Nanoporous surface alloys as highly active and durable oxygen reduction reaction electrocatalysts†
Rongyue
Wang‡
ab,
Caixia
Xu‡
c,
Xuanxuan
Bi
b and
Yi
Ding
*ab
aKey Laboratory for Liquid-Solid Structural Evolution and Processing of Materials, Ministry of Education, Shandong University, Jinan, 250061, China. E-mail: yding@sdu.edu.cn
bSchool of Chemistry and Chemical Engineering, Shandong University, Jinan, 250100, China
cSchool of Chemistry and Chemical Engineering, University of Jinan, Jinan, 250022, China
First published on 27th October 2011
Abstract
It is of critical importance to design and fabricate highly active and durable oxygen reduction reaction (ORR) catalysts for the application of proton exchange membrane fuel cells (PEMFCs). By a simple two-step dealloying process, the active components in a Pt/Ni/Al ternary alloy were sequentially leached out in a highly controllable manner, generating a novel nanoporous surface alloy structure. Characterized by an open bicontinuous spongy morphology, the resulting nanostructure is interconnected by ∼3 nm diameter ligaments which are comprised of a Pt/Ni alloy core and a nearly pure Pt surface. In the absence of any catalyst support, these nanoporous surface alloys show much enhanced durability and electrocatalytic activity for ORR as compared to the commercial Pt/C catalyst. At a high potential, such as 0.9 V versusRHE, nanoporous Pt/Ni surface alloys show a remarkable specific activity of 1.23 mA cm−2. These nanomaterials thus hold great potential as cathode catalysts in PEMFCs in terms of facile preparation, clean catalyst surface, and enhanced ORR activity and durability.
Broader contextProton exchange membrane fuel cells (PEMFCs) are highly efficient green energy power sources for automobile transportation and portable electronics. However, the lack of highly active and durable cathode catalysts for oxygen reduction reaction (ORR) significantly inhibits the widespread application of PEMFCs. At present, the state-of-the-art commercial cathode catalysts for PEMFCs are in the form of carbon supported Pt nanoparticles. Despite the high dispersion of Pt particles, the mass specific activity of Pt/C should be increased at least four times in order to meet the application demand. Moreover, as the Pt nanoparticles only have weak interactions with the carbon support, they tend to aggregate to lose surface area and performance during the long-term operation. Recently, nanoporous metals made by dealloying have attracted increasing attention due to their unique 3-D bi-continuous porous structures and potential applications in sensor, actuator, catalysis and electrocatalysis. Here we show that a unique nanoporous Pt/Ni surface alloy can be made by a simple yet delicate two-step dealloying process, which shows remarkably high activity for ORR. More importantly, by effectively bypassing the common destabilization mechanisms involved in most nanoparticle-based catalysts, these novel nanoporous electrocatalysts show much improved durability. |
1. Introduction
Proton exchange membrane fuel cells (PEMFCs) have attracted considerable research interests because of their great application potential as alternative energy sources in the fields of automotive systems and portable electronics.1,2 To achieve the point of commercial application, many aspects need to be assessed, in particular the design and processing of highly effective catalysts for oxygen reduction reaction (ORR) occurring at the cathode of PEMFCs.3 Although significant advances have been made for developing non-Pt catalysts in recent years, Pt-based catalysts still represent the highest ORR activity.4–6 Currently, the state-of-the-art commercial ORR catalyst is in a form of fine Pt nanoparticles (∼3 nm) dispersed on carbon supports (Pt/C).3 While the mass activity of this type of cathode catalyst is still far from satisfactory, an even more serious problem for these materials is their poor durability. Due to the corrosion of the carbon support and the weak interaction between the catalyst and support, the Pt nanoparticles usually undergo aggregation, dissolution and Oswald ripening which would induce enormous loss of the electrochemical surface area (ECSA) and fuel cell performance during long-term operation.7–9While most research has been focusing on improving the performance of Pt/C-like nanoparticle based supported structures,9–12 more recently supportless Pt nanostructures were found to exhibit interesting properties with evidently alleviated ECSA loss.13–15 For example, Yan et al.13 demonstrated that supportless Pt and PtPd nanotube structures had improved activity and durability for ORR. Sun et al.14 reported that multi-armed nanowire structures showed much enhanced durability as compared with Pt/C and the durability could be further improved by eliminating the carbon support. Yu et al.15 prepared free standing Pt nanowire membrane and demonstrated their implementation as highly stable ORR catalysts. While these studies have shed light on some fundamental issues associated with durable ORR catalysts, it is noted that most of these reported catalysts show little mass specific activity enhancement as compared with Pt/C catalysts, mostly because of their relatively large feature dimension (typically 5 nm or above).16 It is thus highly desirable to fabricate new nanocatalysts of practical value which exhibit high intrinsic catalytic activity and durability, while their Pt utilization should be at least comparable to the current ones. Alloying with transition metals has been demonstrated to be an effective strategy to improve the ORR performance of Pt by changing its electronic structure as well as correlating the ligand effect and strain effect.17–21 Recent fundamental studies on single crystal surfaces have demonstrated that specific activity could be improved by nearly an order of magnitude on fine tuned Pt near surface structure with a pure Pt surface and alloy sub-surface configuration, usually termed as a surface alloy.17,18,20 In the present work, we report on the design and fabrication of an interesting type of nanoporous surface alloys by a simple two-step dealloying process under mild conditions. By sequentially leaching away the active components in a Pt/Ni/Al ternary alloy, the resulting nanostructure shows a unique nanoporous surface alloy structure with a nearly pure Pt layer covering the underlying Pt/Ni alloy ligaments of typical dimensions around 3 nm. By effectively bypassing the common problems of nanoparticle aggregation and loss of contact to carbon black support involved in the traditional methodology, these novel nanostructured alloy electrocatalysts show evidently improved durability as well as up to 6-fold enhancement in catalytic activity toward ORR as compared to the Pt/C catalyst.
2. Experimental section
Pt/Ni/Al alloy foils were made by refining pure Pt, Ni, and Al (99.95%) in an arc furnace, followed by melt-spinning under N2-protected atmosphere. The atomic content of Al is controlled at ∼80%. NaOH was obtained from Shanghai Sinopharm Chemical Reagent Co., Ltd. of China with an analytic purity and used as received. Nanoporous Pt/Ni (NP-Pt1Ni1) alloys were first fabricated by dealloying Pt/Ni/Al alloy foils in 0.5 M NaOH solution at room temperature for 48 h. The NP-Pt1Ni1 alloys were further treated in dilute HNO3 (0.7 M) at room temperature for 30 min to prepare nanoporous surface alloys (NP-Pt6Ni1). The products were washed several times with ultra-pure water (18.2 MΩ) and dried at room temperature. The Johnson Matthey Pt/C (20 wt%) catalyst was purchased from Alfa Aesar.Powder X-ray diffraction (XRD) data were collected on a Bruker D8 Advance X-ray diffractometer using Cu Kα radiation (λ = 1.5418 Å). The measurements were done in reflection geometry at a scan rate of 0.04° s−1. The structure and chemical composition of all samples were characterized on a JEOL JSM-6700F field emission scanning electron microscope (SEM), equipped with an Oxford INCA X-sight energy dispersive X-ray spectrometer (EDS). Transmission electron microscopy (TEM) images were obtained with a JEM-2100 high-resolution transmission electron microscope (200 kV). To prepare the TEM samples, catalysts were sonicated and suspended in ethanol solution and were drop-cast onto carbon-coated copper grids, followed by solvent evaporation in air at room temperature.
All electrochemical measurements were performed with a CHI 760C electrochemical workstation using a conventional three electrode cell with Pt foil as a counter electrode and reversible hydrogen electrode (RHE) as the reference electrode. All electrochemical surface areas (ECSAs) of the catalysts were calculated by integrating the charge associated with H adsorption/desorption on Pt surface. ORR activities were measured by using a PAR model 636 rotating disk electrode system in an O2-saturated 0.1 M HClO4 solution at 30 °C. The NP-Pt1Ni1 and NP-Pt6Ni1 catalyst suspensions were diluted to ∼0.5 mg mL−1 by sonicating the catalyst sample, carbon powder, ethanol, and Nafion solution (0.05 wt%) for 30 min, in which the mass ratio of the catalyst sample and carbon powder was about 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2. For the Pt/C catalyst, the dispersion (about 1 mg mL−1) was prepared by sonicating the catalyst, ethanol, and Nafion solution (0.05 wt%) for 5 min. A proper amount of the catalyst suspension was dropped onto a pre-polished 5 mm diameter glassy carbon electrode, and the loadings of Pt for Pt/C catalyst, NP-Pt1Ni1, and NP-Pt6Ni1 were determined to be 16.3, 27.9, and 14.7 μg cm−2 by inductively coupled plasma atomic emission spectroscopy (ICP-AES). Prior to the electrochemical measurements, the electrolytes were deoxygenated by bubbling N2 for 30 min. After activation in 0.1 M HClO4 by cyclic voltammetry (CV) between 0.04 and 1.3 V for 10 cycles at a scan rate of 50 mV s−1, the ORR experiment was performed in O2 saturated fresh 0.1 M HClO4 at a scan rate of 10 mV s−1.
:2. For the Pt/C catalyst, the dispersion (about 1 mg mL−1) was prepared by sonicating the catalyst, ethanol, and Nafion solution (0.05 wt%) for 5 min. A proper amount of the catalyst suspension was dropped onto a pre-polished 5 mm diameter glassy carbon electrode, and the loadings of Pt for Pt/C catalyst, NP-Pt1Ni1, and NP-Pt6Ni1 were determined to be 16.3, 27.9, and 14.7 μg cm−2 by inductively coupled plasma atomic emission spectroscopy (ICP-AES). Prior to the electrochemical measurements, the electrolytes were deoxygenated by bubbling N2 for 30 min. After activation in 0.1 M HClO4 by cyclic voltammetry (CV) between 0.04 and 1.3 V for 10 cycles at a scan rate of 50 mV s−1, the ORR experiment was performed in O2 saturated fresh 0.1 M HClO4 at a scan rate of 10 mV s−1.
3. Results and discussion
It is well known that selective dissolution of active species from an alloy frequently results in the formation of a porous structure with nanosized ligaments and pores.22 While the nano-ligaments are formed from the local aggregation of gradually released noble metal atoms, they generally inherit the same crystallographic structure as the original grains of the precursor alloys, thus exhibiting an interesting single crystal and porous domain structure.23 Unlike the traditional nanoparticle based electrocatalysts, nanoporous metals hold a unique combination of a highly conductive network and a highly accessible open nanoporosity, which is particularly advantageous for electrocatalysis.24–28 Moreover, recent study by Erlebacher et al. demonstrated that the catalytic activity of nanoporous structure could be further enhanced by incorporating ionic liquids to improve the dissolution of oxygen in electrolyte.29 Considering that the highest ORR activity was observed from Pt/Ni alloy surfaces,18 here we chose nanoporous Pt/Ni alloys as our targeted cathode catalysts.To make nanoporous alloy electrocatalysts, we selected Al-based ternary alloys as the source precursors due to the more active property and rich supply of Al. Although in principle, binary source alloys can also be used to achieve a similar goal,29–31 the ternary route favors the formation of porous structures with higher porosity and better composition control.32 The content of Ni in the resulting nanoporous alloys was controlled at ∼50 at%, which can be realized by simply adjusting the feed ratio of the ternary alloy precursors during the alloy-refining process (see Fig. S1†). Because the formation of these alloy nanostructures are not based on chemical co-reduction of respective source metal ions, this process can achieve a nearly 100% yield with essentially no precious metal loss. More importantly, this process can in principle pre-decide the chemical composition of the resulting nanoporous metals or alloys, and by carefully controlling the processing conditions, porous nanoalloys with almost any composition (i.e., ranging from pure NP-Pt33 through NP-Pt/Ni alloys to NP-Ni34) may be fabricated using this simple process. This is also in sharp contrast to the traditional approach of chemical synthesis, where the feed ratio of metal salts does not guarantee the same nominal composition in the final alloy sample, mainly due to the different reducing capacities of individual elements.
The morphology of as-dealloyed NP-Pt/Ni samples was first investigated by SEM. As shown in Fig. S2, the prepared NP-Pt/Ni alloy has a uniform three dimensional (3D) bi-continuous porous structure with fine ligaments and relatively larger pore channels. The presence of these pore channels is beneficial to the fast mass transportation during the catalytic reaction. The ligament size is estimated to be less than 5 nm. The relative ratio between Pt and Ni determined by EDS (Fig. S3†) was about 1:1 (denoted as NP-Pt1Ni1), which is highly consistent with pristine alloy. It can be concluded that the bimetallic composition remains nearly unchanged before and after dealloying, indicating an excellent control of the compositions of the resulting nano-alloys with this method. Moreover, the prepared NP-Pt1Ni1 alloy could be further dealloyed to form a surface alloy structure with Pt enriched surface and Pt/Ni core. It was reported that the core/shell structure with Pt enriched surface and alloy core exhibited an even more enhanced activity toward ORR than the alloy counterpart.21,35,36 To prepare such a structure, the freshly prepared NP-Pt1Ni1 alloy was further treated in dilute HNO3 solution for 30 min and the final composition of the resulting sample has an overall Pt:Ni ratio close to 6:1 (denoted as NP-Pt6Ni1, Fig. S4†).
XRD was used to examine the crystal structure of NP-Pt1Ni1 and NP-Pt6Ni1 samples. As shown in Fig. S5,† the NP-Pt1Ni1 and NP-Pt6Ni1 samples show broad diffraction peaks with 2θ values at around 41.2, 46.9, 69.7 and 40.7, 46.7, 69, respectively, which can be assigned to the (111), (200), (220) diffractions for a face centered cubic (fcc) structure. The broad diffraction features are mainly due to the large surface stress developed during dealloying.37 There are no extra diffraction peaks for possible phases such as Ni (or their oxides) and ordered Pt-M alloys,38 suggesting the formation of disordered single-phase Pt/Ni alloy. As compared to the standard diffraction pattern of Pt, all diffraction peaks shift to higher angles due to the substitution of smaller Ni metal atoms. The NP-Pt6Ni1 sample shows smaller diffraction angles than that of the NP-Pt1Ni1 sample which reveals the lower Ni content upon further dealloying.
Detailed structural information of NP-Pt1Ni1 and NP-Pt6Ni1 alloy was obtained by TEM analyses. The sharp contrast between the dark skeletons and inner bright regions in TEM images (Fig. 1a and 1b) provides clear evidence for the formation of porous structures. It is noted that there is no distinguishable difference for the morphology and structural dimension before and after post-dealloying in dilute HNO3, although the composition was changed. The residual Ni atoms are thought to mostly locate in the core region of ligaments, forming a Pt enriched surface.29 Similar to other nanoporous metals, NP-Pt/Ni alloys possess a certain degree of single crystalline grain structure, although their ligaments are typically 3 nm in diameter. It could be clearly seen from the high resolution TEM (HRTEM) images and the corresponding Fourier filtered images that the lattice fringes are continuous across the pores, albeit with the existence of many defects and lattice distortion (see Fig. 1c and 1d). The lattice spacing was measured to be 0.21 (Fig. S6†) and 0.23 nm for NP-Pt1Ni1 and NP-Pt6Ni1, which corresponds to the (111) crystal plane of Pt/Ni alloys. The slight lattice spacing difference of these two samples should be attributed to the composition difference. The Fourier transformed image (inset in Fig. 1d) further demonstrate that the entire macroscopic sample is associated within a single network structure, with excellent structural integrity and thus superior electron conductivity. Meanwhile, the interconnected hollow void embedded in the network skeleton is favorable for molecular mobilization along the channels. These structural features form a nearly ideal multifunctional platform for electrocatalysis applications.
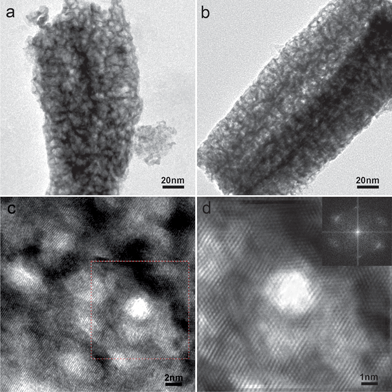 | ||
| Fig. 1 (a, b) TEM images of NP-Pt1Ni1 and NP-Pt6Ni1. (c) HRTEM images of NP-Pt6Ni1. (d) Fourier filtered HRTEM image of the highlighted part in Fig. 1c. The Fourier transformed image of the square site in Fig. 1c is shown as an inset. | ||
Fig. 2a–c shows the voltammetric behaviors of NP-Pt1Ni1 and NP-Pt6Ni1 alloys in 0.1 M HClO4 solution along with those of commercial Pt/C catalyst for comparison. All curves are composed of three characteristic potential regions, namely hydrogen under-potential adsorption/desorption (H-UPD) at 0–0.4 V, a double layer at 0.4–0.65 V, and the formation of metal oxides and their reduction at higher potentials. The absence of the oxidation peak associated with pure Ni indicates that the prepared samples are in true alloy states, consistent with the XRD result illustrated above. Compared with the NP-Pt1Ni1 alloy, the NP-Pt6Ni1 sample shows more characteristic Pt oxidation and H-UPD curves which indicates that this sample has a surface state close to pure Pt. Notably, compared with Pt/C, the redox waves for nanoporous alloys were shifted to significantly more positive potentials, suggesting the delayed formation and weakening of Pt-oxygenated species after alloying with Ni, a desired feature for a good ORR catalyst. The ECSA of NP-Pt1Ni1, NP-Pt6Ni1, and Pt/C were measured to be 30, 52.8, and 75 m2 g−1 by using the H-UPD method.
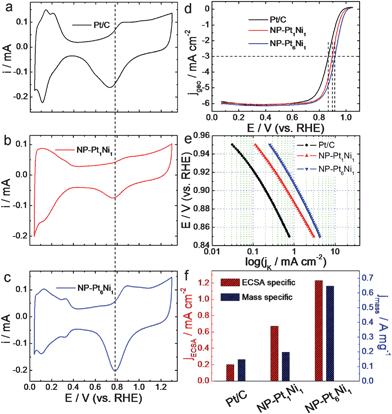 | ||
| Fig. 2 (a, b, c) CV curves of Pt/C, NP-Pt1Ni1, and NP-Pt6Ni1 in N2-purged 0.1 M HClO4 solution. Scan rates are 50 mV s−1. (d) ORR polarization curves for NP-Pt1Ni1 and NP-Pt6Ni1 in O2-saturated 0.1 M HClO4 solution. The ORR polarization curve of Pt/C was also shown for comparison. Scan rates are 10 mV s−1. (e) ECSA specific kinetic current densities (jk) for NP-Pt1Ni1 and NP-Pt6Ni1 at different potentials. The ECSA specific kinetic current density (jk) for Pt/C was also shown for comparison. (f) The ECSA and Pt mass specific kinetic current densities for Pt/C, NP-Pt1Ni1, and NP-Pt6Ni1 at 0.9 V. | ||
Fig. 2d provides the polarization curves for these novel nanostructures for ORR in 0.1 M oxygen-saturated HClO4 solution at 30 °C. The ORR polarization curve of Pt/C was also shown for comparison. All curves exhibit two distinguishable potential regions: a mixed kinetic-diffusion control region between 0.8 and 1.0 V and a well-defined diffusion controlled region below 0.8 V. Intriguingly, although three samples have similar electrochemical surface area (ECSA), NP-Pt1Ni1, and NP-Pt6Ni1 alloys exhibit much higher half-wave potentials at 0.897 and 0.916 V, respectively, which are 31 and 50 mV more positive than that of Pt/C catalyst (∼0.866 V), indicating dramatically improved reaction kinetics for ORR at lower over-potentials. To further compare the ORR activity, ECSA specific kinetic activities were shown in Fig. 2e, in which the electrochemically active surface areas of the Pt electrodes were calculated based on hydrogen desorption on Pt surface in 0.1 M HClO4 solution. It can be clearly found that both NP-Pt1Ni1 and NP-Pt6Ni1 alloys show greatly improved ECSA specific kinetic activities than that of Pt/C catalyst in the whole potential range (0.85–0.95 V). For instance, the current densities at 0.90 V are about 0.67 and 1.23 mA cm−2 (see Fig. 2f) for NP-Pt1Ni1 and NP-Pt6Ni1 alloys, which are enhanced by a factor of 3.4 and 6.2 as compared with that of the Pt/C catalyst (∼0.2 mA cm−2).18 These values also compare favorably with the recently reported results for ionic liquid impregnated nanoporous Pt/Ni alloy (about 1 mA cm−2)29 and 100% truncated octahedral Pt3Ni catalyst (0.85 mA cm−2),39 especially for the NP-Pt6Ni1 sample with a unique surface alloy structure. Shown in Fig. 2f are the mass specific activities which were calculated by normalizing the kinetic currents at 0.9 V to the Pt loadings. The mass specific activity is 0.65 A mg−1 for NP-Pt6Ni1 alloy which is 4.3 times of that for Pt/C catalysts (0.15 A mg−1).
As mentioned above, the stability of an ORR catalyst is vital for its application in PEMFCs. The durability of the NP-Pt1Ni1 and NP-Pt6Ni1 catalysts were thus evaluated by applying continuous potential sweeps between 0.04 and 1.3 V in 0.1 M HClO4 solution for 4000 cycles at 30 °C (140 h testing). From Fig. 3, it is interesting to find that the ECSA of NP-Pt1Ni1 catalyst shows an unexpected increase in the first one hundred cycles to 130% of the pristine surface area. The ECSA increase of NP-Pt1Ni1 catalyst was associated with Ni dissolution in the potential excursion. This phenomenon was also observed in the core/shell nanoparticle structures and was attributed to surface roughening and contaminants removal.40 After approaching the highest point, the ECSA starts to decline, and after 4000 cycles, the ECSA of the NP-Pt1Ni1 catalyst remained a high value of 72.1% of the original state, or 55.4% of the peak value observed during the cycling. However, for NP-Pt6Ni1, its ECSA sees a regular decrease in the whole range of cycling, and 50% of the ECSA remained after 4000 cycles. In comparison, Pt/C underwent a nearly 80% loss in ECSA upon the same potential cycling.13,14 These results proved that supportless NP-Pt/Ni nanoalloys are significantly more stable than the supported Pt/C catalyst.
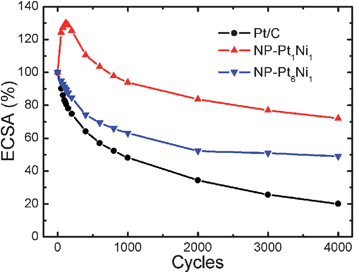 | ||
| Fig. 3 Loss of ECSA of Pt/C, NP-Pt1Ni1, and NP-Pt6Ni1 catalysts as a function of cycling number in N2-purged 0.1 M HClO4 solution at 30 °C (0.04–1.3 V vs.RHE, scan rate 20 mV s−1). | ||
To understand the different durability behaviors of these materials, the morphological evolution of Pt/C and NP-Pt6Ni1 before and after 4000 cycles was investigated by TEM. From Fig. 4a, the pristine Pt/C structure contains 3 nm Pt nanoparticles uniformly dispersed on 50 nm carbon supports, which is typical for this type of commercial catalyst. However, after the durability test, these Pt particles were found to have undergone severe aggregation and growth, and the majority of Pt particles have grown to 10–20 nm (Fig. 4b). Some nanoparticles have even lost contact with the carbon support. It is commonly accepted that the destabilization of Pt/C catalyst is associated with (1) Ostwald ripening and aggregation of the Pt nanoparticles, which were caused by the high surface energy of Pt nanoparticles and the weak interaction between catalysts and carbon supports; (2) loss of contact to support due to carbon corrosion.7,8 In contrast, the supportless nanoporous surface alloys have a different destabilization mechanism. As shown in Fig. 4d, upon potential cycling, the porous structure of NP-Pt6Ni1 demonstrated a much less severe coarsening as compared to Pt/C, and most ligaments still have a diameter smaller than 10 nm. Moreover, the original bicontinuous porous structure was very well retained in the final sample (Fig. 4c & 4d), suggesting the mass transport is governed by a surface-diffusion mechanism.22,29 Because the surface diffusion scaling follows a power law decay which is proportional to a quarter order of time t, the coarsening of a porous structure is expected to be significantly retarded as the ligament size grows larger, especially for small surface diffusivity materials such as Pt.41 This mechanism, along with the uniquely strained Pt-rich surface alloy structure, may explain the excellent stability of NP-Pt/Ni catalysts against coarsening.
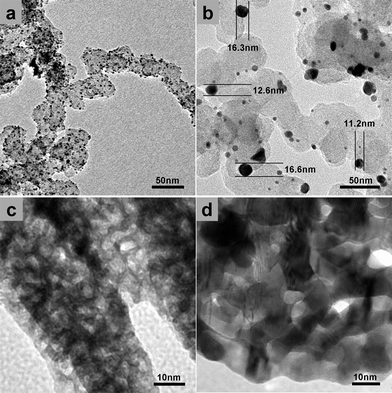 | ||
| Fig. 4 TEM of Pt/C (a) before and (b) after 4000 CV cycles in N2-purged 0.1 M HClO4 solution. TEM of NP-Pt6Ni1 (c) before and (d) after 4000 CV cycles in N2-purged 0.1 M HClO4 solution. | ||
4. Conclusions
In summary, we have demonstrated a simple and straightforward fabrication route to nanoporous Pt/Ni surface alloys with a characteristic structural dimension of ∼3 nm. The resulting nanoporous catalysts exhibited superior ORR activity and long-term durability as compared to the commercial Pt/C catalyst. It is suggested that the surface strain and the alloying effect of Ni in this Pt-skin surface alloy structure may account for the observed performance improvement. In addition, the unique nanoporous structure is suggested to play a vital role in terms of easy transport of electrons and medium molecules along these interconnected skeletons and bi-continuous hollow channels in all three dimensions. The different destabilization mechanism of nanoporous metals also demonstrates their unparalleled long-term durability as compared with other supported nanoparticle-based catalysts. With obvious advantages of high catalytic activity, high stability, low cost, and simple processing, this design and processing concept can be easily extended to other systems, and one will expect that a whole new family of multi-component alloy nanostructures will soon be explored. These high surface-area nanomaterials with designed functions and properties thus hold great promise for potential applications in important energy and environmental science fields.Acknowledgements
This work was supported by the 973 Program Project of China (2012CB932801) and the National Science Foundation of China (90923011, 51001053, 51171092). Y. D. is a Tai-Shan Scholar supported by the Independent Innovation Foundation of Shandong University (IIFSDU), the Research Fund for the Doctoral Program of Higher Education of China (20090131110019) and the Shandong Natural Science Fund for Distinguished Young Scholars (JQ200815). R. W. acknowledges the Scholarship Award for Excellent Doctoral Student granted by the Ministry of Education.References
- B. C. H. Steele and A. Heinzel, Nature, 2001, 414, 345–352 CrossRef CAS.
- M. Z. Jacobson, W. G. Colella and D. M. Golden, Science, 2005, 308, 1901–1905 CrossRef CAS.
- H. A. Gasteiger, S. S. Kocha, B. Sompalli and F. T. Wagner, Appl. Catal., B, 2005, 56, 9–35 CrossRef CAS.
- H. A. Gasteiger and N. M. Markovic, Science, 2009, 324, 48–49 CrossRef CAS.
- A. Morozan, B. Jousselme and S. Palacin, Energy Environ. Sci., 2011, 4, 1238–1254 CAS.
- C. Sealy, Mater. Today, 2008, 11, 65–68 CrossRef CAS.
- R. Borup, J. Meyers, B. Pivovar, Y. S. Kim, R. Mukundan, N. Garland, D. Myers, M. Wilson, F. Garzon, D. Wood, P. Zelenay, K. More, K. Stroh, T. Zawodzinski, J. Boncella, J. E. McGrath, M. Inaba, K. Miyatake, M. Hori, K. Ota, Z. Ogumi, S. Miyata, A. Nishikata, Z. Siroma, Y. Uchimoto, K. Yasuda, K. I. Kimijima and N. Iwashita, Chem. Rev., 2007, 107, 3904–3951 CrossRef CAS.
- K. Hartl, M. Hanzlik and M. Arenz, Energy Environ. Sci., 2011, 4, 234–238 CAS.
- Z. Z. Jiang, Z. B. Wang, Y. Y. Chu, D. M. Gu and G. P. Yin, Energy Environ. Sci., 2011, 4, 728–735 CAS.
- J. Zhang, K. Sasaki, E. Sutter and R. R. Adzic, Science, 2007, 315, 220–222 CrossRef CAS.
- K. P. Gong, D. Su and R. R. Adzic, J. Am. Chem. Soc., 2010, 132, 14364–14366 CrossRef CAS.
- C. Wang, D. van der Vliet, K. L. More, N. J. Zaluzec, S. Peng, S. H. Sun, H. Daimon, G. F. Wang, J. Greeley, J. Pearson, A. P. Paulikas, G. Karapetrov, D. Strmcnik, N. M. Markovic and V. R. Stamenkovic, Nano Lett., 2011, 11, 919–926 CrossRef CAS.
- Z. W. Chen, M. Waje, W. Z. Li and Y. S. Yan, Angew. Chem., Int. Ed., 2007, 46, 4060–4063 CrossRef CAS.
- S. H. Sun, G. X. Zhang, D. S. Geng, Y. G. Chen, R. Y. Li, M. Cai and X. L. Sun, Angew. Chem., Int. Ed., 2011, 50, 422–426 CrossRef CAS.
- H. W. Liang, X. A. Cao, F. Zhou, C. H. Cui, W. J. Zhang and S. H. Yu, Adv. Mater., 2011, 23, 1467–1471 CrossRef CAS.
- C. Wang, D. van der Vilet, K. C. Chang, H. D. You, D. Strmcnik, J. A. Schlueter, N. M. Markovic and V. R. Stamenkovic, J. Phys. Chem. C, 2009, 113, 19365–19368 CAS.
- J. Greeley, I. E. L. Stephens, A. S. Bondarenko, T. P. Johansson, H. A. Hansen, T. F. Jaramillo, J. Rossmeisl, I. Chorkendorff and J. K. Norskov, Nat. Chem., 2009, 1, 552–556 CrossRef CAS.
- V. R. Stamenkovic, B. Fowler, B. S. Mun, G. F. Wang, P. N. Ross, C. A. Lucas and N. M. Markovic, Science, 2007, 315, 493–497 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, M. Arenz, K. J. J. Mayrhofer, C. A. Lucas, G. F. Wang, P. N. Ross and N. M. Markovic, Nat. Mater., 2007, 6, 241–247 CrossRef CAS.
- I. E. L. Stephens, A. S. Bondarenko, F. J. Perez-Alonso, F. Calle-Vallejo, L. Bech, T. P. Johansson, A. K. Jepsen, R. Frydendal, B. P. Knudsen, J. Rossmeisl and I. Chorkendorff, J. Am. Chem. Soc., 2011, 133, 5485–5491 CrossRef CAS.
- P. Strasser, S. Koh, T. Anniyev, J. Greeley, K. More, C. F. Yu, Z. C. Liu, S. Kaya, D. Nordlund, H. Ogasawara, M. F. Toney and A. Nilsson, Nat. Chem., 2010, 2, 454–460 CrossRef CAS.
- J. Erlebacher, M. J. Aziz, A. Karma, N. Dimitrov and K. Sieradzki, Nature, 2001, 410, 450–453 CrossRef CAS.
- Y. Ding, Y. J. Kim and J. Erlebacher, Adv. Mater., 2004, 16, 1897–1900 CrossRef CAS.
- T. Fujita, H. Okada, K. Koyama, K. Watanabe, S. Maekawa and M. W. Chen, Phys. Rev. Lett., 2008, 101, 166601 CrossRef CAS.
- Y. Ding and M. W. Chen, MRS Bull., 2011, 34, 569–576 CrossRef.
- R. Y. Wang, C. Wang, W. B. Cai and Y. Ding, Adv. Mater., 2010, 22, 1845–1848 CrossRef CAS.
- J. S. Yu, Y. Ding, C. X. Xu, A. Inoue, T. Sakurai and M. W. Chen, Chem. Mater., 2008, 20, 4548–4550 CrossRef CAS.
- X. Y. Lang, H. Guo, L. Y. Chen, A. Kudo, J. S. Yu, W. Zhang, A. Inoue and M. W. Chen, J. Phys. Chem. C, 2010, 114, 2600–2603 CAS.
- J. Snyder, T. Fujita, M. W. Chen and J. Erlebacher, Nat. Mater., 2010, 9, 904–907 CrossRef CAS.
- L. F. Liu, E. Pippel, R. Scholz and U. Gosele, Nano Lett., 2009, 9, 4352–4358 CrossRef CAS.
- L. F. Liu, R. Scholz, E. Pippel and U. Gosele, J. Mater. Chem., 2010, 20, 5621–5627 RSC.
- C. X. Xu, R. Y. Wang, M. W. Chen, Y. Zhang and Y. Ding, Phys. Chem. Chem. Phys., 2010, 12, 239–246 RSC.
- Z. H. Zhang, Y. Wang, Z. Qi, W. H. Zhang, J. Y. Qin and J. Frenzel, J. Phys. Chem. C, 2009, 113, 12629–12636 CAS.
- M. Raney, Ind. Eng. Chem., 1940, 32, 1199–1203 CrossRef CAS.
- V. R. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, P. N. Ross and N. M. Markovic, J. Am. Chem. Soc., 2006, 128, 8813–8819 CrossRef CAS.
- S. Chen, P. J. Ferreira, W. C. Sheng, N. Yabuuchi, L. F. Allard and Y. Shao-Horn, J. Am. Chem. Soc., 2008, 130, 13818–13819 CrossRef CAS.
- S. Van Petegem, S. Brandstetter, R. Maass, A. M. Hodge, B. S. El-Dasher, J. Biener, B. Schmitt, C. Borca and H. Van Swygenhoven, Nano Lett., 2009, 9, 1158–1163 CrossRef CAS.
- M. Watanabe, K. Tsurumi, T. Mizukami, T. Nakamura and P. Stonehart, J. Electrochem. Soc., 1994, 141, 2659–2668 CrossRef CAS.
- J. B. Wu, J. L. Zhang, Z. M. Peng, S. C. Yang, F. T. Wagner and H. Yang, J. Am. Chem. Soc., 2010, 132, 4984–4985 CrossRef CAS.
- D. L. Wang, H. L. Xin, Y. C. Yu, H. S. Wang, E. Rus, D. A. Muller and H. D. Abruna, J. Am. Chem. Soc., 2010, 132, 17664–17666 CrossRef CAS.
- J. Erlebacher, J. Electrochem. Soc., 2004, 151, C614–C626 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: EDS, XRD, and TEM results for the prepared nanoporous Pt/Ni surface alloys. See DOI: 10.1039/c1ee02243a |
| ‡ These two authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |