem, 2010, 2, 1265–1270 Search PubMed.
- S. Enthaler, J. Langermann and T. Schmidt, Energy Environ. Sci., 2010, 3, 1207–1217 RSC.
- K. Hirota, K. Kuwata and Y. Nakai, Bull. Chem. Soc. Jpn., 1958, 31, 861–864 CrossRef CAS.
- Y. K. Sun, J. J. Vajo, C. Y. Chan and W. H. Weinberg, J. Vac. Sci. Technol., A, 1988, 6, 854–855 CrossRef.
- D. H. S. Ying and R. J. Madix, J. Catal., 1980, 61, 48–56 CAS.
- N. Aas, Y. Li and M. Bowker, J. Phys.: Condens. Matter, 1991, 3, S281–S286 CrossRef CAS.
- X. D. Peng and M. A. Barteau, Catal. Lett., 1991, 7, 395–402 CrossRef.
- P. A. Dilara and J. M. Vohs, J. Phys. Chem., 1993, 97, 12919–12923 CrossRef CAS.
- V. A. Gercher and D. F. Cox, Surf. Sci., 1994, 312, 106–114 CrossRef CAS.
- J. Stubenrauch, E. Brosha and J. M. Vohs, Catal. Today, 1996, 28, 431–444 CrossRef CAS.
- R. Larsson, M. H. Jamroz and M. A. Borowiak, J. Mol. Catal. A: Chem., 1998, 129, 41–51 CrossRef CAS.
- G. Y. Popova, T. V. Andrushkevich, Y. A. Chesalov and E. S. Stoyanov, Kinet. Catal., 2000, 41, 805–811 CrossRef CAS.
- A. Bandara, J. Kubota and A. Wada, J. Phys. Chem. B, 1997, 101, 361–368 CrossRef CAS.
- J. Rasko, T. Kecskes and J. Kiss, J. Catal., 2004, 224, 261–268 CrossRef CAS.
- T. Shido and Y. Iwasawa, J. Catal., 1993, 141, 71–81 CrossRef CAS.
- G. Jacobs, P. M. Patterson, U. M. Graham, A. C. Crawford and B. H. Davis, Int. J. Hydrogen Energy, 2005, 30, 1265–1276 CrossRef CAS.
- G. Jacobs, P. M. Patterson, U. M. Graham, A. C. Crawford, A. Dozier and B. H. Davis, J. Catal., 2005, 235, 79–91 CrossRef CAS.
- Y. Yasaka, H. Yoshida, C. Wakai, N. Matubayasi and M. Nakahar, J. Phys. Chem. A, 2006, 110, 11082–11090 CrossRef CAS.
- M. Ojeda and E. Iglesia, Angew. Chem., Int. Ed., 2009, 48, 4800–4803 CrossRef.
- D. A. Bulusheva, S. Beloshapkin and J. R. H. Ross, Catal. Today, 2010, 154, 7–12 CrossRef CAS.
- F. Solymosi, Á. Koós, N. Liliom and I. Ugrai, J. Catal., 2011, 279, 213–219 CrossRef CAS.
- G. Halasi, G. Schubert and F. Solymosi, Catal. Lett., 2011, 142, 218–223 Search PubMed.
- D. A. Bulushev, L. Jiaa, S. Beloshapkin and J. R. H. Ross, Chem. Commun., 2012, 48, 4184–4186 RSC.
- R. Williams, R. S. Crandall and A. Bloom, Appl. Phys. Lett., 1978, 33, 381–383 CrossRef.
- H. Wiener, Y. Sasson and J. Blum, J. Mol. Catal., 1986, 35, 277–284 CrossRef CAS.
- X. Zhou, Y. Huang, C. Liu, J. Liao, T. Lu and W. Xing, ChemSusChem, 2010, 3, 1379–1382 CrossRef CAS.
- Y. Huang, X. Zhou, M. Yin, C. Liu and W. Xing, Chem. Mater., 2010, 22, 5122–5128 CrossRef CAS.
- S. W. Ting, S. Cheng, K. Y. Tsang, N. van der Laak and K. Y. Chan, Chem. Commun., 2009, 7333–7335 RSC.
- K. Tedsree, T. Li, S. Jones, C. W. A. Chan, K. M. K. Yu, P. A. J. Bagot, E. A. Marquis, G. D. W. Smith and S. C. E. Tsang, Nat. Nanotechnol., 2011, 6, 302–307 CrossRef CAS.
- K. Tedsree, C. W. A. Chan, S. Jones, Q. Cuan, W. K. Li, X. Q. Gong and S. C. E. Tsang, Science, 2011, 332, 224–228 CrossRef CAS.
- X. Gu, Z. H. Lu, H. L. Jiang, T. Akita and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11822–11825 CrossRef CAS.
- M. Yadav, T. Akita, N. Tsumori and Q. Xu, J. Mater. Chem., 2012, 22, 12582–12586 RSC.
- Y. Zhao, L. Deng, S. Ya Tang, D. M. Lai, B. Liao, Y. Fu and Q. X. Guo, Energy Fuels, 2011, 25, 3693–3697 CrossRef CAS.
- Q. Y. Bi, X. L. Du, Y. M. Liu, Y. Cao, H. Y. He and K. N. Fan, J. Am. Chem. Soc., 2012, 134, 8926–8933 CrossRef CAS.
- G. A. Olah, A. Goeppert and G. K. S. Prakash, Beyond Oil and Gas: The Methanol Economy, Wiley-VCH, Weinheim, 2006 Search PubMed.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1995, 95, 259–272 CrossRef CAS.
- P. G. Jessop, F. Joó and C.-C. Tai, Coord. Chem. Rev., 2004, 248, 2425–2442 CrossRef CAS.
- P. G. Jessop, in Handbook of Homogeneous Hydrogenation, ed. J. G. de Vries and C. J. Cornelis, Wiley-VCH, Weinheim, 2007, pp. 489–511 Search PubMed.
- P. Munshi, A. D. Main, J. C. Linehan, C. C. Tai and P. G. Jessop, J. Am. Chem. Soc., 2002, 124, 7963–7971 CrossRef.
- J. Elek, L. Nádasdi, G. Papp, G. Laurenczy and F. Joó, Appl. Catal., A, 2003, 255, 59–67 CrossRef CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Nature, 1994, 368, 231–233 CrossRef CAS.
- R. Fornika, H. Görls, B. Seemann and W. Leitner, J. Chem. Soc., Chem. Commun., 1995, 1479–1481 RSC.
- R. Tanaka, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2009, 131, 14168–14169 CrossRef CAS.
- Y. Himeda, N. Onozawa-Komatsuzaki, H. Sugihara and K. Kasuga, Organometallics, 2007, 26, 702–712 CrossRef CAS.
- J. F. Hull, Y. Himeda, W. H. Wang, B. Hashiguchi, R. Periana, D. J. Szalda, J. T. Muckerman and E. Fujita, Nat. Chem., 2012, 4, 383–388 CrossRef CAS.
- Y. Himeda, Eur. J. Inorg. Chem., 2007, 3927–3941 CrossRef CAS.
- H. Hayashi, S. Ogo and S. Fukuzumi, Chem. Commun., 2004, 2714–2715 RSC.
- S. Ogo, R. Kabe, H. Hayashi, R. Harada and S. Fukuzumi, Dalton Trans., 2006, 4657–4663 RSC.
- Y. Maenaka, T. Suenobu and S. Fukuzumi, Energy Environ. Sci., 2012, 5, 7360–7363 RSC.
- G. C. Chinchen, P. J. Denny, J. R. Jennings, M. S. Spencer and K. C. Waugh, Appl. Catal., 1988, 36, 1–65 Search PubMed.
- A. Kiennemann, J. Idriss, J. P. Hindermann, J. C. Lavalley, A. Vallet, P. Chaumette and P. Courty, Appl. Catal., 1990, 59, 165–184 Search PubMed.
- M. Bowker, R. A. Hadden, H. Houghton, J. N. Hyland and K. C. Waugh, J. Catal., 1988, 109, 263–273 CrossRef CAS.
- P. A. Taylor, P. B. Rasmussen, C. V. Ovesen, P. Stoltze and I. Chorkendorff, Surf. Sci., 1992, 261, 191–206 CrossRef CAS.
- H. Nakano, I. Nakamura, T. Fujitani and J. Nakamura, J. Phys. Chem. B, 2001, 105, 1355–1365 CrossRef CAS.
- M. W. Farlow and H. Adkins, J. Am. Chem. Soc., 1935, 57, 2222–2223 CrossRef CAS.
- D. Preti, C. Resta, S. Squarcialupi and G. Fachinetti, Angew. Chem., Int. Ed., 2011, 50, 12551–12554 CrossRef CAS.
- H. Wiener, J. Blum, H. Feilchenfeld, Y. Sasson and N. Zalmanov, J. Catal., 1988, 110, 184–190 CrossRef CAS.
- Y. Zhang, J. Fei, Y. Yu and X. Zheng, Catal. Commun., 2004, 5, 643–646 CrossRef CAS.
- Z. Zhang, Y. Xie, W. Li, S. Hu, J. Song, T. Zhiang and B. Han, Angew. Chem., Int. Ed., 2008, 47, 1127–1129 CrossRef CAS.
- T. Inoue, A. Fujishima, S. Konishi and K. Honda, Nature, 1979, 277, 637–638.
- B. Aurian-Blajeni, M. Halmann and J. Manassen, Sol. Energy, 1980, 25, 165–170 CrossRef CAS.
- K. R. Thampi, J. Kiwi and M. Grazel, Nature, 1987, 327, 506–508 CrossRef CAS.
- H. Fujiwara, H. Hosokawa, K. Kurakoshi, Y. Wada, S. Yanagida, T. Okada and H. Kobayashi, J. Phys. Chem. B, 1997, 101, 8270–8278 CrossRef CAS.
- Y. Amao, ChemCatChem, 2011, 3, 458–474 Search PubMed.
- D. Mandler and I. Willner, J. Chem. Soc., Perkin Trans. 1, 1988, 2, 997–1003 Search PubMed.
- I. Willner and D. Mandler, J. Am. Chem. Soc., 1989, 111, 1330–1336 CrossRef CAS.
- I. Willner, N. Lapidot, A. Riklin, R. Kasher, E. Zahavy and E. Katz, J. Am. Chem. Soc., 1994, 116, 1428–1441 CrossRef CAS.
- I. Willner, I. Willner and N. Lapidot, J. Am. Chem. Soc., 1990, 112, 6438–6439 CrossRef CAS.
- M. Kodaka and Y. Kubota, J. Chem. Soc., Perkin Trans. 1, 1999, 2, 891–894 Search PubMed.
- R. Miyatani and Y. Amao, Biotechnol. Lett., 2002, 24, 1931–1934 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Mol. Catal. B: Enzym., 2004, 27, 121–125 CrossRef CAS.
- R. Miyatani and Y. Amao, J. Jpn. Pet. Inst., 2004, 47, 27–31 Search PubMed.
- I. Tsujisho, M. Toyoda and Y. Amao, Catal. Commun., 2006, 7, 173–176 CrossRef CAS.
- S. Sato, T. Morikawa, S. Saeki, T. Kajino and T. Motohiro, Angew. Chem., Int. Ed., 2010, 49, 5101–5105 CAS.
- T. Arai, S. Tajima, S. Sato, K. Uemura, T. Morikawa and T. Kajino, Chem. Commun., 2011, 47, 12664–12666 RSC.
- S. Sato, T. Arai, T. Morikawa, K. Uemura, T. M. Suzuki, H. Tanaka and T. Kajino, J. Am. Chem. Soc., 2011, 133, 15240–15243 CrossRef CAS.
- T. Arai, S. Sato, K. Uemura, T. Morikawa, T. Kajino and T. Motohiro, Chem. Commun., 2010, 46, 6944–6946 RSC.
- T. M. Suzuki, H. Tanaka, T. Morikawa, M. Iwaki, S. Sato, S. Saeki, M. Inoue, T. Kajino and T. Motohiro, Chem. Commun., 2011, 47, 8673–8675 RSC.
- R. P. Linstead and S. L. S. Thomas, J. Chem. Soc., 1940, 1127–1134 RSC.
- N. F. Grunenfelder and T. H. Schucan, Int. J. Hydrogen Energy, 1989, 14, 579–586 CrossRef.
- J. K. Ali, E. J. Newson and D. W. T. Rippin, Chem. Eng. Sci., 1994, 13, 2129–2134 Search PubMed.
- N. Meng, S. Shinoda and Y. Saito, Int. J. Hydrogen Energy, 1997, 22, 361–367 CrossRef CAS.
- C. M. Jensen, Proceedings of the U.S. DOE Hydrogen Program Review, Herndon, VA, 1997 Search PubMed.
- S. Hodoshima, S. Takaiwa, A. Shono, K. Satoh and Y. Saito, Appl. Catal., A, 2005, 282, 235–242 Search PubMed.
- G. P. Pez, A. Scott, A. Cooper and H. Cheng, US Pat., 7101530, 2006.
- A. Cooper and G. P. Pez, Proceedings of the US DOE hydrogen Program review, Crystal City, VA, 2006 Search PubMed.
- D. E. Schwarz, T. M. Cameron, P. J. Hay, B. L. Scott, W. Tumas and D. L. Thorn, Chem. Commun., 2005, 5919–5921 RSC.
- Y. Okada, E. Sasaki, E. Watanabe, S. Hyodo and H. Nishijima, Int. J. Hydrogen Energy, 2006, 31, 1348–1356 CrossRef CAS.
- A. Moores, M. Poyatos, Y. Luo and R. H. Crabtree, New J. Chem., 2006, 30, 1675–1678 RSC.
- R. B. Biniwalea, S. Rayalua, S. Devottaa and M. Ichikawab, Int. J. Hydrogen Energy, 2008, 33, 360–365 CrossRef CAS.
- R. H. Crabtree, Energy Environ. Sci., 2008, 1, 134–138 RSC.
- Y. Cui, S. Kwok, A. Bucholtz, B. Davis, R. A. Whitney and P. G. Jessop, New J. Chem., 2008, 32, 1027–1037 RSC.
- D. Teichmann, W. Arlt, P. Wasserscheid and R. Freymann, Energy Environ. Sci., 2011, 4, 2767–2773 RSC.
| This journal is © The Royal Society of Chemistry 2012 |