Modifying the barriers for oxygen-vacancy migration in fluorite-structured CeO2 electrolytes through strain: a computer simulation study†
Roger A.
De Souza
*,
Amr
Ramadan
and
Stefanie
Hörner
Institute of Physical Chemistry, RWTH Aachen University, D-52056, Aachen, Germany. E-mail: desouza@pc.rwth-aachen.de
First published on 7th December 2011
Abstract
Static lattice simulation techniques were used to examine the effect of strain on oxygen-vacancy migration in the fluorite-structured oxygen-ion conducting electrolyte CeO2. Activation energies for vacancy migration, ΔEmig, were calculated as a function of isotropic and biaxial strain. In both cases, significant modification of the energetic barriers for oxygen-vacancy migration was found. Analysis of the data yields the activation volumes, ΔVmig, and activation enthalpies, ΔHmig. Simple comparisons based on the calculated data suggest that a biaxial, tensile strain of 4% may increase the in-plane conductivity at T = 500 K by close to four orders of magnitude. Enhancement of the oxygen-ion conductivity of an oxide heterostructure through space-charge effects is also discussed.
Broader contextResearchers have over decades optimised the compositions of AO2–M2O3 fluorite-structured solid solutions for their application as oxygen-ion conducting electrolytes in Solid Oxide Fuel Cells. Solid solutions based on CeO2, in particular, are of great interest for intermediate temperature SOFC technology (operating at ca. 600 °C), which offers several benefits over the standard technology (operating at ca. 900 °C), including cheaper fabrication, improved durability and more robust construction. Recently, mechanical strain has been proposed as a new means of enhancing the ionic conductivity of such materials, and not by the factors of 2–5 typical of composition optimisation but by many orders of magnitude. There is, however, much debate as to whether the reported enhancement of 108 is a real effect or an experimental artefact. Our atomistic simulations suggest that strain can increase the ionic conductivity by 3–4 orders of magnitude, but they militate against the reported enhancement of 108. Strained AO2–M2O3 electrolytes may be employed in micro-SOFC technology. |
Introduction
Despite decades of searching for superior alternatives, fluorite-structured solid solutions, such as ZrO2–M2O3 and CeO2–M2O3, remain the best available oxygen-ion conducting electrolytes for Solid Oxide Fuel Cell (SOFC) applications. Such materials combine high and exclusively ionic conductivity, over a suitably wide range of oxygen partial pressures, not only with chemical and mechanical stability, but also with thermal and chemical compatibility with other SOFC components.1–11Concurrent with the search for superior electrolytes, the past decades have also seen the optimisation of the AO2–M2O3 fluorite materials' compositions, not only with regard to the type and amount of the M2O3 substituent, in order to maximise both the concentration and mobility of the ionic charge carriers (oxygen vacancies),4–11 but also with regard to lowering the level of SiO2 and/or the addition of silica scavengers, in order to avoid the formation of highly resistive, wetting grain-boundary phases.12
In an attempt to increase the conductivity beyond this composition-optimised level, researchers have turned their attention to the manipulation of the microstructure. Primarily two strategies are being pursued: (a) increasing the density of homo-interfaces (grain boundaries) drastically, by moving from microcrystalline to nanocrystalline samples; and (b) creating specific hetero-interfaces, by employing epitaxial thin-film geometries. The rationale behind such work is that the ionic conductivity can be raised either through enhanced charge-carrier mobility in the interface core (a structural effect), through charge-carrier accumulation in the bulk regions adjacent to the interface core (a space-charge effect), or through a reduced activation enthalpy for charge-carrier migration in the bulk phase (a strain effect). In general, regardless of the strategy pursued, the results obtained have been rather modest:13–24 in some cases a small increase was observed; in other cases, a small decrease. One major exception is the report by Garcia-Barriocanal et al.25 of colossal ionic conductivity in ultrathin (1 nm) YSZ layers (ZrO2 with 8 mol% Y2O3), sandwiched epitaxially between thicker layers of SrTiO3. Such heterostructures were found to exhibit conductivities that were higher than bulk YSZ samples by a factor of 108. This enormous increase was ascribed by the authors to the huge (7%) tensile strain in the YSZ films and disorder of oxygen ions at the interface.
Doubts have been raised, however, as to the nature of the charge carriers in these samples:26–29 Is the measured conductivity due to ionic charge carriers in YSZ or due to electronic charge carriers in SrTiO3? At present, the balance of evidence is strongly in favour of the latter. In particular Cavallaro et al.29 measured the pO2 dependence of the total conductivity of a SrTiO3|YSZ|SrTiO3 heterostructure and found σ ∝ pO20.20. Such an exponent is not consistent with ionic transport in YSZ, and in fact is characteristic of electron-hole conduction in Fe-doped SrTiO3.30
Nevertheless, the fundamental issue remains as to how much the ionic conductivity of a fluorite-structured oxide can be enhanced through lattice strain. Recent simulation studies28,31 suggest that biaxial strain can increase the ionic conductivity by a factor of 104–106. But, as large as these predicted enhancement factors are, they are still only a small fraction of the reported 108 enhancement; thus, if these simulation studies are to be believed, they provide additional evidence that the experimentally measured conductivity is electronic.
The questions hanging over the recent simulation studies28,31 concern both the simulation methodology and the simulated data. First, being based on computationally expensive density-functional-theory (DFT) calculations, the simulations were restricted to small simulation cells, and one may question if lattice relaxation was accurately captured in the migration calculations. Second, the calculations refer to constant volume and not constant pressure; as will be shown in this study, there are significant differences between the two for the conditions of interest. Third, in neither study it is shown that the simulations can reproduce experimental transport data, such as the vacancy migration enthalpy in unstrained YSZ; this casts doubts on the accuracy of the reported degrees of enhancement.‡
In the present study, static atomistic simulations, based on empirical pair-potentials (EPP), were used to determine the effect of strain on the energetic barriers for oxygen-vacancy migration in a fluorite-structured lattice. By explicitly relaxing several hundreds of ions surrounding the migrating ion, such simulation methods are able to model long-range relaxations effectively. Besides, by carrying out such calculations one can ascertain how well methods that employ empirical pair-potentials perform at large deviations from the equilibrium lattice parameter. We took CeO2 as a representative fluorite-structured electrolyte because the (unstrained) material, in contrast to ZrO2, adopts cubic symmetry for M2O3 substituent levels from zero upwards. As a first step towards a deeper and thorough understanding of strain effects, we focus on bulk material (as opposed to an interface) and we do not take account of defect–defect interactions, that is, we assume that the oxygen vacancies introduced by the M2O3 substituent are mobile and non-interacting. Another issue we do not address directly is that of lattice stability. We assume that fluorite-structured ceria is mechanically and thermodynamically stable throughout the entire range of strains examined. Araki and Arai24 predict on the basis on classical molecular dynamics simulations that YSZ fractures at a uniaxial tensile strain of 3.7%. From experiment32,33 it is known that CeO2 transforms from a cubic fluorite-type structure to an orthorhombic cotunnite-type (PbCl2) structure at a pressure ptr ≈ 31 GPa. In our static atomistic simulations, the fluorite lattice is prevented from exhibiting fracture and from undergoing phase transitions. Our principal concerns are the systematic investigation of the effect of strain on the migration energetics and the consequences for the ionic conductivity.
Simulation methods
The computational methodologies used in this work are well established.34–36 The constituent ions of the solid are treated as classical particles that bear an electrical charge corresponding to their formal oxidation number Z. The ions interact with each other through long-range Coulombic forces and short-range forces that account for electron cloud overlap (Pauli repulsion) and dispersion (van der Waals) interactions: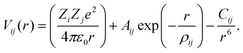 | (1) |
The calculation of defect energies employed the standard procedure of partitioning the crystal into two regions: a spherical inner region with the defect at its centre (region I) and an outer region that extends to infinity (region II). In region I, which contained between 300 and 400 ions, the positions of the individual ions are relaxed explicitly, under the perturbation of the defect, to zero net force. For the remainder of the crystal (region II), the forces due to the defect are relatively weak, and therefore the response can be treated by a quasi-continuum approximation (Mott–Littleton38). An interfacial region is also introduced to provide a smooth transition between the two regions.
Various sets of empirical parameters for CeO2 are available in the literature.39–41 In this study, the parameter set reported by Balducci et al.41 was used, since it yields a migration energy of an oxygen vacancy in cubic unstrained, undoped CeO2, ΔEmig = 0.6 eV, close to that found experimentally.42,43 The parameters for these empirical pair-potentials (EPP) are summarised in Table 1. All calculations were carried out with the GULP code.44
| Interaction ij | A ij /eV | ρij/Å | C ij /eV Å6 |
|---|---|---|---|
| O2−⋯O2− | 22764.30 | 0.1490 | 27.89 |
| Ce4+⋯O2− | 1986.83 | 0.3511 | 20.40 |
| Ion | Y/e | k/eV Å−2 |
|---|---|---|
| O2− | −2.077 | 27.290 |
| Ce4+ | 7.7 | 291.75 |
Results and discussion
Vacancy migration in isotropically strained CeO2
The lattice energy of fluorite-structured ceria (corresponding to a unit cell of Ce4O8) is plotted in Fig. 1(a) as a function of unit cell volume. The data are described well by a 3rd order Birch–Murnaghan equation of state,45 with bulk modulus BEPP = 261 GPa and bulk modulus derivative B′EPP = 3.78. The calculated bulk modulus is ca. 15% higher than experimental values33,46 (Bexpt = 220–230 GPa); DFT calculations with standard functionals yield values ca. 15% lower than experiment (BDFT ≈ 180 GPa).46–49 From the data in Fig. 1(a) we calculated the equivalent applied pressure according to p = −∂ Elatt/∂ Vlatt; and plotting normalised cell volume against pressure in Fig. 1(b), we find good agreement between our theoretical data and experimental data obtained by Duclos et al.33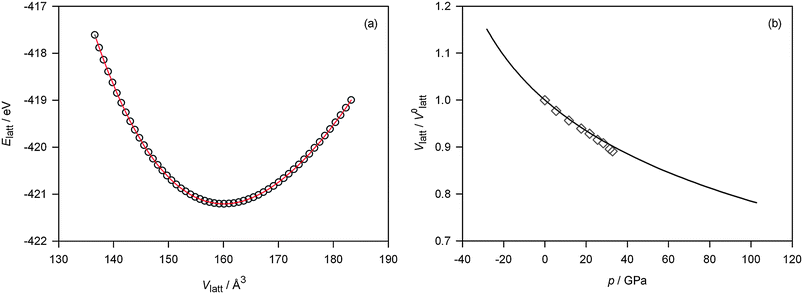 | ||
| Fig. 1 Fluorite-structured CeO2: (a) Calculated lattice energy Elatt as a function of lattice volume Vlatt (open circles); fit to a 3rd order Birch–Murnaghan equation of state (solid line). (b) Normalised lattice volume Vlatt/V0latt as a function of pressure p, calculated from the data shown in (a) (solid line), compared with experimental data from Duclos et al.33 (open diamonds). | ||
On the other hand, preliminary calculations (not shown), based solely on consideration of lattice enthalpies, Hlatt = Elatt(p = 0) + pVlatt, yield a pressure ptr ≈ 95 GPa for the fluorite to cotunnite transition, significantly higher than experiment (ptr ≈ 31 GPa).32,33 Fortunately the calculated transition pressure is close to the highest pressure examined in this study (see Fig. 1b), and hence the migration energies obtained below refer to a hypothetical cubic structure above the transition pressure. The discrepancy is probably due in part to the use of empirical pair-potentials and in part to the simplification we made, when varying the volume of the orthorhombic structure, of maintaining constant a/b and b/c ratios. It is an open question whether there are other phase transitions, either to be found experimentally or to be predicted theoretically with this set of empirical pair-potentials, in particular at negative p. (Negative pressures correspond to isotropic tensile strain—a situation that is of course difficult to achieve experimentally.) As previously noted we assume in the following that fluorite-structured CeO2 neither fractures nor undergoes any phase transitions within the range of strains/pressures investigated.
Oxygen transport in cubic fluorite-structured oxides takes place by oxygen ions jumping along the six equivalent <100> directions of the cubic structure into adjacent vacant sites. The variation in the migration energy of an oxygen vacancy in cubic CeO2 with isotropic strain is shown in Fig. 2. Compressing the lattice increases ΔEmig, whilst dilating the lattice has the opposite effect. The magnitude of the effect is surprisingly large. If the lattice is subjected to sufficient tensile strain (ε ≈ +0.05, in this case), the migration energy decreases in fact to zero. Further lattice dilation (not shown) yields negative migration energies, i.e., the migrating ion prefers the saddle-point configuration to the initial configuration. This is indicative of a lattice instability.
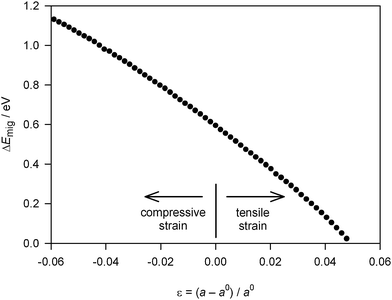 | ||
| Fig. 2 Activation energy of oxygen-vacancy migration, ΔEmig, in fluorite-structured CeO2 as a function of isotropic strain ε. (a0 is the lattice constant at zero strain.) | ||
In executing a jump, a migrating oxygen ion has to push past two cations, as shown in Fig. 3. The radius of a sphere that just passes through this aperture without disturbing the lattice, rcrit, is far smaller than the radius of an oxygen ion, rO. And the smaller the critical radius, the more the lattice has to be perturbed in order for the oxygen ion to migrate, and hence the higher the migration energy. The critical radius rcrit for an AO2 fluorite-structured oxide can be expressed50 in terms of the cubic lattice parameter a and the ionic radius of the cation rA
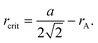 | (2) |
 | ||
| Fig. 3 (a) Cubic fluorite-structured lattice of CeO2; the <100> migration paths within the unit cell are shown in grey. (b) View from an oxygen vacancy towards a neighbouring oxygen ion along the migration path (the ions are drawn proportional to their Shannon ionic radii51). The two cations present a steric hindrance to the migration of the oxygen ion, as rO > rcrit. | ||
Although the behaviour shown in Fig. 2 is qualitatively consistent with the critical-radius model—lattice compression, for instance, leads to smaller rcrit, and thus to higher ΔEmig—, one cannot make a quantitative comparison between ΔEmig and rcrit because rA in eqn (2) [and rO of the migrating ion] will also vary with a. Furthermore it is unclear how to apportion a change in a to changes in rA and rO in an unambiguous and physically reasonable fashion.
The activation volume for vacancy migration, ΔVmig, describes the change in lattice volume, as the migrating ion passes through the saddle-point configuration. It is defined as the first derivative of the Gibbs energy of migration, ΔGmig, with respect to pressure p,
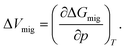 | (3) |
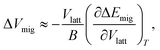 | (4) |
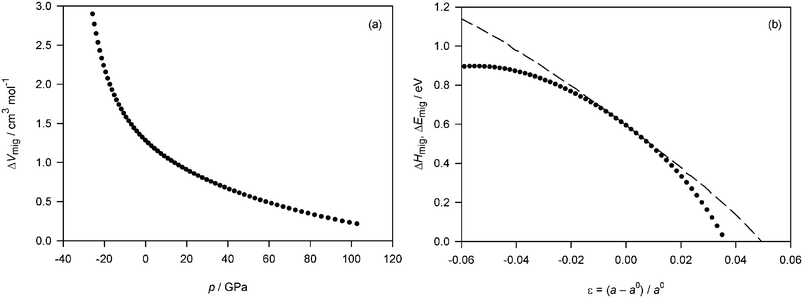 | ||
| Fig. 4 Fluorite-structured CeO2 subjected to isotropic strain: (a)activation volume for oxygen-vacancy migration, ΔVmig, as a function of pressure p; (b) the activation enthalpy of migration, ΔHmig, (symbols) and the activation energy of migration, ΔEmig, (dashed line) as a function of isotropic strain ε. | ||
In Fig. 4(b) we plot the migration enthalpy, calculated from ΔHmig = ΔEmig(p = 0) + pΔVmig, against isotropic strain. For small strains, there is little difference between the activation energy of migration and the activation enthalpy of migration. For larger strains, significant differences appear; in particular the same decrease in activation energy can be achieved at smaller dilatative strains, e.g., ΔHmig goes to zero at ε ≈ +0.035, whereas ΔEmig goes to zero at ε ≈ +0.05.
Vacancy migration in biaxially strained CeO2
Upon subjecting cubic fluorite-structured CeO2 to a biaxial strain along [100] and [010], the unit cell becomes tetragonal, a = b ≠ c. For an isotropic medium, c is given by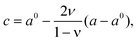 | (5) |
Since biaxial strain breaks the fluorite structure's cubic symmetry, the six equivalent, orthogonal migration paths for an oxygen vacancy in the cubic structure become four equivalent orthogonal migration directions in the ab plane of the tetragonal cell (in-plane migration), and two anti-parallel migration directions perpendicular to the ab plane (out-of-plane migration). The activation energies for vacancy migration along these two non-equivalent paths, ΔEoutmig and ΔEinmig, are shown in Fig. 5(a).
![(a)
Activation energies for vacancy migration in CeO2 biaxially strained along [100] and [010]. (b) Inverse separation of Ce ions in biaxially strained fluorite-structured CeO2, d−1Ce–Ce, as a function of biaxial strain ε.](/image/article/2012/EE/c2ee02508f/c2ee02508f-f5.gif) | ||
| Fig. 5 (a) Activation energies for vacancy migration in CeO2 biaxially strained along [100] and [010]. (b) Inverse separation of Ce ions in biaxially strained fluorite-structured CeO2, d−1Ce–Ce, as a function of biaxial strain ε. | ||
For biaxial strains |ε| < 0.02, the behaviour of ΔEoutmig and ΔEinmig can easily be rationalised within the critical-radius model. As shown in Fig. 5(b), compressive biaxial strain in the ab plane closes the out-of-plane migration aperture, and to a lesser extent, the in-plane migration aperture (the inverse Ce–Ce separation increases), and the migration energies increase correspondingly. Small tensile biaxial strains have the opposite effect, as they open the migration apertures, albeit to differing degrees. For larger biaxial strains, there are significant deviations from the expected behaviour: in particular, at large tensile strains [rhs of Fig. 5(a)], ΔEoutmig goes through a minimum and ΔEinmig decreases to zero rather rapidly. The reasons for the unexpected behaviour are unclear. It may represent the limits of the critical radius model (a simple static picture!) in describing the dynamics of the migration process. It may simply be due to the use of empirical pair potentials, i.e., the possible failure of pair potentials to capture accurately the atomic interactions at large deviations from equilibrium values. It may also be evidence of lattice instability at large ε.
For a system characterised by the stress tensor σ, the Gibbs free energy of migration can be written as53
| ΔGmig = ΔEmig − T ΔSmig − σ·ΔVmig, | (6) |
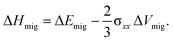 | (7) |
The biaxial stress σxx is given by
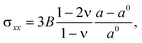 | (8) |
![Fluorite-structured CeO2 subjected to biaxial strain along [100] and [010]: (a)activation volume for oxygen-vacancy migration, ΔVoutmig and ΔVinmig, as a function of biaxial stress σxx; (b) the activation enthalpies of migration, ΔHoutmig and ΔHinmig, (symbols) and the activation energies of migration, ΔEoutmig and ΔEinmig, (dashed lines) as a function of biaxial strain ε.](/image/article/2012/EE/c2ee02508f/c2ee02508f-f6.gif) | ||
| Fig. 6 Fluorite-structured CeO2 subjected to biaxial strain along [100] and [010]: (a)activation volume for oxygen-vacancy migration, ΔVoutmig and ΔVinmig, as a function of biaxial stress σxx; (b) the activation enthalpies of migration, ΔHoutmig and ΔHinmig, (symbols) and the activation energies of migration, ΔEoutmig and ΔEinmig, (dashed lines) as a function of biaxial strain ε. | ||
The activation volumes are different for the two paths, and show different dependences on biaxial stress (with huge stresses needed to produce a significant effect); more importantly, one or other of the migration volumes becomes negative at the extreme biaxial stresses, which is highly suggestive of lattice instabilities.
Fig. 6(b) is a plot of the migration enthalpies calculated from eqn (7) against biaxial strain. As seen for the isotropic case in Fig. 4(b), there is no significant difference between ΔEmig and ΔHmig for small strains, but deviations do appear for larger strains.
Comparison with experiment
Enhancement of σion through strain
Describing quantitatively the ionic conductivity of AO2–M2O3 solid solutions as a function of temperature and amount of M2O3 requires three effects to be taken into account:48,56oxygen vacancies interacting with the aliovalent substituent cations; oxygen vacancies interacting with one another (i.e. there is a repulsive vacancy–vacancy interaction); and the migration barriers for oxygen vacancies being modified by aliovalent substituent cations. Determining the effect of strain on all these effects is left for future study. Here, in order to estimate the degree to which the ionic conductivity of a fluorite-structured oxide can be enhanced through strain, we perform a simple calculation based on the migration of non-interacting oxygen vacancies in a fluorite solid solution. In this case, the ionic conductivity can be written as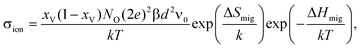 | (9) |
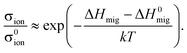 | (10) |
Let us consider, then, a system comprising a thin film CeO2-based electrolyte on a substrate with a lattice mismatch of, say, 4%. Let us assume that no misfit dislocations form at the interface; that the film is epitaxial, uniformly strained and continuous over macroscopic length scales; and that the entire mismatch between the expitaxial thin film and the substrate is taken up exclusively by the film, i.e., there is no relaxation of the substrate in the vicinity of the interface. Consequently, from the data shown in Fig. 6(b) one can predict that a biaxial strain of ε = + 0.04 will increase the in-plane ionic conductivity at T = 500 K by σion/σ0ion ≈ 103.7, that is, by several orders of magnitude. For two reasons we refrain from predicting a maximum possible enhancement through strain. First, there are various indications that CeO2 is unstable at large biaxial strains, with ΔHinmig for ε > +0.05 [see Fig. 6(b)]taking (physically unreasonable) negative values. This brings us to the second reason: since the lowest possible migration enthalpy is zero, the maximum possible enhancement depends only on the calculated migration enthalpy in the absence of strain (here, ΔH0mig = 0.59 eV). If this set of empirical potentials had predicted a lower (higher) value for ΔH0mig, the maximum possible enhancement would of course also be lower (higher). The main conclusion, though, is that, in line with another simulation study,28 a strain of +0.04 is predicted to enhance the ionic conductivity of a fluorite-structured electrolyte at T = 500 K by around four orders of magnitude.
We now apply our treatment to the experimentally examined SrTiO3|YSZ|SrTiO3 heterostructure.25 The measured activation enthalpy of conduction was found to decrease from ΔH0mig = 1.1 eV at ε = 0 to ΔHmig = 0.64 eV at ε = 0.07. According to eqn (10), the resulting enhancement in ionic conductivity at T = 500 K is σion/σ0ion ≈ 104.6. The enhancement can be increased to σion/σ0ion ≈ 105.6 by including a large possible variation in ΔSmig with strain (see Appendix). Hence, the discrepancy between this predicted value and the measured enhancement of 108 indicates, that the measured conductivity is electronic;26–29 that one of the pre-exponential factors exhibits an extremely strong dependence on strain (‘extremely strong’ because σ is linearly proportional to the various pre-exponential factors, and we require changes of several orders of magnitude); or that another effect is active (space charge has been suggested,31 and in principle charge carrier concentrations in space-charge layers may be orders of magnitude different from bulk values). We discuss the latter two options below.
The missing orders of magnitude
Of the pre-exponential factors in eqn (9), the candidate most likely to exhibit an increase with strain is xV (the increase in d2 with ε is trivial and small, and ν0 is likely to decrease with increasing ε). In the simple model of mobile, non-interacting vacancies generated by an M2O3 substituent, xV is by definition unaffected by strain. But in a more realistic model of the ionic conductivity containing interactions between substituent cations and oxygen vacancies, interactions between oxygen vacancies themselves and modification of vacancy migration barriers by substituent cations, strain may, through changes in these defect-defect interactions, cause the effective value of xV to increase. It remains to be seen, however, whether substantial increases are possible.Lastly we consider whether charge carrier accumulation in space-charge layers can provide the missing orders of magnitude. Before we address this question, however, we need to confront the more fundamental question of why charge carriers should accumulate in YSZ. If we cannot provide an answer to the latter, more fundamental question, the first question becomes irrelevant.
Is there, then, a space-charge effect through which the oxygen vacancy concentration in a thin YSZ film can be increased beyond the level defined by the yttrium concentration? Yes, there is: If the standard chemical potential for oxygen vacancies (as building units), 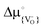 , is lower in YSZ than in SrTiO3, and the interface does not trap oxygen vacancies (i.e.
, is lower in YSZ than in SrTiO3, and the interface does not trap oxygen vacancies (i.e.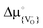 at the SrTiO3|YSZ interface is not lower than in either SrTiO3 or YSZ),57 then oxygen vacancies will transfer from the SrTiO3 substrate to the YSZ layer, generating space-charge layers in both materials. The situation is analogous to the case of bringing two semiconductors into contact: there will be transfer of charge carriers from one semiconductor to the other until the electrochemical potential of electrons (Fermi level) is equal in both systems. In the present case two oxides containing oxygen vacancies are brought into contact, and there will be transfer of oxygen vacancies from one to the other until the electrochemical potential of oxygen vacancies is equal in both systems. It must be noted that the transfer of oxygen vacancies from SrTiO3 to YSZ is conditional upon
at the SrTiO3|YSZ interface is not lower than in either SrTiO3 or YSZ),57 then oxygen vacancies will transfer from the SrTiO3 substrate to the YSZ layer, generating space-charge layers in both materials. The situation is analogous to the case of bringing two semiconductors into contact: there will be transfer of charge carriers from one semiconductor to the other until the electrochemical potential of electrons (Fermi level) is equal in both systems. In the present case two oxides containing oxygen vacancies are brought into contact, and there will be transfer of oxygen vacancies from one to the other until the electrochemical potential of oxygen vacancies is equal in both systems. It must be noted that the transfer of oxygen vacancies from SrTiO3 to YSZ is conditional upon 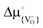 being lower in YSZ than in SrTiO3, and at present there is no experimental or theoretical evidence for or against this condition. Thus, although it is more or less certain that vacancy re-distribution will take place, since the
being lower in YSZ than in SrTiO3, and at present there is no experimental or theoretical evidence for or against this condition. Thus, although it is more or less certain that vacancy re-distribution will take place, since the 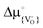 values in SrTiO3 and YSZ are unlikely to be equal, it remains to be seen whether vacancies transfer from SrTiO3 to YSZ or vice versa.
values in SrTiO3 and YSZ are unlikely to be equal, it remains to be seen whether vacancies transfer from SrTiO3 to YSZ or vice versa.
Bearing this in mind and turning now to the original question we note that, since the YSZ solid solution used in the experiments contains 8 mol% Y2O3, the site fraction of oxygen vacancies is already rather high, xV = 0.04. As a consequence, an increase in conductivity of even one order of magnitude is not possible. For the extremely simplified case of non-interacting vacancies migrating on a uniform lattice, the conductivity varies [see eqn (9)] according to xV(1 − xV), that is, maximum conductivity is obtained for half the sublattice being empty (i.e. occupied with vacancies). Thus, the maximum possible enhancement in conductivity through accumulation of oxygen vacancies in YSZ (with 8 mol% Y2O3) is
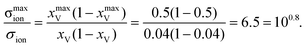 | (11) |
And this maximum enhancement requires the oxygen-ion sublattice of the entire 1 nm film to be half-filled, that is, the YSZ film would have the composition (Zr0.84Y0.16O1)1.84+; neither experiment nor theory provides any support for such major changes in the oxygen-ion sublattice. Upon inclusion of defect–defect interactions, the degree of enhancement through space-charge accumulation will change, but the changes are expected to be minor; furthermore they may not necessarily be beneficial. It is conceivable that, on account of the vacancy–vacancy interaction being repulsive, a transfer of vacancies from SrTiO3 to YSZ leads to a decrease in the electrolyte's conductivity.
Concluding remarks
We have employed static lattice simulations to examine the effect of isotropic and biaxial strain on the migration thermodynamics of oxygen vacancies in fluorite-structured ceria. We find that strain can alter the migration thermodynamics considerably, and we confirm the possibility of enhancing the ionic conductivity of an AO2–M2O3 solid solution by orders of magnitude through mechanical strain. In addition we demonstrated that static atomistic simulations, based on empirical pair-potentials (EPP), are capable of providing physically reasonable results on strained systems. Our work emphasises the need for further experimental and theoretical studies of electrolyte heterostructures: experimental studies should take account of extraneous resistances58 and should isolate the ionic contribution to the total measured conductivity, for example, by performing oxygen isotope diffusion experiments;59 and theory should focus on the strained electrolyte's mechanical and thermodynamic stability and on quantitative prediction of the ionic conductivity of an AO2–M2O3 solid solution at the strain-producing interface. We conclude that strain-induced enhancement of eight orders of magnitude25 appears to be beyond the limit of what is physically reasonable and that space-charge accumulation is unlikely to enhance the conductivity of an AO2–M2O3 solid solution, essentially because there are already so many charge carriers present.Appendix
In deriving eqn (10) we neglected, in particular, any possible variation in the activation entropy of migration, ΔSmig, with strain. Below we verify that, even though ΔSmig/k appears as the argument of an exponential term, the effect provides at most a small correction.Korte et al.21 used the Maxwell relation,
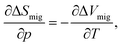 | (A1) |
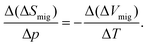 | (A2) |
Let us assume, then, that ΔVmig can increase by 1 cm3 mol−1 (i.e. an increase of the order of ΔVmig itself) for a temperature increase of ΔT = 600 K. The change in isotropic pressure in straining biaxially a YSZ film by 7% can be calculated from data given by Korte et al.21 to be Δp = −12 GPa. Thus, from eqn (A2) we obtain a limit for the increase in the activation entropy of migration: Δ(ΔSmig) ≈ 2.4 k. This yields, for YSZ at T = 500 K, an enhancement in in-plane ionic conductivity of
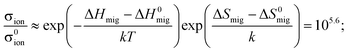 | (A3) |
Acknowledgements
RDS thanks A. Ramadan and S. Hörner for coding most of the calculations presented here, and C. Korte and J. Janek for illuminating discussions.References
- E. Baur and H. Preis, Z. Elektrochem., 1937, 43, 727–732 CAS.
- C. Wagner, Naturwissenschaften, 1943, 31, 265–268 CrossRef CAS.
- H. L. Tuller and A. S. Nowick, J. Electrochem. Soc., 1975, 122, 255–259 CrossRef CAS.
- H. Inaba and G. Tagawa, Solid State Ionics, 1996, 83, 1–16 CrossRef CAS.
- B. C. H. Steele, Mater. Sci. Eng., B, 1992, 13, 79–87 CrossRef.
- J. C. Boivin and G. Mairesse, Chem. Mater., 1998, 10, 2870–2888 CrossRef CAS.
- J. B. Goodenough, Annu. Rev. Mater. Res., 2003, 33, 91–128 CrossRef CAS.
- V. V. Kharton, F. M. B. Marques and A. Atkinson, Solid State Ionics, 2004, 174, 135–149 CrossRef CAS.
- A. Lashtabeg and S. J. Skinner, J. Mater. Chem., 2006, 16, 3161–3170 RSC.
- L. Malavasi, C. A. J. Fischer and M. S. Islam, Chem. Soc. Rev., 2010, 39, 4370–4387 RSC.
- A. Orera and P. R. Slater, Chem. Mater., 2010, 22, 675–690 CrossRef CAS.
- J.-H. Lee, Monatsh. Chem., 2009, 140, 1081–1094 CrossRef CAS.
- S. Azad, O. A. Marina, C. M. Wang, L. Saraf, V. Shutthanandan, D. E. McCready, A. El-Azab, J. E. Jaffe, M. H. Engelhard, C. H. F. Peden and S. Thevuthasan, Appl. Phys. Lett., 2005, 86, 131906 CrossRef.
- I. Kosacki, T. Suzuki, V. Petrovsky and H. U. Anderson, Solid State Ionics, 2000, 136–137, 1225–1233 CrossRef CAS.
- C. Korte, A. Peters, J. Janek, D. Hesse and N. Zakharov, Phys. Chem. Chem. Phys., 2008, 10, 4623–4635 RSC.
- A. Karthikeyan, C.-L. Chang and S. Ramanathan, Appl. Phys. Lett., 2006, 89, 183116 CrossRef.
- R. A. De Souza, M. J. Pietrowski, U. Anselmi-Tamburini, S. Kim, Z. A. Munir and M. Martin, Phys. Chem. Chem. Phys., 2008, 10, 2067–2072 RSC.
- N. H. Perry, S. Kim and T. O. Mason, J. Mater. Sci., 2008, 43, 4684–4692 CrossRef CAS.
- C. Peters, A. Weber, B. Butz, D. Gerthsen and E. Ivers-Tiffee, J. Am. Ceram. Soc., 2009, 92, 2017–2024 CrossRef CAS.
- X. X. Guo and J. Maier, Adv. Mater., 2009, 21, 2619–2631 CrossRef CAS.
- C. Korte, N. Schichtel, D. Hesse and J. Janek, Monatsh. Chem., 2009, 140, 1069–1080 CrossRef CAS.
- K. Suzuki, M. Kubo, Y. Oumi, R. Miura, H. Takaba, A. Fahmi, A. Chatterjee, K. Teraishi and A. Miyamoto, Appl. Phys. Lett., 1998, 73, 1502–1504 CrossRef CAS.
- T. X. T. Sayle, S. C. Parker and D. C. Sayle, J. Mater. Chem., 2006, 16, 1067–1081 RSC.
- W. Araki and Y. Arai, Solid State Ionics, 2011, 190, 75–81 CrossRef CAS.
- J. Garcia-Barriocanal, A. Rivera-Calzada, M. Varela, Z. Sefrioui, E. Iborra, C. Leon, S. J. Pennycook and J. Santamaria, Science, 2008, 321, 676–680 CrossRef CAS.
- X. Guo, Science, 2009, 324, 465 CrossRef CAS.
- N. Schichtel, C. Korte, D. Hesse and J. Janek, Phys. Chem. Chem. Phys., 2009, 11, 3043–3048 RSC.
- A. Kushima and B. Yildiz, J. Mater. Chem., 2010, 20, 4809–4819 RSC.
- A. Cavallaro, M. Burriel, J. Roqueta, A. Apostolidis, A. Bernardi, A. Tarancón, R. Srinivasan, S. N. Cook, H. L. Fraser, J. A. Kilner, D. W. McComb and J. Santiso, Solid State Ionics, 2010, 181, 592–601 CrossRef CAS.
- Z. Zhang, W. Sigle, R. A. De Souza, W. Kurtz, J. Maier and M. Rühle, Acta Mater., 2005, 53, 5007–5015 CrossRef CAS.
- T. J. Pennycook, M. J. Beck, K. Varga, M. Varela, S. J. Pennycook and S. T. Pantelides, Phys. Rev. Lett., 2010, 104, 115901 CrossRef.
- G. A. Kourouklis, A. Jayaraman and G. P. Espinosa, Phys. Rev. B, 1988, 37, 4250–4253 CrossRef CAS.
- S. J. Duclos, Y. K. Vohra, A. L. Ruoff, A. Jayaraman and G. P. Espinosa, Phys. Rev. B, 1988, 38, 7755–7758 CrossRef CAS.
- C. R. A. Catlow and W. C. Mackrodt, Computer simulation of solids, Lecture Notes in Physics 166, Springer, Berlin, 1982 Search PubMed.
- C. R. A. Catlow, in Solid State Chemistry: Techniques, ed. A. K. Cheetham and P. Day, Clarendon Press, Oxford, 1987, pp. 231–277 Search PubMed.
- J. H. Harding, Rep. Prog. Phys., 1990, 53, 1403–1466 CrossRef CAS.
- B. G. Dick and A. W. Overhauser, Phys. Rev., 1958, 112, 90–103 CrossRef.
- N. F. Mott and M. J. Littleton, Trans. Faraday Soc., 1938, 34, 485–499 RSC.
- V. Butler, C. R. A. Catlow, B. E. F. Fender and J. H. Harding, Solid State Ionics, 1983, 8, 109–113 CrossRef CAS.
- S. Vyas, R. W. Grimes, D. H. Gay and A. L. Rohl, J. Chem. Soc., Faraday Trans., 1998, 94, 427–434 RSC.
- G. Balducci, M. S. Islam, J. Kapar, P. Fornasiero and M. Graziani, Chem. Mater., 2000, 12, 677–681 CrossRef CAS.
- K. Fuda, K. Kishio, S. Yamauchi and K. Fueki, J. Phys. Chem. Solids, 1985, 46, 1141–1146 CrossRef CAS.
- S. B. Adler and J. W. Smith, J. Chem. Soc., Faraday Trans., 1993, 89, 3123–3128 RSC.
- J. D. Gale, J. Chem. Soc., Faraday Trans., 1997, 93, 629–637 RSC.
- F. J. Birch, Phys. Rev., 1947, 71, 809–824 CrossRef CAS.
- L. Gerward, J. Staun Olsen, L. Petit, G. Vaitheeswaran, V. Kanchana and A. Svanee, J. Alloys Compd., 2005, 400, 56–61 CrossRef CAS.
- P. J. Hay, R. L. Martin, J. Uddin and G. E. Scuseria, J. Chem. Phys., 2006, 125, 034712 CrossRef.
- M. Nakayama and M. Martin, Phys. Chem. Chem. Phys., 2009, 11, 3241–3249 RSC.
- J. Kullgren, C. W. M. Castleton, C. Müller, D. M. Ramo and K. Hermansson, J. Chem. Phys., 2010, 132, 054110 CrossRef.
- J. A. Kilner and R. J. Brook, Solid State Ionics, 1982, 6, 237–252 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- M. J. Gillan, Philos. Mag. A, 1981, 43, 301–312 CrossRef CAS.
- M. J. Aziz, Y. Zhao, H.-J. Gossmann, S. Mitha, S. P. Smith and D. Schiferl, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 054101 CrossRef.
- E. T. Park and J.-H. Park, Proc. 3rd Intnl. Meeting Pac. Rim Ceram. Soc., Kyungju, Korea, 1998, http://www.osti.gov/servlets/purl/656718-lsmLVa/webviewable/ Search PubMed.
- J.-C. M'Peko and M. F. de Souza, Appl. Phys. Lett., 2003, 83, 737–739 CrossRef CAS.
- M. Martin, J. Electroceram., 2006, 17, 765–773 CrossRef CAS.
- R. A. De Souza, Phys. Chem. Chem. Phys., 2009, 11, 9939–9969 RSC.
- H.-R. Kim, J.-C. Kim, K.-R. Lee, H.-I. Ji, H.-W. Lee, J.-H. Lee and J.-W. Son, Phys. Chem. Chem. Phys., 2011, 13, 6133–6137 RSC.
- J. M. Perkins, S. Fearn, S. N. Cook, R. Srinivasan, C. M. Rouleau, H. M. Christen, G. D. West, R. J. H. Morris, H. L. Fraser, S. J. Skinner, J. A. Kilner and D. W. McComb, Adv. Funct. Mater., 2010, 20, 2664–2674 CrossRef CAS.
Footnotes |
| † Dedicated to Prof. Harry L. Tuller on the occasion of his 65th birthday |
‡ The degree of enhancement reported by Pennycook et al.31 is particularly open to question. From their quantum molecular dynamics simulations of oxygen vacancies in strained ZrO2 they calculated a migration energy of 0.4 eV. This value is compared, inconsistently, with the experimentally determined activation enthalpy for oxygen vacancy migration in unstrained YSZ,25 to give an enhancement of 4 × 106. If the experimental migration enthalpies in both strained and unstrained YSZ25 had been correctly reproduced, a much lower enhancement of <![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 4 × 104 would have been obtained (see later). The enhancement predicted by Kushima and Yildiz28 is far less contentious. 4 × 104 would have been obtained (see later). The enhancement predicted by Kushima and Yildiz28 is far less contentious. |
| This journal is © The Royal Society of Chemistry 2012 |