Understanding and recent development of carbon coating on LiFePO4 cathode materials for lithium-ion batteries
Jiajun
Wang
and
Xueliang
Sun
*
Dept. of Mechanical and Materials Engineering, University of Western Ontario, London, Ontario N6A 5B9, Canada. E-mail: xsun@eng.uwo.ca; Fax: +1-519-661-3020; Tel: +1-519-661-2111 ext. 87759
First published on 11th November 2011
Abstract
Olivine-structured LiFePO4 has been the focus of research in developing low cost, high performance cathode materials for lithium ion batteries. Various processes have been developed to synthesize LiFePO4 or C/LiFePO4 (carbon coating on LiFePO4), and some of them are being used to mass produce C/LiFePO4 at the commercial or pilot scale. Due to the low intrinsic electronic and ionic conductivities of LiFePO4, the decrease of particle size and the nano-layer of carbon coating on LiFePO4 particle surfaces are necessary to achieve a high electrochemical performance. Significant progress has been made in understanding and controlling phase purity, particle size and carbon coating of the C/LiFePO4 composite material in the past. However, there are still many challenges in achieving a high quality product with high consistency. In this review, we summarize some of the recent progress and advances based on selected reports from peer-reviewed journal publications. Several typical synthesis methods and the effect of carbon coating quality on the properties of C/LiFePO4 composite are reviewed. An insight into the future research and further development of C/LiFePO4 composite is also discussed.
 Jiajun Wang | Jiajun Wang is presently a post-doctoral fellow at Prof. Xueliang (Andy) Sun's Nanomaterials and Energy Group at the University of Western Ontario, Canada. He received his PhD in electrochemistry from Harbin Institute of Technology (Harbin, China) in 2008 working on synthesis and application of highly stable electrode materials for fuel cell systems. His current research focuses on development of advanced C/LiFePO4 cathode for lithium-ion batteries, collaborating with Phostech Lithium Inc. as well as the study of novel electrode materials for lithium-air batteries. He has broad research interest and experience in nanomaterial fabrication and characterization, electrochemistry, and clean energy techniques. He has authored and co-authored over 20 refereed journal publications and has around 300 citations to his work. |
 Xueliang Sun | Professor Xueliang (Andy) Sun is a Canada Research Chair and Associate Professor at the University of Western Ontario, Canada. Dr Sun received his PhD in Materials Chemistry under the direction of Prof. George Thompson in 1999 at the University of Manchester, UK, followed by work as a postdoctoral fellow under the direction of Prof. Keith Mitchell at the University of British Columbia, Canada and as a Research Associate under the direction of Prof. Jean-Pol Dodelet at l'Institut national de la recherche scientifique (INRS), Canada. His current research interests are associated with synthesis of nanomaterials for electrochemical energy storage and conversion. His focus is on design and synthesis of various one-dimensional nanostructures such as nanotubes and nanowires as well as their composites with nanoparticles and thin films as electrocatalysis and catalyst support in fuel cells and as anode and cathodes in lithium ion batteries and Li-air batteries. |
Broader contextThe threats arising from limited oil storage and global warming have forced us to look for alternative energy storage and conversion systems. In addition, the rapid pace and progress of the portable electronics and electric vehicles technique demand a powerful energy storage system with high energy density and high safety as well as low cost. Following two decades of development, rechargeable lithium-ion batteries (LIB) are considered as the most promising energy storage technology. Nowadays, consumer demand is motivating tremendous efforts for high capacity, high power density, fast recharging rate, and great cycling performance. However, these demands bring many challenges to material stability, especially when they are used in electric vehicles (EV). Compared with other cathode materials, olivine-type LiFePO4 draws extensive attention because of its high theoretical capacity, environmental acceptability, and low cost, especially superior thermal stability and safety. The major drawback for LiFePO4 is the intrinsically poor conductivity, which is currently being overcome by coating them with conductive carbon. Carbon coating is a critical component in C/LiFePO4 composites. A deep and comprehensive understanding of carbon coating and its effects as well as the synthesis process to control carbon coating on LiFePO4 will contribute to the LIB industry and accelerate the commercialization of EV. |
1. Introduction
Lithium-ion rechargeable batteries are the most promising power system that can offer a higher operative voltage and energy density compared to other rechargeable battery systems such as the widely used nickel metal hydride (NiMH) batteries being used in commercial hybrid electric vehicles (HEV). In recent years, there has been a dramatic increase in research and commercialization activities of lithium ion batteries for large scale energy storage and for on-board energy storage in electric vehicles (EV) and plug-in hybrid electric vehicles (PHEV).1,2 Challenges remain in making low cost, high-performance and high-safety lithium ion batteries for vehicle applications.3,4Among the components in lithium-ion batteries, the cathode material has attracted much attention because it has a significant impact on battery capacity, cycle life, safety and cost structure.5 Due to its high capacity LiCoO2 has been widely used in small batteries for portable electronics since its introduction to the market by Sony Inc. in 1991. However, its use in large size batteries has been vetoed by safety concerns. In addition to the safety risks, the high cost, toxicity, and environmental concerns prohibit the large scale applications of LiCoO2 material in HEV, PHEV or EV. Other materials including lithium nickel manganese cobalt (NMC), lithium nickel cobalt aluminium (NCA), lithium manganese spinel and olivine lithium iron phosphate are being investigated and commercialized for various applications. Each one has advantages and disadvantages.6–9LiFePO4 is of particular interest for use in large size batteries, due to its intrinsic structural and chemical stability that leads to safe and long cycle life batteries. Besides, olivine LiFePO4 is composed of low cost and environmentally benign Fe and PO4 moieties, which is an important merit for large scale applications. One of the main obstacles for practical applications of LiFePO4 is its poor rate capability, which can be attributed to slow kinetics of lithium-ion diffusion coefficient (10−14 to 10−16 cm2 s−1) and the poor electronic conductivity (<10−9 s cm−1).10,11
Since the pioneering work of John Goodenough and coworkers,12–15 numerous works have been done to investigate the synthesis, structure, defects and physical, chemical and electrochemical properties of LiFePO4. Some review articles or book/chapters were published concerning the synthesis and properties of lithium iron phosphate.16–18 In the present work, focus is given to the C/LiFePO4 and the impact of carbon precursors on the synthesis and electrochemical performance of C/LiFePO4. An insight into the future research and development of C/LiFePO4 composites is also discussed.
2. The structure and physical/chemical properties of pristine LiFePO4
The olivine structure of LiFePO4 crystal consists of a polyoxyanionic framework containing LiO6 octahedra, FeO6 octahedra and PO4 tetrahedra. Strong P–O covalent bonds in (PO4)3−polyanion stabilize the oxygen when fully charged and avoid O2 release at high states of charge making LiFePO4 an excellent, stable and safe cathode material (Fig. 1).15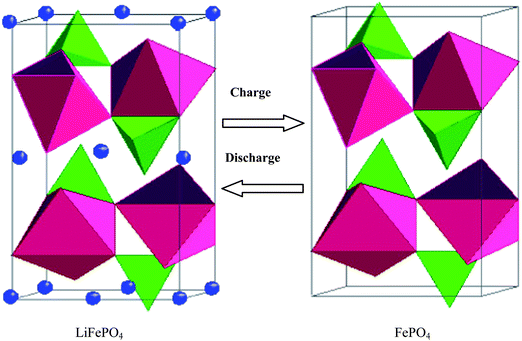 | ||
| Fig. 1 The olivine structure is stable during Li-ions insertion and extraction. | ||
The cation arrangement in LiFePO4 differs from that in layered LiCoO2 or spinel LiMn2O4. The divalent Fe2+ ions occupy the corner-shared octahedra. The P5+ is located in tetrahedral sites, and Li+ resides in chains of edge-shared octahedra. The skeleton of PO4 polyanions is very stable thermally, but the corner-shared FeO6 octahedra of LiFePO4 are separated by the oxygen atoms of the PO43− tetrahedra and cannot form a continuous FeO6 network, which results in the poor electronic conductivity of LiFePO4.20 At room temperature, the electronic conductivity of pristine LiFePO4 is only 10−9 to 10−10 S cm−1,21 which is much lower than those of LiCoO2 (∼10−3 S cm−1) and LiMn2O4 (2 × 10−5 to 5 × 10−5 S cm−1).22,23
Atomistic modeling and first-principle calculations indicate that the lowest Li migration energy is found for the pathway along the [010] channel, with a nonlinear, curved trajectory between adjacent Li sites (Fig. 2).24,25 Although the theoretical calculation showed that the intrinsic ionic diffusion coefficient is as high as 10−8 (LiFePO4) to 10−7 (FePO4) cm2 s−1, the one-dimensional channels are easily blocked by defects and impurities because blockages in one-dimensional paths are different from those in two-dimensional and three-dimensional paths where Li ions can move around the blocked sites.26 The ionic diffusion coefficients in LiFePO4 are therefore lower than the theoretical value. For example, the diffusion coefficients were reported by Takeda et al. in a wide range of 10−14 to 10−12 cm2 s−1 with the potentiostatic intermittent titration technique, 10−15 to 10−12 cm2 s−1 with electrochemical impedance spectroscopy, and 10−14 cm2 s−1 with cyclic voltammetry, respectively.27 A lower diffusion coefficient (10−15 to 10−12 cm2 s−1) was also found in another report using the similar techniques.28 Nevertheless, it should be mentioned that it is very difficult to accurately measure the diffusion coefficient of Li-ion in LiFePO4 by these methods due to a very flat charge–discharge potential induced by the phase transformation between LiFePO4 and FePO4.29
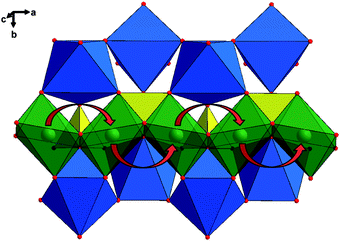 | ||
| Fig. 2 Structure of LiFePO4 depicting the curved trajectory of Li ion transport along the b-axis, shown with red arrows. The iron octahedra are shown in blue, the phosphate tetrahedral in yellow, and the lithium ions in green. Reproduced with permission from ref. 24. Copyright 2010 American Chemical Society. | ||
Apparently, the low electronic and low ionic conductivities dictate the slow kinetics of charge and discharge. Various models are proposed to explain the mechanism and kinetics of charge and discharge, like the shrinking core model, domino-cascade model and the recent spinodal decomposition model.30–32 Debates are still going on concerning the rate limiting step in the charge and discharge processes. Irrespective of those debates, various approaches towards mitigating the slow ionic and electronic conductivities are proposed and explored.
3. Various approaches to improve the performance of LiFePO4
3.1 Size reduction
Size reduction to nanoscale dimension has been pointed out by John Goodenough et al. as one of the effective methods to solve the kinetic problems of LiFePO4.13 Nanosize pristine LiFePO4 has been synthesized by hydrothermal reaction under mild conditions, which indeed shows desirable electrochemical properties when the nanoparticles are well connected with the current collectors by using a large amount of carbon particle additives.33–35 The carbon particles play the role of forming a conductive network in the cathode coating to improve the electron transport.Many other studies have been conducted to mitigate lithium diffusion limitations in LiFePO4 by reducing particle size to shorten the diffusion length. Recent studies also show that ionic diffusion constant depends on particle size with diffusion in nanosized particles being much faster than in micrometre-sized particles or bulk (Fig. 3).36 However, the higher surface area arising from the smaller nanoparticle size also results in other undesirable effects such as low material packing density in the cathode and potential electrode/electrolyte reactions.37,38 An alternative effective method to improve the lithium-ion diffusion is using a higher operating temperature. However, not only the lithium-ion diffusion rate increases at high temperature, but also many undesirable reactions occur at the same time.39 Besides the benefit of short diffusion length in the nano-sized particles, it was reported that coexisting crystalline phases have greater mutual solubility for lithium (reduced miscibility gap) in nanoscale LiFePO4.40 At higher temperatures of 100–200 °C, a solid solution is formed. In this case, a fast diffusion is expected during charge and discharge since no phase boundaries are involved.41
![(a) Crystal structure of LiFePO4 illustrating 1D Li+ diffusion channels oriented along the [010] direction. (b) Schematic illustration of Li+ diffusion impeded by immobile point defects in 1D channels. Reproduced with permission from ref. 35. Copyright 2010 American Chemical Society.](/image/article/2012/EE/c1ee01263k/c1ee01263k-f3.gif) | ||
| Fig. 3 (a) Crystal structure of LiFePO4 illustrating 1D Li+ diffusion channels oriented along the [010] direction. (b) Schematic illustration of Li+ diffusion impeded by immobile point defects in 1D channels. Reproduced with permission from ref. 35. Copyright 2010 American Chemical Society. | ||
Although the size reduction strategy has been employed to improve the performance of LiFePO4 in many studies, recent reports indicate that the specific capacity of LiFePO4 has no clear dependence on the particle size in the range of 50–400 nm.42,43 For example, with the introduction of highly conductive carbon, some LiFePO4 materials with a larger particle size (150–300 nm) exhibited better performance than the materials of smaller size (30–100 nm). This is because Li-ion transport in these LiFePO4 particles is not diffusion-controlled, and the contribution of size reduction to the capacity of LiFePO4 is therefore limited.42
It has to be pointed out that the nano-sized particles do not necessarily always give excellent electrochemical performance. For instance, it was found that in the hydrothermal synthesis anti-site defects may form, which block lithium ion transportation, causing slow kinetics.44 Therefore, an optimum particle size is crucial to high-performance of LiFePO4, considering these adverse effects of nanosize LiFePO4 in the practical applications. A particle size range of 200–400 nm was suggested in some reports for the high quality powder-based applications.42
3.2 Conductive surface coating
Coating of the LiFePO4 surface with Cu, Ag, carbon, or conducting polymers is very effective in improving the electronic conductivity of the powders.45 Although metal additives may effectively improve the conductivity of LiFePO4, it is difficult to achieve a uniform metal dispersion on the surface of LiFePO4.45 Metal oxidation to form insulating films or soluble ions that might interfere with the negative electrode cyclability is also to be taken into account while carbon is well known for its stability and compatibility and use in practical composite cathode. Furthermore, use of a high-cost metal additive is not suitable for large-scale applications of the LiFePO4 products. The conductive polymers such as polypyrrole generally have poor mechanical properties and instability.46 Although recently poly(3,4-ethylenedioxythiophene) (PEDOT) chemically grown on partially delitiated LiFePO4 seems to lead to good cyclability,47 by comparison, carbon coating has been particularly attractive with respect to its high conductivity using carbon concentrations as low as 0.5–2 wt%,50–53 its low cost, and simplicity of introduction during or after the LiFePO4 synthesis and most importantly its proven use as a conductivity additive in composite electrodes and its chemical stability in the battery. A combination of carbon coating and fine particle size can improve the rate performance and material utilization for LiFePO454–56 and has been commercialized.13,56 To make high capacity, low cost LiFePO4 batteries with a suitable size, it is desirable to reduce the amount of carbon and improve the carbon coating quality. Although numerous reports in the literature have focused on C/LiFePO4 composite materials and great progress has been achieved in this field in the recent years (Fig. 4),57,58 the state-of-art understanding of the role of carbon in the C/LiFePO4 composite material is still scarce.3.3 Lattice engineering
Doping with supervalent cations in LiFePO4 was first mentioned by Goodenough–Armand and reported by Chiang's group as an efficient method to improve the bulk conductivity and an increase of eight orders of magnitude in electronic conductivity was shown.15,63 Later on, Ravet et al. showed that the electronic conductivity increase may come from the residual carbon from the organic precursors being used.64 Nazar's group reported that a nano-network of metal-rich phosphides might also contribute to the enhanced conductivity.65 Islam et al. investigated the doping behavior with atomistic simulation techniques and indicated that supervalent doping (e.g., Ga3+, Ti4+, Nb5+) on either Li or M sites is energetically unfavorable and does not result in a large increase in electronic (small polaron) species.66 Clearly, the effectiveness of doping supervalent elements has been under debate since the beginning, and it is still not proven to this day.In addition to the supervalent cation doping, it has been reported that anion doping (e.g. F−, Cl−) was also effective in enhancing the electrochemical performance of C/LiFePO4. For example, F-substitution (LiFe(PO4)0.9F0.3/C) may improve the electric conductivity of LiFePO4 and alleviate the polarization under high current densities.67 Also the high ionicity of F− may contribute to the diffusion of lithium ion in the F-doped LiFePO4.68 However, the detailed mechanism and role of such anions in the LiFePO4 crystal structure are still not clear.
Similar to the controversy of supervalent cation doping, there are still many debates about whether these anions are actually doped into the LiFePO4 crystal lattice, whether the increased performance of LiFePO4 is due to the presence of carbon, and whether the anion-substitution in LiFePO4 will affect the stability of PO4 polyanions. For the time being, there is still no convincing evidence or answers for these questions.
Recent work by Chiang's group showed that doping can reduce the lithium miscibility gap as stated by Armand–Goodenough in 1997 for modified olivine,15 ease phase transformation and expand Li diffusion channels.69 However, given that Li site doping contributes much more to the increase of electronic conductivity than Fe site doping,70,71Li site doping may affect the activation energy of Li ion transportation and block the one-dimensional Li ion migration path,72,73 which restrict the wide use of this technique in LiFePO4. Up to now, to alleviate the poor electronic conductivity problem, the carbon coating technique is therefore still generally introduced to prepare C/LiFePO4 composites.
3.4 Surface engineering of chemical composition
Another interesting report was published recently in Nature by G. Ceder's group concerning surface modifications by using non-stoichiometric composition generating lithium phosphate species containing iron.74 They achieved a rate capability equivalent to full battery discharge in 10–20 s under ambient temperature. The phenomenon was explained by a fast ion-conducting polyphosphate surface phase which increases lithium ion diffusion across the surface towards the (010) facet of LiFePO4 and thus enhances the rate capability. However, the hypothesis was commented by J. B. Goodenough et al.,75 and the debate still continues,76 since it is difficult to discriminate between the impact of nano-sized particles in the presence of carbon coating on powder and the exact contribution of an additional lithium phosphate phase.Other non-carbon second phase (e.g. metal oxides, polymers) coatings were also reported to modify the LiFePO4 surface. Metal oxide has been used to improve the performance of LiFePO4. V2O3 possesses a very high electronic conductivity of over 103 S cm−1 at room temperature, thus it was used as the second phase to modify C/LiFePO4 to enhance its conductivity.77 It was also found that some other metal oxides (CeO2, ZrO2, TiO2) can not only enhance the discharge capacity but also improve the thermal stability and cycle performance.78,79 The improved cycling performance is due to the metal oxide layer working as a protecting layer against HF attack from the electrolyte.80 However, similar to metal additives, the uniform coating and high cost of metal oxides are still big challenges for its real application. By using the atomic layer deposition (ALD) technique, ultra-thin and uniform metal oxide films have been achieved in our recent work.81,82 Dramatic electrochemical performance improvement has been demonstrated with only 2 atomic layers of Al2O3 on LiCoO2 by other researchers.83,84ALD is therefore believed to be an important technique for surface coatings on LiFePO4.80
Various approaches that have been attempted to improve the electrochemical performance of LiFePO4 are illustrated in Table 1.
| Methods | Catalogue | Advantages | Disadvantages | Performance | References |
|---|---|---|---|---|---|
| a PPy: polypyrrole, PANI: polyaniline. | |||||
| Size reduction | Micro (10 μm) | High tap density | Low rate performance | C/LFP exhibits a specific capacity of 71 mA h·g−1 at 1 C | 59 |
| Sub-micro (0.5–1.0 μm) | Medium tap density | Medium performance | C/LFP exhibits a specific capacity of 129 mA h·g−1 at 1 C | 59 | |
| Sub-micro (0.5μm) | Medium tap density | Medium performance | C-free LFP has a poor specific capacity of 72 mA h·g−1 at 1 C | 60 | |
| Nano-size (100–200 nm) | High rate performance | Low tap density | C-free LFP exhibits a capacity of 147 mA h·g−1 at 5 C | 61 | |
| Nano-size (20 nm) | High rate performance | Low tap density | C-free LFP exhibits a specific capacity of 163 mA h·g−1 at 1 C | 62 | |
| Conductive surface coating | Metal (Cu/Ag) | High conductivity | High cost non-uniform | Cu or Ag (1 wt%)-LiFePO4 exhibits a capacity of (140 mA h·g−1 at 0.2 C rate) | 45 |
| Polymer (PPy/PANI)a | High conductivity | Poor mechanical properties | C/LFP/16 wt% PPy and C/LFP/7 wt% PANI exhibit a specific capacity of ∼145 and ∼140 mA h·g−1 at 1 C respectively | 48 | |
| Carbon | High conductivity | No improving intrinsic conductivity | Phostech C/LFP (0.5–1.0 μm) has a capacity of 150 mA h·g−1 | 49 | |
| Lattice engineering | Supervalent cation doping | — | No convincing evidences | Li1−XMxFePO4 (M = Zr, Nb, Mg) increases its conductivity by a factor of ∼108 and has a capacity of ∼150 mA h·g−1 at 0.1 C | 63 |
| Anion doping | — | Possible structure change | LiFe (PO4)1−XCl3X/C (x = 0.02) delivered 138 mA h·g−1 at 1 C LiFe(PO4)0.9F0.3/C shows110 mA h·g−1 at 10 C | 67,68 | |
| Surface engineering | Ion-conducting polyphosphate | — | Mechanism debates | LiFe0.9P0.95O4−δ shows a super high capacity of 170 (20 C) and 130 mA h·g−1 (50 C) | 74 |
| Metal oxide coating | Improving cycle performance | High cost, non-uniform | ZrO2/LFP exhibits 146 mA h·g−1 at 0.1 C | 77 | |
| V2O3/LFP exhibits 143.5 mA h·g−1 at 0.2 C | 78 | ||||
| TiO2 and CeO2 improve the stability of LFP | 79 |
4. Understanding of carbon coating on LiFePO4
4.1 Synthesis of LiFePO4
Various synthetic methods have been explored to produce LiFePO4 powder and have been reviewed in many review articles.17,42,85 It is not the subject of this work to revisit all the synthetic methods, but we will instead discuss the synthetic methods in connection with carbon coating.Generally speaking, all solid state or wet processes that have been reported so far for inorganic synthesis were used to make LiFePO4, such as solid state reaction starting from all kinds of solid or liquid precursors86–88 or wet chemistry processes including hydrothermal,89 solvothermal,90 ion-thermal,91 sol–gel,92coprecipitation,93 or spray pyrolysis.94
In the solid state reaction, carbon or carbon precursors are normally mixed with iron, phosphorus (or phosphate) and lithium sources. After reaction at an elevated temperature, LiFePO4 is formed with carbon deposition on the particle surface. Mechanochemical activation has been reported to increase the reaction kinetics and lead to fine particles.88
Compared with the solid-state-based methods, the solution-based methods allow the precursors at the molecular level in solution to be homogeneously mixed. The carbon precursor may be incorporated into the precursors depending on the needs. For instance, in the sol–gel process, carbon precursors are usually added or be part of the chelating agent.92 In this case, only LiFePO4 precursor is formed at low temperatures during sol-gel process, and a further annealing process at higher temperatures (above 600 °C) is still required to make LiFePO4. Under these high temperatures conditions, carbon deposition on LiFePO4 surface can effectively suppress particle growth.55 On the other hand, in the hydrothermal or solvothermal process, LiFePO4 nanoparticles can be formed under pressure in an autoclave at low temperature (<200 °C). The particle size and morphology can be tailored by the pH, temperature and surfactant. In this case, normally carbon precursors are not necessarily be added. However, a post-treatment of the LiFePO4 nanoparticles with organic precursors and calcination at elevated temperature are usually performed to increase the conduction. Details of carbon coating associated with each process will be discussed in Section 4.5.
4.2 Carbon addition to enhance the conductivity of LiFePO4
To sustain fast charge and discharge in the battery, it is essential to have fast transport of electrons and lithium ions. In the commercial process of battery manufacturing, highly conductive carbon additives like carbon black, graphite or carbon fibers are often added to active materials with a binder to enhance the conductivity of the composite cathode or anode coating. When the particle size of LiFePO4 becomes small (down to the nano or submicron size), a large proportion of carbon additive is required to connect all active materials, which causes low loading of active materials. Effective dispersion of carbon additives with active LiFePO4 is also a challenge. Mechanofusion technology has been used to dry coating carbon nanoparticles on the LiFePO4 surface. Surfactant addition to the coating slurry was also commonly used to help disperse carbon particles.Carbon can also be added to the active materials during the synthesis process to improve the conductivity of the active materials. Various trials to add carbon particles to the reaction precursors have been made in the solid state and wet processes. The advantage is that one can add highly conductive carbon like graphite in combination with high surface area conductive carbon black. The challenge is to disperse the carbon well with the precursors. Usually a significant percentage of carbon (5–20 wt%) is required to reach the percolation point of high conductivity especially with submicron LiFePO4 particles.53
The most effective way to increase the conductivity with a minimal dead weight penalty is to apply a carbon coating on the LiFePO4 particle surface by pyrolysing solid-, liquid- or gas-phase organic precursors or a thin layer of polymers on the LiFePO4 surface, as reported by Ravet et al.50,51,54–56 In such a process a Pyrolytic Carbon Deposit (PCD) can be formed simultaneously during LiFePO4 synthesis or after the synthesis in a second step. For instance, the pristine LiFePO4 or its precursor can be coated with organic or polymeric materials in solution and then be heated to an elevated temperature to carbonize the organic layer forming a thin layer of carbon.
The poor conductivity can be improved by creating such a conductive carbon coating layer, sometimes at the nano-scale, on the LiFePO4 particles with a minimal amount of adherent carbon (usually less that 2 wt%).50,51,55,56 The electrons can be spread to the entire surface of the particles through this carbon layer during charge/discharge and therefore improves the kinetics and reversibility of the lithium insertion/de-insertion cycles. This method of carbon coating can also suppress particle growth during or after synthesis and improve product purity by eliminating any Fe3+ species using the reducing gases generated from the pyrolysis.55,95,96 These will be further discussed in the following sections.
4.3 Effect of carbon on the tap density of C/LiFePO4
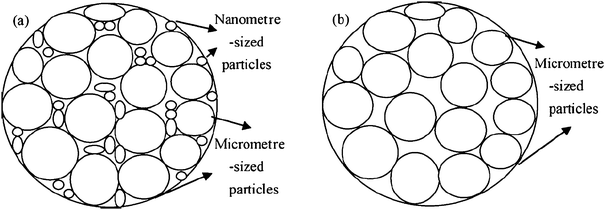
While carbon coating improves the conductivity of LiFePO4, a monotonic decrease of tap density with carbon content is observed. This is understandable since carbon has much lower specific density than LiFePO4 without taking into account possible carbon deposit porosity.97 Chang et al. investigated the effect of carbon content on the tap density of LiFePO4 synthesized using self-produced high-density FePO4 as a precursor,98 and glucose as a carbon source, and found that the tap density decreases from 2.15 to 1.66 g cm−3 when the carbon content increases from 3 wt% to 9 wt%. An optimum carbon content of 7 wt% was suggested, which gives a tap density of 1.80 g cm−3, and a superior electrochemical performance than other compositions. Clearly, the 7% of carbon is dead mass, leading to 7% capacity loss, which is not efficient for developing high-performance commercial materials.Tap density of C/LiFePO4 is not only determined by the carbon content, but also determined by the particle size, particle morphology and particle size distribution. A spherical C/LiFePO4 material synthesized by using the precipitation method gave a tap density of 1.75 g cm−3,99 which is remarkably higher than the non-spherical LiFePO4 powders (tap-density 1.0–1.3 g cm−3) of similar size. Another method being reported recently to improve C/LiFePO4 tap density is to prepare agglomerate particles. For example, a tap density of 1.1 g cm−3 was achieved by preparing agglomerate C/LiFePO4 composite via the spark-plasma-sintering (SPS) technique.100 More recently, further improvement has been reported by using a two-step drying method to increase the tap densities from 1.37 to 1.80 g cm−3.97 This improvement could be attributable to the net structure formed by adding a second drying step in the process, resulting in agglomerations of the colloid particles. Because of the denser structure, the grains of the particles are more prone to grow and crystallize in the drying process. Another report shows that composite synthesized with two kinds of Fe3+ precursors (consisting of nanometre-sized and micrometre-sized particles) exhibited less vacancy than that prepared with single Fe3+ precursor (composed mainly of micrometre-sized particles) and led to high tap density which increases from 1.19 to 1.40 g cm−3 (Fig. 5).101
 | ||
| Fig. 4 (a) TEM image and (b) high-resolution TEM image of single-crystalline C/LiFePO4 nanowires (reproduced with the permission from ref. 57, Copyright 2011 Wiley), (c) TEM image and (d) high-resolution TEM image of C/LiFePO4 nanoplate (reproduced from ref. 58 with permission; Copyright Springer 2010). | ||
 | ||
| Fig. 5 The possible packing models of the samples (a) LFP1 and (b) LFP2. Reprinted from ref. 101 with permission. Copyright 2010 Elsevier. | ||
In addition, tap density of the C/LiFePO4 is well related to the experimental conditions and methods. The calcination temperature in the coprecipitation method is an important influencing factor for tap density of C/LiFePO4 as it affects the morphology and crystal structure of the powders.102 Sun et al. reported that C/LiFePO4 prepared at 800 °C exhibited high crystallinity as well as a higher tap density of 1.09 g cm−3 compared with the one prepared with other temperatures (650, 700, 750, and 850 °C). In further work, the tap density of C/LiFePO4 (∼3 wt% carbon amount) was increased to 1.5 g cm−3 by using self-prepared FePO4 as precursor and a pitch as carbon source.103 This highly dense C/LiFePO4 material consisting of nanoscale particles (200–300 nm) and pores (100–200 nm) shows a 2.5 times greater volumetric energy density with similar rate capability compared with nano LiFePO4. More recently, to improve the uniformity of carbon coating combining with high tap density,104 they reported a double carbon coating on microscale nanoporous LiFePO4. Carbon coated FePO4 was first prepared viacoprecipitation using sucrose as the first carbon source, and the resulting C/FePO4 was further mixed with Li2CO3 (lithium source) and pitch (the second carbon source) and calcined at high temperature (Fig. 6). This material still has a high tap density of 1.5 g cm−3 with about 0.5 wt% carbon amount and a superior specific capacity, rate capacity, and volumetric energy density. Meanwhile, in another work, to make better dispersion of carbon on LiFePO4 through the coating of its precursor, polyvinylpyrrolidone (PVP) was also employed as the carbon source to reduce the polarity of water and a smooth and homogeneous carbon precursor coating was achieved on micro-scale LiFePO4.105 The 6 μm C/LiFePO4 microsphere with a higher tap density (1.6 g cm−3) exhibits an excellent rate capability and delivers a volumetric capacity of 225 mA h cm−3.
 | ||
| Fig. 6 SEM images obtained from a focused ion beam cut of LiFePO4 at low (a) and high (b) magnification. Cross-sectional TEM image of LiFePO4 (c) and the corresponding EELS image (d) (where red: C, blue: Fe, and green: P). Reprinted from ref. 104 with permission. Copyright 2010 Wiley. | ||
4.4 Effect of carbon on the properties of C/LiFePO4 composite
Carbon coatings greatly enhance the specific capacity, rate capacity and cycling performance of LiFePO4. Cyclic voltammetry (CV) was used to evaluate the kinetics of lithium intercalation and de-intercalation.50,52,106 Each of the CV curves consists of an oxidation peak and a reduction peak, corresponding to the charge–discharge reaction of the Fe2+/Fe3+ redox couple. The midpoint between the oxidation and reduction peaks, which corresponds to the redox potential of the LiFePO4 electrode, is about 3.45 vs.Li/Li+. In pure LiFePO4 materials without carbon coating, a larger separation between redox peaks appears, indicating a reduced reversible behavior which is mainly related to the sluggish diffusion step. Coating olivine particles with a carbon layer can greatly improve reversible performance for the insertion and extraction of Li ions and their kinetics. This is because the pyrolytic carbon in the product suppresses sintering and particle growth during or after synthesis, leading to fine particles which shorten the distance of the transport passages in the solid-phase, and improves the insertion and extraction of Li ions. The separation potential between redox peaks was also reduced further with the increasing content of carbon.98,106 The peak current of C/LiFePO4 nanocomposite materials increases with carbon content also, which means better electrode reaction kinetics and better rate performance of the LiFePO4 cathode material.Great improvement of specific capacity can be achieved on LiFePO4 after carbon coating, which is due to the better reversible performance for Li ion insertion and extraction and the electrode kinetics (Fig. 7).107,108 A high conductivity as a result of the carbon coating can also lead to a reduction of the cell impedance (Fig. 8).109 A further decrease of the charge transfer resistance can be obtained with the increase of carbon content. Thus the rate capacity of LiFePO4 can be greatly enhanced by carbon coating.107,110
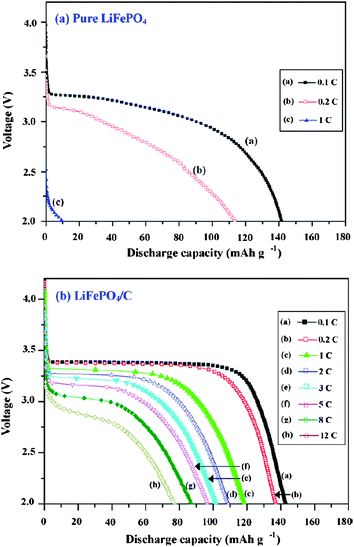 | ||
| Fig. 7 The typical charge and discharge curves of (a) LiFePO4 (pure) and (b) LiFePO4/C cells. Reprinted from ref. 107 with permission. Copyright 2008 Elsevier. | ||
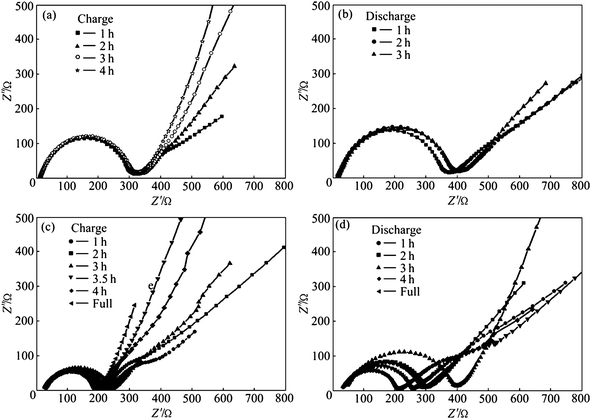 | ||
| Fig. 8 Electrochemical impedance spectra of cathodes: (a) Non-modified LiFePO4; (b) non-modified LiFePO4; (c) CNTs-modified LiFePO4; and (d) CNTs-modified LiFePO4. Reprinted from ref. 109 with permission. Copyright 2010 Elsevier. | ||
Besides the better specific capacity and rate performance, better cycling performance of LiFePO4 can be obtained by carbon coatings. A good cycle performance of less than 5% discharge capacity loss over 1100 cycles at a low discharge rate (0.6 C rate) was reported in a C/LiFePO4 nanocomposite with a core–shell structure.111 The cycling stability of C/LiFePO4 at a high discharge rate is very critical for practical applications. A recent report reveals that the C/LiFePO4 composite also shows a greatly enhanced cyclic stability at 4 C, which is retained as high as 92% after 1000 cycles.112 In another report,113LiFePO4/carbon composite was synthesized by a simple high-energy ball milling combined with the spray-drying method, the C/LiFePO4 composite obtained shows a greatly enhanced rate performance and an excellent cyclic stability at room temperature with 92% retention of its original discharge capacity beyond 2400 cycles (10 C rate). In addition, the cycling performance was affected by the synthesis conditions,114 although the material synthesized at a lower sintering temperature had higher discharge capacity due to a lower degree of crystallization and plenty of nano-sized microstructure. The lower sintering temperature also leads to the instability of the crystal structure and the solution of active materials, which results in the degradation of discharge capacity over long-time cycling. Therefore, synthesizing the material with high crystallization and moderate particle size in the presence of the carbon source is an effective way to improve the cycling performance of C/LiFePO4 cathode materials.
In this section, the details of carbon coating including carbon sources, carbon content, carbon thickness, carbon structure, carbon distribution and morphology, and carbon surface area and porosity are reviewed.
However, it is difficult to define the optimum carbon content without discussing the carbon structure, the carbon distribution, morphology as well as the LiFePO4 effective surface developed, especially with nano-scale particles. If the carbon is not homogeneously distributed on the LiFePO4 particle surfaces, as in most cases, it is even harder to define the optimum amount of carbon content (wt%) and carbon film thickness. The carbon film thickness is generally related to the carbon content, and high carbon content can generally contribute to high conductivity but also means thick carbon coating (for instance larger than 3–5 nm) which might restrict the easy penetration of lithium ions. Less carbon means thin carbon coating, low negative impact on tap density, but it is normally linked with non-uniform coatings or not enough surface conductivity.
Zaghib et al. investigated the electrochemical properties of LiFePO4 cathodes containing different carbon contents from 0 to 12% of conductive additive (carbon black or a mixture of carbon black and graphite) and found that the specific capacity increases with increasing carbon content and does not show a maximum value until 12% of carbon content,117 but it does show decreased volumetric capacity of electrode. Another study reported an optimum range of carbon content from 3.5 to 10.3%, and the performance rapidly decreased with further increase in the carbon content.118 The rapid decrease of performance can be attributed to the following possible reasons: (1) amorphous carbon phase dilutes the density of the crystalline LiFePO4 phase, (2) excess carbon suppresses formation of the crystalline LiFePO4 phase, and (3) high carbon content combined with high temperature (800 °C) reduces Fe and P to form inactive Fe2P. Compared with the range of carbon amount, an exact optimum 5 wt% was reported which shows a best performance of C/LiFePO4 in a battery.119 A similar optimum carbon content (4.6%) was also reported using a different preparation method.120 However, the optimum carbon content is different in various studies. Lu et al. added high surface area carbon to LiFePO4 and indicated that C/LiFePO4 with 8.0 wt% carbon exhibited the best performance.121 Chang et al. suggested that 7 wt% carbon may be an optimum value for the C/LiFePO4 material with regard to the tap density because the greater carbon content will result in the lower tap density,122 but the discharge capacity increases little if the carbon content is over 7 wt%.
The optimum value reported for carbon content varies in the literature. This is because the synthesis process including precursors, experimental conditions and methods strongly influences the physical characteristics of the formed carbon material, resulting in the different electrochemical behavior of C/LiFePO4 materials. For example, 90% of the theoretical capacity at the 0.5 C rate in C/LiFePO4 composites at room temperature has been achieved by using 15 wt% carbon gels as a carbon source.53 The same capacity value was obtained by reducing the carbon content to nearly 3 wt% in other reports using a different carbon incorporation method.116 Thus the optimum carbon content can be just considered as a reference value in these studies since in fact total carbon in the electrode has to be taken into account which contains the carbon coating, other particulate carbon introduced during LiFePO4 synthesis, and any other carbon added to the binder during the composite electrode formulation. Considering the impact of carbon on tap density and volumetric capacity, a lower carbon content is critical for real applications. A small amount of in situcarbon (∼1.5 wt%) derived from ethylene glycol was reported to decrease the particle size of LiFePO4 and to improve significantly the rate capability of LiFePO4.123,124 The obtained C/LiFePO4 material shows a high-rate capacity with 150 mA h g−1 at C/25 rate and around 100 mA h g−1 at 5 C rate. Although the capacity is not the highest one among the reported studies, the lowest carbon content reported in the literature makes it more promising in industrial production and application.
During the carbon coating process, the carbon is usually derived from organic precursors that are added to the reaction mixture before firing. During firing, the organic material is decomposed and carbonized in an inert or reducing atmosphere. Therefore, carbon thickness also can be controlled by the addition of the amount of carbon precursors. It is easily understood that the carbon coating thickness increases with the amount of carbon precursor. For example, a carbon coating thickness of about 1–2 nm was obtained by Dominko et al. from 3.2 wt% citrate anion as a carbon source, while it increased to about 10 nm when 12.3 wt% citrate anion was used (Fig. 9).125 In addition, although there are no reports showing direct evidence for the relationship between carbon thickness and carbon precursor, it is not difficult to understand this correlation because different carbon precursors have various optimum decomposed temperatures and contribute to different amounts of carbon which generally lead to different carbon thickness. Moreover, carbon thickness is also related to the LiFePO4 particle size and surface area. Smaller-sized LiFePO4 particles with higher surface areas generally get thinner carbon layers when carbon amount and physical properties are fixed because more surface area is coated due to the high surface area of small-sized LiFePO4.
![HRTEM images of tiny edges of LiFePO4/C composite particles. (A) Crystal planes in the [200] direction are ending in about 1 nm thick carbon layer (the source of carbon was solely the citrate anion), (B) crystal planes in the [101] direction are ending in about 2–3 nm thick carbon layer (the source of carbon was citrate anion and 1.7 wt% of HEC) and (C) crystal planes in the [211] direction are ending in about 10 nm thick carbon layer (the sources of carbon were citrate anion and 20.4 wt% of HEC). Reprinted from ref. 125 with permission. Copyright 2005 The Electrochemical Society.](/image/article/2012/EE/c1ee01263k/c1ee01263k-f9.gif) | ||
| Fig. 9 HRTEM images of tiny edges of LiFePO4/C composite particles. (A) Crystal planes in the [200] direction are ending in about 1 nm thick carbon layer (the source of carbon was solely the citrate anion), (B) crystal planes in the [101] direction are ending in about 2–3 nm thick carbon layer (the source of carbon was citrate anion and 1.7 wt% of HEC) and (C) crystal planes in the [211] direction are ending in about 10 nm thick carbon layer (the sources of carbon were citrate anion and 20.4 wt% of HEC). Reprinted from ref. 125 with permission. Copyright 2005 The Electrochemical Society. | ||
With regard to the effect of carbon thickness on the C/LiFePO4 performance, Cho et al. indicated that the thickness of carbon and conductivity increases with the carbon content.126 By controlling the thickness of carbon (from a few nanometres to a few tens of nanometres where the carbon content was controlled between 1.25 and 2.54 wt%), the sample with the carbon thickness of 4–8 nm exhibited the best discharge capacity due to the easy diffusion of lithium ion and uniform coatings. It is evident that an optimum carbon coating thickness is desirable to balance the conductivity and easy lithium ion penetration, but similar to the optimum carbon amount, it is also difficult to find an identical value among different studies. For example, core–shell C/LiFePO4 composites synthesized via a chemical vapor deposition assisted solid-state reaction show 3 nm carbon coating, which exhibits a high specific capacity of 114.6 mA h g−1 at 5 C rate.127
Because the synthesis method and precursors used vary in different reports in the literature, it is meaningless to compare these optimum carbon content values and thicknesses directly. However, from the aforementioned studies, it is obvious that a thin carbon film with low carbon content is important for LiFePO4 in order to have easy diffusion of lithium ion and high tap density. However, it is still a challenge to make thin and uniform carbon coatings on LiFePO4.
The structure of the carbon deposit can be tuned by using additives during C/LiFePO4 synthesis. The choice of an appropriate carbon source can directly affect the carbon structure. A high amount of graphite carbon is observed for the C/LiFePO4 composite prepared with sucrose as an additional source of carbon.133 The incorporation of naphthalenetetracarboxylic dianhydride during synthesis also results in a more graphitic carbon coating and improves utilization of LiFePO4 in lithium cells.129 Besides, the addition of a small amount of functionalized aromatic or ring-forming compounds before the final heating process also enriches graphitized carbon in the residual carbon.128,129,134,135 Recently, a polyaromatic compound, e.g.polystyrene, with a lower sp3/sp2 peak ratio contributes to more highly graphite-like carbon formation for LiFePO4 during polymer pyrolysis and the resultant C/LiFePO4 exhibited a superior capacity.136
Although optimizing the carbon structure can be achieved by choosing an appropriate carbon precursor, it is still difficult to produce highly graphitic coatings by the pyrolysis at relatively low temperatures (600–800 °C) used for synthesis of LiFePO4 because carbon is difficult to graphitize at these low temperatures without catalysts. It is well known that some iron compounds can catalyze the formation of graphite at relatively low temperatures. Raman studies indicate that such an effect can occur at a pyrolysis temperature of 700 °C, which is much lower than the pyrolysis process without catalysts.95,96
Furthermore, carbon nanotubes, which consist of curled graphene sheets, can be made at temperatures as low as 600–700 °C using organic or polymeric carbon sources and iron compounds as catalysts. Thus, addition of graphitization catalysts during the preparation process may result in carbon with lower D/G ratios or more graphite carbon material. Doeff et al. realized it by adding small amounts of ferrocene (graphitization catalysts) into pyromellitic acid (carbon sources),131 and the C/LiFePO4 material obtained shows dramatically improved rate performance even with an overall carbon content of below 2 wt%.
Apart from carbon sources, the preparation method has an important impact on the formation of carbon structure. Similar to carbon nanotube growth, a novel chemical vapor deposition (CVD) assisted solid-state route was reported that a graphite shell with the thickness of 3–5 nm was formed on LiFePO4 using polyvinyl alcohol (PVA) and benzene vapor as the reducing agent and carbon source (Fig. 10).127 The discharge capacity of C/LiFePO4 with a graphite shell is much higher than that of the LiFePO4 composite without a graphite shell. Such a chemical vapor deposition-assisted synthesis method may be more promising for synthesis of C/LiFePO4 composite material.
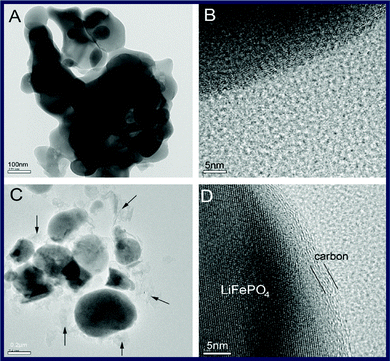 | ||
| Fig. 10 TEM micrographs of C/LiFePO4 composites (A and B) without CVD and (C and D) with CVD. Reprinted from ref. 127 with permission. Copyright 2009 Elsevier. | ||
Another way to increase the graphite carbon content is by direct addition of highly graphitic carbon material to LiFePO4. Carbon nanotube is a promising material due to the highly graphite content and superior conductivity.137,138 Liu et al. investigated the effect of carbon nanotubes on the electrochemical performance of LiFePO4 batteries.139 Compared with the LiFePO4 batteries with carbon black additive, those with carbon nanotube additive show better electrochemical performance with a capacity retention ratio of 99.2% after 50 cycles at 1/3 C rate. This is because the carbon nanotubes functioned as a link to connect the LiFePO4 particles and enabled active materials to transport lithium ions and electrons at fast rates, resulting in the disappearance of Li-ion migration resistance through the SEI film and the decrease of charge transfer resistance (Fig. 11).107,140–143 Besides carbon nanotubes, Carbon Nano-Fibers (CNFs) have been used in replacement of amorphous carbon.144 The addition of functionalized Carbon Nano-Fiber to LiFePO4 also shows better electrochemical performance compared to the addition of acetylene black to LiFePO4, even though the coating is not homogeneous. The improved performance is probably due to the high electronic conductivity of the cathode material due to the CNF addition and the efficient contact between electrochemical active particles and the electronic conducting CNFs.
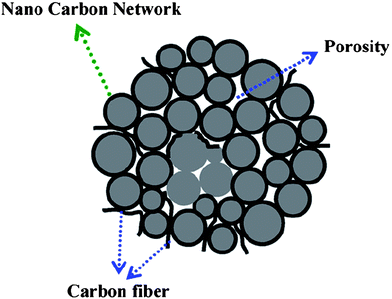 | ||
| Fig. 11 The schematic representation of LiFePO4/C powders with a continuous nanocarbon network. Reprinted from ref. 107 with permission. Copyright 2008 Elsevier. | ||
Recently, as a novel carbon material, graphene has shown its great potential in LiFePO4 applications due to its unique structure, superior electronic conductivity, and high surface area. Su et al. directly mixed graphene with a commercial LiFePO4 material to form the graphene-based conductive network. The conducting network was constructed flexibly through a “plane-to-point” mode due to the soft two-dimensional structure of graphene, compared to the “point to point” conducting mode in the conventional carbon-based conductive additive.145 The obtained LiFePO4/graphene composite exhibits an excellent electrochemical performance. In addition to direct mixture, more studies were performed by introducing graphene during the LiFePO4 synthesis process. Ding et al. prepared LiFePO4/graphene composites by a coprecipitation method and achieved considerable improvement in capacity delivery and cycle performance of LiFePO4.146 However, the rate performance was not significantly improved because LiFePO4 nanoparticles were only loosely loaded on graphene sheets and limit the enhancement in electronic conductivity. Zhou et al. recently wrapped LiFePO4 nanoparticles with a 3D graphene network and formed spherical graphene-modified LiFePO4 by spray-drying and annealing processes.147 The conductive network can facilitate electron transport effectively, while the presence of abundant voids between the LiFePO4 nanoparticles and graphene sheets was beneficial for Li+ diffusion. The composite cathode material thus delivers a superior rate performance of 70 mA h g−1 at 60 C discharge rate. The effective three-dimensional conducting network with bridging graphene nanosheets was also created by a hydrothermal route in another report.148 Consequently, graphene may offer great potential for applications in future high-performance LiFePO4.
There are mainly two methods for carbon introduction to LiFePO4. One is direct addition of various particulate carbon materials by mechanical means, which allows the choice of high surface area carbon or high-graphitized carbon material, but no available techniques can lead to uniform coating on the surface of LiFePO4 since carbon is neither soluble nor fusible. The other method is in situ deposition by using the pyrolysis of a carbon precursor during the synthesis of LiFePO4, which allows the achievement of uniform coating of the precursor on LiFePO4. Kim et al. compared C/LiFePO4 composite materials with two different carbon addition methods.149 One is carbon powder direct introduction and the other is using sucrose as the carbon precursor. The sucrose precursor method leads to the formation of a thin, porous and more uniform carbon coating around the particles and results in a slightly higher discharge capacity compared to the other sample.
In spite of this, these approaches based on the thermal decomposition of carbon-containing precursors can still only produce LiFePO4 particles with a partial coating of carbon. A uniform carbon coating is still highly desirable. Recently, an in situpolymerization method was developed by Wang et al. for the synthesis of a C/LiFePO4 composite formed from a highly crystalline LiFePO4 core with a size of about 20–40 nm and a uniform semi-graphitic carbon shell with a thickness of about 1–2 nm (Fig. 12).111Aniline was oxidization polymerized on the outer surface of FePO4 with the presence of Fe3+ and then Fe3+ ions were reduced to Fe2+ during the process. After the subsequent heat treatment, the polymer shell was transformed into a carbon shell that restricts the in situ crystallite growth of LiFePO4.
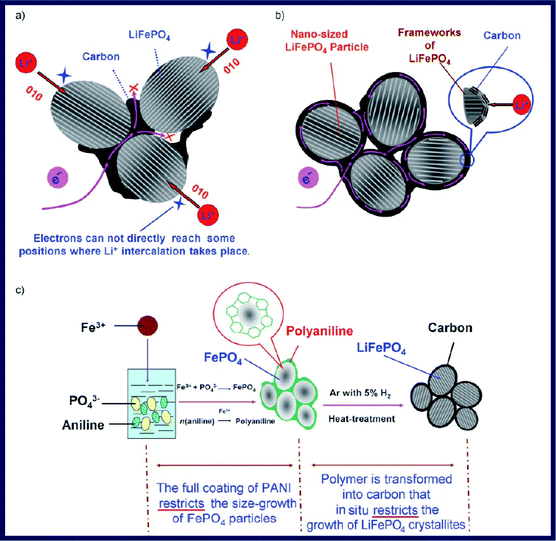 | ||
| Fig. 12 (a) Electron-transfer pathway for LiFePO4 particles partially coated with carbon. (b) Designed ideal structure for LiFePO4 particles with typical nano-size and a complete carbon coating. (c) Preparation process for the C/LiFePO4 composite including an in situpolymerization reaction and two typical restriction processes. Reproduced from ref. 111 with permission. Copyright 2008 Wiley. | ||
Although much work has been done to form a uniform film of carbon on LiFePO4, it is still a major challenge to achieve homogeneously uniform carbon coverage especially on irregular shaped particles and agglomerates, which are more difficult to cover completely and would require more carbon than spherical ones. In view of that, introduction of LiFePO4 into a carbon network and matrix may be a compromised method and has attracted some interest.107,150–153 A continuous, dispersive nano-carbon network was reported by Chen and demonstrated high rate capability (≥12 C) and long cycle life (≥700 cycles at a 3 C discharge rate).107 The use of a nanoporous carbon matrix serves as a mixed conducting 3D nano-network, enabling both Li ions and electrons to migrate and reach each active particle, hence realizing the full potential of nanoactive materials. Wu embedded LiFePO4 nanoparticles in a nanoporous carbon matrix.152 In this nanocomposite, the electronically conducting nanoporous carbon matrix with mesopores filled by electrolyte can be considered not only as a mixed conducting 3D network that allows both Li ion and electron to migrate and reach each LiFePO4 nanoparticle, but also as an electrolyte container for high rate charging/discharging. The nanocomposite electrode shows a superior high power/energy capability. Sides et al. designed a new type of template-prepared nanostructured LiFePO4 electrode consisting of monodispersed nanofibers of the LiFePO4 electrode material mixed with an electronically conductive carbon matrix.153 This unique nanocomposite morphology allows these electrodes to have a capacity of 150 mA h g−1 at a rate of 5 C and maintains a substantial fraction of the theoretical capacity even at rates exceeding 50 C. This new nanocomposite electrode shows such excellent rate capabilities because the nanofiber morphology mitigates the problem of slow Li ion transport in the solid state, and the conductive carbon matrix overcomes the inherently poor electronic conductivity of LiFePO4.
Lu et al. studied LiFePO4 cathode material coated with high surface area carbon (BET surface area, 2099 m2 g−1) by the pyrolysis of peanut shells under argon and treatment with a proprietary porogenic agent,111 with the goal of altering the pore structure and increasing the surface area of the pyrolysis carbon. The high surface area carbon increases the overall conductivity of C/LiFePO4 composite from 10−8 to 10−4 S cm−1 and increases the specific area of the composite resulting in improved electrochemical performance including specific capacity, cycling properties and rate capability.
The introduction of hydrolyzed sugar gives a porous carbon structure on LiFePO4 because the sugar (C12H22O11) is hydrolyzed to glucose (C6H12O6) and fructose (C6H12O6), which ultimately oxidizes to gluconic acid or a polyhydroxyl acid. The polyhydroxyl similar to polymer has cross-link property, which would decompose to porous structure of carbon. The resultant LiFePO4/C composite can achieve high specific capacity (143 mA h g−1) after the 100 cycles with 1 C charge/discharge rate at 50 °C.155 Wang et al. reported that the decomposition of polyethylene glycol led to a porous structure of carbon on LiFePO4.156 The satisfactory initial discharge capacity (139 mA h g−1 at 1 C) and superior rate capacity therefore can be obtained. Recently, the yeast cells as a template and cementation agent and glucose as carbon source is adopted to prepare the porous carbon coated LiFePO4 composite.157 The C/LiFePO4, with the specific surface area of 98.3 m2 g−1, exhibits an initial discharge capacity of 147 mA h g−1 and superior cycling stability.
Along with carbon sources, preparation methods also affect the surface area and porosity. The sol–gel method based on citric acid is a widely used method to prepare porous C/LiFePO4 composite materials.158,159 Perfectly interconnected pores are formed due to vigorous gas evolution (mainly CO and CO2) during degradation of a citrate precursor. The superposition of a continuous thin carbon film (electron conductor) on pores (ion conductor when filled with electrolyte) represents a unique architecture in which the electrons and ions are simultaneously supplied to the site of insertion in the particle interior. Superior electrochemical performance may be obtained while preserving a high tap density.
Organic carboxylic acids are also a common carbon source. Citric acid may be the most popular carboxylic acid for carbon sources and has attracted much interest due to the low pyrolysis temperature (as low as 450 °C).163 This feature ensures a low synthesis temperature of C/LiFePO4 composite with a high surface area and a small particle size. Zhang et al. synthesized C/LiFePO4 by using citric acid as a carbon source and showed high discharge capacities of ∼153 and 92 mA h g−1 at 1 C and 20 C rates respectively.164 Yan et al. also reported a superior performance of C/LiFePO4 synthesized by a solid-state reaction using citric acid as a carbon source.163 It delivers an initial discharge capacity of 128 mA h g−1 at 4 C, which is retained as high as 92% after 1000 cycles. In addition, at −20 °C, the composite exhibits a discharge capacity of 110 mA h g−1 at 0.1 C. In addition to citric acid, Fey attempted to coat LiFePO4 by incorporating various organic carboxylic acids as carbon sources.165,166 These acids include: (a) mono-acid containing a ring structure (salicylic acid, malonic acid), (b) straight-chain diacids (sebacic acid, adipic acid, ascorbic acid), and (c) tri-acids (citric acid). The use of carboxylic acid as a carbon source increases the overall conductivity (∼10−4 S cm−1) of the material. The best cell performance was delivered by the sample coated with 1.9 wt% carbon using 60 wt% malonic acid as the carbon source.
In addition to sugar and organic carboxylic acids, various polymeric precursors were also studied. It was found that highly graphitized carbons are formed during pyrolysis of functionalized aromatic or ring-forming compounds on LiFePO4.166,167 A polyaromatic compound, e.g.polystyrene, with more functionalized aromatic groups displayed improved performance because its decomposition temperature was close to the temperature of the LiFePO4 phase transformation, which resulted in fine particle size and uniform carbon distribution on the composite surface.168
At room temperature, the conductive Fe2P has an electronic conductivity of 1.5 S cm−1, whereas the semiconducting amorphous carbon formed at the intermediate processing temperatures (typically, 600–750 °C) displays conductivity on the order of 10−3 S cm−1.169,170 It is believed that iron phosphides play a more important role in improvement of the bulk conductivity of LiFePO4. Xu et al. also indicated that the carbon amount, morphology, and particle size are directly related to the formation of Fe2P.172 This Fe2P phase among the C/LiFePO4 composite increases the electronic conductivity and evidently improves the electrochemical performance of the composite, whereas too much iron phosphides will lower the discharge capacity of the electrode since they are inert for the de-insertion/insertion of lithium ion.173 Therefore, an optimum amount of iron phosphides may be important for C/LiFePO4 with high performance. In practical preparation processes, the coating carbon can also be considered as the reductive additive under high temperature with Ar gas, and the presence of this reducing circumstance may promote the formation of these highly conductive iron phosphides, all of which should be considered in synthesis of high-performance C/LiFePO4 composite.
4.5 Synthesis process to control carbon coating of C-LiFePO4 composites
It is well known that an inherent difficulty in the conventional solid-state process for synthesis of LiFePO4 is achieving a homogeneous mixture of the precursors. Trivalent ions in the precursors have extremely low diffusion coefficients in the solid state. Thus, it is hard to avoid parallel reactions and inhomogeneities. Some impurity phases therefore appear and by-products remain. To cope with it, melt casting provides us a promising method to make highly pure LiFePO4. Instead of the conventional solid-state process, synthesis in the molten state is an ideal liquid-phase reaction, so high purity and high tap density for the material can be expected. However, one usually can get ingot or large particles by atomization and further grinding to micron and nano-size is needed, which can be achieved by various milling technologies.
In a typical synthesis process, a variety of “low cost” precursors will be melted at 1000 °C followed by cooling leading to a high purity LiFePO4 material with excellent crystallinity and large crystals. The phase purity, crystallinity, and microstructures of the final product depend on the reducing conditions and the solidification process of the melt. Particle size reduction of the resulting LiFePO4 ingot was achieved by a continuous wet milling procedure, which reduces the particle size of the LiFePO4 crystals to 200 nm. C/LiFePO4 samples were prepared by pyrolyzing a variety of carbon precursors to LiFePO4 at 700 °C. Applying carbon coating of the LiFePO4 particles made by melt casting/grinding shows some similarity to that of hydrothermal particles and clear differences are high crystallinity and high purity as well as simplicity of the process. In addition, Zhou et al. also synthesized well-crystallized LiFePO4 by the KCl molten salt method.175Sucrose was used as a conductive precursor and contributed to 3 wt% carbon in the final product. The obtained C/LiFePO4 composite has a high tap density of 1.55 g cm−3 and shows good electrochemical activity.
Solid-state reaction based methods. Solid-state synthesis is a conventional method for preparing C/LiFePO4 which includes several successive steps of intimate grinding and annealing of the stoichiometric mixture of starting materials. In a typical synthesis process, the starting materials containing lithium sources, phosphorus source, and iron(II) precursors were first pretreated at low temperature to prepare precursors and then crystallized at high temperature under an inert or reducing atmosphere to prevent oxidization of iron(II).
Temperature is an important factor influencing the synthesis process of C/LiFePO4. Impurities and an ill-defined crystalline structure are often found in the final products if the sintering temperature is low. However, large particle size and broad particle size distribution of LiFePO4 powder are usually obtained in a solid-state method due to the high sintering temperature. Therefore, there is an optimum temperature for solid-state synthesis of carbon coated LiFePO4 for every different precursor combination.176–178 Zhang et al. reported that the optimized temperature was 800 °C,176 and the capacity of the LiFePO4 was decreased as the temperature was higher or lower than 800 °C. This is because high temperature induces growth and sintering of LiFePO4 particles. However, most of other researchers used a lower temperature ranging from 600 °C to 700 °C to get smaller-sized products. In addition to its impact on particle size, temperature also affects the shape of the grains. An obvious anisotropic growth occurred at a high temperature and the growth rate along the (100) crystal plane was more rapid than that of (111) crystal.179 However, the anisotropic growth of the grain is restrained at 650 °C and spherical carbon-coated LiFePO4 was obtained, which had excellent electrochemical performance.
Conventional solid state methods use iron(II) precursors as starting materials, which increase the preparation cost of the LiFePO4/C composite. In a variant of these conventional solid-state method, Fe(III) salts can be used as raw materials in the presence of a reductant, reducing the material cost and simplifying the manufacturing process. As a reductant, pure carbon can be added directly during the carbothermal reduction process to make C/LiFePO4.180 Organic carbon precursors can also serve as the reductants during the pyrolysis process to reduce the Fe3+ precursor (FePO4·2H2O),51,54,56 which will also combine the carbon coating process and reducing process and a reduction process from hydrogen containing pyrolysis gases in one step.95,96 An advantage of this combined process is to benefit from the pyrolytic carbon deposit and a low reduction temperature to better control LiFePO4 sintering and particle growth.55 A comparison between pure carbon (acetylene black) and carbon precursor (glucose) as carbon sources has been made and the C/LiFePO4 synthesized using glucose as carbon source showed a higher electrochemical capacity and the lower capacity fading,181 which is due to a uniform carbon coating on LiFePO4 through in situpyrolysis of the glucose precursor.182
Temperature still significantly affects the morphologies and performance of synthesized C/LiFePO4 composites in conventional solid-state and carbothermal processes. Both the crystallization degree and particle size increase remarkably with the synthesis temperature.183 The material synthesized at lower sintering temperature had lower crystallization degree and plenty of nano-sized particles, leading to its high electrochemical activity and high initial discharge capacity. However, the lower sintering temperature also leads to the instability of the crystal structure and the solution of active materials, which results in the degradation of discharge capacity on long-time cycling. Thus, synthesizing the material with perfect crystallization degree and moderate particle size at an optimum temperature in a reduction process is very important to improve the cycling performance of C/LiFePO4.
Although the solid-state reaction with or without reduction has been widely adopted for synthesis of crystalline olivine-phase LiFePO4, it is a complex process, including in some cases repeated grinding to gain homogeneous particles and several steps at high temperature to impede formation of the impurity phase. These repeated heating treatments result in undesirable particle growth and agglomeration, presenting a major drawback to practical implementation of this synthesis method.
Recently, the microwave irradiation method has been proposed for the preparation of many functional materials due to its prominent advantages such as heating the material at the molecular level, uniformity, low cost and especially shorter reaction time. Thus, a product with small and uniform particle size can easily be obtained due to the uniform heating and avoiding long reaction time at high temperatures. Zhan et al. developed a rapid synthesis method of highly crystalline C/LiFePO4 composite by microwave heating in under 4 min with glucose as the carbon source and reducing agent.184 Compared with the pure solid-state method, the sample obtained with ball-milling followed by microwave irradiation method has a smaller average particle size (160–600 nm) and more uniform size distribution due to the rapid reaction. Cycling tests show that the sample prepared by a microwave method can deliver 150 mA h g−1 at 17 mA g−1 (0.1 C).
Wet chemical methods. In comparison with conventional solid-state reaction methods, solution-based methods offer advantages of homogeneous mixture of precursors at the molecular level which ensure higher phase purity of LiFePO4 and low reaction temperature to better control of particle size and morphology such as small size and porous structure achieved via a sol–gel method.185 The material prepared with the sol–gel process usually exhibits a better electrochemical rate performance due to a larger number of surface apertures, a smaller mesoporous volume, and a more interlaced pore system compared with the solid-state method. The sol–gel method also offers a wide choice of carbon sources like citric acid,186sucrose,187lauric acid,188ethylene glycol,189 and polymer.190 Yang and Xu synthesized a C/LiFePO4 composite of a particle size of 200–300 nm by a sol–gel method.191 The composite demonstrated specific capacities of around 150 mA h g−1 at discharge rates of 2 C. A smaller size of 20–30 nm C/LiFePO4 composite was achieved by a sol–gel process using citric acid as a complexing agent and a carbon source.192 The electronic conductivity of the nano-sized C/LiFePO4 composites is greatly improved, reaching a value of 2.46 × 10−3 S cm−1 at room temperature. Furthermore, the rate capability of the synthesized C/LiFePO4 composite is much improved because of its small grain size and good electronic conductivity compared with other studies.
In addition to the sol–gel method, the hydrothermal (or solvothermal) method is also one of the common wet chemical methods for in situ preparation of C/LiFePO4 composite. By assistance with the microwave technique, the hydrothermal process can be finished within a few minutes, which provides a rapid, one-spot method for synthesis of C/LiFePO4. Manthiram et al. synthesized the C/LiFePO4 nanocomposite via a microwave-assisted hydrothermal method and the material has a more uniform particle size (220–225 nm) and exhibits high capacity with excellent cyclability and rate capability.193 They also compared the microwave-assisted hydrothermal (employing water as a solvent) and solvothermal (employing tetraethyleneglycol as a solvent) synthesis approach and found that the microwave solvothermal process employing a tetraethyleneglycol as a solvent offers much smaller particle size than the microwave-hydrothermal process employing water as a solvent (Fig. 13).194 Thus, microwave-solvothermal methods may be more promising for synthesis of high performance C/LiFePO4.
 | ||
| Fig. 13 (a) TEM and (b) high resolution TEM images of the LiFePO4/C nanocomposite obtained by an ex situcarbon coating of the MW-ST LiFePO4 nanorods by heating with sucrose at 700 °C for 1 h in a flowing 2% H2/98% Ar atmosphere. (c) TEM and (d) high resolution TEM images of the LiFePO4/C nanocomposite obtained by an in situcarbon coating with glucose during the MW-HT process, followed by heating at 700 °C for 1 h in a flowing 2% H2/98% Ar atmosphere. Reproduced from ref. 194 with permission. Copyright 2008 American Chemical Society. | ||
Gas state method-spray pyrolysis. It is well known that the electrochemical performance of the electrode is strongly affected by the powder properties, such as the mean size, the morphology, the specific surface area, the crystallinity, and the composition of the material.195Spray pyrolysis is thought to be an effective technique for producing cathode powders with fine size and regular morphology as well as high crystallinity and phase purity.196–198 Even if the post-annealing of as-prepared powders is required to obtain the desired materials, a shorter annealing time of the as-prepared powders can be expected in comparison with conventional solid-state preparation methods. In contrast, several long sintering and regrinding procedures are sometimes needed to obtain the final product in the conventional solid-state reaction method, resulting in a severe increase of particle size.
Kang et al. prepared carbon-mixed LiFePO4 by spray pyrolysis from a colloidal spray solution.199 The nano-sized material affected the morphologies, mean particle sizes, and electrochemical properties of the LiFePO4 cathode powders. Micro-sized LiFePO4 powder with narrow size distributions can be obtained and carbon coating improved the initial discharge capacity and the cycle performance of the LiFePO4 powders. The similar micro-sized carbon included LiFePO4 powders also prepared by spray pyrolysis from the spray solution with sucrose.200,201 However, the electrochemical performance of the above-prepared materials was not sufficient for large-scale application because the size of the LiFePO4 particles was sub-micron or micron-sized.202 By a combination of spray pyrolysis with wet ball-milling, LiFePO4 nanoparticles with a mean diameter of 58 nm were successfully prepared.202,203 The sample delivered a superior performance with the first discharge capacities of 100 mA h g−1 at charge–discharge rates of 10 C.
Recently, ultrasonic spray pyrolysis has been shown to be an effective technique for producing fine ceramic particles with homogeneous chemical composition in a short time.204 With the assistance of an ultrasonic technique, the precursors were dispersed and mixed more homogeneously. The purity of the products is higher, and the composition of the powders is easy to control. Moreover, non-agglomerated powders were obtained. Therefore, ultrasonic and ball-milling assisted spray pyrolysis are attractive methods for fabrication of high-density samples because in these methods well-crystallized and homogeneous small particles can be prepared with the spherical shape and pure phase.
More details about the effects of the synthesis process and the coated carbon on the performance of C/LiFePO4 are summarized in Table 2.
| Preparation method | Carbon | Electrochemical performance | References | |||
|---|---|---|---|---|---|---|
| Precursors | Content | Thickness | Structure (ID/IG) | |||
| a LFP: LiFePO4;. b PS: polystyrene. c PEO: polyethylene oxide. d PB: polybutadiene. e SBS: styrene–butadiene–styrene. | ||||||
| Solid-state reaction | Citric acid | — | — | 0.53 | Initial discharge capacity of 128 mA h g−1 (4 C) and was retained as high as 92% after 1000 cycles | 112 |
| Solid-state reaction | Benzene | — | 3 nm | 0.75–0.77 | Initial discharge capacity of 155.4 (0.1 C) and 135.8 mA h g−1 (1 C) | 127 |
| Solid-state reaction | Peanut shells | 0, 6, 8, 10, 12 wt% | — | 0.928–0.962 | LFP a (8.0 wt% carbon) exhibits an initial discharge of 138 and 128 mA h g−1 at (0.2 C) after 170 cycles | 121 |
| Solid-state reaction | Glucose | 3,5,7,9 wt% | — | — | LFP (7 wt% carbon) has discharge capacity of 167.0 mA h g−1 (0.1 C) and 98.3 mA h g−1 (5 C) | 69 |
| Solid-state reaction | PS b + malonic acid | 0.02–2.54 wt% | 2–25 nm | ∼0.95 | LFP (2.28 wt% carbon, 4–8 nm) delivers maximum discharge capacity of 151 mA h g−1 (0.2 C) and sustains 415 cycles at 80% of capacity retention | 19 |
| Coprecipitation | PEO c, PS, PBd and SBSe | ∼2 wt% | — | 0.99–1.14 | PS sample delivers the largest charge capacity of 147 mA h g−1 at 0.5 C | 136 |
| Sol–gel | Acetylene black, sucrose and glucose | 5–30 wt% | — | — | Initial discharge capacity was 125.3 (acetylene black), 133.3 (sucrose) and 155.0 (glucose) mA h g−1 at 0.1 C | 161 |
| Sol–gel | Citrate | 3.2–12.7 wt% | 1–10 nm | — | LFP (3.2 wt%, 1–2 nm carbon thickness) exhibits best performance of 150 mA h g−1 (0.2 C) and 117 mA h g−1 (5 C) | 125 |
| Sol–gel | Citric acid, sucrose | — | 2.4–4 nm | 1.5 | Discharge capacities of 153, 120, 112, and 94 mA h g−1 at 0.1, 1, 3, and 5 C rates, respectively | 133 |
| Microwave | Glucose | 0, 2.8, 7.4, 12.1, 17.2 wt% | — | — | LFP (12.1% carbon) exhibits the best discharge capacity of 147 mA h g−1 and good cycling stability (more than 141 mA h g−1 at the 20th cycle) at 0.1 C | 157 |
| Hydrothermal | Carbon black | 0, 3, 5, 10 wt% | — | — | LFP (5% carbon) was 128 mA h g−1 at 1st cycle and 127 mA h g−1 after 30 cycles (0.1 mA cm−2), respectively | 119 |
| Microwave | Polyethylene glycol | 3.9%, 4.6%, 5.2%, 7.3% | — | — | LFP (4.6% carbon) has a largest initial specific capacity of 152 mA h g−1 at 0.2 C | 120 |
| Solvothermal | Carbon nanotubes | 8 wt%, 5.2%, 7.3% | — | — | LFP has discharge capacity of ∼161 mA h g−1 (0.1 C) of 152 mA h g−1 (0.2 C) and 59 mA h g−1 (20 C) | 151 |
4.6 Thin-film electrode technique for interface and mechanism study
Carbon coating techniques on LiFePO4 have been developed and commercialized. However, the role of carbon is still not completely understood in spite of its importance in the rate capability and cycle stability of LiFePO4 electrodes. Geometrically, well-defined thin films are suitable for fundamental research since there are no binder and conductive additives included.205 This is especially true for poorly conductive active materials because the thickness can be reduced to a value at which the poor electronic conductivity does not significantly affect the electrochemical behavior. Therefore, the intrinsic properties of LiFePO4 can be investigated by using the carbon-free thin film. Nowadays, various techniques have been used to prepare LiFePO4 thin films, i.e., pulsed laser deposition (PLD),206–208 radio-frequency magnetron sputtering deposition (RF-MSD),209,210 and electrostatic spray deposition.211 Among them, PLD and RF-MSD are the more common methods for preparation of LiFePO4 thin film electrodes. Chung employed the pulse laser deposition technique to deposit the C/LiFePO4 composite thin films on the Si/SiO2/Ti/Pt substrates (Fig. 14).212 The effects of carbon content and post-deposition annealing treatments on the electrochemical performances of the as-deposited film electrodes were carefully investigated. The results showed that the co-deposited carbon was homogeneously distributed throughout the LiFePO4 films and 2 mol% carbon was the optimized amount for the highest capacity. | ||
| Fig. 14 (a and c) Plane-view and (b and d) cross-section SEM images of the LiFePO4-C thin films. (a and b) Before and (c and d) after annealing treatment at 600 °C for 6 h. Reproduced from ref. 212 with permission. Copyright 2008 American Chemical Society. | ||
Thin film technology was also of particular interest to provide insights on the mechanism study. Li-ion diffusion kinetics was studied by Xie in detail,213 who prepared LiFePO4 thin films using radio frequency (RF) magnetron sputtering. The Li-ion diffusion kinetics has proved to be affected by the incorporation of carbon.214 Compared with the C/LiFePO4-binder composite electrodes, thin film electrodes seem to be more appropriate for the measurement of the chemical diffusion coefficient since they are conducting additive and binder free, and have a well-defined geometry and good contact with the current collector.
In addition to the help in understanding the carbon coating process and mechanism, LiFePO4 thin film electrodes with thickness ranging from a few nanometres to hundreds of nanometres are also essential for solid-state microbatteries, especially in low power applications, as the active or standby power sources for microelectronics. It has been demonstrated that LiFePO4 thin films can be used as a promising cathode film for lithium microbatteries by depositing the LiFePO4 film with RF-MSD.215 Similarly, C/LiFePO4 thin films with a thickness of 1 μm were also deposited on a stainless steel substrate by RF-MSD.210 The presence of carbon enhances the electronic conductivity, resulting in a high rate capability.
Thin film technology (with no conducting additive) has thus thrown some light on the intrinsic sensing properties of LiFePO4 thin films as well as provided insights on their sensing response mechanism. Of most importance, the role of carbon may be investigated in detail by coating a thin carbon film on the LiFePO4 film. Carbon features including carbon content, structure, thickness, etc. can be investigated accurately. By combining with material characterization techniques such as scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray photoelectron spectroscopy (XPS), Raman, and synchrotron based in situ X-ray diffraction,216–218 more detailed information about carbon and even the interface between carbon and LiFePO4 film can be obtained, which will be very helpful for the development of high performance LiFePO4.
5. Promising synthesis methods of C/LiFePO4 for future large-scale production
For industrial applications, large-scale and high-quality C/LiFePO4 combining with low processing cost and easy manufacturing are the primary requirements for the synthesis methods. Based on these factors, there are several promising methods for mass production including solid-state reaction, hydrothermal method, and molten state method (Table 3).| Methods | Advantages | Disadvantages | Size | C addition | Grade | References |
|---|---|---|---|---|---|---|
| a NPs: nanoparticles; NWs: nanowires; NRs: nanorods; PCD: Pyrolytic Carbon Deposit. | ||||||
| Solid-state based | Low cost, process simplicity | Difficult to control particle growth | Sub-micro or micro-size | In situ coating or post-addition | Energy | 219,220 |
| Hydrothermal method | Nanosize, morphology control (NPs, NWs, NRs, etc.) | Waste liquid. Strict reaction conditions (temperature, pH, time, etc.) | Nano-size | In situ coating or post-addition | Power | 221,222,223 |
| Molten state | Simplicity, low cost, common precursors, high crystallinity | No effective morphology controls | Nano-, sub-micro, micro-size | Post-addition (PCD process) | Both | 59,174 |
As the conventional method for mass production of most lithium-ion battery cathode materials, the solid-state method is still the most widely used process for mass production of C/LiFePO4 due to process simplicity in synthesis and carbon coating, in spite of the drawback of particle size control.
The hydrothermal/solvothermal methods have great potential for nano-sized C/LiFePO4 in large-scale production. With the addition of carbon precursors, this method can prepare and manufacture high surface area C/LiFePO4 powders with improved rate performance. The obtained C/LiFePO4 products are more suitable for high power application. However, the challenges for hydrothermal/solvothermal methods are process complexity and high cost.
By combining the advantages of the solid-state reaction (high crystallinity) with the hydrothermal method (uniform chemical mixture and high-purity phase), the molten-state method provides a simple, reliable, cheap, less wasteful technique to produce high quality LiFePO4 for large-scale battery applications. The synthesis within a molten state combines ideal liquid-phase reaction kinetics and short dwell times to provide a product with high material density using a simple process method. This simple synthetic method can use the various common precursors. The melting of carbon-coated LiFePO4 powder at 1000 °C followed by its cooling leads to a high purity LiFePO4 material with excellent crystallinity and large crystals. With the proper milling procedures, the melting products can be made into various sizes from micro-, sub-micro-, to nanosize. With the following pyrolytic carbon deposit process,51 various-grade C/LiFePO4 can be achieved to fulfil the needs of current and future battery markets (Fig. 15).59,174 With the increasing demands for electric vehicles, a simple synthetic method using commodity precursors or even natural sources is required for the large-scale synthesis of LiFePO4. It is believed that the molten synthetic method is a viable alternative to the current methods of synthesis.
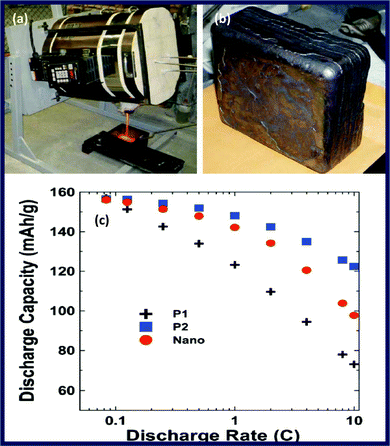 | ||
| Fig. 15 (a and b) Photograph of large-scale LiFePO4 casting from synthesis and the resultant 10 kg ingot in Phostech Lithium Inc. (c) Comparison of commercial energy grade LiFePO4 (P1, cross), power grade LiFePO4 (P2, squares), and nanomilled melt-cast LiFePO4 samples (circles). Reproduced from ref. 59 and 174 with permission. Copyright 2010. The Electrochemical Society. | ||
6. Conclusions and remarks
Although C/LiFePO4 materials have been successfully commercialized and the contributions of carbon to LiFePO4 in improving conductivity and rate capacity have been widely accepted, some fundamental questions still remain and need to be investigated in detail.Uniform and full carbon coatings throughout the LiFePO4 powder are an ideal structure, which ensures LiFePO4 particles transfer electrons through all directions and alleviate the polarization phenomenon. Carbon coatings must also be either porous or very thin to allow easy penetration of lithium ions. Therefore, it is very important to seek a simple and efficient method for synthesis of LiFePO4 with uniform carbon coatings.
Optimizing the carbon structure is the key to obtain good performance of LiFePO4. While highly graphitic carbon material can greatly enhance the electrochemical behavior of LiFePO4, it is difficult to produce highly graphitic coatings at relatively low temperatures used for synthesis of LiFePO4. Direct introduction of carbon nanotubes or graphitic carbon or in situgrowth of these graphite materials on LiFePO4 will be an effective method. An in-depth study of the surface chemistry of LiFePO4 and its influence on carbon coating will direct the optimization of the nature of the carbon coating.
A high tap density of C/LiFePO4 material is considered to be an important factor for practical applications. The introduction of carbon, however, decreases the energy density of the material significantly, especially for the volumetric energy density. An effective route to improve the material's tap density is to design novel structures, including spherical, aggregated and especially microspheral structures composed of tightly compacted nanocrystallites.
The role of carbon is still not completely understood in spite of the fact that its importance in the rate capability and cycle stability of LiFePO4 electrodes has been proven. Thin film electrodes are of great interest because they can serve as a simplified model for elaborating the detailed electrochemical process of their counterpart active materials. Carbon can be coated on the surface of LiFePO4 thin-film electrodes and the details of carbon effect, lithium-ion penetration through carbon layer, and the interface between LiFePO4 and carbon can be investigated to develop a better understanding.
With regard to the large-scale industrial production, a reliable, low-cost, highly effective synthetic method for C/LiFePO4 is still highly desirable. The solid-state method has already been widely adopted by industry for mass production, and it will remain one of the most used processes for manufacturing C/LiFePO4 due to process simplicity and low cost. The wet process including hydrothermal, solvothermal has great potential for nano-sized C/LiFePO4 production because it can moderate the size and morphology of C/LiFePO4. Although currently this method has some drawbacks such as strict reaction conditions, it is still a very promising synthetic approach for future power-grade cathode materials. Compared with the above two methods, the molten state method combines the advantages from solid-state reaction and hydrothermal methods and provides a simple, reliable, cheap, less wasteful technique to produce well defined LiFePO4 for large-scale battery applications. With the proper procedures, the melting products can be made into various sizes from micro- to nano-size and fulfil various needs of energy-grade and power-grade products for current and future battery markets. This novel method might become a very competitive candidate for future C/LiFePO4 production.
Acknowledgements
This work is supported by Natural Sciences and Engineering Research Council of Canada (NSERC), Phostech Lithium Inc., Canada Research Chair (CRC) Program, the University of Western Ontario, and MITACS Elevate Strategic Fellowship Program. The authors express their sincere thanks to Dr Michel Gauthier and Dr Guoxian Liang for the fruitful discussion on the organizing and drafting of this work. The authors are also in debt to David Tweddell and Dr Henry Huang for their kind help and discussion.Notes and references
- O. K. Park, Y. Cho, S. Lee, H.-C. Yoo, H.-K. Song and J. Cho, Energy Environ. Sci., 2011, 4, 1621–1633 CAS.
- G. Jeong, Y.-U. Kim, H. Kim, Y.-J. Kim and H.-J. Sohn, Energy Environ. Sci., 2011, 4, 1986–2002 CAS.
- B. Scrosati, J. Hassoun and Y.-K. Sun, Energy Environ. Sci., 2011, 4, 3287–3295 CAS.
- V. Etacheri, R. Marom, R. Elazari, G. Salitra and D. Aurbach, Energy Environ. Sci., 2011, 4, 3243–3262 CAS.
- X. Zhi, G. Liang, L. Wang, X. Qu, J. Zhang and J. Cui, J. Power Sources, 2009, 189, 779–782 CrossRef CAS.
- M. S. Whittingham, Chem. Rev., 2004, 104, 4271–4302 CrossRef CAS.
- C. H. Chen, J. Liu, M. E. Stoll, G. Henriksen, D. R. Vissers and K. Amine, J. Power Sources, 2004, 128, 278–285 CrossRef CAS.
- M. M. Thackeray, W. I. F. David, P. G. Bruce and J. B. Goodenough, Mater. Res. Bull., 1983, 18, 461–472 CrossRef CAS.
- J. B. Goodenough and Y. Kim, Chem. Mater., 2010, 22, 587–603 CrossRef CAS.
- P. P. Prosini, M. Lisi, D. Zane and M. Pasquali, Solid State Ionics, 2002, 148, 45–51 CrossRef CAS.
- R. Amin, P. Balaya and J. Maier, Electrochem. Solid-State Lett., 2007, 10, A13–A16 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188–1194 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, ECS Meeting Abstract, No. 58, Los Angeles, CA., May 5–10, 1996, vol. 96-1 Search PubMed.
- J. B. Goodenough, A. K. Padhi, K. S. Nanjundaswamy and C. Masquelier. US Pat., 5910382, 1999.
- A. Michel, J. B. Goodenough, A. K. Padhi, K. S. Nanjundaswamy and C. Masquelier. US Pat., 6514640, 2003.
- J.-M. Tarascon and M. Armand, Nature, 2001, 414, 359–367 CrossRef CAS.
- L. X. Yuan, Z. H. Wang, W. X. Zhang, X. L. Hu, J. T. Chen, Y. H. Huang and J. B. Goodenough, Energy Environ. Sci., 2011, 4, 269–284 CAS.
- Y. Wang, P. He and H. Zhou, Energy Environ. Sci., 2011, 4, 805–817 CAS.
- Y. D. Cho, G. T. K. Fey and H. M. Kao, J. Power Sources, 2009, 189, 256–262 CrossRef CAS.
- S. Okada, S. Sawa, M. Egashira, J. Yamaki, M. Tabuchi, H. Kageyama, T. Konishi and A. Yoshino, J. Power Sources, 2001, 97–98, 430–432 CrossRef CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123–126 CrossRef CAS.
- J. Molenda, A. Stoklosa and T. Bak, Solid State Ionics, 1989, 36, 53–58 CrossRef CAS.
- Y. Shimakawas, T. Numata and J. Tabuchi, J. Solid State Chem., 1997, 131, 138–143 CrossRef.
- B. L. Ellis, K. T. Lee and L. F. Nazar, Chem. Mater., 2010, 22, 691–714 CrossRef CAS.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085–5092 CrossRef CAS.
- H. Fang, Z. Pan, L. Li, Y. Yang, G. Yan, G. Li and S. Wei, Electrochem. Commun., 2008, 10, 1071–1073 CrossRef CAS.
- J. Xie, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda and O. Yamamoto, Electrochim. Acta, 2009, 54, 4631–4637 CrossRef CAS.
- C. Wang and J. Hong, Electrochem. Solid-State Lett., 2007, 10, A65–A69 CrossRef CAS.
- V. Srinivasan and J. Newman, Electrochem. Solid-State Lett., 2006, 9, A110–A114 CrossRef CAS.
- C. Delmas, M. Maccario, L. Croguennec, F. L. Cras and F. Weill, Nat. Mater., 2008, 7, 665–671 CrossRef CAS.
- C. Delacourt, P. Poizot, J.-M. Tarason and C. Masquelier, Nat. Mater., 2005, 4, 254–260 CrossRef CAS.
- S. Yang, Y. Song, K. Ngala, P. Y. Zavalij and M. S. Whittingham, J. Power Sources, 2003, 119, 239–246 CrossRef.
- G. Sun, G. Sun, E. Jin, H. B. Gu and Q. Jiang, J. Appl. Electrochem., 2011, 41, 99–106 CrossRef CAS.
- K. Saravanan, P. Balaya, M. V. Reddy, B. V. R. Chowdari and J. J. Vittal, Energy Environ. Sci., 2010, 3, 457–464 CAS.
- R. Malik, D. Burch, M. Bazant and G. Ceder, Nano Lett., 2010, 10, 4123–4127 CrossRef CAS.
- A. S. Arico, P. G. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijc, Nat. Mater., 2005, 4, 366–377 CrossRef CAS.
- P. G. Bruce, B. Scrosati and J. M. Tarascon, Angew. Chem., Int. Ed., 2008, 47, 2930–2946 CrossRef CAS.
- A. S. Andersson, J. O. Thomas, B. Kalska and L. Haggstrom, Electrochem. Solid-State Lett., 2000, 3, 66–68 CrossRef CAS.
- C. Delacourt, PhD thesis, University Picardie—Jules Verne, 2005.
- N. Meethong, Y. H. Kao, M. Tang, H.-Y. Huang, W. C. Carter and Y.-M. Chiang, Chem. Mater., 2008, 20, 6189–6198 CrossRef CAS.
- N. Meethong, H. Y. S. Huang, W. C. Carter and Y.-M. Chiang, Electrochem. Solid-State Lett., 2007, 10, A134–A138 CrossRef CAS.
- W. J. Zhang, J. Power Sources, 2011, 196, 2962–2970 CrossRef CAS.
- W. J. Zhang, J. Electrochem. Soc., 2010, 157, A1040–A1046 CrossRef CAS.
- S. F. Yang, P. Y. Zavalij and M. S. Whittingham, Electrochem. Commun., 2001, 3, 505–508 CrossRef CAS.
- F. Croce, A. D. Epifanio, J. Hassoun, A. D. T. Olczac and B. Scrosati, Electrochem. Solid-State Lett., 2002, 5, A47–A50 CrossRef CAS.
- A. Fedorková, R. Oriňáková, A. Oriňák, H.-D. Wiemhöfer, D. Kaniansky and M. Winter, J. Solid State Electrochem., 2009, 14, 2173–2178 CrossRef.
- D. Lepage, C. Michot, G. Liang, M. Gauthier and S. B. Schougaard, Angew. Chem., Int. Ed., 2011, 50, 6884–6887 CrossRef CAS.
- Y. H. Huang and J. B. Goodenough, Chem. Mater., 2008, 20, 7237–7241 CrossRef CAS.
- http://www.phostechlithium.com/ .
- N. Ravet, J. B. Goodenough, S. Besner, M. Simoneau, P. Hovington and M. Armand, The Electrochemical Society Meeting, Honolulu, HI, Oct. 17–22, 1999, vol. 99–2, Abstract 127 Search PubMed.
- N. Ravet, S. Besner, M. Simoneau, A. Vallée, M. Armand and J. F. Magnan, US Pat., 6855273, 2005; 6962666, 2005; and 7344659, 2008.
- N. Ravet, Y. Chouinard, J. F. Magnan, S. Besner, M. Gauthier and M. Armand, J. Power Sources, 2001, 97–98, 503–507 CrossRef CAS.
- H. Huang, S. C. Yin and L. F. Nazar, Electrochem. Solid-State Lett., 2001, 4, A170–A172 CrossRef CAS.
- N. Ravet, J.-F. Magnan, M. Gauthier and M. Armand, International Conference on Materials for Advanced Technologies (ICMAT 2001), MRS Singapore, July 1–6, 2001 Search PubMed.
- M. Armand, M. Gauthier, J. F. Magnan and N. Ravet, PCT WO 02/27823 and WO 01/27824.
- M. Gauthier, D. Geoffroy, M. Armand and N. Ravet, The 20th International Seminar on Primary and Secondary Batteries, Florida Educational Seminar Inc., Boca Raton, Florida, USA, March 17–20, 2003, http://www.powersources.net/, mailto:info@powersources.net or from the authors Search PubMed.
- C. Zhu, Y. Yu, L. Gu, K. Weichert and J. Maier, Angew. Chem., Int. Ed., 2011, 50, 6278–9282 CrossRef CAS.
- K. Saravanan, J. J. Vittal, M. V. Reddy, B. V. R. Chowdari and P. Balaya, J. Solid State Electrochem., 2010, 14, 1755–1760 CrossRef CAS.
- D. D. McNeil, L. Devigne, C. Michot, I. Rodigues, G. Liang and M. Gauthier, J. Electrochem. Soc., 2010, 157, A463–A468 CrossRef.
- R. Dominko, M. Gaberscek, J. Drofenik, M. Bele, S. Pejovnik and J. Jamnik, J. Power Sources, 2003, 119–121, 770–773 CrossRef CAS.
- C. Delacourt, P. Poizot, S. Levasseur and C. Masqueliera, Electrochem. Solid-State Lett., 2006, 9, A352–A355 CrossRef CAS.
- D.-H. Kim and J. Kim, Electrochem. Solid-State Lett., 2006, 9, A439–A442 CrossRef CAS.
- S. Y. Chung, J. T. Bloking and Y. M. Chiang, Nat. Mater., 2002, 1, 123–128 CrossRef CAS.
- N. Ravet, A. Abouimrane and M. Armand, Nat. Mater., 2003, 2, 702 CrossRef CAS.
- P. Subramanya Herle, B. L. Ellis, N. Coombs and L. F. Nazar, Nat. Mater., 2004, 3, 147–152 CrossRef.
- C. A. J. Fisher, V. M. Hart Prieto and M. S. Islam, Chem. Mater., 2008, 20, 5907–5915 CrossRef CAS.
- L. Yang, L. Jiao, Y. Miao and H. Yuan, J. Solid State Electrochem., 2009, 13, 1541–1544 CrossRef CAS.
- X. Z. Liao, Y. S. He, Z. F. Ma, X. M. Zhang and L. Wang, J. Power Sources, 2007, 174, 720–725 CrossRef CAS.
- N. Meethong, Y. H. Kao, S. A. Speakman and Y.-M. Chiang, Adv. Funct. Mater., 2009, 19, 1060–1070 CrossRef CAS.
- D. Y. Wang, H. Li, S. S. Shi, X. J. Huang and L. Q. Chen, Electrochim. Acta, 2005, 50, 2955–2958 CrossRef CAS.
- J. Hong, C. S. Wang, X. Chen, S. Upreti and M. S. Whittingham, Electrochem. Solid-State Lett., 2009, 12, A33–A38 CrossRef CAS.
- C. Y. Ouyang, D. Y. Wang, S. Q. Shi, Z. X. Wang, H. Li, X. J. Huang and L. Q. Chen, Chin. Phys. Lett., 2006, 23, 61–65 CrossRef CAS.
- D. Wang, C. Ouyang, T. Drezen, I. Exnar, A. Kay, N. H. Kwon, P. Gouerec, J. H. Miners, M. Wang and M. Gratzel, J. Electrochem. Soc., 2010, 157, A225–A229 CrossRef CAS.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 CrossRef CAS.
- K. Zaghib, J. B. Goodenough, A. Mauger and C. Julien, J. Power Sources, 2009, 194, 1021–1023 CrossRef CAS.
- G. Ceder and B. Kang, J. Power Sources, 2009, 194, 1024–1028 CrossRef CAS.
- Y. Jin, C. P. Yang, X. H. Rui, T. Cheng and C. H. Chen, J. Power Sources, 2011, 196, 5623–5630 CrossRef CAS.
- H.-H. Chang, C.-C. Chang, C.-Y. Su, H.-C. Wu, M.-H. Yang and N.-L. Wu, J. Power Sources, 2008, 185, 466–472 CrossRef CAS.
- H. Liu, G. X. Wang, D. Wexler, J. Z. Wang and H. K. Liu, Electrochem. Commun., 2008, 10, 165–169 CrossRef CAS.
- Z. Chen, Y. Qin, K. Amine and Y.-K. Sun, J. Mater. Chem., 2010, 20, 7606–7612 RSC.
- X. Meng, D. Geng, J. Liu, R. Li and X. Sun, Nanotechnology, 2011, 22, 165602 CrossRef.
- X. Meng, Y. Zhong, Y. Sun, M. N. Banis, R. Li and X. Sun, Carbon, 2011, 49, 1133–1144 CrossRef CAS.
- Y. S. Jung, A. S. Cavanagh, L. A. Riley, S.-H. Kang, A. C. Dillon, M. D. Groner, S. M. George and S.-H. Lee, Adv. Mater., 2010, 22, 2172–2176 CrossRef CAS.
- I. D. Scott, Y. S. Jung, A. S. Cavanagh, Y. Yan, A. C. Dillon, S. M. George and S.-H. Lee, Nano Lett., 2011, 11, 414–418 CrossRef CAS.
- O. Toprakci, H. A. K. Toprakci, L. Ji and X. Zhang, KONA Powder and Particle J., 2010, 8, 50–73 Search PubMed.
- H. H. Chang, C. C. Chang, H. C. Wu, Z. Z. Guo, M. H. Yang, Y. P. Chiang, H. S. Sheu and N. L. Wu, J. Power Sources, 2006, 158, 550–556 CrossRef CAS.
- A. F. Liu, Z. H. Hu, Z. B. Wen, L. Lei and J. An, Ionics, 2010, 16, 311–316 CrossRef CAS.
- J. K. Kim, G. Cheruvally, J. W. Choi, J. U. Kim, J. H. Ahn, G. B. Cho, K. W. Kim and H. J. Ahn, J. Power Sources, 2007, 166, 211–218 CrossRef CAS.
- H. F. Xiang, D. W. Zhang, Y. Jin, C. H. Chen, J. S. Wu and H. H. Wang, J. Mater. Sci., 2011, 46, 4906–4912 CrossRef CAS.
- H. Yang, X. L. Wu, M. H. Cao and Y. G. Guo, J. Phys. Chem. C, 2009, 113, 3345–3351 CAS.
- N. Recham, L. Dupont, M. Courty, K. Djellab, D. Larcher, M. Armand and J.-M. Tarascon, Chem. Mater., 2009, 21, 1096–1107 CrossRef CAS.
- F. Yu, J. J. Zhang, Y. F. Yang and G. Z. Song, J. Power Sources, 2010, 195, 6873–6878 CrossRef CAS.
- K. S. Park, J. T. Son, H. T. Chung, S. J. Kim, C. H. Lee and H. G. Kim, Electrochem. Commun., 2003, 5, 839–842 CrossRef CAS.
- T. N. L. Doan and I. Taniguchi, J. Power Sources, 2011, 196, 1399–1408 CrossRef CAS.
- A. A. Salah, A. Mauger, K. Zaghib, J. B. Goodenough, N. Ravet, M. Gauthier, F. Gendron and C. M. Julien, J. Electrochem. Soc., 2006, 153, A1692–A1701 CrossRef.
- N. Ravet, M. Gauthier, K. Zaghib, J. B. Goodenough, A. Mauger, F. Gendron and C. M. Julien, Chem. Mater., 2007, 19, 2595–2602 CrossRef CAS.
- Z. Chen and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1184–A1189 CrossRef CAS.
- Z. R. Chang, H. J. Lv, H. W. Tang, H. J. Li, X. Z. Yuan and H. Wang, Electrochim. Acta, 2009, 54, 4595–4599 CrossRef CAS.
- Z. Liu, X. Zhang and L. Hong, J. Appl. Electrochem., 2009, 39, 2433–2438 CrossRef CAS.
- T. Takeuchi, M. Tabuchi, A. Nakashima, T. Nakamura, Y. Miwa, H. Kageyama and K. Tatsumi, J. Power Sources, 2005, 146, 575–579 CrossRef CAS.
- M. E. Zhong and Z. T. Zhou, Mater. Chem. Phys., 2010, 119, 428–431 CrossRef CAS.
- S. W. Oh, H. J. Bang, S. T. Myung, Y. C. Bae, S. M. Lee and Y. K. Sun, J. Electrochem. Soc., 2008, 155, A414–A420 CrossRef CAS.
- S. W. Oh, S. T. Myung, H. J. Bang, C. S. Yoon, K. Amine and Y. K. Sun, Electrochem. Solid-State Lett., 2009, 12, A181–A185 CrossRef CAS.
- S. W. Oh, S. T. Myung, S. M. Oh, K. H. Oh, K. Amine, B. Scrosati and Y. K. Sun, Adv. Mater., 2010, 22, 4842–4845 CrossRef CAS.
- S. W. Oh, S. T. Myung, S. M. Oh, C. S. Yoon, K. Amine and Y. K. Sun, Electrochim. Acta, 2010, 55, 1193–1199 CrossRef CAS.
- H. Liu, C. Li, H. P. Zhang, L. J. Fu, Y. P. Wu and H. Q. Wu, J. Power Sources, 2006, 159, 717–720 CrossRef CAS.
- J. M. Chen, C. H. Hsu, Y. R. Lin, M. H. Hsiaoa and G. T. K. Fey, J. Power Sources, 2008, 184, 498–502 CrossRef CAS.
- B. Jin, E. M. Jin, K. H. Park and H. B. Gu, Electrochem. Commun., 2008, 10, 1537–1540 CrossRef CAS.
- Z. Y. Chen, H. L. Zhu, W. Zhu, J. L. Zhang and Q. F. Li, Trans. Nonferrous Met. Soc. China, 2010, 20, 614–618 CrossRef CAS.
- D. Zane, M. Carewska, S. Scaccia, F. Cardellini and P. P. Prosin, Electrochim. Acta, 2004, 49, 4259–4271 CrossRef CAS.
- Y. Wang, Y. Wang, E. Hosono, K. Wang and H. Zhou, Angew. Chem., Int. Ed., 2008, 47, 7461–7465 CrossRef CAS.
- X. Yan, G. Yang, J. Liu, Y. Ge, H. Xie, X. Pan and R. Wang, Electrochim. Acta, 2009, 54, 5770–5774 CrossRef CAS.
- J. Liu, J. Wang, X. Yan, X. Zhang, G. Yang, A. F. Jalbout and R. Wang, Electrochim. Acta, 2009, 54, 5656–5659 CrossRef CAS.
- X. Zhi, G. Liang, L. Wang, X. Ou, J. Zhang and J. Cui, J. Power Sources, 2009, 189, 779–782 CrossRef CAS.
- P. P. Prosini, D. Zane and M. Pasquali, Electrochim. Acta, 2001, 46, 3517–3523 CrossRef CAS.
- Z. Chen and J. R. Dahn, J. Electrochem. Soc., 2002, 149, A1184–A1189 CrossRef CAS.
- K. Zaghib, J. Shim, A. Guerfi, P. Charest and K. A. Striebel, Electrochem. Solid-State Lett., 2005, 8, A207–A210 CrossRef CAS.
- S. S. Zhang, J. L. Allen, K. Xua and T. R. Jow, J. Power Sources, 2005, 147, 234–240 CrossRef CAS.
- E. M. Jin, B. Jin, D. K. Jun, K. H. Park, H. B. Gu and K. W. Kim, J. Power Sources, 2008, 178, 801–806 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong and M. Liu, Electrochim. Acta, 2009, 54, 3206–3210 CrossRef CAS.
- C. Z. Lu, G. Ting-Kuo Fey and H. M. Kao, J. Power Sources, 2009, 189, 155–162 CrossRef CAS.
- Z. R. Chang, H. J. Lv, H. W. Tang, H. J. Li, X. Z. Yuan and H. Wang, Electrochim. Acta, 2009, 54, 4595–4599 CrossRef CAS.
- H. Gabrisch, J. D. Wilcox and M. M. Doeff, Electrochem. Solid-State Lett., 2006, 9, A360–A363 CrossRef CAS.
- Y. Lin, M. X. Gao, D. Zhu, Y. F. Liu and H. G. Pan, J. Power Sources, 2008, 184, 444–448 CrossRef CAS.
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, S. Pejovnik and J. Jamnik, J. Electrochem. Soc., 2005, 152, A607–A610 CrossRef CAS.
- Y. D. Cho, G. T. K. Fey and H. M. Kao, J. Power Sources, 2009, 189, 256–262 CrossRef CAS.
- B. Zhao, Y. Jiang, H. Zhang, H. Tao, M. Zhong and Z. Jiao, J. Power Sources, 2009, 189, 462–466 CrossRef CAS.
- Y. Hu, M. M. Doeff, R. Kostecki and R. Finones, J. Electrochem. Soc., 2004, 151, A1279–A1285 CrossRef CAS.
- M. M. Doeff, Y. Hu, F. McLarnon and R. Kostecki, Electrochem. Solid-State Lett., 2003, 6, A207–A209 CrossRef CAS.
- J. D. Wilcox, M. M. Doeff, M. Marcinek and R. Kostec, J. Electrochem. Soc., 2007, 154, A389–A395 CrossRef CAS.
- M. M. Doeff, J. D. Wilcox, R. Kostecki and G. Lau, J. Power Sources, 2006, 163, 180–184 CrossRef CAS.
- H. C. Shin, W. I. I. CHo and H. Jang, Electrochim. Acta, 2006, 52, 1472–1476 CrossRef CAS.
- J. K. Kim, J. W. Choi, G. S. Chauhan, J. H. Ahn, G. C. Hwang, J. B. Choi and H. J. Ahn, Electrochim. Acta, 2008, 53, 8258–8264 CrossRef CAS.
- T. Nakamura, Y. Miwa, M. Tabuchi and Y. Yamada, J. Electrochem. Soc., 2006, 153, A1108–A1114 CrossRef CAS.
- C. W. Ong, Y. K. Lin and J. S. Chen, J. Electrochem. Soc., 2007, 154, A527–A533 CrossRef CAS.
- Y. H. Nien, J. R. Carey and J. S. Chen, J. Power Sources, 2009, 193, 822–827 CrossRef CAS.
- J. Chen, J. Z. Wang, A. I. Minett, Y. Liu, C. Lynam, H. Liu and G. G. Wallace, Energy Environ. Sci., 2009, 2, 393–396 CAS.
- H. Zhang, G. Cao and Y. Yang, Energy Environ. Sci., 2009, 2, 932–943 CAS.
- Y. Liu, X. Li, H. Guo, Z. Wang, W. Peng, Y. Yang and R. Liang, J. Power Sources, 2008, 184, 522–526 CrossRef CAS.
- L. Wang, Y. Huang, R. Jiang and D. Jia, J. Electrochem. Soc., 2007, 154, A1015–A1019 CrossRef CAS.
- J. J. Chen and M. S. Whittingham, Electrochem. Commun., 2006, 8, 855–858 CrossRef CAS.
- Y. Zhou, J. Wang, Y. Hu, R. O'Hayre and Z. Shao, Chem. Commun., 2010, 46, 7151–7153 RSC.
- X. L. Li, F. Y. Kang, X. D. Bai and W. C. Shen, Electrochem. Commun., 2007, 9, 663–666 CrossRef CAS.
- M. S. Bhuvaneswari, N. N. Bramnik, D. Ensling, H. Ehrenberg and W. Jaegermann, J. Power Sources, 2008, 180, 553–560 CrossRef CAS.
- F. Y. Su, C. You, Y. B. He, W. Lv, W. Cui, F. Jin, B. Li, Q. H. Yang and F. Kang, J. Mater. Chem., 2010, 20, 9644–9650 RSC.
- Y. Ding, Y. Jiang, F. Xu, J. Yin, H. Ren, Q. Zhuo, Z. Long and P. Zhang, Electrochem. Commun., 2010, 12, 10–13 CrossRef CAS.
- X. Zhou, F. Wang, Y. Zhu and Z. Liu, J. Mater. Chem., 2011, 21, 3353–3358 RSC.
- L. Wang, H. Wang, Z. Liu, C. Xiao, S. Dong, P. Han, Z. Zhang, X. Zhang, C. Bi and G. Cui, Solid State Ionics, 2010, 181, 1685–1689 CrossRef CAS.
- J. K. Kim, G. Cheruvally, J. H. Ahn and H. J. Ahn, J. Phys. Chem. Solids, 2008, 29, 1257–1260 CrossRef.
- Y. Liu, C. Cao and J. Li, Electrochim. Acta, 2010, 55, 3921–3926 CrossRef CAS.
- T. Muraliganth, A. V. Murugan and A. Manthiram, J. Mater. Chem., 2008, 18, 5661–5668 RSC.
- X. L. Wu, L. Y. Jiang, F. F. Cao, Y. G. Guo and L. J. Wan, Adv. Mater., 2009, 21, 2710–2714 CrossRef CAS.
- C. R. Sides, F. Croce, V. Y. Young, C. R. Martin and B. Scrosati, Electrochem. Solid-State Lett., 2005, 8, A484–A487 CrossRef CAS.
- C. M. Doherty, R. A. Caruso, B. M. Smarsly, P. Adelhelm and C. J. Drummond, Chem. Mater., 2009, 21, 5300–5306 CrossRef CAS.
- M. R. Yang, W. H. Ke and S. H. Wu, J. Power Sources, 2005, 146, 539–543 CrossRef CAS.
- L. N. Wang, X. C. Zhan, Z. G. Zhang and K. L. Zhang, J. Alloys Compd., 2008, 456, 461–465 CrossRef CAS.
- W. Zhou, W. He, Z. Li, H. Zhao and S. Yan, J. Solid State Electrochem., 2009, 13, 1819–1823 CrossRef CAS.
- M. Gaberscek, R. Dominko, M. Bele, M. Remskar, D. Hanzel and J. Jamnik, Solid State Ionics, 2005, 176, 1801–1805 CrossRef CAS.
- F. Yu, J. Zhang, Y. Yang and G. Song, J. Power Sources, 2009, 189, 794–797 CrossRef CAS.
- K. Wang, R. Cai, T. Yuan, X. Yu, R. Ran and Z. Shao, Electrochim. Acta, 2009, 54, 2861–2868 CrossRef CAS.
- Z. Chen, H. Zhu, S. Ji, R. Fakir and V. Linkov, Solid State Ionics, 2008, 179, 1810–1815 CrossRef CAS.
- C. Lai, Q. Xu, H. Ge, G. Zhou and J. Xie, Solid State Ionics, 2008, 179, 1736–1739 CrossRef CAS.
- X. Yan, G. Yang, J. Liu, Y. Ge, H. Xie, X. Pan and R. Wang, Electrochim. Acta, 2009, 54, 5770–5774 CrossRef CAS.
- D. Zhang, X. Yu, Y. Wang, R. Cai, Z. Shao, X. Z. Liao and Z. F. Ma, J. Electrochem. Soc., 2009, 156, A802–A808 CrossRef CAS.
- G. Ting-Kuo Fey, T. L. Lu, F. Y. Wu and W. H. Li, J. Solid State Electrochem., 2008, 12, 825–833 CrossRef CAS.
- G. Ting-Kuo Fey and T. L. Lu, J. Power Sources, 2008, 178, 807–814 CrossRef.
- C. W. Ong, Y. K. Lin and J. S. Chen, J. Electrochem. Soc., 2007, 154, A527–A533 CrossRef CAS.
- Y. H. Nien, J. R. Carey and J. S. Chen, J. Power Sources, 2009, 193, 822–827 CrossRef CAS.
- P. Subramanya-Herle, B. Ellis, N. Coombs and L. F. Nazar, Nat. Mater., 2004, 3, 147–152 CrossRef.
- Y. H. Rho, L. F. Nazar, L. Perry and D. Ryan, J. Electrochem. Soc., 2007, 154, A283–A289 CrossRef CAS.
- G. Lewis and C. E. Myers, J. Phys. Chem., 1963, 67, 1289–1292 CrossRef CAS.
- Y. Xu, Y. Lu, L. Yan, Z. Yang and R. Yang, J. Power Sources, 2006, 160, 570–576 CrossRef CAS.
- Y. Lin, M. X. Gao, D. Zhu, Y. F. Liu and H. G. Pan, J. Power Sources, 2008, 184, 444–448 CrossRef CAS.
- M. Gauthier, C. Michot, N. Ravet, J. Dufour, G. Liang, J. Wontcheu, L. Gauthier and D. D. McNeil, J. Electrochem. Soc., 2010, 157, A453–A462 CrossRef CAS.
- J. F. Ni, H. H. Zhou, J. T. Chen and X. X. Zhang, Mater. Lett., 2007, 61, 1260–1264 CrossRef CAS.
- S. S. Zhang, J. L. Allen, K. Xu and T. R. Jow, J. Power Sources, 2005, 147, 234–240 CrossRef CAS.
- G. T. K. Fey, T. L. Lu, F. Y. Wu and W. H. Li, J. Solid State Electrochem., 2008, 12, 825–833 CrossRef.
- H. Liu, C. Li, H. P. Zhang, L. J. Fu, Y. P. Wu and H. Q. Wu, J. Power Sources, 2006, 159, 717–720 CrossRef CAS.
- Y. Z. Dong, Y. M. Zhao, Y. H. Chen, Z. F. He and Q. Kuang, Mater. Chem. Phys., 2009, 115, 245–250 CrossRef CAS.
- J. Barker, M. Y. Saidi and J. L. Swoyer, Electrochem. Solid-State Lett., 2003, 6, A53–A55 CrossRef CAS.
- H. P. Liu, Z. X. Wang, X. H. Li, H. J. Guo, W. J. Peng, Y. H. Zhang and Q. Y. Hu, J. Power Sources, 2008, 184, 469–472 CrossRef CAS.
- L. Wang, G. C. Liang, X. Q. Ou, X. K. Zhi, J. P. Zhang and J. Y. Cui, J. Power Sources, 2009, 189, 423–428 CrossRef CAS.
- X. Zhi, G. Liang, L. Wang, X. Ou, J. Zhang and J. Cui, J. Power Sources, 2009, 189, 779–782 CrossRef CAS.
- X. F. Guo, H. Zhan and Y. H. Zhou, Solid State Ionics, 2009, 180, 386–391 CrossRef CAS.
- S. Beninati, L. Damen and M. Mastragostino, J. Power Sources, 2009, 194, 1094–1098 CrossRef CAS.
- K. F. Hsu, S. Y. Tsay and B. J. Hwang, J. Mater. Chem., 2004, 14, 2690–2695 RSC.
- J. K. Kim, J. W. Choi, G. S. Chauhan, J. H. Ahn, G. C. Hwang, J. B. Choi and H. J. Ahn, Electrochim. Acta, 2008, 53, 8258–8264 CrossRef CAS.
- D. Choi and P. N. Kumta, J. Power Sources, 2007, 163, 1064–1069 CrossRef CAS.
- Y. Lin, M. X. Gao, D. Zhu, Y. F. Liu and H. G. Pan, J. Power Sources, 2008, 184, 444–448 CrossRef CAS.
- N. J. Yun, H. W. Ha, K. H. Jeong, H. Y. Park and K. Kim, J. Power Sources, 2006, 160, 1361–1368 CrossRef CAS.
- J. S. Yang and J. J. Xu, Electrochem. Solid-State Lett., 2004, 7, A515–A518 CrossRef CAS.
- K. F. Hsu, S. Y. Tsay and B. J. Hwang, J. Mater. Chem., 2004, 14, 2690–2695 RSC.
- A. Vadivel Murugan, T. Muraliganth and A. Manthiram, J. Electrochem. Soc., 2009, 156, A79–A83 CrossRef CAS.
- A. Vadivel Murugan, T. Muraliganth and A. Manthiram, J. Phys. Chem. C, 2008, 112, 14665–14671 Search PubMed.
- Y. Li, C. Wan, Y. Wu, C. Jiang and Y. Zhu, J. Power Sources, 2000, 85, 294–298 CrossRef CAS.
- K. Matsuda and I. Taniguchi, J. Power Sources, 2004, 132, 156–160 CrossRef CAS.
- S. H. Park, S. W. Oh and Y. K. Sun, J. Power Sources, 2005, 146, 622–625 CrossRef CAS.
- S. H. Park, S. W. Oh, S. T. Myung, Y. C. Kang and Y. K. Sun, Solid State Ionics, 2005, 176, 481–486 CrossRef CAS.
- S. H. Ju and Y. C. Kang, Mater. Chem. Phys., 2008, 107, 328–333 CrossRef CAS.
- S. L. Bewlay, K. Konstantinov, G. X. Wang, S. X. Dou and H. K. Liu, Mater. Lett., 2004, 58, 1788–1791 CrossRef CAS.
- K. Konstantinov, S. L. Bewlay, G. X. Wang, M. Lindsay, J. Z. Wang, H. K. Liu, S. X. Dou and J. H. Ahn, Electrochim. Acta, 2004, 50, 421–426 CrossRef CAS.
- M. Konarova and I. Taniguchi, Powder Technol., 2009, 191, 111–116 CrossRef CAS.
- M. Konarova and I. Taniguchi, J. Power Sources, 2009, 194, 1029–1035 CrossRef CAS.
- M. R. Yang, T. H. Teng and S. H. Wu, J. Power Sources, 2006, 159, 307–311 CrossRef CAS.
- K. Tang, J. Sun, X. Yu, H. Li and X. Huang, Electrochim. Acta, 2009, 54, 6565–6569 CrossRef CAS.
- C. Yada, Y. Iriyama, S. K. Jeong, T. Abe, M. Inaba and Z. Ogumi, J. Power Sources, 2005, 146, 559–564 CrossRef CAS.
- S. W. Song, R. P. Reade, R. Kostecki and K. Striebel, J. Electrochem. Soc., 2006, 153, A12–A16 CrossRef CAS.
- F. Sauvage, E. Baudrin, L. Gengembre and J. M. Tarascon, Solid State Ionics, 2005, 176, 1869–1876 CrossRef CAS.
- K. F. Chiu, J. Electrochem. Soc., 2007, 154, A129–A133 CrossRef CAS.
- J. Hong, C. S. Wang, N. J. Dudney and M. J. Lance, J. Electrochem. Soc., 2007, 154, A805–A809 CrossRef CAS.
- J. Ma and Q. Z. Qin, J. Power Sources, 2005, 148, 66–71 CrossRef CAS.
- Z. G. Lu, M. F. Lo and C. Y. Chung, J. Phys. Chem. C, 2008, 112, 7069–7078 CAS.
- J. Xie, N. Imanishi, T. Zhang, A. Hirano, Y. Takeda and O. Yamamoto, Electrochim. Acta, 2009, 54, 4631–4637 CrossRef CAS.
- H. Liu, C. Li, H. P. Zhang, L. J. Fu, Y. P. Wu and H. Q. Wu, J. Power Sources, 2006, 159, 717–720 CrossRef CAS.
- X. J. Zhu, L. B. Cheng, C. G. Wang, Z. P. Guo, P. Zhang, G. D. Du and H. K. Liu, J. Phys. Chem. C, 2009, 113, 14518–14522 CAS.
- M. Balasubramanian, X. Sun, X. Q. Yang and J. McBreen, J. Power Sources, 2001, 92, 1–8 CrossRef CAS.
- W. S. Yoon, K. Y. Chung, J. Mcbreen, K. Zaghib and X. Q. Yang, Electrochem. Solid-State Lett., 2006, 9, A415–A417 CrossRef CAS.
- H. C. Shin, K. W. Nam, W. Y. Chang, B. W. Cho, W. S. Yoon, X. Q. Yang and K. Y. Chung, Electrochim. Acta, 2011, 56, 1182–1189 CrossRef CAS.
- D. Jugovic and D. Uskokovic, J. Power Sources, 2009, 190, 538–544 CrossRef CAS.
- H. C. Kang, D. K. Jun, B. Jin, E. M. Jin, K. H. Park, H. B. Gu and K. W. Kim, J. Power Sources, 2008, 179, 340–346 CrossRef CAS.
- X. Qin, X. H. Wang, H. M. Xiang, J. Xie, J. J. Li and Y. C. Zhou, J. Phys. Chem. C, 2010, 114, 16806–16812 CAS.
- G. Yang, H. M. Ji, H. D. Liu, K. F. Huo, J. J. Fu and P. K. Chu, J. Nanosci. Nanotechnol., 2010, 10, 980–986 CrossRef CAS.
- H. Nakano, K. Dokko, S. Koizumi, H. Tannai and K. Kanamura, J. Electrochem. Soc., 2008, 155, A909–A914 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |