 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chiral Ag(I) and Pt(II) complexes of ditopic NHC ligands: synthesis, structural and spectroscopic properties†‡
Paul
Marshall
a,
Robert L.
Jenkins
a,
William
Clegg
b,
Ross W.
Harrington
b,
Samantha K.
Callear
c,
Simon J.
Coles
c,
Ian A.
Fallis
a and
Athanasia
Dervisi
*a
aSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: dervisia@cardiff.ac.uk; Fax: +44 (0) 29 20874030; Tel: +44 (0) 29 20874081
bSchool of Chemistry, Newcastle University, Newcastle upon Tyne NE1 7RU, UK. E-mail: bill.clegg@newcastle.ac.uk; Tel: +44 (0) 191 222 6649
cSchool of Chemistry, University of Southampton, Highfield, Southampton, SO17 1BJ, UK. E-mail: s.j.coles@soton.ac.uk; Fax: +44 (0) 2380 596 723; Tel: +44 (0) 2380 596 722
First published on 7th September 2012
Abstract
The butyl and isopropyl derivatives (4I, 5Br) of chiral pool derived bis-imidazolium dehydrohexitol salts have been prepared. The ditopic N-heterocyclic carbenes 4 and 5 form dinuclear Ag(I) and Pt(II) complexes. All compounds were fully characterised by multinuclear NMR spectroscopy. The bis-imidazolium salt 4I and platinum complexes cis-[Pt2(μ-2)(dmso)2Cl4] and cis-[Pt2(μ-4)(dmso)2Cl4] were characterised by X-ray crystallography. In the case of the Pt(II) complexes, the carbene ring is positioned in a sterically preferred orientation, approximately perpendicular to the platinum coordination plane. The 1H, 13C, 15N and 195Pt NMR spectra of the platinum complexes show the presence of rotamers due to hindered rotation about the carbene–metal bond.
Introduction
N-Heterocyclic carbene (NHC) ligands and their transition metal complexes have established themselves as a cornerstone of modern coordination chemistry and are now used in a wide range of applications. Platinum(II)–NHC complexes in particular catalyse a number of important reactions, such as hydrosilylations,1 hydroaminations,2 tandem hydroboration-cross-coupling reactions3 and diboration of unsaturated molecules.4 Platinum(II)–NHC complexes have also seen recent activity in medicinal applications as possible chemotherapeutic agents.5 Thus efficient syntheses of platinum–NHC complexes are of considerable current interest. Synthetic routes to Pt–NHC complexes include direct reaction of the carbene precursor in the presence of base,1e,6 or of the free carbene, with suitable platinum salts.7 Alternatively, transmetalation of Ag–NHC complexes with a platinum source is also known.8Silver(I) carbenes are extensively used as ligand transfer agents for the majority of imidazolium and other azolium salts, forming a part of a convenient, and sometimes the only, route for the synthesis of metal carbene complexes.9 Ag(I)–NHC complexes have also found many medicinal applications, mainly due to their activity as antimicrobials.10 Other potential uses include high-end materials applications, such as luminescent chemosensors.11 Silver–carbene complexes are structurally diverse, displaying a broad range of coordination motifs, ranging from simple two-coordinate linear molecules9e,12 to helicates,13 polymers,14 rings,15 cages16 and clusters.17
Based on our previously reported chiral pool derived dehydrohexitol framework precursor 1,18 we recently described an efficient method leading to the bis-carbene precursor imidazolium salts 2X (Scheme 1) with 2,5-exo stereochemistry.19 The endo and exo stereochemistry refers to the substitution with respect to the bent V-shaped core of the fused dehydrohexitol ring. Here we report the synthesis of the isopropyl- and butyl-imidazole derivatives (4X and 5X) following an alternative synthetic route and expand on the coordination chemistry for this family of bridging NHC ligands, to include examples of their Pt(II) complexes.
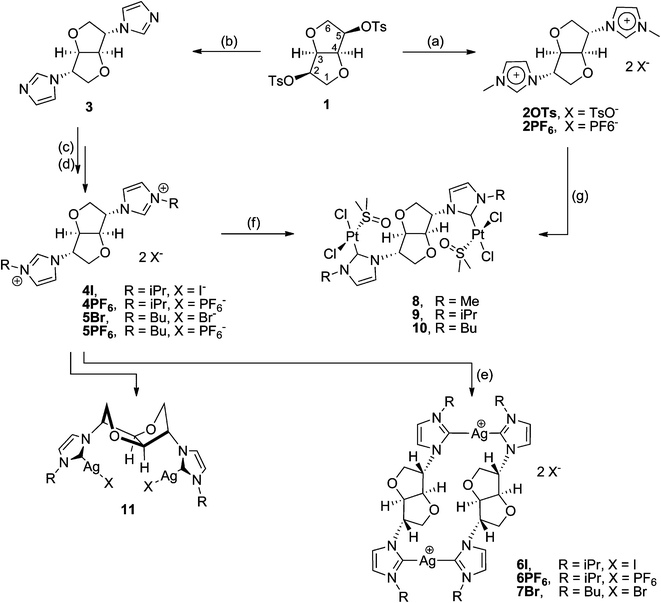 | ||
| Scheme 1 Reagents and conditions. (a) N-Methylimidazole, 140 °C; (b) Cs2CO3, dmf, 100 °C; (c) 4I: excess isopropylbromide, CH3CN, reflux, 5Br: excess butylbromide, dmf, 100 °C; (d) excess KPF6, H2O; (e) Ag2O, CH2Cl2, room temperature, 2 days; (f/g) K2PtCl4 or PtCl2, NaOAc, dmso, 70 °C, 2 days. | ||
Discussion
The 2,5-exo-bisimidazole 3 was prepared from the reaction of ditosylate 1 with imidazole in dmf in the presence of Cs2CO3 (Scheme 1). Subsequent quaternerisation with the corresponding alkyl halide afforded the bis-isopropyl- and butyl-imidazolium salts 4I and 5Br. Following counterion metathesis with potassium hexafluorophosphate in water, 4PF66 is furnished as a crystalline white solid and 5PF66 as a colourless viscous ionic liquid. The imidazolium salts have the expected exo stereochemistry at the 2 and 5 positions of the bicyclic ring as in the parent hexitol, L-iditol. The dicationic salts are readily soluble in water and polar organic solvents such as lower alcohols, acetone, acetonitrile and dmso. Single crystals of 4I suitable for X-ray studies were grown from an acetonitrile solution of the salt, and the structure of the cation is shown in Fig. 1.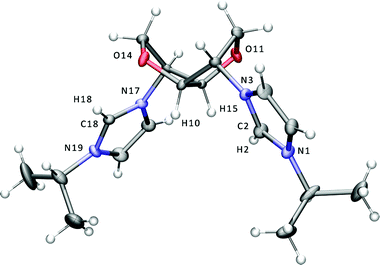 | ||
| Fig. 1 Displacement ellipsoid plot at the 50% probability level of the bis-imidazolium cation of 4I. The iodide anions are omitted for clarity. Selected bond lengths (Å) and angles (°): N1–C2 1.333(5), C2–N3 1.341(5), N17–C18 1.331(5), C18–N19 1.329(5), N1–C2–N3 109.3(4), N17–C18–N19 109.5(3). | ||
Silver(I) complexes
The silver(I) complexes of carbenes 4 and 5 were prepared from the reaction of Ag2O with the corresponding imidazolium salts, 4I, 4PF66 and 5Br. NHC carbenes 4 and 5 form the bridged metallacyclic dinuclear silver complexes, [Ag2(μ-NHC)2][X]2 shown in Scheme 1, irrespective of the metal to ligand ratio and type of counterion used. The analogous methyl–imidazolyl complex [Ag2(μ-2)2][PF6]2 and its solid-state structure has been reported previously by us.19 Unfortunately, the isopropyl and butyl derivatives, 6I and 7Br, did not afford good crystals of sufficient quality to allow for their X-ray structure determinations. As was the case for NHC ligand 2, the neutral acyclic bis-monocarbene complexes (11) were not observed. Electrospray mass spectra show maximal mass peaks at m/z 1001 and 1009 for the silver metallacycle complexes, 6I and 7Br respectively, corresponding to loss of one of the counterions from the parent compound, [Ag(μ-NHC)]2[X]2. In the 13C NMR spectra of the silver complexes the carbene resonance appears as a sharp singlet at δ 180.6 ppm for 6I and δ 181.6 ppm for 7Br. Coupling to 107Ag or 109Ag was unobserved in both cases. Silver–carbene complexes have been used previously in transmetalation reactions with platinum(II) salts to afford the corresponding Pt(NHC) complexes.8b,20 In the case of the silver complex 7Br reaction with Pt(cod)Cl2 afforded mixtures of Pt(II)-containing species and partially protonated carbene ligand.Platinum(II) complexes
In order to avoid time- and resource-consuming routes, the platinum complexes 8–10 were prepared directly from the reaction of the corresponding ligand salts (2PF66, 4PF66 and 5PF66) with PtCl2 or K2PtCl4 in quantitative yields (Scheme 1). Reactions were carried out in the presence of NaOAc in dmso, under aerobic conditions using unpurified technical grade solvent. Reactions in other solvents, e.g. dma (N,N′-dimethylacetamide), afforded complex mixtures of intractable products. The above synthetic procedure afforded the bis-monocarbene complexes, cis-[Pt2(μ-NHC)(dmso)2Cl4], regardless of the metal to ligand ratio used.7,8,20 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 metal to ligand reactions afforded the corresponding cis-[Pt2(μ-NHC)(dmso)2Cl4] complexes, with the excess imidazolium salt remaining unreacted. Complexes 8–10 are air- and moisture-stable and can be stored for prolonged times in air without any visible decomposition. They are soluble in chlorinated solvents, acetonitrile and dmso.
:1 metal to ligand reactions afforded the corresponding cis-[Pt2(μ-NHC)(dmso)2Cl4] complexes, with the excess imidazolium salt remaining unreacted. Complexes 8–10 are air- and moisture-stable and can be stored for prolonged times in air without any visible decomposition. They are soluble in chlorinated solvents, acetonitrile and dmso.
Solution NMR measurements
Relatively small changes are observed in the 1H NMR spectra for the hydrogen atoms of the proligands of 2, 4 and 5 upon coordination to silver(I). More pronounced deshielding of the proton resonances, however, is seen for the platinum complexes, most likely due to the divalent metal. Notably, there are large downfield shifts of the methine protons (H2/5) adjacent to the imidazolyl nitrogen atoms from ca. δ 5.1 ppm (CD3CN) in the imidazolium salts to δ 5.9–6.1 ppm (CD3CN) in the coordinated ligands. A large downfield shift is also observed for the isopropyl methine proton of 9, from δ 4.73 ppm in 4I to 5.63 ppm (CD3CN).Restricted rotation about the C–Pt bond
The 1H and 13C NMR spectra of the platinum complexes, 8–10, reveal more than one isomers present in solution which have been attributed to rotamers arising from restricted rotation about the Pt–CNHC bond. High metal–carbene rotation barriers (>92 kJ mol−1) have been observed for Rh(I)– and Ir(I)–NHC systems and attributed mainly to steric effects.21 Similarly, for Pt(NHC)(dmso)Cl2-type complexes Rourke et al. have reported barriers to rotation in excess of 85 kJ mol−1.8a
In the 195Pt NMR spectra of complexes 8–10 two signals were observed in each case, centred at ca. −3500 ppm (referenced to Na2PtCl6).22 Presumably, some of the rotamer signals remain unresolved in the corresponding 195Pt NMR spectra. For comparison, the δ195Pt reported for the related carbene complexes, [Pt(NHC)(dmso)Me2], are in the region of −3900 to −4000 ppm.20 The effect of the different N-imidazole substituents (Me, iPr, Bu) of complexes 8–10 on the 195Pt NMR chemical shifts (δ195Pt) is negligible. This is in agreement with reports for other PtCl2 systems, such as those of chelating dipyridyl ligands.23 Due to solubility limitations, different solvents were used for each complex during these measurements (dmso-d6 for 8, CD3CN for 9 and CD2Cl2 for 10). Contrary to previous observations, where large Δδ195Pt of ca. 400 ppm were observed with changes in the solvent from CD2Cl2 to dmso-d6, there were no significant solvent effects observed for complexes 8–10.24
1H–15N HMBC spectra were also collected for the platinum complexes. In most cases analyses of the cross-peaks in the two-dimensional spectra provided a means of differentiating and assigning the two non-symmetrically substituted imidazolyl 15N resonances. Relatively small differences in the chemical shifts of the imidazole derived nitrogens were observed (Δδ15N), with the largest Δδ15N observed for complex 9 at 16 ppm and the smallest for complex 10. In this case, the two 15N resonances centred at 204.5 ppm remained unresolved. Although the signals for NO and NR in the 15N dimension of the spectra for 8–10 were not resolved further, the presence of rotamers is implied after inspection of the cross-section of the peaks in the 2D-spectra (where NO represents the nitrogen atom next to the oxolane ring and NR the one next to the corresponding alkyl substituent).
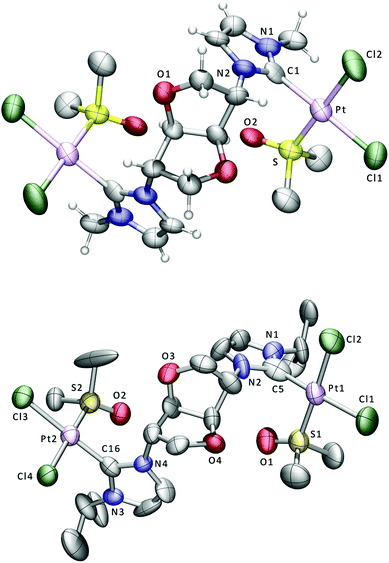 | ||
| Fig. 2 Displacement ellipsoid plots at the 50% probability level of the platinum complexes 8 (above) and 9; for clarity only some of the H atoms are shown. Selected bond lengths (Å) and angles (°) for 8: Pt–Cl(1) 2.361(8), Pt–Cl(2) 2.305(8), Pt–S 2.205(7), Pt–C(1) 1.98(2), Cl(1)–Pt–Cl(2) 90.0(3), Cl(1)–Pt–S 90.4(3), Cl(1)–Pt–C(1) 176.1(8), Cl(2)–Pt–S 178.9(3), Cl(2)–Pt–C(1) 88.4(6), S–Pt–C(1) 91.2(6). Selected bond lengths (Å) and angles (°) for 9: Pt(1)–Cl(1) 2.335(4), Pt(1)–Cl(2) 2.328(4), Pt(1)–S(1) 2.189(4), Pt(1)–C(5) 1.948(14), Pt(2)–Cl(3) 2.338(4), Pt(2)–Cl(4) 2.306(4), Pt(2)–S(2) 2.190(4), Pt(2)–C(16) 1.88(2), Cl(1)–Pt(1)–Cl(2) 90.86(13), Cl(1)–Pt(1)–S(1) 90.88(14), Cl(1)–Pt(1)–C(5) 177.1(4), Cl(2)–Pt(1)–S(1) 175.60(14), Cl(2)–Pt(1)–C(5) 88.3(4), S(1)–Pt(1)–C(5) 90.2(4), Cl(3)–Pt(2)–Cl(4) 90.62(18), Cl(3)–Pt(2)–S(2) 91.02(18), Cl(3)–Pt(2)–C(16) 176.0(5), Cl(4)–Pt(2)–S(2) 178.04(16), Cl(4)–Pt(2)–C(16) 88.3(5), S(2)–Pt(2)–C(16) 90.1(5). | ||
Experimental
General remarks
All manipulations were performed using standard glassware under aerobic conditions, except where otherwise noted. Solvents of analytical grade and deuterated solvents for NMR measurements were used as received. Literature methods were employed for the synthesis of 119 and Ag2O.26 All other reagents were used as received. Ether means diethyl ether and dmf dimethylformamide unless specified otherwise. NMR spectra were obtained on Bruker Avance AMX 250, 400, 500, 600 (QCI Quadruple Resonance CryoProbe) or Jeol Eclipse 300 spectrometers. The chemical shifts are given as dimensionless δ values and are frequency referenced relative to TMS for 1H and 13C and Na2PtCl6 for 195Pt. For 1H–15N heteronuclear multiple-bond correlation (HMBC) experiments, CH3NO2 was used as the standard for the 15N chemical shifts (δ = 381.7 vs. liquid NH3).27 In the HMBC experimental, NR stands for the alkyl substituted nitrogen of the imidazole ring and NO for the one next to the dioxolane ring.Coupling constants J are given in hertz (Hz) as positive values regardless of their real individual signs. The multiplicity of the signals is indicated as s, d, or m for singlets, doublets, or multiplets, respectively. The abbreviation br is given for broadened signals. Mass spectra and high-resolution mass spectra were obtained in electrospray (ES) mode unless otherwise reported, on a Waters Q-Tof micromass spectrometer. IR spectra were measured on a JASCO 660plus FT-IR spectrometer from 4000 to 600 cm−1.
For the 1H and 13C NMR spectra of the platinum complexes, 8–10 reported below, signals for all 3 observed isomers are reported with individual peaks not assigned to a specific isomer.
X-ray crystallography
Data for 4I were measured at 120 K on a Bruker Nonius KappaCCD diffractometer at the window of a Bruker Nonius FR591 rotating anode (λMo-Kα = 0.71073 Å) driven by COLLECT and processed by DENZO software.28 The structure was determined in SHELXS-97 and refined using SHELXL-97.29 Data for 8 and 9 were obtained from very small and weakly diffracting crystals by use of synchrotron radiation (λ = 0.6889 Å) at 120 K, with a Crystal Logics kappa diffractometer and Rigaku Saturn 724+ CCD detector at beamline I19 of Diamond Light Source; software was Rigaku CrystalClear,30 and Bruker APEX231 and SHELXTL.29 Key crystallographic data are shown in Table 1.| Compound | 4I | 8 | 9 |
|---|---|---|---|
| Chemical formula | C18H28N4O22+·2I− | C18H30Cl4N4O4Pt2S2·CH2Cl2 | C22H38Cl4N4O4Pt2S2 |
| Formula mass | 586.2 | 1047.5 | 1018.66 |
| Crystal system | Orthorhombic | Orthorhombic | Tetragonal |
| a/Å | 8.9301(2) | 15.032(12) | 19.248(6) |
| b/Å | 10.8793(2) | 15.651(13) | |
| c/Å | 23.2605(4) | 6.587(5) | 23.080(14) |
| Unit cell volume/Å3 | 2259.83(8) | 1550(2) | 8551(6) |
| Space group | P212121 | P21212 | P41212 |
| Z | 4 | 2 | 8 |
| μ/mm−1 | 2.80 | 8.90 | 6.34 |
| No. of reflections measured | 19225 |
10347 |
67854 |
| No. of independent reflections | 5180 | 2790 | 7575 |
| R int | 0.0479 | 0.1691 | 0.1014 |
| Final R values (F, F2 > 2σ) | 0.0313 | 0.0797 | 0.0530 |
| Final R values (F2, all data) | 0.0723 | 0.2144 | 0.1457 |
| Goodness of fit on F2 | 1.045 | 1.063 | 0.982 |
| Difference map extremes, e Å−3 | 1.06, −0.85 | 3.22, −1.55 | 1.02, −0.80 |
In addition to the intended compound 9 (with 8 identical molecules in the tetragonal unit cell, all of the same chirality), the structure also contains highly disordered solvent and/or other components that could not be modelled as discrete atoms and could not be identified from the observed electron density distribution. This has been treated with the SQUEEZE option of the program PLATON.32 The refined structural model contains only the ordered part of the crystal structure and the unidentified disordered components are not included in any of the tabulated or deposited values. The crystal structure of 8 contains disordered dichloromethane solvent molecules, which it was possible to model with discrete atom positions. For all three structures, the correct enantiomer was confirmed on the basis of significant anomalous scattering effects.33
Crystallographic data for all compounds have been deposited with the Cambridge Crystallographic Data Centre as supplementary publications CCDC 893546–893548.
Conclusions
Short synthetic routes to the chiral, ether-functionalized NHC precursors, 2OTs, 4I and 5Br have been developed. The bridging ligands 2, 4 and 5 form either cationic dinuclear cyclic structures with Ag(I) or neutral linear complexes in the case of Pt(II). The robust nature of the Pt(II) complexes is indicated by their synthesis being achieved under aerobic conditions using bench grade solvents. For the cis-Pt(dmso)Cl2 complexes, hindered rotation of the carbene ligand about the metal–NHC bond is evident in their 1H, 13C, 15N and 195Pt NMR spectra, where more than one rotamers can be observed.Acknowledgements
We thank the EPSRC for funding for the National Crystallography Service (Southampton and Newcastle Universities), and Diamond Light Source for access to beamline I19.Notes and references
- (a) C. X. Lu, S. J. Gu, W. Z. Chen and H. Y. Qiu, Dalton Trans., 2010, 39, 4198–4204 RSC; (b) D. Troegel and J. Stohrer, Coord. Chem. Rev., 2011, 255, 1440–1459 CrossRef CAS; (c) I. E. Marko, S. Sterin, O. Buisine, G. Mignani, P. Branlard, B. Tinant and J. P. Declercq, Science, 2002, 298, 204–206 CrossRef CAS; (d) G. F. Silbestri, J. C. Flores and E. de Jesus, Organometallics, 2012, 31, 3355–3360 CrossRef CAS; (e) J. J. Hu, F. W. Li and T. S. A. Hor, Organometallics, 2009, 28, 1212–1220 CrossRef CAS.
- R. Zhang, Q. Xu, L. Y. Mei, S. K. Li and M. Shi, Tetrahedron, 2012, 68, 3172–3178 CrossRef CAS.
- V. Lillo, J. A. Mata, A. M. Segarra, E. Peris and E. Fernandez, Chem. Commun., 2007, 2184–2186 RSC.
- V. Lillo, J. Mata, J. Ramirez, E. Peris and E. Fernandez, Organometallics, 2006, 25, 5829–5831 CrossRef CAS.
- S. Das Adhikary, D. Bose, P. Mitra, K. Das Saha, V. Bertolasi and J. Dinda, New J. Chem., 2012, 36, 759–767 RSC.
- Y. Unger, A. Zeller, M. A. Taige and T. Strassner, Dalton Trans., 2009, 4786–4794 RSC.
- S. Fantasia, J. L. Petersen, H. Jacobsen, L. Cavallo and S. P. Nolan, Organometallics, 2007, 26, 5880–5889 CrossRef CAS.
- (a) C. P. Newman, R. J. Deeth, G. J. Clarkson and J. P. Rourke, Organometallics, 2007, 26, 6225–6233 CrossRef CAS; (b) D. Meyer, A. Zeller and T. Strassner, J. Organomet. Chem., 2012, 701, 56–61 CrossRef CAS.
- (a) A. J. Arduengo, H. V. R. Dias, J. C. Calabrese and F. Davidson, Organometallics, 1993, 12, 3405–3409 CrossRef CAS; (b) O. Guerret, S. Sole, H. Gornitzka, M. Teichert, G. Trinquier and G. Bertrand, J. Am. Chem. Soc., 1997, 119, 6668–6669 CrossRef CAS; (c) H. M. J. Wang and I. J. B. Lin, Organometallics, 1998, 17, 972–975 CrossRef CAS; (d) M. Boronat, A. Corma, C. Gonzalez-Arellano, M. Iglesias and F. Sanchez, Organometallics, 2010, 29, 134–141 CrossRef CAS; (e) H. V. Huynh, C. H. Yeo and Y. X. Chew, Organometallics, 2010, 29, 1479–1486 CrossRef CAS.
- (a) Y. L. Li, K. Hindi, K. M. Watts, J. B. Taylor, K. Zhang, Z. C. Li, D. A. Hunstad, C. L. Cannon, W. J. Youngs and K. L. Wooley, Chem. Commun., 2010, 46, 121–123 RSC; (b) S. Patil, J. Claffey, A. Deally, M. Hogan, B. Gleeson, L. M. M. Mendez, H. Muller-Bunz, F. Paradisi and M. Tacke, Eur. J. Inorg. Chem., 2010, 1020–1031 CrossRef CAS; (c) A. Kascatan-Nebioglu, M. J. Panzner, C. A. Tessier, C. L. Cannon and W. J. Youngs, Coord. Chem. Rev., 2007, 251, 884–895 CrossRef CAS; (d) A. Melaiye, R. S. Simons, A. Milsted, F. Pingitore, C. Wesdemiotis, C. A. Tessier and W. J. Youngs, J. Med. Chem., 2004, 47, 973–977 CrossRef CAS; (e) A. Kascatan-Nebioglu, A. Melaiye, K. Hindi, S. Durmus, M. J. Panzner, L. A. Hogue, R. J. Mallett, C. E. Hovis, M. Coughenour, S. D. Crosby, A. Milsted, D. L. Ely, C. A. Tessier, C. L. Cannon and W. J. Youngs, J. Med. Chem., 2006, 49, 6811–6818 CrossRef CAS; (f) J. C. Garrison and W. J. Youngs, Chem. Rev., 2005, 105, 3978–4008 CrossRef CAS.
- J. C. Y. Lin, R. T. W. Huang, C. S. Lee, A. Bhattacharyya, W. S. Hwang and I. J. B. Lin, Chem. Rev., 2009, 109, 3561–3598 CrossRef CAS.
- L. Ray, M. M. Shaikh and P. Ghosh, Inorg. Chem., 2007, 47, 230–240 CrossRef.
- V. J. Catalano, M. A. Malwitz and A. O. Etogo, Inorg. Chem., 2004, 43, 5714–5724 CrossRef CAS.
- A. B. Powell, C. W. Bielawski and A. H. Cowley, J. Am. Chem. Soc., 2009, 131, 18232–18233 CrossRef CAS.
- V. J. Catalano and M. A. Malwitz, Inorg. Chem., 2003, 42, 5483–5485 CrossRef CAS.
- A. Rit, T. Pape and F. E. Hahn, J. Am. Chem. Soc., 2010, 132, 4572–4573 CrossRef CAS.
- B. Liu, W. Z. Chen and S. W. Jin, Organometallics, 2007, 26, 3660–3667 CrossRef CAS.
- (a) C. Carcedo, A. Dervisi, I. A. Fallis, L. Ooi and K. M. A. Malik, Chem. Commun., 2004, 1236–1237 RSC; (b) A. Dervisi, C. Carcedo and L.-l. Ooi, Adv. Synth. Catal., 2006, 348, 175–183 CrossRef CAS.
- C. Carcedo, J. C. Knight, S. J. A. Pope, I. A. Fallis and A. Dervisi, Organometallics, 2011, 30, 2553–2562 CrossRef CAS.
- G. L. Petretto, M. A. Wang, A. Zucca and J. P. Rourke, Dalton Trans., 2010, 39, 7822–7825 RSC.
- (a) S. Burling, S. Douglas, M. F. Mahon, D. Nama, P. S. Pregosin and M. K. Whittlesey, Organometallics, 2006, 25, 2642–2648 CrossRef CAS; (b) A. R. Chianese, X. W. Li, M. C. Janzen, J. W. Faller and R. H. Crabtree, Organometallics, 2003, 22, 1663–1667 CrossRef CAS; (c) M. Iglesias, D. J. Beetstra, A. Stasch, P. N. Horton, M. B. Hursthouse, S. J. Coles, K. J. Cavell, A. Dervisi and I. A. Fallis, Organometallics, 2007, 26, 4800–4809 CrossRef CAS; (d) L. Busetto, M. C. Cassani, C. Femoni, M. Mancinelli, A. Mazzanti, R. Mazzoni and G. Solinas, Organometallics, 2011, 30, 5258–5272 CrossRef CAS.
- B. M. Still, P. G. A. Kumar, J. R. Aldrich-Wright and W. S. Price, Chem. Soc. Rev., 2007, 36, 665–686 RSC.
- (a) T. Pawlak, L. Pazderski, J. Sitkowski, L. Kozerski and E. Szlyk, Magn. Reson. Chem., 2011, 49, 59–64 CrossRef CAS; (b) L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szlyk, Magn. Reson. Chem., 2010, 48, 417–426 CrossRef CAS; (c) L. Pazderski, T. Pawlak, J. Sitkowski, L. Kozerski and E. Szlyk, Magn. Reson. Chem., 2009, 47, 932–941 CrossRef CAS; (d) L. Pazderski, J. Tousek, J. Sitkowski, L. Kozerski and E. Szlyk, Magn. Reson. Chem., 2009, 47, 658–665 CrossRef CAS; (e) L. Pazderski, E. Szlyk, J. Sitkowski, B. Kamienski, L. Kozerski, J. Tousek and R. Marek, Magn. Reson. Chem., 2006, 44, 163–170 CrossRef CAS.
- L. Pazderski, E. Szłyk, J. Sitkowski, B. Kamieński, L. Kozerski, J. Toušek and R. Marek, Magn. Reson. Chem., 2006, 44, 163–170 CrossRef CAS.
- D. Brissy, M. Skander, P. Retailleau and A. Marinetti, Organometallics, 2007, 26, 5782–5785 CrossRef CAS.
- (a) C. V. W. Donald and E. Janssen, Org. Synth., 1963, Coll. Vol. 4, 547 Search PubMed; (b) C. V. W. Donald and E. Janssen, Org. Synth., 1956, 36, 46 Search PubMed.
- E. Procházková, M. Šála, R. Nencka and M. Dračínský, Magn. Reson. Chem., 2012, 50, 181–186 CrossRef.
- Z. M. W. Otwinowski, Methods in Enzymology, Academic Press, New York, 1997 Search PubMed.
- M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef.
- R. C. CrystalClear Software for Data Collection, The Woodlands, TX, 2008 Search PubMed.
- B. A. I. APEX2 Software for Data Processing, Madison, WI, 2008 Search PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A: Found. Crystallogr., 1983, 39, 876 CrossRef.
Footnotes |
| † Dedicated to Professor David Cole-Hamilton on the occasion of his retirement and for his outstanding contribution to transition metal catalysis. |
| ‡ Electronic supplementary information (ESI) available: X-ray crystallographic data as CIF files for the compounds 4I, 8 and 9; figures showing NMR spectra. CCDC 893546–893548. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c2dt31740k |
| This journal is © The Royal Society of Chemistry 2012 |