Ti-based compounds as anode materials for Li-ion batteries
Guan-Nan
Zhu
,
Yong-Gang
Wang
and
Yong-Yao
Xia
*
Department of Chemistry and Shanghai Key Laboratory of Molecular Catalysis and Innovative Materials, Institute of New Energy, Fudan University, Shanghai 200433, China. E-mail: yyxia@fudan.edu.cn; Fax: (+86)-21-51630318; Tel: (+86)-21-51630318
First published on 23rd February 2012
Abstract
Li-ion batteries are one of the most promising electrochemical power sources to be widely used in portable electronics, electric vehicles, and stationary energy storage systems. Ti-based materials have been intensively investigated as important anodes for Li-ion batteries due to their high safety and excellent cycling stability. The present work reviews the latest advances in the exploration and development of Ti-based compounds, such as Li4Ti5O12, Li2Ti3O7, TiO2-B and H2Ti3O7, as high performance anode materials for Li-ion batteries. The relationship between the preparation, composition, structure, morphology and electrochemical performance are summarized and analyzed. Further, the related advancements and challenges in practical energy applications are discussed.
Broader contextLi-ion battery (LIB) technology (typically using carbon materials as the anode) powers most of today's portable electronic devices, but faces serious challenges over the years to come if it is to take over the hybrid electric vehicles and stationary power sources for smart-grid application. This is partly because the carbon material suffers from great volume change and severe safety concerns have been raised over lithium dendrites. Ti-based compounds, especially Li4Ti5O12 with zero structural change, have been demonstrated as the most promising anode materials for large-sized LIBs since they exhibit excellent cycling reversibility and a high operating voltage to ensure improved safety. However, the rate capability of these Ti-based materials are relatively low because of a large polarization at high charge–discharge rates. Consequently, many researches have been developed to enhance its electrical conductivity by ion doping and surface modification, and ionic diffusivity by designing various nanosized materials. In the present review, we give an overview of the latest advancements in Ti-based anodes categorized by Li–Ti–O, Na–Ti–O and H–Ti–O systems, highlighting the importance and necessity of electrode designs including morphological tailoring and surface modification in improving the electrochemical performance. Also, the prospects and challenges in practical application are analyzed, aiming to give deep insights into the material science and electrochemical field. |
Introduction
With growing concerns over increased oil prices, the stretched energy situation and environmental issues, the demand for developing sustainable and green energy sources are at the top of the agenda in today's energy-based society. Although a variety of renewable energy technologies have been designed and presented such as wind and solar power, few of them are widely utilized owing to the significant fluctuation and unpredictable changes in the weather. Li-ion batteries, considered to be the most important energy storage and conversion technology with prominent advantages of high energy and power density as well as long cycling life, have witnessed large-scale application in portable electronic devices, communication facilities, stationary energy storage systems and ever-enlarging markets of electric vehicles (EV/HEV/PHEVs).The electrochemical performance of Li-ion batteries strongly depends on the electrode properties, and a prudent selection of electrode materials including anodes and cathodes plays a central role. The conventional Li-ion batteries use carbon-based materials (typically graphite) as anode materials, which have the advantages of low cost, high abundance and outstanding kinetics.1 However, there are two disadvantages that greatly limit their practical applications for large-sized batteries. For example graphite, including natural and artificial graphite, suffers greatly from volume expansion and shrinkage during Li-ion intercalation and extraction that fatigues the graphite particles causing cracking, leading to a loss in electrical contact between the resulting particles and thereby decreasing capacity;2,3 and another limitation comes from the safety concerns arising from lithium dendrite formation during the overcharge process owing to a low Li-intercalation potential of the graphite anode approaching 0 V (vs. Li/Li+).4,5
Herein, a fundamental solution is to find alternatives to carbon-based materials with good safety and cycling stability. Ti-based oxides such as Li4Ti5O12 (2Li2O·5TiO2), Li2Ti3O7 (Li2O·3TiO2), Li2Ti6O13 (Li2O·6TiO2), H2Ti3O7 (H2O·3TiO2) and TiO2-B as presented in Fig. 1, have been considered as potential alternative materials to traditional carbon-based anodes, since they exhibit excellent Li-ion insertion/extraction reversibility with small structural change and a much higher operating voltage ranging from 3 to 1 V (vs. Li/Li+) ensuring a better safety of the battery by avoiding the problem of lithium dendrites.6–8
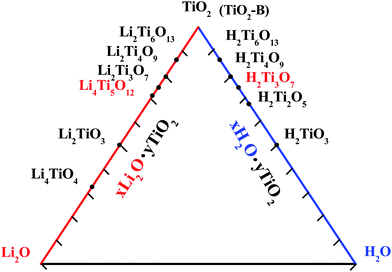 | ||
| Fig. 1 The phase diagram of Ti-based compounds including Li–Ti–O and H–Ti–O systems. | ||
Despite these advantages, the rate capability of Ti-based oxides are relatively low because of a large polarization at high charge–discharge rates resulting from the poor electrical conductivity and sluggish lithium-ion diffusion.9–13 In an attempt to overcome these drawbacks, many approaches have been developed to improve the battery performance. In the present review, smart material design strategies towards several typical materials in the Li–Ti–O, Na–Ti–O and H–Ti–O systems, with respect to the composition, morphology, and surface modification in order to enhance the ionic diffusion and electronic conductivity, have been extensively summarized and analyzed.
1. Li4Ti5O12
The spinel Li4Ti5O12 material, as an alternative anode to carbon, has been extensively studied as one of the most promising anode candidates for rechargeable Li-ion batteries. Li4Ti5O12 can accommodate three Li ions with a theoretical specific capacity of 175 mAh g−1 at a flat potential of 1.55 V, corresponding to a chemical formula of Li7Ti5O12. Structures of both Li4Ti5O12 and Li7Ti5O12 are illustrated in Fig. 2(a) and (b).17 The intercalation process occurred along varied lithium diffusion paths like 8a-16c-8a or 8a-16c-48f-16d (Fig. 2(c)) at room temperature inside the original formula unit of (Li)8a[Li1/3Ti5/3]16d(O4)32e, resulting in the end composition of [Li2]16c[Li1/3Ti5/3]16d(O4)32e.18 A negligible volume change of only 0.2% is caused between both end members of Li4Ti5O12 and Li7Ti5O12 accompanied by a change of the lattice axis from 8.3595 Å to 8.3538 Å, and as a result it is a “zero strain” material with excellent cycling stability.19–21 Besides, it has a high potential around 1.5 V (vs. Li/Li+), which avoids the battery short circuit triggered by the formation of lithium dendrites to achieve safe operation. Combined with virtues of low cost and environmental friendliness, the spinel Li4Ti5O12 material has been demonstrated as the most promising anode material in practical energy application.6–8,22,23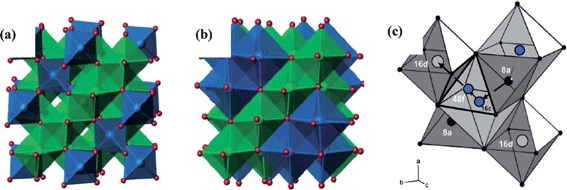 | ||
| Fig. 2 (a) Li4Ti5O12 spinel structure type: blue tetrahedra represent lithium, and green octahedra represent disordered lithium and titanium (ref. 17); (b) Li7Ti5O12 rock salt structure type: blue octahedra represent lithium, and green octahedra represent disordered lithium and titanium (ref. 17); (c) Schematic presentation of possible diffusion pathways for Li4Ti5O12: grey and black spheres denote titanium ions and Li ions, respectively; blue spheres show temporarily occupied sites (ref. 18). | ||
However, the Li4Ti5O12 material has an inherently insulating property due to the empty Ti 3d state with a band energy of about 2 eV,24,25 which seriously hinders its high rate performance. The Li-ion intercalation of Li4Ti5O12 involves three process as follows: (i) the solvated Li ions diffuse from the bulk electrolyte solution; (ii) a charge-transfer reaction occurs at the interface between the Li4Ti5O12 surface and the electrolyte, accompanied by accepting an electron; and (iii) the Li ions diffuse into the bulk Li4Ti5O12.9 Accordingly, two general methods are typically adopted to improve the rate capability: one is to accelerate charge-transfer reaction by enhancing ionic diffusion and electronic conductivity via surface modification or ion doping, and the other is to reduce the Li-ion diffusion distance in the bulk phase by designing nanoscale Li4Ti5O12 materials. Both methods will be discussed in detail in the following sections.
1.1 Morphology tailoring and size control of Li4Ti5O12 materials for enhancement in rate capability
Compared with traditional bulk Li4Ti5O12 materials on the microscale, novel nanostructured materials have gained more attention as high-rate anodes for rechargeable Li-ion batteries. Owing to the small size and large surface area, the rate-limiting diffusion pathway is reduced and a better contact between the electrode material and the electrolyte in assurance of high lithium flux is also achieved, both contributing to an improvement of the rate capability.26–41 A further cause for such enhancement has been investigated from a structural view. Compared with its bulk counterpart, the nanosized Li4Ti5O12 material displays an obvious increased solid solution or single-phase intercalation/extraction in the charge/discharge curve, which implies the mixed 8a/16c site occupation in contrast to the separated one in the two-phase region.42–44 Based on theoretical assumption and experimental verification, the disordered coexistence of 8a and 16c domains observed in the solid solution region is considered to be beneficial for Li-ion mobility or ionic conductivity and thus makes a major contribution to the enhancement of power performance.43–45In this section, various morphologies of nanostructured Li4Ti5O12, including 0D nanoparticles, 1D nanowires and nanotubes, and other special structural designs are presented and evaluated.
The soft chemistry method, such as sol–gel, liquid phase-based and sonochemical process, is one of the most commonly used ways to prepared nanosized Li4Ti5O12 particles, which can range from tens to hundreds of nanometers.46–49 For this method, a careful control over the hydrolytic rate and amount of titanium salt as starting precursor is essential to ensure good phase purity of the resultant particles. Moreover, as a low-temperature preparation method, it is difficult to obtain optimal crystallinity of the resulting particles that plays a decisive role in cycling stability for electrode materials.
The molten salt method has been developed to prepare highly crystalline Li4Ti5O12 nanoparticles. The molten salt flux produces a liquid/solid reaction interface, thus providing a large effective reaction area to accelerate the Li4Ti5O12 growth in a relatively short time.50–52 Our group utilized LiCl as the molten salt or high-temperature flux, TiO2 and Li2CO3 as the reactant to successfully prepare highly crystalline nanosized Li4Ti5O12 with a narrow particle size distribution of ∼100 nm. An investigation of the influencing parameters came to the conclusion that the flux amount and annealing time were two primary factors influencing the size control of resulting Li4Ti5O12 nanoparticles.51 Unfortunately, impurity generation seems inevitable owing to interference of molten salts in the ratio of Li/Ti.50–52
The irradiation-based means typical of the microwave method is a relatively new solution to prepare nanosized Li4Ti5O12. The deep penetration and direct energy transfer of microwaves into the reactants facilitate preparation via a fast route under lower temperature, which could effectively restrict particle growth.53–55 Still, the feasibility of such a method is hampered by problems of phase purity and crystallinity.
Despite a uniform morphology and narrow size distribution, nanoparticles often encounter a common problem of dense aggregation, which greatly prohibits the electrolyte from penetrating into electrochemically active particles. Preparing well-dispersed particles by formation of porous structures could alleviate large agglomerations by addition of special agents like surfactants56 or organic acids.36,40 For example, one nanocrystalline Li4Ti5O12 with a flaky morphology and highly porous structure has been prepared, in which the interconnected porous network was accumulated by the primary crystallites of varying size between 20 and 50 nm. The nanoparticles provide short Li-ion diffusion paths and the porous feature favors electrolyte percolation. As a consequence, the electrodes yield attractive rate performances by maintaining a capacity of 140 mAh g−1 and 70 mAh g−1 at 10 C and 100 C rates, respectively.36
However, even if the particles are porous and made of small crystallites, the nature of easy agglomeration for the nanoparticles prevents uniform mixing with the polymer binder and conducting agent to form a homogeneous electrode.37 Furthermore, the problem of high interparticle contact resistance remains unsolved. A fundamental strategy targeting the two problems will be discussed in section 1.2.1.
Template-based methods are one common way to prepare hollow structured Li4Ti5O12. By selecting carbon spheres as the hard template due of a high thermal stability and simple pre-modification,57–59 monodisperse Li4Ti5O12 hollow spheres with an average outer diameter of 1.0 μm and a mean wall thickness of 60 nm were successfully synthesized, which, compared with Li4Ti5O12 dense particles, exhibited a good rate capability by retaining a specific capacity of 100 mAh g−1 at 10 C rate.59 In another example, a self-assembled poly(methyl methacrylate) colloidal crystal template was used to prepare unique 3D Li4Ti5O12 (Fig. 3) with a hierarchical pore structure by a combination of macro- and mesopores, which gave a markedly improved rate capability.17 Apart from template-based methods, a hydrothermal route was explored to prepare mesoporous Li4Ti5O12 hollow microspheres with the shell consisting of nanosheets, which delivered an impressive specific discharge capacity of 131 mAh g−1 at the very high rate of 50 C.60
 | ||
| Fig. 3 (a) SEM micrograph and (b) TEM micrograph of 3D Li4Ti5O12 (ref. 17). | ||
 | ||
| Fig. 4 (a) SEM image and (b) TEM image of the 0D–1D heterogeneous Li4Ti5O12 nanostructure (ref. 64). | ||
The case of mesoporous Li4Ti5O12 microspheres well demonstrate this design concept.65 As Fig. 5 shows, the monodisperse primary nanoparticles of about 20 nm in size induced mesopores by uniform accumulation, which retained common features of nanosized materials; and the secondary sub-micron spheres consisting of packed primary nanoparticles ensured a high tap density. Such material maintained a high capacity of 114 mAh g−1 at 30 C as well as good cycling stability by retaining 125 mAh g−1 at 20 C after 200 cycles, which was comparable or even superior to Li4Ti5O12 materials with single morphology like Li4Ti5O12 hollow spheres59 or nanotubes.63 Likewise, hierarchically porous Li4Ti5O12 microspheres assembled by well-crystalline nanoparticles prepared by a simpler hydrothermal route also exhibited attractive rate capability and cycling performance.66 In another case, the novel Li4Ti5O12 microspheres composed of sawtooth-like nanosheets exhibited nano-/micro-hierarchical structure and demonstrated a high capacity retention of 76% at the rate of 57 C.67
 | ||
| Fig. 5 SEM images of (a) several and (b) single mesoporous Li4Ti5O12 microspheres (ref. 65). | ||
Furthermore, Amine et al. prepared a “MSNP-LTO” material shown in Fig. 6 which stands for “micron secondary nano primary Li4Ti5O12”, to explore the potential of such a configuration as an anode for HEV application.68 The MSNP-LTO/LMO (LiMn2O4) system was then fabricated to conduct extensive electrochemical tests and demonstrated prominent advantages in the rate capability, calendar life, heat generation and low-temperature behavior.
 | ||
| Fig. 6 SEM images under (a) low and (b) high magnification of MSNP-LTO, showing secondary and primary particles, respectively (ref. 68). | ||
Despite great advances in the preparation of Li4Ti5O12 nanomaterials, several challenges still remain in the following aspects: First, it is often difficult to match nano-dimension with optimal crystallinity of the resulting materials by a low-temperature method, although the molten salt method can take both sides into account by compromising phase purity. Second, the interparticle contact resistance remains a main rate-limiting factor, which is attributed to large agglomeration of nanomaterials. Finally, considerable efforts are still needed to solve the issue of low packing density in a more facile way.
1.2 Surface modification of Li4Ti5O12 materials for enhancement in rate capability
In terms of overcoming the detrimental drawback of poor electronic conductivity of the Li4Ti5O12 material, surface modification by means of carbon, metal and inorganic/organic compounds is considered to be one of the most promising ways.| Li4Ti5O12 + xLi+ + xe− ↔ Li4+xTi5O12 (0 < x ≤ 3) |
The reaction occurs only when equivalent ions and electrons are available simultaneously. In order to optimize the rate capability, it is necessary for a comprehensive consideration of both electronic conductivity and ionic diffusivity. The electronic conductivity is an evaluation of the ability to accept electrons while ion diffusivity is a reflection of the speed of Li-ion diffusion. Li4Ti5O12 has a serious drawback of poor electronic conductivity that adversely affects its rate capability. Both carbon mixing (see section 1.2.2) and carbon coating methods have been demonstrated to be very useful in solving this problem. Our group in 2007 for the first time employed “carbon coating” via the CVD (chemical vapor deposition) method,9 by which the active Li4Ti5O12 particles could be covered with well-distributed and continuous carbon layer. The difference in the reaction pathway between the carbon simply mixed and a carbon-coated LTO electrode is reflected in the charge-transfer interface reaction, and can be explained by a simple model shown in Fig. 7. For the carbon-mixed electrode, most regions on a particle surface are in direct contact with the electrolyte and thus facilitate the Li-ion transfer from the electrolyte to the LTO particle surface, but the reaction can only occur on limited spots in which the electronically conductive path is built. In the case of the carbon-coated electrode, carbon as the electronic conductive phase forms the coating layer around each active particle so that each spot on the particle surface is able to accept electrons and crystal defects present in the coated carbon layer allow the Li-ions to pass through the carbon layer.70 By providing enough effective reaction areas, the carbon-coated Li4Ti5O12 electrode presents a much higher electronic conductivity of 2.05 S cm−1 compared with the raw Li4Ti5O12 electrode of ∼10−13 S cm−1.14
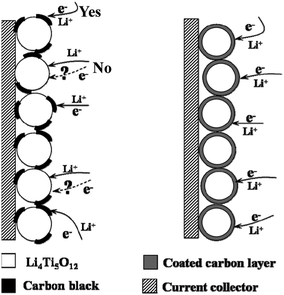 | ||
| Fig. 7 Schematic representations of electrochemical reaction paths on carbon-mixed (left) and carbon-coated Li4Ti5O12 electrodes (right) (ref. 9). | ||
On the one hand, the carbon coating method could dramatically improve the electronic conductivity; on the other hand, the coated carbon layer hinders the Li ions from diffusing into the bulk Li4Ti5O12 to some extent. Therefore, our group took further insights into the condition of “carbon coating” in order to highlight the effects of carbon structure (graphitic or amorphous formation) and thickness on the ionic diffusion, which could be quantified by measurement of apparent diffusion coefficient (DLi)10 or Warburg coefficient (σw).45 When the treating temperature increases from 700 to 900 °C, the coated carbon presents a more clearly layered and compact structure that is typical of high graphitization, and as a consequence, DLi decreases, indicating that more graphitized structure with fewer defects or cracks would reduce accessible Li-ion diffusion paths. Meanwhile, the optimal thickness of the carbon layer should be as thin as possible while preserving its integrity and uniformity, and a thinner or thicker coating would adversely affect the electronic conductivity or DLi, respectively. The best rate capability could be obtained by a simultaneous optimization of the electronic conductivity and ionic diffusivity, which corresponds to 2.16 S cm−1 and 1.3 × 10−11 cm2 s−1, respectively.10
Besides improvement of electronic conductivity, it is also easy to detect other potential use of the “carbon coating” method based on the model presented in Fig. 7. First, the uniform and integrated carbon layer over each active Li4Ti5O12 particle serves as a “protective barrier” to separate one from another, which avoids aggregation of particles to greatly reduce the interparticle contact resistance, and moreover, the insulating particles armed with carbon layers become fine electronic conductors, both contributing to the rate capability. Besides, “carbon coating” is essentially a high-temperature treating method, which is a strong guarantee for preparation of well-crystalline materials. Clearly, its application in nanomaterial preparation will be expected to solve the troubles puzzling traditional nanomaterials.
Very recently, our research group developed a brand-new carbon pre-coating way to successfully prepare nanostructured Li4Ti5O12 spinel materials with different morphologies including nanorods, hollow spheres and nanoparticles,11 demonstrating the pre-coated carbon layer over TiO2 before the chemical reaction with the Li salt could effectively prevent the final Li4Ti5O12 from growing large. From Fig. 8(c) and (d), the uniformity and shape of the carbon layer was well retained when experiencing a huge local structural change from tetragonal TiO2 to cubic Li4Ti5O12, but a relatively small absolute volume variation on the nanoscale,15,16 which rendered the preservation of the rod-like morphology (Fig. 8(a) and (b)). Thus, the coated carbon layer acted as a buffer layer to successfully confine the dimensions of the resultant Li4Ti5O12 to the nanoscale as well as to establish electrically conductive networks between the insulating Li4Ti5O12 particles, achieving a dual optimization of ionic diffusion and electronic conductivity. Based on these features, the Li4Ti5O12 nanorods of about 50 nm in diameter with a uniform 5 nm-thick carbon coverage demonstrated an outstanding rate capability that even at the high rate of 20 C, it still maintained 77% (∼120 mAh g−1) of the capacity at 0.5 C rate. Also, it is worth noting that the electronically conductive paths built by carbon layers could be well maintained during extensive cycles, largely benefited from the zero-volume expansion of the Li4Ti5O12 material during charge/discharge, and this serves a powerful proof for the suitability of the “carbon coating” method in improving the electronic conductivity of the Li4Ti5O12 material.
 | ||
| Fig. 8 TEM images of (a) carbon-coated TiO2 rod precursors and (b) carbon-coated Li4Ti5O12 nanorod materials; HRTEM images of the carbon layer on (c) the TiO2 rod and (d) the Li4Ti5O12 nanorod (ref. 11). | ||
Apart from CVD method, in situ pyrolysis of the carbon source was also applied to realize carbon coating. For example, Wang et al. used PANI (polyaniline)-coated TiO2 and CH3COOLi as precursors to prepare the carbon-coated Li4Ti5O12 nanoparticles in a tailored size of 50–70 nm with double surface modification of conductive Ti(III) and carbon, showing a high power performance.45 Deng et al. prepared Li4Ti5O12/C composites by a one-step solid-state reaction method using four commonly used organic compounds or polymers as the carbon source, i.e., polyacrylate acid (PAA), citric acid (CA), maleic acid (MA) and polyvinyl alcohol (PVA). The as-prepared Li4Ti5O12/C showed the maximum discharge capacity of 168.6 mAh g−1 at 0.2 C, and a capacity of 132.7 mAh g−1 at 10 C rate.71 Very recently, Lee and his coworkers reported a mesostructured spinel Li4Ti5O12 carbon nanocomposite (denoted as Meso-LTO-C) with large and uniform pores (>15 nm) simply synthesized via block copolymer self-assembly using polyisoprene-block-poly(ethylene oxide) (PI-b-PEO) with an sp2-hybridized carbon-containing hydrophobic block. The in situ pyrolyzed carbon not only maintains the porous mesostructure, but also improves the electronic conductivity. The Meso-LTO-C/Li cell exhibits a reversible capacity of 115 mAh g−1 at 10 C rate with 90% capacity retention after 500 cycles.72
Despite considerable enhancement for the rate capability by the “carbon coating” method, it remains a challenge to develop a facile way to simultaneously solve the intrinsic insulation, slow lithium-ion diffusion as well as low volumetric energy density of the Li4Ti5O12 anode material. One satisfactory solution is to prepare micro-/nano-scale hybrid materials coupled with high electronic conductivity.73,74
Our group proposed a facile way to prepare a novel carbon-coated nanosized Li4Ti5O12 nanoporous micro-sphere material (referred to as CN-LTO-NMS).74 The synthesized process was developed based on the “carbon pre-coating” idea presented above in combination with a spray dry process. As the SEM and TEM images show in Fig. 9(A), the carbon distributed uniformly on/in the micron-size spheres but also covered continuously over each nanosized particle with a layer in thickness of about 5 nm, which was believed to establish a well-developed electrically conductive network through the whole material; the nanosized primary particles of about 200 nm ensured fast Li-ion diffusion in the CN-LTO-NMS bulk phase; the interconnected nanopores could form open channels to provide more favorable paths for penetration of the electrolyte, which also favored high rate capability. Besides, the micron-size spherical particles of 10–20 μm induced a large tap density of 0.82 g cm−3 and consequently enhanced the volumetric energy density. Based on the electrochemical performance results in Fig. 9(B), the “CN-LTO-NMS” material demonstrated an amazing rate capability by maintaining 79% of the capacity at 20 C (vs. 0.2 C) (Fig. 9(B)-(a)) as well as excellent cycling stability with a capacity retention of 95% after 1000 cycles at 1 C (vs. 0.2 C) (Fig. 9(B)-(b)).
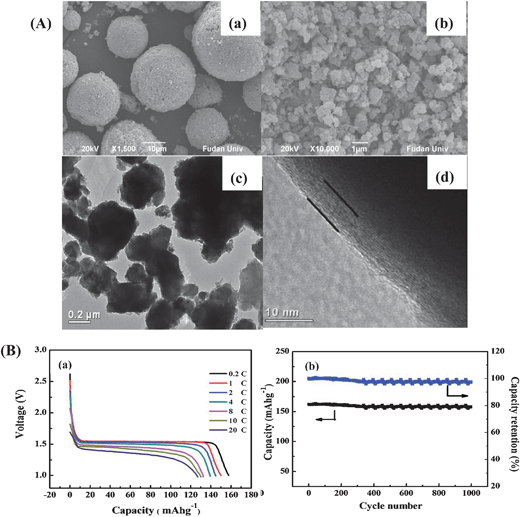 | ||
| Fig. 9 (A) SEM images of CN-LTO-NMS for (a) the micron-size secondary particles and (b) nanosized primary particles; (c) TEM image of CN-LTO-NMS for carbon-coated nanosized particles and (d) HRTEM image for a single carbon-coated nanosized particle, in which the “line” indicates the coated carbon layer. (B) Rate capability under different current rates (from right to left): 0.2, 1, 2, 4, 8, 10 and 20 C (a) and cycling performance for 1000 cycles at 1 C (b) of Li/CN-LTO-NMS half cell (ref. 74). | ||
The concept of preparing nano/micro hybrid materials is also applied in other electrode materials such as LiFePO475,76 and is expected to extend to more electrode materials, which will promote the extensive and broad applications of Li-ion batteries in situations requiring high power, good safety, and long cycle life, such as EV/HEV/PHEVs markets.
A novel nanocable design that combines dual features of the electronic conductive core and ionic conductive shell has been developed. In a typical nanocable, the end of its electronic conductive core is exposed in order to be connected by conductive agents and further to form a 3D nanoscale network.82,83 Based on such design, MWNT@Li4Ti5O12 core/sheath coaxial nanocables with rich hierarchical pores were successfully synthesized and are displayed in Fig. 10 (“MWNT” means multi-walled carbon nanotubes). They showed an impressive rate capability by delivering a discharge capacity of 148, 136, 123, 96, 68 mAh g−1 at the current rate of 5, 10, 20, 40 and 60 C.82
 | ||
| Fig. 10 FESEM images of MWNT@Li4Ti5O12 coaxial nanocables, the arrows indicate the feature of the core/sheath nanocable structure (a) and (b). TEM and HRTEM images of MWNT@Li4Ti5O12 coaxial nanocables (c) and (d), respectively (ref. 82). | ||
Recently, graphene, a new two-dimensional ultra-thin carbon material, has been extensively investigated for holding promise to form a composite with many materials due to its superior electrical conductivity, extremely high surface area of over 2600 m2 g−1, chemical tolerance and structural flexibility.84–91 Since graphene could create a conductive network to effectively hinder aggregation and growth of Li4Ti5O12, the Li4Ti5O12/graphene hybrid nanomaterial is readily synthesized to show fast ionic and electronic transport.92–94 Shen et al. reported a Li4Ti5O12/GNS (graphene nanosheet) nanocomposite. As illustrated in Fig. 11, the Li4Ti5O12 nanoparticles with a size of 30 nm were uniformly anchored on/in GNS matrix by an in situ synthesis, where GNS was the key factor in controlling the size of Li4Ti5O12 particles by acting as the nucleation site for deposits through interaction with metal ions. Such composite displayed an impressive stable discharge capacity of about 90 mAh g−1 at the high rate of 60 C after 100 cycles, which was attributed to rapid electron transport and tailored Li4Ti5O12 particle size.94 PAS (polyacene) obtained from the pyrolysis of the organic compound PF (phenol formaldehyde) is also used in the formation of carbon-based composite.95–97

As an illustration, “surface nitridation” could induce a dense conductive layer well adhered to the surface of Li4Ti5O12 by forming chemical bonding between N and the surface atoms. When Li4Ti5O12 was subjected to thermal nitridation in NH3 atmosphere, a core/shell structure with TiN and Li2CO3 species on the surface and Li4Ti5O12 in the bulk could be formed without lattice parameter change. The introduction of a mixed-valent intermediate phase “Li4+δTi5O12” and a conductive layer “TiN” significantly improved the electronic conductivity of the bulk Li4Ti5O12, and consequently, the N-modified Li4Ti5O12 could retain a capacity of 120 mAh g−1 at the rate of 10 C.102
Furthermore, the composition of coating layer could be tuned by selecting ionic liquid with different components as the a carbon precursor.103–106 For example, by choosing the ionic liquid 1-ethyl-3-methylimidazolium dicyanamide (EMIm-dca) as the precursor, which is composed of C, N, and H elements, an amorphous layer containing carbon from the pyrolysis of ionic and newly formed metallic conductive TiNx interphase was formed on the surface of porous Li4Ti5O12, leading to a mixed conductive 3D network. Such composite showed better rate performance than porous Li4Ti5O12 coated with carbon without N, highlighting the advantage of the formed interphase of a Ti–N–C-like compound in enhancing the electric conductivity as well as the surface stability of Li4Ti5O12.106
Generally, the means of forming a new conductive phase has an advantage of generating a strong interaction between the conductive layer and the active material to ensure a better electronic contact. It is worth noting that the discharge capacity would suffer from the reaction on the surface of active Li4Ti5O12 phase, therefore, surface modification has to be performed to a suitable extent to achieve desirable conductivity with an acceptable capacity penalty.
1.3 Ion doping of Li4Ti5O12 materials for enhancement in rate capability
Ion doping is another way to improve the electronic conductivity of Li4Ti5O12 materials, which forces transformation of partial Ti4+ into Ti3+ to generate mixing Ti3+/Ti4+ as charge compensation and thus increase the concentration of electrons.107–116 A variety of cations and anions including Mg2+, Al3+, Ni3+, Mn3+, Cr3+, Co3+, Fe3+, Ga3+, La3+, Zr4+, Mo4+, V4+, V5+, F− and Br− are doped in Li+, Ti4+ and O2− sites and related investigations in structural properties, ion valence and distribution and conductive characteristics are also performed.103–136 Despite a favorable influence on the electrical conductivity for the doped compounds, the doping ions could enter the Li4Ti5O12 matrix to strongly affect its local and average structure like lattice distortion and further change the electrochemical performances such as specific capacity and cycling stability.Capsoni et al. performed an extensive investigation for Cr and Ni doping of Li4Ti5O12, giving deep insight into the key features related with cation doping issues. The occurrence of Cr3+ insertion into the octahedral 16d site of the Li4Ti5O12 matrix induced a decrease of lattice parameter from 8.3573 to 8.3548 Å; the non-homogeneous distribution of Cr dopant in the structure led to axial distortions. Thus, Cr substitution would strongly affect the spinel cubic structure, which made a sharp contrast to the case of Ni doping. With respect to the conductivity behavior, only Cr doping leads to an increase of conductivity compared with the undoped sample while Ni addition does not. This was attributed to the localized Ni 3d bands causing electrons to be not easily excited.117 In another work, the Cr3+ substitution was reported to be capable of promoting the cycling stability118 similar to the case of Al3+ doping,119,120 while a Ni3+ doped sample showed faster capacity fade during cycles.
As the doped can could partly enter the lattice structure of matrix Li4Ti5O12, it is reasonable to foresee that the doping behavior should have an influence on the morphology and particle size of doped Li4Ti5O12.120–122 For example, Zr4+ doping was found to reduce the particle size to less than 100 nm and effectively alleviate particle agglomeration, contributing to the improvement in rate capability. The optimal composition of Li4Ti4.9Zr0.1O12 obtained a desirable rate capability by maintaining a discharge capacity of 143, 132 and 118 mAh g−1 at 5, 10, and 20 C.122
Some special ion doping could alter electrochemical behaviors of Li4Ti5O12 by discharging the anode down to 0 V, and such deep-discharging anodes help to widen the voltage window and increase the specific capacity of Li-ion batteries.123,124 A typical example is the V-doped sample of Li4Ti4.95V0.05O12. It exhibited lower electrode polarization due to V5+ doping and a stable cycling behavior by retaining a discharge capacity of 218.4 mAh g−1 after 50 cycles at the potential window of 2–0 V, which could be ascribed to the stabilizing effect on the spinel structure by forming cation–oxygen bonding to retain the local symmetry during cycling.125
Ion doping by anions such as Br− and F− is also reported, but findings related with the electrochemical performances are far from reaching common ground and further studies are needed.117,126,127
With respect to the method of ion doping, further insights into structural alternations, interface characteristics, interactions between the doping ions and matrix as well as the resulting ion distribution are necessary to evaluate the complicated effects of ion doping on the electrochemical properties of doped Li4Ti5O12, where more sophisticated techniques such as electron paramagnetic resonance (EPR), Li nuclear magnetic resonance magic-angle spinning (NMR-MAS) and micro-Raman spectroscopy are expected to play an active role in detecting the correlation of structural characteristics and electrochemical performances.117
2. Other compounds in the Li–Ti–O system
2.1 Li2Ti3O7
In addition to the spinel-type Li4Ti5O12, the well-known highly Li-ion conducting ramsdellite-type Li2Ti3O7 is also an attractive anode candidate for Li-ion batteries in the lithium titanium oxides (Li–Ti–O) system.137 As Fig. 12 shows, Li2Ti3O7 belongs to space group of Pnma formed by the ‘framework’ structural part consisting of LiO6 and/or TiO6 octahedron linkage and the ‘tunnel’ part being one-dimensional channels,138–140 which has made it well known for being a fast Li-ion conductor. Different from the two phase coexistence reaction occurring in the case of Li4Ti5O12, the intercalation/extraction of Li2Ti3O7 proceeds in a solid solution manner or one-phase intercalation mechanism, which involves a series of phase transitions as a function of Li content. Theoretically, 2.28 lithium ions can be electrochemically intercalated in Li2Ti3O7 to give a specific capacity of 235 mAh g−1. Besides, it has a good structural stability during cycling due to a small volume change of approximately 2%.140–143 Accordingly, it satisfies several requirements of large capacity, high structural durability and long cycle life for Li-ion batteries.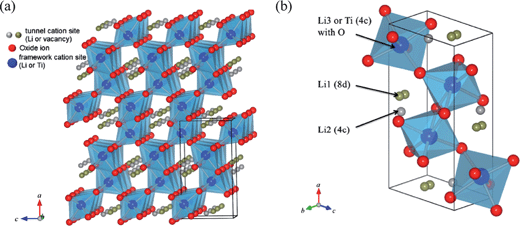 | ||
| Fig. 12 Schematic description of the ramsdellite-type Li2+xTi3O7 crystal structure: (a) projection onto (0 1 0) plane and (b) Li sites on a unit cell (ref. 140). | ||
The ion doping issue is also investigated for ramsdellite-type Li2Ti3O7. As the results point out, substituting less charged metal elements in titanium sites could create wider channels so that more vacancies are available for lithium and the ramsdellite lattice structure is also stabilized, thus improving the specific capacity and cycling performance.143–145 Still, more efforts are needed to reveal the relationship between macroscopic electrochemical behaviors and atomic level structural information before and after doping.146
The development of a low temperature synthetic technique of ion exchange in molten salt provides a possibility of preparing a layered-structure Li2Ti3O7, which belongs to monoclinic system, space group P21/m, and has the Na2Ti3O7-type layered structure constructed by TiO6 octahedra that the interlayer spaces are occupied by Li ions.147,148 It is interesting to investigate the layered structure related electrochemical intercalation. The layered Li2Ti3O7 displayed a flat potential plateau at approximately 1.6 V (vs. Li/Li+) at the initial cycle but after that, the profile gradually adopted a single sloping curve in the voltage range of 2.1–1.6 V similar to that of ramsdellite-type Li2Ti3O7, the cause of which was still unclear since the framework structure was kept intact indicated by XRD. A discharge capacity of 136 mAh g−1 was reached for the first cycle and only half was retained for the following cycles, where a better Na+/Li+ exchange was reported to be favorable for the reversibility of sample. However, the lack of substantial cycling data makes it impossible to determine the durability of the monoclinic structure during repeated charge/discharge process.149
2.2 Li2Ti6O13
Using the ionic exchange method, another lithium titanium oxide in the Li–Ti–O system, Li2Ti6O13 with the monoclinic C2/m space group, can be prepared from tunnel-structured Na2Ti6O13. It nearly maintained an unchanged basic [Ti6O13]2− framework in the parent Na2Ti6O13 yet with shifting of Li occupation site in the tunnel space.150–154 Interestingly, the total Li-ion conductivity in monoclinic Li2Ti6O13, estimated to be σtotal = 5.60 × 10−6 S cm−1 at room temperature, is comparable to the good Li-ion conducting ramsdellite-type Li2Ti3O7.154The electrochemical properties and mechanism have also been investigated recently.154–158 It delivered a discharge capacity of above 200 mAh g−1 with a flat plateau at about 1.5 V (vs. Li/Li+), however, the following Li extraction profile deviated to a regime with a sloping feature between 2.1 and 1.6 V and the charge capacity was drastically reduced to 160158 or even 90 mAh g−1,154 which was retained after the two initial cycles. The significant capacity loss of more than 30% was probably caused by the degradation of the host Li2Ti6O13 structure or irreversible phase transition.154
3. TiO2 polymorphs
To date, TiO2 polymorphs including anatase, rutile and TiO2-B (bronze), have been reported for lithium electrochemical reactivity. Li-reaction with the TiO2 polymorphs is conveniently expressed as| TiO2 + xLi+ + xe− ↔ LixTiO2 |
3.1 Rutile and anatase TiO2
As the most thermodynamically stable polymorph of TiO2, rutile in its bulk crystalline form can only accommodate negligible Li (<0.1 Li per TiO2 unit) at room temperature.159–161 However, the charge/discharge properties of rutile with a nanostructured morphology could be greatly improved. For example, Liu and co-workers recently reported that mesoporous rutile could accommodate more than 0.7 Li+ (235 mAh g−1) during the first discharge with a reversible capacity of 0.55 Li+ (185 mAh g−1) between 1 and 3 V vs. Li+/Li.162In comparison with the rutile polymorph, the uptake of Li+ appears more facile in the anatase lattice. It delivers a reversible capacity as maximum as x = 0.5 in bulk anatase with a flat voltage profile at 1.7 V vs. Li+/Li,163–165 indicating a classic bi-phase electrochemical reaction process of the Li-insertion/extraction. Similar to the rutile structure, decreasing the particle size into the nano-regime (<100 nm) alternates the electrochemical reactions and reactivity with Li+, giving a reversible capacity as large as 200 mAh g−1.166–172 The Li-interaction with the nanostructures appears to deviate from the two phase equilibrium phenomenon in the bulk materials, but behaving more like solid solution.166 The more detailed information of different types of TiO2 as anode for LIB, especially for rutile and anatase TiO2, can be found in the previous literatures.157
3.2 TiO2-B
TiO2-B, possessing a higher specific capacity than other compounds in the Li–Ti–O system or other polymorphs of titania like anatase or rutile, has gained tremendous attention for its use as a desirable host for Li-ion intercalation.173–176 The crystal structure of TiO2-B is monoclinic (space group C2/m) composed of edge and corner-sharing TiO6 octahedra (Fig. 13). One of its most appealing structural features lies in the characteristic parallel channels running along the [010] orientation. Such a unique feature has been shown to be favorable for the incorporation and diffusion of Li ions in the channels theoretically177–180 and experimentally,173–176 which is important for its use as an anode in Li-ion batteries. To examine the intrinsic Li-ion mobility in TiO2-B, three Li diffusion paths with respect to their energy barriers were calculated based on DFT study: (010) (path ii) < (001) (path i) < (100) (path iii), demonstrating that the favored pathway for facile Li diffusion is along the b-axis channel in the [010] direction and suggesting anisotropic transport behavior in TiO2-B single crystals.179 Moreover, the kinetics of lithium storage in TiO2-B are found to be governed by the mixing effects of a pseudo-capacitive faradic process and diffusion-limited solid-state reaction, which are further dependent on microstructures of different TiO2-B materials.181,182 Therefore, it is especially important for morphology tailoring and structural design to take full advantage of the intrinsic facile lithium diffusion path.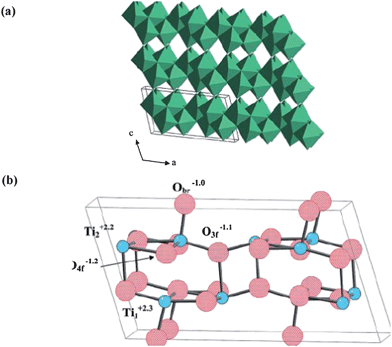 | ||
| Fig. 13 Bulk crystal structure of TiO2-B: (a) ribbons of edge-sharing TiO6 octahedra, connected in the ‘c’ direction by corner-sharing; (b) ball-and-stick model of the unit cell with lattice sites and calculated Bader charges as labeled (ref. 179). | ||
Hydrothermal method is one common approach to prepare different morphologies of TiO2-B by controlling the hydrothermal temperature, treatment time, base solution and the following heat treatment. Armstrong et al. prepared TiO2-B nanowires with a typical diameter of 20–40 nm and a length of 2–10 μm, which reaches a maximum composition of Li0.91TiO2-B to deliver an initial discharge capacity of 305 mAh g−1. Besides, the capacity of the TiO2-B nanowires after 50 cycles could be maintained twice that of TiO2-B nanoparticles with an average particle size comparable to the diameter of TiO2-B nanowires, illustrating the importance of controlling the dimensions of nanostructured materials for optimization of electrochemical performance.183–185 Furthermore, Armstrong et al. compared TiO2-B nanotubes with nanowires to probe effects of the high surface area on the electrochemical performance. They found that nanotubes with a larger surface-to-volume ratio delivered a higher initial discharge capacity but a larger irreversibility, suggesting an strong correlation between a high surface area and side reactions at the electrode/electrolyte interface; moreover, a greater voltage separation between charge and discharge curves was observed to indicate the inferior electrochemical kinetics, which is possibly related with structural defects.181,182 The temperature of heat treatment following the hydrothermal process needs careful control to retain the morphology and phase purity of high-crystalline TiO2-B.186–188 A systematic thermal transformation study revealed that nanostructured H2Ti3O7 could convert into TiO2-B upon annealing in the temperature range of 120–400 °C without losing its nanotubular morphology, accompanied by a decrease in interlayer spacing of nanotube walls.186 Similarly, nanorod titanates can transform to rod-shaped TiO2-B at 450 °C, except that a higher temperature was necessary for phase transformation from TiO2-B to anatase and rutile.187 Both cases reveal the intrinsic metastable characteristic of TiO2-B.
Under a milder condition using reflux (T ∼ 120 °C, P = 1 bar), TiO2-B nanoribbons with a small thickness of 6 nm were prepared. This material displayed a reversible capacity of 200 mAh g−1 at 3 C rate, and a good cyclability with a capacity loss of 5% over 500 cycles at 3 C rate. It could maintain 100 mAhg−1 at 15 C rate for a 50% carbon black loaded electrode, highlighting the importance of the electronic conductivity for the rate capability of TiO2-B active material.174
The sol–gel method to prepare TiO2-B involves unique selection of reactants and process design. For example, a novel sol–gel method using mixtures of surfactants and ionic liquids at the low temperature of 100 °C was proposed towards the synthesis of nanocrystalline TiO2-B.189 In another case, a porous 2D TiO2-B nanosheet with different traces of anatase was prepared, via a simple sol–gel process using TTIP and formic acid as precursors followed by heat treatment above 250 °C. The initial discharging capacity can reach as high as 357 mAh g−1, while the reversibility and retention of capacity needs improvement by optimization of the preparation conditions.190
The performance of TiO2-B electrodes at high discharge/charge rates is undesirable partly due to the aggregation tendency of TiO2-B nanomaterials, which leads to an increase in interparticle contact resistance.191 Preparing hybrid nanostructured electrodes is one effective solution to address this problem. TiO2-B@carbon composite nanowires with a novel core/shell architecture was presented in Fig. 14, where the TiO2-B nanofiber core provided open channels for facile Li+ transport and an amorphous carbon shell possessed high Li+ capacity as well as suppressed the aggregation of nanofibers. The composite nanowires extended the cut-off voltage window to a wider 3–0 V from 3–1 V (vs. Li/Li+), common for TiO2-B materials, and exhibited a high reversible capacity of 560 mAh g−1 after 100 cycles at the current density of 30 mAg−1 as well as an excellent rate capability by maintaining 200 mAh g−1 when cycled at the current density of 750 mAg−1.192
 | ||
| Fig. 14 FESEM images of (a) TiO2-B@carbon composite nanowires and (b) single TiO2-B@carbon composite nanowire with porous surface structure; (c) TEM image and SAED pattern (inset) of a single TiO2-B@carbon composite nanowire; (d) HRTEM image of a section of a single TiO2-B@carbon composite nanowire (d) (ref. 192). | ||
Storage devices incorporating TiO2-B anodes are also reported. For example, the Li-ion battery consisting of TiO2-B nanowires as the anode and LiFePO4 or LiNi0.5Mn1.5O4 as the cathode, demonstrated superior safety and inherent overcharge protection compared to using graphite as the anode.173 Moreover, a hybrid supercapacitor fabricated using TiO2-B nanowires as the anode and CNTs as the cathode delivered an enhanced energy density at a rate of 10 C, double the value of the CNTs–CNTs supercapacitor, while maintaining desirable cycling stability.193
However, few similar studies for TiO2-B have been reported, which is most likely due to its metastable thermodynamics as described above. Most strategies for enhancement of electronic conductivity involve high-temperature heat treatment, which are not feasible in the case of TiO2-B since such methodology would fail to retain the phase purity of TiO2-B due to the thermal transformation. In this perspective, low-temperature surface modification or composite preparation may offer potential ways in improving the insulating property of TiO2-B. Also, extensive studies targeting morphology tailoring in order to reduce material agglomeration and increase structural robustness are equally important to optimize the electrochemical properties.
4. A family of alkali titanium oxides and their derivatives
4.1 Compounds in the Na–Ti–O system
Besides lithium titanates in the Li–Ti–O system, another series of alkali titanates represented by a general formula of A2TinO2n+1 (n = 3–9, A is an alkaline metal) have been reported, aiming to explore their potential as electrode materials in view of the layered structures provided by the atomic arrangement of O2− and Ti4+. Interestingly, the series of layered compounds have a typical ‘block’ structure and further display a structural evolution owing to varied ‘block’ configuration, size and arrangement.202In a typical Na–Ti–O system, the structure-related electrochemical properties of Na2Ti3O7 and Na2Ti6O13 are mostly discussed. Na2Ti3O7 consists of TiO6 octahedra, which share edges and corners. It delivered an initial capacity of 44 mAh g−1 with a sloping profile between 1.6 and 1.0 V (vs. Li/Li+), which was gradually decreased to less than 30 mAh g−1 at the 10th cycle.149 For comparison, Na2Ti6O13 delivered a large initial discharge capacity of 140 mAh g−1 but also suffered from a considerable capacity loss of about 20% during the charge while the shape of voltage profile was maintained, indicating a possible side reaction with the electrolyte. Meanwhile, K2Ti6O13 showed a similar electrochemical behavior except for having a different charge/discharge curve.203,204
4.2 Compounds in the H–Ti–O system
Despite the favorable open structure, alkali-based titanium oxides show a restricted reversible capacity. A possible explanation comes from the large radius of alkali atom, which may have an adverse effect in the facile lithium transport or accessibility of intercalated Li sites.147–150 Hydrogen titanium oxides (H–Ti–O), as one important series of derivatives of alkali titanium oxides by substituting the alkali element for H, were originally applied in photocatalysis,205 sensors206 and solar cells207 and currently attract much attention for application in Li-ion batteries.208–212 Structurally, one H–Ti–O compound could transform to another via thermally-induced structural transformation from loss of the interlayer water with a condensed layer, and a complete dehydration results in the formation of the most condensed phase TiO2.186,187H2Ti3O7 is the best studied among the H–Ti–O family. It inherits the Ti–O skeleton from the parent Na2Ti3O7 but the positions of H+ show quite a different distribution in the interlayer.212 Current studies are mainly focused on the morphology-related electrochemical properties of H2Ti3O7.
Nanotubular H2Ti3O7, with an outer diameter of several nanometers and a high surface area of more than 300 m2 g−1, often delivered a large initial discharge capacity of about 300 mAh g−1 with a curve consisting of a potential plateau and slope, but suffered a significant capacity loss ranging from 30% to 50% accompanied by a disappearance of the plateau. The detrimental effect of Li2O on the surface and inside the layer caused by the reaction of the structural binding water with lithium was blamed for the huge capacity loss.208,209 Similar to the case of TiO2-B, it was also proposed that the electrochemical lithium storage of H2Ti3O7 was controlled by the mixed process of both the pseudocapacitive effect and diffusion-limited reaction.208
In comparison, H2Ti3O7 nanowires with an average diameter of about 100 nm and a smaller surface area of 152.6 m2 g−1 showed a better reversibility with the sloping feature of the voltage profile kept unchanged.210 The rate capability of H2Ti3O7 nanowires was also reported.210,211 The result of a stabilized capacity of about 100 mAh g−1 at a high rate of 40 Ag−1 after 200 cycles was impressive,211 providing a possibility for H2Ti3O7 nanostructures in high-rate applications. Unfortunately, extensive cycling performance upon hundreds of cycles is seldom discussed and further research is still necessary to evaluate the structural durability.
5. Summery and outlook
Ti-based oxides have demonstrated their potential as suitable anode candidates for Li-ion batteries due to attributes of environmental friendliness, high safety and excellent cycling stability. Li–Ti–O compounds have capacity of about 200 mAh g−1 with a charge/discharge plateau of about 1.5 V, and typically exhibit a good cycling stability. Substantial efforts have been focused on the improvement of the rate capability resulting from the intrinsic low electronic conductivity and slow lithium diffusion. Introducing composite nanostructures by morphology design and grain control, which integrate with an electron-conductive phase (ion doping or incorporation of new electrically conductive phases) at the nanoscale, have shown remarkable rate performance enhancement, presumably because of both shorter Li-ion diffusion distances and improved electron transport. Among the Li–Ti–O systems, Li4Ti5O12 is ready for practical application in areas of portable electronics, stationary power sources for renewable sources and electrically-powered vehicles (EV/HEV/PHEVs) by introduction of diversified aim-specific products. For example, a “Nanosafe” Li-ion battery using Li4Ti5O12 as the anode and LiMn2O4 as the cathode developed by Altair Engineering Inc., has given a fabulous performance in smoothing out the fluctuation of grid applications by presenting an ultra-high energy conversion efficiency of more than 91% during the charge/discharge process. Battery materials capable of ultra-high charging and discharging are indispensable components to promote the popularity of electric vehicles. EnerDel and Think Nordic Corporation co-developed a type of HEV, where the electrically-powered system using Li4Ti5O12 as the anode was applied to features such as reducing engine idling and recuperating the surplus energy generated by a decelerating vehicle for use in motor assist functions. For high-rate applications, Toshiba Corporation developed a battery branded as “SCiB”, which employs Li4Ti5O12 as the anode and LiCoO2 as the cathode, which has exhibited excellent cycling stability with a capacity loss of less than 10% after 3000 cycles and remarkable rate capability by fast charging capacity of more than 90% within 5 min. Furthermore, a Li-ion battery comprising a Li4Ti5O12 anode and LiMPO4 (M = Fe, Mn) cathode also demonstrates a possible use in load levelling applications owing to its impressive stability upon extensive cycling and excellent safety features.213TiO2 polymorphs, including anatase, rutile and TiO2-B (bronze), can deliver a theoretical capacity as large as 335 mAh g−1. Li-insertion activity for TiO2 polymorphs is not only dependent on the crystal size, but also on particle morphology and crystallographic orientation. For example, with a much higher diffusivity along the c-axis than in the ab-plane, rutile nanorods grown along [001] direction demonstrated a substantially improved Li-insertion activity. TiO2-B appears to have higher Li-insertion electrochemistry due to the prominent advantage of its open layered structure. Similar to these Li–Ti–O compounds, nanoarchitectured design and feasibility of morphology control for optimization of the electrochemical performance are highlighted. Moreover, a way to improve its cycling stability is another concerning issue for these TiO2 polymorphs.
Despite great progress either in the lab research or practical application, challenges for Ti-based materials still remain. For example, gas evolution usually occurs during the cycling and storage, which is most likely due to the large surface area of nanosized particles and good catalysis of Ti-based compounds, but the detailed mechanism has not been well understood until very recently. The difficulty in controlling the state of charge (SOC) should be solved due to the flat charge/discharge curve of some Ti-based compounds when combined with cathode materials with flat charge/discharge plateaus. Besides various targeting material designs to tailor the material for a certain purpose, the overall optimization of the battery plays a decisive role in its practical application, which needs a good compatibility between each component such as the anode, cathode and electrolyte, coupled with sophisticated assembling techniques to permit fast, reversible charging/discharging with high capacity.
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (20925312), the State Key Basic Research Program of PRC (2011CB935903), and Shanghai Science & Technology Committee (10JC1401500, 08DZ2270500).References
- S. Yang, X. Feng, L. Zhi, Q. Cao, J. Maier and K. Müllen, Adv. Mater., 2010, 22, 838 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, Y. Hara and Y. Adachi, J. Electrochem. Soc., 2000, 147, 1245 CrossRef CAS.
- M. Yoshio, H. Wang and K. Fukuda, Angew. Chem., 2003, 115, 4335 CrossRef.
- K. S. Park, A. Benayad, D. J. Kang and S. G. Doo, J. Am. Chem. Soc., 2008, 130, 14930 CrossRef CAS.
- M. Yoshio, H. Wang, K. Fukuda, T. Umeno, T. Abe and Z. Ogumi, J. Mater. Chem., 2004, 14, 1754 RSC.
- E. Ferg, R. J. Gummow, A. Dekock and M. M. Thackeray, J. Electrochem. Soc., 1994, 141, L147 CrossRef CAS.
- A. Deschanvres, B. Raveau and Z. Sekkal, Mater. Res. Bull., 1971, 6, 699 CrossRef CAS.
- F. Ronci, P. Reale, B. Scrosati, P. Panero, V. Rossi Albertini, P. Perfetti, M. di Michiel and J. M. Merino, J. Phys. Chem. B, 2002, 106, 3082 CrossRef CAS.
- L. Cheng, X. L. Li, H. J. Liu, H. M. Xiong, P. W. Zhang and Y. Y. Xia, J. Electrochem. Soc., 2007, 154, A692 CrossRef CAS.
- G. N. Zhu, C. X. Wang and Y. Y. Xia, J. Electrochem. Soc., 2011, 158, A102 CrossRef CAS.
- L. Cheng, J. Yan, G. N. Zhu, J. Y. Luo, C. X. Wang and Y. Y. Xia, J. Mater. Chem., 2010, 20, 595 RSC.
- J. Wang, X. M. Liu, H. Yang and X. D. Shen, J. Alloys Compd., 2011, 509, 712 CrossRef CAS.
- H. Liu, Y. Feng, K. Wang and J. Xie, J. Phys. Chem. Solids, 2008, 69, 2037 CrossRef CAS.
- C. H. Chen, J. T. Vaughey, A. N. Jansen, D. W. Dees, A. J. Kahaian, T. Goacher and M. M. Thackeray, J. Electrochem. Soc., 2001, 148, A102 CrossRef CAS.
- E. M. Sorensen, S. J. Barry, H. K. Jung, J. R. Rondinelli, J. T. Vaughey and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 482 CrossRef CAS.
- J. Kim and J. Cho, Electrochem. Solid-State Lett., 2007, 10, A81 CrossRef CAS.
- E. M. Sorensen, S. J. Barry, H. K Jung, J. R. Rondinelli, J. T. Vaughey and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 482 CrossRef CAS.
- M. Wilkening, R. Amade, W. Iwaniak and P. Heitjans, Phys. Chem. Chem. Phys., 2007, 9, 1239 RSC.
- S. Scharner, W. Weppner and P. Schmid-Beurmann, J. Electrochem. Soc., 1999, 146, 857 CrossRef CAS.
- M. Wagemaker, D. R. Simon, E. M. Kelder, J. Schoonman, C. Ringpfeil, U. Haake, D. Lutzenkirchen-Hecht, R. Frahm and F. M. Mulder, Adv. Mater., 2006, 18, 3169 CrossRef CAS.
- J. F. Colin, V. Godbole and P. Novák, Electrochem. Commun., 2010, 12, 804 CrossRef CAS.
- J. Jiang and J. R. Dhan, J. Electrochem. Soc., 2006, 153, A310 CrossRef CAS.
- I. Belharouak, Y. K. Sun, W. Lu and K. Amine, J. Electrochem. Soc., 2007, 154, A1083 CrossRef CAS.
- C. Y. Ouyang, Z. Y. Zhong and M. S. Lei, Electrochem. Commun., 2007, 9, 1107 CrossRef CAS.
- T. Brousse, P. Fragnaud, R. Marchand, D. M. Schleich, O. Bohnke and K. West, J. Power Sources, 1997, 68, 412 CrossRef CAS.
- G. G. Amatucci, F. Badway, A. Du Pasquier and T. Zheng, J. Electrochem. Soc., 2001, 148, A930 CrossRef CAS.
- K. M. Colbow, J. R. Dahn and R. R. Haering, J. Power Sources, 1989, 26, 397 CrossRef CAS.
- E. Ferg, R. J. Gummow, A.d. Kock and M. M. Thackeray, J. Electrochem. Soc., 1994, 141, L147 CrossRef CAS.
- T. Ohzuku, A. Ueda and N. Yamamoto, J. Electrochem. Soc., 1995, 142, 1431 CrossRef CAS.
- S. I. Pyun, S. W. Kim and H. C. Shin, J. Power Sources, 1999, 81–82, 248 CrossRef CAS.
- K. Zaghib, M. Simoneau, M. Armand and M. Gauthier, J. Power Sources, 1999, 81–82, 300 CrossRef CAS.
- K. Zaghib, M. Armand and M. Gauthier, J. Electrochem. Soc., 1998, 145, 3135 CrossRef CAS.
- K. C. Hsiao, S. C. Liao and J. M. Chen, Electrochim. Acta, 2008, 53, 7242 CrossRef CAS.
- N. S. Ergang, J. C. Lytle, H. Yan and A. J. Stein, J. Electrochem. Soc., 2005, 152, A1989 CrossRef CAS.
- E. M. Sorensen, S. J. Barry, H. K. Jung, J. R. Rondinelli, J. T. Vaughey and K. R. Poeppelmeier, Chem. Mater., 2006, 18, 482 CrossRef CAS.
- A. S. Prakash, P. Manikandan, K. Ramesha, M. Sathiya, J. M. Tarascon and A. K. Shukla, Chem. Mater., 2010, 22, 2857 CrossRef CAS.
- P. Liu, E. Sherman and M. Verbrugge, J. Solid State Electrochem., 2009, 14, 585 CrossRef.
- A. S. Aricò, P. Bruce, B. Scrosati, J. M. Tarascon and W. V. Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef.
- M. Green, E. Fielder, B. Scrosati, M. Wachtler and J. S. Moreno, Electrochem. Solid-State Lett., 2003, 6, A75 CrossRef CAS.
- C. Y. Lin and J. G. Duh, J. Alloys Compd., 2011, 509, 3682 CrossRef CAS.
- Y. J. Hao, Q. Y. Lai, J. Z. Lu, D. Q. Liu and X. Y. Ji, J. Alloys Compd., 2007, 439, 330 CrossRef CAS.
- W. J. H. Borghols, M. Wagemaker, U. Lafont, E. M. Kelder and F. M. Mulder, J. Am. Chem. Soc., 2009, 131, 17786 CrossRef CAS.
- M. Wagemaker, D. R. Simon, E. M. Kelder, J. Schoonman, C. Ringpfeil, U. Haake, D. Lützenkirchen-Hecht, R. Frahm and F. M. Mulder, Adv. Mater., 2006, 18, 3169 CrossRef CAS.
- M. Wagemaker, E. R. H. van Eck, A. P. M. Kentgens and F. M. Mulder, J. Phys. Chem. B, 2009, 113, 224 CrossRef CAS.
- Y. G. Wang, H. M. Liu, K. X. Wang, H. Eiji, Y. R. Wang and H. S. Zhou, J. Mater. Chem., 2009, 19, 6789 RSC.
- Y. J. Hao, Q. Y. Lai, D. Q. Liu, Z. Xu and X. Y. Ji, Mater. Chem. Phys., 2005, 94, 382 CrossRef CAS.
- S. S. Lee, K. T. Byun, J. P. Park, S. K. Kim, H. Y. Kwaka and I. W. Shim, Dalton Trans., 2007, 4182 RSC.
- L. Kavana and M. Grătzel, Electrochem. Solid-State Lett., 2002, 5, A39 CrossRef.
- T. Yuan, K. Wang, R. Cai, R. Ran and Z. P. Shao, J. Alloys Compd., 2009, 477, 665 CrossRef CAS.
- P. Afanasiev and C. Geantet, Coord. Chem. Rev., 1998, 178–180, 1725 CrossRef CAS.
- L. Cheng, H. J. Liu, J. J. Zhang, H. M. Xiong and Y. Y. Xia, J. Electrochem. Soc., 2006, 153, A1472 CrossRef CAS.
- Y. Bai, F. Wang, F. Wu, C. Wu and L. Y. Bao, Electrochim. Acta, 2008, 54, 322 CrossRef CAS.
- L. H. Yang, C. Dong and J. Guo, J. Power Sources, 2008, 175, 575 CrossRef CAS.
- J. Li, Y. L. Jin, X. G. Zhang and H. Yang, Solid State Ionics, 2007, 178, 1590 CrossRef CAS.
- G. Yang, G. Wang and W. Hou, J. Phys. Chem. B, 2005, 109, 11186 CrossRef CAS.
- C. H. Jiang, M. Ichihara, I. Honma and H. S. Zhou, Electrochim. Acta, 2007, 52, 6470 CrossRef CAS.
- Y. S. Lin and J. G. Duh, J. Power Sources, 2011, 196, 10698 CrossRef CAS.
- C. H. Jiang, Y. Zhou, I. Honma, T. Kudo and H. S. Zhou, J. Power Sources, 2007, 166, 514 CrossRef CAS.
- N. D. He, B. S. Wang and J. J. Huang, J. Solid State Electrochem., 2009, 14, 1241 CrossRef.
- Y. F. Tang, L. Yang, S. H. Fang and Z. Qiu, Electrochim. Acta, 2009, 54, 6244 CrossRef CAS.
- K. S. W. Sing, D. H. Everrett, R. A. W. Haul, L. Moscou, R. A. Pierotti, J. Rouquerol and T. Siemieniewska, Pure Appl. Chem., 1985, 57, 603 CrossRef CAS.
- J. R. Li, Z. L. Tang and Z. T. Zhang, Electrochem. Commun., 2005, 7, 894 CrossRef CAS.
- S. C. Lee, S. M. Lee, J. W. Lee, J. B. Lee, S. M. Lee, S. S. Han, H. C. Lee and H. J. Kim, J. Phys. Chem. C, 2009, 113, 18420 CAS.
- D. K. Lee, H. W. Shim, J. S. An, C. M. Cho, I. S. Cho, K. S. Hong and D. W. Kim, Nanoscale Res. Lett., 2010, 5, 1585 CrossRef CAS.
- Y. F. Tang, L. Yang, Z. Qiu and J. S. Huang, J. Mater. Chem., 2009, 19, 5980 RSC.
- L. F. Shen, C. Z. Yuan, H. J. Luo, X. G. Zhang, K. Xu and Y. Y. Xia, J. Mater. Chem., 2010, 20, 6998 RSC.
- J. Z. Chen, L. Yang, S. H. Fang and Y. F. Tang, Electrochim. Acta, 2010, 55, 6596 CrossRef CAS.
- K. Amine, I. Belharouak, Z. H. Chen, T. Tran, H. Yumoto, N. Ota, S. T. Myung and Y. K. Sun, Adv. Mater., 2010, 22, 3052 CrossRef CAS.
- S. H. Yu, A. Pucci, T. Herntrich, M. G. Willinger, S. H. Baek, Y. E. Sung and N. Pinna, J. Mater. Chem., 2011, 21, 806 RSC.
- Z. Xiong, S. Shi, C. Ouyang, M. Lei, L. Hu, Y. Ji, Z. Wang and L. Chen, Phys. Lett. A, 2005, 337, 247 CrossRef CAS.
- X. B. Hu, Z. J. Lin, K. R. Yang, Y. J. Huai and Z. H. Deng, Electrochim. Acta, 2011, 56, 5046 CrossRef CAS.
- E. Kang, Y. S. Jung, G. H. Kim, J. Y. Chun, U. Wiesner, A. C. Dillon, J. K. Kim and J. Lee, Adv. Funct. Mater., 2011, 21, 4349 CrossRef CAS.
- H. G. Jung, S. T. Myung, C. S. Yoon, S. B. Son, K. H. Oh, K. Amine, B. Scrosati and Y. K. Sun, Energy Environ. Sci., 2011, 4, 1345 CAS.
- G. N. Zhu, H. J. Liu, J. H. Zhuang, C. X. Wang, Y. G. Wang and Y. Y. Xia, Energy Environ. Sci., 2011, 4, 4016 CAS.
- H. G. Jung, S. T. Myung, C. S. Yoon, S. B. Son, K. H. Oh, K. Amine, B. Scrosati and Y. K. Sun, Energy Environ. Sci., 2011, 4, 1345 CAS.
- H. Yang, X. L. Wu, M. H. Cao and Y. G. Guo, J. Phys. Chem. C, 2009, 113, 3345 CAS.
- A. Thess, R. Lee, P. Nikolaev, H. Dai, P. Petit, J. Robert, C. Xu, Y. H. Lee, S. G. Kim, A. Rinzler, D. T. Colbert, G. Scuseria, D. Tomanek, J. E. Fischer and R. Smalley, Science, 1996, 273, 483 CAS.
- B. J. Landi, R. P. Raffaelle, M. J. Heben, J. L. Alleman, W. VanDerveer and T. Gennett, Nano Lett., 2002, 2, 1329 CrossRef CAS.
- R. P. Raffaelle, B. J. Landi, J. D. Harris, S. G. Bailey and A. F. Hepp, Mater. Sci. Eng., B, 2005, 116, 233 CrossRef.
- X. Li, M. Z. Qu and Z. L. Yu, Solid State Ionics, 2010, 181, 635 CrossRef CAS.
- J. Huang and Z. Jiang, Electrochim. Acta, 2008, 53, 7756 CrossRef CAS.
- L. F. Shen, C. Z. Yuan, H. J. Luo, X. G. Zhang, K. Xua and F. Zhang, J. Mater. Chem., 2011, 21, 761 RSC.
- K. Naoi, S. Ishimoto, Y. Isobe and S. Aoyagi, J. Power Sources, 2010, 195, 6250 CrossRef CAS.
- G. X. Wang, X. P. Shen, J. Yao and J. Park, Carbon, 2009, 47, 2049 CrossRef CAS.
- D. Y. Pan, S. Wang, B. Zhao, M. H. Wu, H. J. Zhang, Y. Wang and Z. Jiao, Chem. Mater., 2009, 21, 3136 CrossRef CAS.
- Z. S. Wu, W. C. Ren, L. Wen, L. B. Gao, J. P. Zhao, Z. P. Chen, G. G. Zhou, F. Li and H. M. Cheng, ACS Nano, 2010, 4, 3187 CrossRef CAS.
- D. H. Wang, D. W. Choi, J. Li, Z. G. Yang, Z. M. Nie, R. Kou, D. H. Hu, C. M. Wang, L. V. Saraf, J. G. Zhang, I. A. Aksay and J. Liu, ACS Nano, 2009, 3, 907 CrossRef CAS.
- Z. B. Lei, N. Christov and X. S. Zhao, Energy Environ. Sci., 2011, 4, 1866 CAS.
- X. J. Zhu, Y. W. Zhu, S. Murali, M. D. Stollers and R. S. Ruoff, ACS Nano, 2011, 5, 3333 CrossRef CAS.
- Q. L. Hao, H. L. Wang, X. J. Yang, L. Lu and X. Wang, Nano Res., 2010, 4, 323 CrossRef.
- J. T. Zhang, J. W. Jiang and X. S. Zhao, J. Phys. Chem. C, 2011, 115, 6448 CAS.
- H. K. Kim, S. M. Bak and K. B. Kim, Electrochem. Commun., 2010, 12, 1768 CrossRef CAS.
- N. Zhu, W. Liu, M. Q. Xue, Z. Xie, D. Zhao, M. N. Zhang, J. T. Chen and T. B. Cao, Electrochim. Acta, 2010, 55, 5813 CrossRef CAS.
- L. F. Shen, C. Z. Yuan, H. J. Luo, X. G. Zhang, S. D. Yang and X. J. Lu, Nanoscale, 2011, 3, 572 RSC.
- H. M. Xie, R. S. Wang, J. R. Ying, L. Y. Zhang, A. F. Jalbout, H. Y. Yu, G. L. Yang, X. M. Pan and Z. M. Su, Adv. Mater., 2006, 18, 2609 CrossRef CAS.
- L. Q. Sun, R. H. Cui, A. F. Jalbout, M. J. Li, X. M. Pan, R. S. Wang and H. M. Xie, J. Power Sources, 2009, 189, 522 CrossRef CAS.
- H. Yu, X. Zhang, A. F. Jalbout, X. Yan, X. Pan, H. G. Xie and R. Wang, Electrochim. Acta, 2008, 53, 4200 CrossRef CAS.
- S. Huang, Z. Wen, X. Zhu and Z. Gu, Electrochem. Commun., 2004, 6, 1093 CrossRef CAS.
- S. Huang, Z. Wen, J. Zhang, Z. Gu and X. Xu, Solid State Ionics, 2006, 177, 851 CrossRef CAS.
- S. Huang, Z. Wen, J. Zhang and X. Yang, Electrochim. Acta, 2007, 52, 3704 CrossRef CAS.
- S. Huang, Z. Wen, B. Lin, J. Han and X. Xu, J. Alloys Compd., 2008, 457, 400 CrossRef CAS.
- J. S. Lee, X. Q. Wang, H. M. Luo, G. A. Baker and S. Dai, J. Am. Chem. Soc., 2009, 131, 4596 CrossRef CAS.
- J. S. Lee, X. Q. Wang, H. M. Luo and S. Dai, Adv. Mater., 2009, 22, 1004 Search PubMed.
- J. P. Paraknowitsch, J. Zhang, D. S. Su, A. Thomas and M. Antonietti, Adv. Mater., 2010, 22, 87 CrossRef CAS.
- J. P. Paraknowitsch, A. Thomas and M. Antonietti, J. Mater. Chem., 2010, 20, 6746 RSC.
- L. Zhao, Y. S. Hu, H. Li, Z. X. Wang and L. Q. Chen, Adv. Mater., 2011, 23, 1385 CrossRef CAS.
- V. S. Hernandez, L. M. Torres Martinez, G. C. Mather and A. R. West, J. Mater. Chem., 1996, 6, 1533 RSC.
- M. Nakayama, Y. Ishida, H. Ikuta and M. Wakihara, Solid State Ionics, 1999, 117, 265 CrossRef CAS.
- A. D. Robertson, L. Trevino, H. Tukamoto and J. T. S. Irvine, J. Power Sources, 1999, 81–82, 352 CrossRef CAS.
- A. D. Robertson, H. Turamoto and J. T. S. Irvine, J. Electrochem. Soc., 1999, 146, 3958 CrossRef CAS.
- T. Ohzuku, K. Tatsumi, N. Matoba and K. Sawai, J. Electrochem. Soc., 2000, 147, 3592 CrossRef CAS.
- P. Reale, S. Panero, F. Ronci, V. Rossi Albertini and B. Scrosati, Chem. Mater., 2003, 15, 3437 CrossRef CAS.
- N. Jovic, B. Antic, A. Kremenovic, A. Spasojevic-de Bire and V. Spasojevic, Phys. Status Solidi A, 2003, 198, 18 CrossRef CAS.
- I. A. Leonidov, O. N. Leonidova, R. F. Samigullina and M. V. Patrakeev, J. Struct. Chem., 2004, 45, 262 CrossRef CAS.
- S. Huang, Z. Wen, X. Zhu and Z. Lin, J. Electrochem. Soc., 2005, 152, A186 CrossRef CAS.
- M. Ganesan, Ionics, 2007, 14, 395 CrossRef.
- D. Capsoni, M. Bini, V. Massarotti, P. Mustarelli, S. Ferrari, G. Chiodelli, M. C. Mozzati and P. Galinetto, J. Phys. Chem. C, 2009, 113, 19664 CAS.
- B. Zhang, H. D. Du, B. H. Li and F. Y. Kang, Electrochem. Solid-State Lett., 2010, 13, A36 CrossRef CAS.
- S. Huang, Z. Wen, X. Zhu and Z. Lin, J. Power Sources, 2007, 165, 408 CrossRef CAS.
- H. Zhao, Y. Li, Z. Zhu, J. Lin, Z. Tian and R. Wang, Electrochim. Acta, 2008, 53, 7079 CrossRef CAS.
- Y. J. Hao, Q. Y. Lai, J. Z. Lu and X. Y. Ji, Ionics, 2007, 13, 369 CrossRef CAS.
- X. Li, M. Qu and Z. Yu, J. Alloys Compd., 2009, 487, L12 CrossRef CAS.
- T. F. Yi, Y. Xie, J. Shu, Z. H. Wang, C. B. Yue, R. S. Zhu and H. B. Qiao, J. Electrochem. Soc., 2011, 158, A266 CrossRef CAS.
- R. Dominko, L. Dupont, M. Gaberscek, J. Jamnik and E. Baudrin, J. Power Sources, 2007, 174, 1172 CrossRef CAS.
- T. F. Yi, J. Shu, Y. R. Zhu, X. D. Zhu, R. S. Zhu and A. N. Zhou, J. Power Sources, 2010, 195, 285 CrossRef CAS.
- X. H. Kang, H. Utsunomiya, T. Achiha, Y. Ohzawa, T. Nakajima, Z. Mazej, B. Žemva and M. Endo, J. Electrochem. Soc., 2010, 157, A437 CrossRef CAS.
- Y. Qi, Y. Huang, D. Jia, S. J. Bao and Z. P. Guo, Electrochim. Acta, 2009, 54, 4772 CrossRef CAS.
- D. Capsoni, M. Bini, V. Massarotti, P. Mustarelli, G. Chiodelli, C. B. Azzoni, M. C. Mozzati, L. Linati and S. Ferrari, Chem. Mater., 2008, 20, 4291 CrossRef CAS.
- C. H. Chen, J. T. Vaughey, A. N. Jansen, D. W. Dees, A. J. Kahaian, T. Goacher and M. M. Thackeray, J. Electrochem. Soc., 2001, 148, A102 CrossRef CAS.
- S. Huang, Z. Wen, Z. Gu and X. Zhu, Electrochim. Acta, 2005, 50, 4057 CrossRef CAS.
- J. L. Allen, T. R. Jow and J. Wolfenstine, J. Power Sources, 2006, 159, 1340 CrossRef CAS.
- Z. Zhong, Electrochem. Solid-State Lett., 2007, 10, A267 CrossRef CAS.
- J. Wolfenstine and J. L. Allen, J. Power Sources, 2008, 180, 582 CrossRef CAS.
- T. F. Yi, J. Shu, Y. R. Zhu, X. D. Zhu, C. B. Yue, A. N. Zhou and R. S. Zhu, Electrochim. Acta, 2009, 54, 7464 CrossRef CAS.
- S. Z. Ji, J. Y. Zhang, W. W. Wang, Y. Huang, Z. R. Feng, Z. T. Zhang and Z. L. Tang, Mater. Chem. Phys., 2010, 123, 510 CrossRef CAS.
- R. Cai, T. Yuan, R. Ran, X. Q. Liu and Z. P. Shao, Int. J. Energy Res., 2011, 35, 68 CrossRef CAS.
- S. Hayashi and H. Hatano, J. Chem. Soc. Jpn., 1994, 102, 379 Search PubMed.
- I. Abrahams, P. G. Bruce, W. I. F. David and A. R. West, J. Solid State Chem., 1989, 78, 170 CrossRef CAS.
- L. Aldon, M. Van Thournout, P. Strobel, O. Isnard, J. Olivier-Fourcade and J. C. Jumas, Solid State Ionics, 2006, 177, 1185 CrossRef CAS.
- W. Cho, T. Kashiwagi, W. Ra, M. Nakayama, M. Wakihara, Y. Kobayashi and H. Miyashiro, Electrochim. Acta, 2009, 54, 1842 CrossRef CAS.
- M. E. Arroyo de Dompablo, E. Moran, A. Varez and F. Garcia-Alvarado, Mater. Res. Bull., 1997, 32, 993 CrossRef.
- M. E. Arroyo de Dompable, A. Varez and F. Garcia-Alvarado, J. Solid State Chem., 2000, 153, 132 CrossRef.
- F. Garcia-Alvarado, M. E. Arroyo de Dompablo, E. Moran, M. T. Gutierrez, A. Kuhn and A. Varez, J. Power Sources, 1999, 81–82, 85 CrossRef CAS.
- M. Van Thournout, A. Picard, M. Womes, J. Olivier-Fourcade and J. C. Jumas, J. Phys. Chem. Solids, 2006, 67, 1355 CrossRef CAS.
- M. Van Thournout, L. Aldon, M. Womes, B. Ducourant, J. Olivier - Fourcade, C. Tessier and S. Levasseur, J. Power Sources, 2007, 174, 1270 CrossRef CAS.
- W. Cho, T. Kashiwagi, W. Ra, Masanobu Nakayama, M. Wakihara, Y. Kobayashi and H. Miyashiro, Electrochim. Acta, 2009, 54, 1842 CrossRef CAS.
- O. V. Yakubovich and V. V. kireev, Crystallogr. Rep., 2003, 48, 24 CrossRef CAS.
- S. Kikkawa, F. Yasuda and M. Koizumi, Mater. Res. Bull., 1985, 20, 1221 CrossRef CAS.
- K. Chiba, N. Kijima, Y. Takahashi, Y. Idemoto and J. Akimoto, Solid State Ionics, 2008, 178, 1725 CrossRef CAS.
- W. A. England, J. B. Goodenough and P. J. J. Wiseman, J. Solid State Chem., 1983, 49, 289 CrossRef CAS.
- L. M. Torres-Martínez, I. Juárez-Ramírez, K. D. Ángel-Sánchez, L. Garza-Tovar, A. Cruz-López and G. D. Ángel, J. Sol-Gel Sci. Technol., 2008, 47, 158 CrossRef.
- W. A. England, J. B. Goodenough and P. J. Wiseman, J. Solid State Chem., 1983, 49, 289 CrossRef CAS.
- A. Orera, M. T. Azcondo, F. García-Alvarado, J. Sanz, I. Sobrados, J. Rodríguez - Carvajal and U. Amador, Inorg. Chem., 2009, 48, 7659 CrossRef CAS.
- K. Kataoka, J. Awaka, N. Kijima, H. Hayakawa, K. Ohshima and J. Akimoto, Chem. Mater., 2011, 23, 2344 CrossRef CAS.
- R. Dominko, E. Baudrin, P. Umek, D. Arcon, M. Gaberscek and J. Jamnik, Electrochem. Commun., 2006, 8, 673 CrossRef CAS.
- S. Y. Yin, L. Song, X. Y. Wang, Y. H. Huang, K. L. Zhang and Y. X. Zhang, Electrochem. Commun., 2009, 11, 1251 CrossRef CAS.
- Z. Yang, D. Choi, S. Kerisit, K. M. Rosso, D. Wang, J. Zhang, G. Graff and J. Liu, J. Power Sources, 2009, 192, 588 CrossRef CAS.
- J. C. Pérez-Flores, A. Kuhn and F. García-Alvarado, J. Power Sources, 2011, 196, 1378 CrossRef.
- T. Ohzuku, Z. Takehara and S. Yoshizawa, Electrochim. Acta, 1979, 24, 219 CrossRef CAS.
- F. Bonino, L. Busani, M. Lazzari, M. Manstretta, B. Rivolta and B. Scrosati, J. Power Sources, 1981, 6, 261 CrossRef CAS.
- L. Kavan, D. Fattakhova and P. Krtil, J. Electrochem. Soc., 1999, 146, 1375 CrossRef CAS.
- D. Wang, D. Choi, Z. Yang, V. V. Viswanathan, Z. Nie, C. Wang, Y. Song, J. Zhang and J. Liu, Chem. Mater., 2008, 20, 3435 CrossRef CAS.
- J. Li, Z. Tang and Z. Zhang, Electrochem. Solid-State Lett., 2005, 8, A316 CrossRef CAS.
- R. van de Krol, A. Goossens and E. A. Meulenkamp, J. Electrochem. Soc., 1999, 146, 3150 CrossRef CAS.
- D. W. Murphy, R. J. Cava, S. M. Zahurak and A. Santoro, Solid State Ionics, 1983, 9–10, 413–418 CrossRef CAS.
- M. Wagemaker, W. J. H. Borghols and F. M. Mulder, J. Am. Chem. Soc., 2007, 129, 4323 CrossRef CAS.
- L. Kavan, M. Kalbac, M. Zukalova, I. Exnar, V. Lorenzen, R. Nesper and M. Graetzel, Chem. Mater., 2004, 16, 477 CrossRef CAS.
- C. H. Sun, X. H. Yang, J. S. Chen, Z. Li, X. W. Lou, C. Z. Li, S. C. Smith, G. Q. Lu and H. G. Yang, Chem. Commun., 2010, 46, 6129 RSC.
- X. P. Gao, Y. Lan, H. Y. Zhu, J. W. Liu, Y. P. Ge, F. Wu and D. Y. Song, Electrochem. Solid-State Lett., 2005, 8, A26 CrossRef CAS.
- J. Xu, C. Jia, B. Cao and W. F. Zhang, Electrochim. Acta, 2007, 52, 8044 CrossRef CAS.
- J. Kim and J. Cho, J. Electrochem. Soc., 2007, 154, A542 CrossRef CAS.
- Y. Ren, L. J. Hardwick and P. G. Bruce, Angew. Chem., 2010, 122, 2624 Search PubMed.
- G. Armstrong, A. R. Armstrong, P. G. Bruce, P. Reale and B. Scrosati, Adv. Mater., 2006, 18, 2597 CrossRef CAS.
- G. L. Xiang, T. Y. Li, J. Zhuang and X. Wang, Chem. Commun., 2010, 46, 6801 RSC.
- Q. J. Li, J. W. Zhang, B. B. Liu, M. Li, R. Liu, X. L. Li, H. L. Ma, S. D. Yu, L. Wang, Y. G. Zou, Z. P. Li, B. Zou, T. Cui and G. T. Zou, Inorg. Chem., 2008, 47, 9870 CrossRef CAS.
- M. Inaba, Y. Oba, F. Niina, Y. Murota, Y. Ogino, A. Tasaka and K. Hirota, J. Power Sources, 2009, 189, 580 CrossRef CAS.
- D. Panduwinata and J. D. Gale, J. Mater. Chem., 2009, 19, 3931 RSC.
- M. Zukalova, M. Kalbac, L. Kavan, I. Exnar and M. Graetzel, Chem. Mater., 2005, 17, 1248 CrossRef CAS.
- C. Arrouvel, S. C. Parker and M. S. Islam, Chem. Mater., 2009, 21, 4778 CrossRef CAS.
- A. R. Armstrong, C. Arrouvel, V. Gentili, S. C. Parker, M. S. Islam and P. G. Bruce, Chem. Mater., 2010, 22, 6426 CrossRef CAS.
- G. Armstrong, A. R. Armstrong, J. Canales and P. G. Bruce, Chem. Commun., 2005, 2454 RSC.
- G. Armstrong, A. R. Armstrong, J. Canales and P. G. Bruce, Electrochem. Solid-State Lett., 2006, 9, A139 CrossRef CAS.
- A. R. Armstrong, G. Armstrong, J. Canales, R. Garcia and P. G. Bruce, Adv. Mater., 2005, 17, 862 CrossRef CAS.
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, J. Power Sources, 2005, 146, 501 CrossRef CAS.
- A. R. Armstrong, G. Armstrong, J. Canales and P. G. Bruce, Angew. Chem., Int. Ed., 2004, 43, 2286 CrossRef CAS.
- E. Morgado Jr, P. M. Jardim, B. A. Marinkovic, F. C. Rizzo, M. A. S. de Abreu, J. L. Zotin and A. S. Araújo, Nanotechnology, 2007, 18, 495710 CrossRef.
- G. N. Zhu, C. X. Wang and Y. Y. Xia, J. Power Sources, 2011, 196, 2848 CrossRef CAS.
- S. Pavasupree, Y. Suzuki, S. Yoshikawa and R. Kawahata, J. Solid State Chem., 2005, 178, 3110 CrossRef CAS.
- H. Kaper, S. Sallard, I. Djerdj, M. Antonietti and B. M. Smarsly, Chem. Mater., 2010, 22, 3502 CrossRef CAS.
- M. C. Tsai, J. C. Chang, H. S. Sheu, H. T. Chiu and C. Y. Lee, Chem. Mater., 2009, 21, 499 CrossRef CAS.
- D. H. Lee, D. W. Kim and J. G. Park, Cryst. Growth Des., 2008, 8, 4506 CAS.
- Z. X. Yang, G. D. Du, Z. P. Guo, X. B. Yu, Z. X. Chen, T. L. Guo and H. K. Liu, J. Mater. Chem., 2011, 21, 8591 RSC.
- Q. Wang, Z. H. Wen and J. Li, Adv. Funct. Mater., 2006, 16, 2141 CrossRef CAS.
- W. J. Zhou, H. Liu, R. I. Boughton, G. J. Du, J. J. Lin, J. Y. Wang and D. Liu, J. Mater. Chem., 2010, 20, 5993 RSC.
- S. H. Nam, H. S. Shim, Y. S. Kim, M. A. Dar, J. G. Kim and W. B. Kim, ACS Appl. Mater. Interfaces, 2010, 2, 2046 CAS.
- H. Kim, M. G. Kim, T. J. Shin, H. J. Shin and J. Cho, Electrochem. Commun., 2008, 10, 1669 CrossRef CAS.
- Y. G. Guo, Y. S. Hu, W. Sigle and J. Maier, Adv. Mater., 2007, 19, 2087 CrossRef CAS.
- Y. C. Qiu, K. Y. Yan, S. H. Yang, L. M. Jin, H. Deng and W. S. Li, ACS Nano, 2010, 4, 6515 CrossRef CAS.
- S. K. Das, M. Patel and A. J. Bhattacharyya, ACS Appl. Mater. Interfaces, 2010, 2, 2091 CAS.
- S. K. Das, S. Darmakolla and Aninda J. Bhattacharyya, J. Mater. Chem., 2010, 20, 1600 RSC.
- F. F. Cao, X. L. Wu, S. Xin, Y. G. Guo and L. J. Wan, J. Phys. Chem. C, 2010, 114, 10308 CAS.
- B. G. Hyde, Solid State Sci., 2003, 5, 15 CrossRef CAS.
- R. Dominko, E. Baudrin, P. Umek, D. Arcon, M. Gaberscek and J. Jamnik, Electrochem. Commun., 2006, 8, 673 CrossRef CAS.
- R. Dominko, L. Dupont, M. Gaberscek, J. Jamnik and E. Baudrin, J. Power Sources, 2007, 174, 1172 CrossRef CAS.
- H. Y. Zhu, X. P. Gao, Y. Lan, D. Y. Song, Y. X. Xi and J. C. Zhao, J. Am. Chem. Soc., 2004, 126, 8380 CrossRef CAS.
- Y. M. Wang, G. J. Du, H. Liu, D. Liu, S. B. Qin, N. Wang, C. G. Hu, X. X. Tao, J. Jiao, J. Y. Wang and Z. L. Wang, Adv. Funct. Mater., 2008, 18, 1131 CrossRef CAS.
- M. D. Wei, Y. Konishi, H. S. Zhou, H. Sugihara and H. Arakawa, J. Electrochem. Soc., 2006, 153, A1232 CrossRef CAS.
- H. Zhang, G. R. Li, L. P. An, T. Y. Yan, X. P. Gao and H. Y. Zhu, J. Phys. Chem. C, 2007, 111, 6143 CAS.
- J. R. Li, Z. L. Tang and Z. T. Zhang, Electrochem. Commun., 2005, 7, 62 CrossRef CAS.
- J. R. Li, Z. L. Tang and Z. T. Zhang, Chem. Mater., 2005, 17, 5848 CrossRef CAS.
- M. D. Wei, K. W. Wei, M. Ichihara and H. S. Zhou, Electrochem. Commun., 2008, 10, 1164 CrossRef CAS.
- S. Zhang, Q. Chen and L. M. Peng, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 014104 CrossRef.
- R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |