 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Influence of counterions on the structure of bis(oxazoline)copper(II) complexes; an EPR and ENDOR investigation†
Mari Elena
Owen
,
Emma
Carter
*,
Graham J.
Hutchings
,
Benjamin D.
Ward
and
Damien M.
Murphy
*
School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff CF10 3AT, UK. E-mail: CarterE4@cardiff.ac.uk; MurphyDM@cardiff.ac.uk
First published on 6th July 2012
Abstract
X- and Q-band EPR and ENDOR spectroscopy was used to study the structure of a series of heteroleptic and homoleptic copper bis(oxazoline) complexes, based on the (−)-2,2′-isopropylidenebis[(4S)-4-phenyl-2-oxazoline] ligand and bearing different counterions (chloride versus triflate); labelled [CuII(1a–c)]. The geometry of the two heteroleptic complexes, [CuII(1a)] and [CuII(1c)], depended on the choice of counterion. Formation of the homoleptic complex was only evident when the CuII(OTf)2 salt was used (CuII(Cl)2 inhibited the transformation from heteroleptic to homoleptic complexes). The hyperfine and quadrupole parameters for the surrounding ligand nuclei were determined by ENDOR. Well resolved 19F and 1H couplings confirmed the presence of both coordinated water and TfO− counterions in [Cu(1a)].
Introduction
Enantioselective asymmetric synthesis involves the preparation of chiral compounds with well defined three-dimensional stereochemistry.1–4 These enantiomerically pure compounds are vital for many applications, for example, in the pharmaceutical industry, for vitamins and flavourings and in nonlinear optical and liquid crystalline materials to name a few. A wide range of synthetic catalysts are now available based on a diverse class of organic ligands which can achieve excellent levels of enantioselectivity for many different reaction types. In several of these cases, the enantioselective reaction is catalyzed by chiral Lewis acid complexes, often based on main group or transition metal salts coordinated to the chiral organic ligand.3Among the many available ligands to promote these asymmetric reactions, the chiral bis(oxazoline) ligands (commonly abbreviated to BOX) have been widely used (Scheme 1).5,6 In particular the CuIIbis(oxazoline) complexes have been successfully used for a diverse range of reactions including the Diels–Alder reaction,7–16 cyclopropanation,17–20 and aziridination.21–23 To date, a vast array of BOX ligands have been developed in order to optimise and tune the catalytic performance.24 The ability of the metal centre, including zinc, nickel, iron and copper, to coordinate through bidentate, tridentate or tetradentate coordination affords opportunities to tune the ligand to the required catalytic reaction.1,24 Upon coordination of the bidentate ligand, an almost planar metallacycle is formed. This, along with the presence of the pendent five-membered rings, are important factors in limiting the flexibility of these ligand systems.4,14 Ligands with a single carbon nucleus bridging between the oxazoline rings are the most commonly employed (Scheme 1), but alternatives have been explored in which adjustments have been made to the nature, size and flexibility of the link between the two oxazoline rings.25
![Structures of the bis(oxazoline) complexes used in this study. Schematic illustration of the [CuII(1a–c)] complexes as identified by EPR/ENDOR.](/image/article/2012/DT/c2dt31273e/c2dt31273e-s1.gif) | ||
| Scheme 1 Structures of the bis(oxazoline) complexes used in this study. Schematic illustration of the [CuII(1a–c)] complexes as identified by EPR/ENDOR. | ||
Since the CuIIBOX complexes are usually generated in situ by reacting the chiral BOX ligand with a suitable CuII salt, the choice of counterion is reported to have a large influence on the resulting enantioselectivities and yields. For example, Fraile et al.,26 demonstrated a significant decrease in selectivity in the reaction of styrene with ethyl diazoacetate when triflate counterions are replaced by chlorides, while Evans et al.,18,21 showed that highly electronegative counterions (Cl−, Br−) are a design prerequisite for efficient asymmetric aziridination. Furthermore Jørgensen,8 reported that the combination of counterion and solvent must be optimised to achieve the highest overall rates and enantioselectivities. This was exemplified by the reported 20-fold greater reactivity and superior enantioselectivity for cationic [CuII(BOX)](SbF6)2 compared to the triflate counterparts.27
Despite the importance of the counterion in modulating the catalytic activity, few experimental techniques can probe such influences in solution. For paramagnetic CuII based BOX complexes, EPR and the related hyperfine techniques such as ENDOR are an ideal method to examine any structural or electronic perturbations to the metal-complex caused by the different counterions. A limited number of papers have been reported on the CW EPR spectra of CuII(BOX) complexes,16,28–31 and these primarily focussed on the oxidation state of the CuII ion, rather than the role of the counterion. One of the first groups to recognise how advanced EPR techniques can be used to probe these counterion (TfO−, SbF6−, Cl−, Br−) effects for the related CuII-bissulfoximine complexes in the Diels–Alder reaction was Bolm and Gescheidt.32,33 Therefore in the current investigation we describe the detailed characterisation of the paramagnetic CuIIBOX complexes (shown in Scheme 1) using EPR and ENDOR spectroscopy, with specific emphasis on the influence of the counterion (TfO− or Cl−) on the structure of the complex in solution.
Experimental section
Chemicals and synthesis
All reagents were purchased from Sigma Aldrich and used as received. The BOX ligand (labelled (1) in Scheme 1, (−)-2,2′-Isopropylidenebis[(4S)-4-phenyl-2-oxazoline], CAS no. 131457-46-0) was used as received. All reactions were performed under atmospheric conditions. The heteroleptic complexes [CuII(1a,c)] were prepared according to literature methods34 by reacting CuII(OTf)2 or CuIICl2 with (1) in tetrahydrofuran–dichloromethane (THF–DCM). The homoleptic complex, [CuII(1b)], was synthesised by stirring (1) with CuII(OTf)2 in THF for 1 h at room temperature. HR ES-MS for [CuII(1b)], found (calc. for C42H44CuN4O4): 731.2635 (731.2659).Spectroscopic measurements
For CW and pulsed EPR/ENDOR measurements, the heteroleptic copper complexes [CuII(1a,c)] (ca. 7 × 10−3 M for EPR, ca. 4 × 10−2 M for ENDOR) were dissolved in either d8-tetrahydrofuran–d2-dichloromethane (abbreviated THF–DCM) or d3-acetonitrile–d2-dichloromethane (abbreviated AcN–DCM) while the homoleptic complex [CuII(1b)] was dissolved in d3-acetonitrile–d2-dichloromethane (AcN–DCM). The choice of solvent considerably affected the solubility, where higher concentrations were required for the CW ENDOR measurements. All X-band EPR spectra were recorded on a Bruker EMX spectrometer operating at 100 kHz field modulation and equipped with a high sensitivity X-band cavity (ER 4119HS). The spectra were recorded at a microwave power of 10 mW at 140 K. The CW Q-band ENDOR spectra were recorded at 10 K on a CW Bruker ESP 300E series spectrometer equipped with an ESP360 DICE ENDOR unit, operating at 12.5 kHz field modulation in a Q-band ENDOR cavity (Bruker ER 5106 QT-E). The ENDOR spectra were obtained using 8 dB RF power from an ENI A-300 RF amplifier and 50 or 200 kHz RF modulation depth and 1 mW microwave power. The pulsed X-band EPR/ENDOR spectra were recorded on a Bruker Elexsys E580 spectrometer equipped with a liquid Helium cryostat from Oxford Inc. The spectra were taken at 10 K, with a repetition rate of 333 kHz. The pulse sequence π–T–π/2–τ–π–τ-echo was used for the Davies ENDOR measurements, using mw pulse lengths of tπ = 256 ns, tπ/2 = 128 ns, and an interpulse time τ of 800 ns. An rf π pulse of variable frequency and a length of 18 μs was applied during time T of 20 μs. EPR simulations were performed using the Sim32 software,35 and ENDOR simulations were performed using the Easyspin package.36DFT calculations
The EPR parameters were calculated via spin-unrestricted density functional computations using the ORCA package37–40 on the reported crystal structures of [CuII(1a)]34 and [CuII(1c)].14 The computations were performed with the B3LYP functional. Basis sets with significant flexibility in the core region were used (ORCA basis sets ‘CoreProp’ (CP(III))41 for copper, and a Barone basis set ‘EPRII’42 for the hydrogen atoms).Results and discussion
CW EPR of [CuII(1a–c)]
The CuII(BOX) complexes are most conveniently prepared by simply stirring a suitable CuII salt with the required BOX ligand in solution. The solvent and the Cu–BOX ratio is then critical in order to form the desired CuII(BOX) complex. This can be easily monitored by EPR spectroscopy, as shown in Fig. 1. A solution of ligand (1) in THF–DCM was stirred with CuII(OTf)2 for 1 h, and the resulting profile of the EPR spectra changes considerably as more of the CuII(OTf)2 progressively coordinates with the BOX ligand (1). Fig. 1a shows the initial EPR spectrum of CuII(OTf)2 in the absence of (1), while Fig. 1b–e shows the resulting spectra after addition of increasing amounts of (1).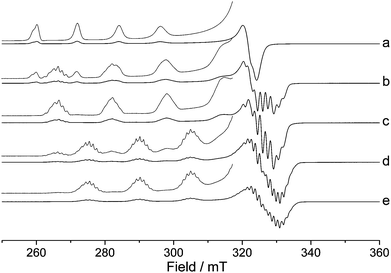 | ||
Fig. 1 X-band CW EPR spectra (140 K) of (a) CuII(OTf)2 dissolved in THF–DCM, containing increasing Cu–BOX (1) ratios; (b) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.5, (c) 1:1, (d) 1:2 and (e) 1:6. Solvent system, 1:1 THF–DCM. :0.5, (c) 1:1, (d) 1:2 and (e) 1:6. Solvent system, 1:1 THF–DCM. | ||
The pronounced superhyperfine couplings observed in Fig. 1b–e, are clearly indicative of CuII coordination to (1). At a Cu–BOX ratio of 1:0.5, a mixture of both CuII(OTf)2 and a CuII coordinated BOX complex is observed (Fig. 1b). At a Cu–BOX ratio of 1:1, a well resolved EPR spectrum is obtained, indicative of the formation of a single CuII(BOX) complex. Finally, as the Cu–BOX ratio increases further (i.e., 1:2 and 1:6, Fig. 1d,e), the shape of the spectra changes further, indicative of the formation of a second CuII(BOX) type complex (1b, vide infra). A similar series of EPR spectra can also be observed for the CuIICl2 salt after reacting with (1) in DCM; see Fig. S1, ESI.† In order to understand the structure of the CuII(BOX) complexes responsible for the spectra shown in Fig. 1b–e, additional Q-band EPR spectra were performed to aid in the simulations and analysis of the spin Hamiltonian parameters.
The Q-band CW EPR spectra for CuII(OTf)2 and CuIICl2 containing different ratios of Cu–BOX ligand are given in Fig. 2 while the corresponding X-band data is given in Fig. 3 along with the spectra of the starting CuII(OTf)2 and CuIICl2 salts for comparison. Since the g-strain effect is larger at Q-band compared to X-band frequency, the resolution of the hyperfine splitting is lost in the g1,2 region of the spectrum. The spin Hamiltonian parameters were extracted by simulation of both the X- and Q-band data, and the resulting parameters are listed in Table 1. The g and ACu tensors for the CuII(OTf)2 and CuIICl2 salts are both axially symmetric, and largely consistent with previous reports43,44 (it should be noted that the profile of these spectra are heavily solvent dependent). The EPR spectrum of CuIICl2 (Fig. 3d) also contains a series of additional lines in the perpendicular region, which arise from the superhyperfine couplings to weakly interacting solvent molecules.
2 (Cu–BOX ratio 1 : 1, in THF–DCM), (b) [CuII(BOX)2] (Cu–BOX ratio 1 : 6, in AcN–DCM), and (c) [CuII(BOX)](Cl)2 (Cu–BOX ratio 1 : 1, in THF–DCM). The corresponding simulations of these [CuII(1a)], [CuII(1b)] and [CuII(1c)] complexes are given in a′–c′.](/image/article/2012/DT/c2dt31273e/c2dt31273e-f2.gif) | ||
| Fig. 2 Q-band CW EPR spectra (50 K) of (a) [CuII(BOX)](OTf)2 (Cu–BOX ratio 1:1, in THF–DCM), (b) [CuII(BOX)2] (Cu–BOX ratio 1:6, in AcN–DCM), and (c) [CuII(BOX)](Cl)2 (Cu–BOX ratio 1:1, in THF–DCM). The corresponding simulations of these [CuII(1a)], [CuII(1b)] and [CuII(1c)] complexes are given in a′–c′. | ||
![X-band CW EPR spectra (140 K) of (a) CuII(OTf)2, (b) [CuII(1a)] (Cu–BOX ratio 1 : 1), (c) [CuII(1b)] (Cu–BOX ratio 1 : 6), (d) CuII(Cl)2 and (e) [CuII(1c)] (Cu–BOX ratio 1 : 1) dissolved in 1 : 1 THF–DCM solvent. The corresponding simulations are given in a′–e′.](/image/article/2012/DT/c2dt31273e/c2dt31273e-f3.gif) | ||
| Fig. 3 X-band CW EPR spectra (140 K) of (a) CuII(OTf)2, (b) [CuII(1a)] (Cu–BOX ratio 1:1), (c) [CuII(1b)] (Cu–BOX ratio 1:6), (d) CuII(Cl)2 and (e) [CuII(1c)] (Cu–BOX ratio 1:1) dissolved in 1:1 THF–DCM solvent. The corresponding simulations are given in a′–e′. | ||
| Complex |
g
1a |
g
2a |
g
3a |
A
1b |
A
2b |
A
3c |
|---|---|---|---|---|---|---|
| All A values given in MHz;a ±0.004;b ±3 MHz;c ±6 MHz. | ||||||
| Cu(OTf)2 | 2.083 | 2.083 | 2.412 | 13.1 | 13.1 | 403.4 |
| CuCl2 | 2.061 | 2.061 | 2.316 | 54.8 | 54.8 | 457.8 |
| [Cu(1a)] | 2.064 | 2.073 | 2.313 | 15.0 | 14.5 | 506.7 |
| DFT | 2.069 | 2.073 | 2.209 | −32.85 | −44.6 | −866.5 |
| [Cu(1b)] | 2.054 | 2.063 | 2.254 | 25.9 | 28.9 | 461.3 |
| [Cu(1c)] | 2.057 | 2.057 | 2.280 | 33.1 | 33.1 | 395.7 |
| DFT | 2.062 | 2.064 | 2.204 | −76.4 | −81.7 | −764.6 |
The EPR spectra of the CuII(BOX) complexes were simulated using slightly rhombic g and ACu tensors (see Table 1). The resolved copper hyperfine splittings are further split due to the hyperfine interaction with two equivalent 14N nuclei in Fig. 3b,e and four equivalent 14N nuclei in Fig. 3c. The g values used in the simulation were extracted more accurately from the Q-band spectra (Fig. 2). Although the g1,2 regions of the X-band spectra are particularly complex, since the Cu and 14N hyperfine couplings are of similar magnitude (Table 1), accurate 14N couplings were determined via the ENDOR measurements (vide infra) and these parameters were used in the EPR simulations.
The g3/A3 values of CuII complexes possessing a dx2–y2 ground state, are usually diagnostic of the coordinating environment.45,46 Therefore the g3/A3 values of 2.313/506.7 MHz and 2.280/395.7 MHz for the complexes responsible for Fig. 3b and e respectively, coupled with the observed hyperfine splittings from two equivalent 14N, are consistent with the presence of the heteroleptic complexes labelled [CuII(BOX)](OTf)2 and [CuII(BOX)]Cl2 (i.e., [CuII(1a)] and [CuII(1c)] in Scheme 1). The altered g3/A3 values of 2.254/461.3 MHz for the CuII complex represented by Fig. 3c, coupled with the four equivalent 14N nuclei clearly resolved in the low field mI = −3/2 Cu hyperfine line, are consistent with a coordinating environment bearing four equivalent nitrogens. This spectrum therefore provides evidence for the presence of the homoleptic complex [CuII(BOX)2] (i.e., [CuII(1b)] in Scheme 1) in solution at the higher Cu–BOX ratios. Whilst homoleptic complexes of this type have been isolated, none have been crystallographically characterised.17
The spin Hamiltonian parameters determined for the two heteroleptic complexes [CuII(1a)] and [CuII(1c)] are notably different. This suggests that the counterions (TfO− and Cl−) must remain coordinated to the CuII centre in solution, in order to alter the observed spin Hamiltonian parameters. Indeed ENDOR spectroscopy reveals the presence of 19F couplings from the TfO− groups, further confirming the presence of the counterion in the coordination sphere (vide infra). Moreover, the different g/A values for [CuII(1a)] and [CuII(1c)] may in part be accounted for by differences in the distortion around the CuII centre caused by the bulky triflate ions relative to the chlorides.
The above EPR results indicate that as the Cu–BOX ratio increases, the heteroleptic and subsequently homoleptic CuIIbis(oxazoline) complexes are formed starting from the CuII(OTf)2 salts (Fig. 1). The analogous trend is not however observed starting from the CuCl2 salt; regardless of the Cu–BOX ratio employed, the homoleptic complex is never formed even when (1) is present in excess (see Fig. S1, ESI†). In other words, the more labile TfO− counterions are easily displaced when an excess of (1) is present in solution, whereas the Cl− counterions remain more strongly coordinated, preventing coordination of a second BOX ligand.
14N ENDOR
In order to extract the hyperfine and nuclear quadrupole principal values of the 14N nuclei from the bis(oxazoline) ligand, X-band Davies ENDOR and Q-band CW ENDOR measurements were conducted on each sample [CuII(1a–c)]. The ENDOR spectra were measured at multiple field positions. The experimental spectra and corresponding simulations at the two frequencies for the heteroleptic [CuII(1a)] complex are shown in Fig. 4 (the simulated parameters are listed in Table 2). The relevant ENDOR spectra for the [CuII(1b,c)] complexes are given in Fig. S2 and S3 in the ESI.† The X-band Davies ENDOR spectra were obtained using soft mw pulses and therefore contain overlapping contributions from 1H, 19F in addition to the strongly coupled 14N nuclei (Fig. 4A). Despite variations in the strength of the mw pulses (so-called hyperfine selective ENDOR), complete suppression of the 1H peaks could not be achieved, so these spectra remain significantly overlapped. Nevertheless the X-band Davies ENDOR spectra are important in order to observe the largest 14N couplings which can sometimes be difficult to detect via CW ENDOR.![(A) X-band Davies ENDOR spectra (10 K) of [Cu(1a)] recorded at the field positions (a) 344.3, (b) 334.5, (c) 330.0, (d) 313.0, (e) 307.0 and (f) 280.6 mT. (B) Q-band CW 14N ENDOR spectra (10 K) of [Cu(1a)] recorded at the field positions (a) 1180.4, (b) 1176.9, (c) 1159.7, (d) 1124.4, (e) 1080.0 and (f) 1030.8 mT. The corresponding simulations (for 14N only) are given in a′–f′. All spectra recorded in a 1 : 1 THF–DCM solvent.](/image/article/2012/DT/c2dt31273e/c2dt31273e-f4.gif) | ||
| Fig. 4 (A) X-band Davies ENDOR spectra (10 K) of [Cu(1a)] recorded at the field positions (a) 344.3, (b) 334.5, (c) 330.0, (d) 313.0, (e) 307.0 and (f) 280.6 mT. (B) Q-band CW 14N ENDOR spectra (10 K) of [Cu(1a)] recorded at the field positions (a) 1180.4, (b) 1176.9, (c) 1159.7, (d) 1124.4, (e) 1080.0 and (f) 1030.8 mT. The corresponding simulations (for 14N only) are given in a′–f′. All spectra recorded in a 1:1 THF–DCM solvent. | ||
| Complex |
A
1a |
A 2 | A 3 |
P
1b |
P 2 | P 3 | e2qQ/hc | ηd |
|---|---|---|---|---|---|---|---|---|
| All values are given in MHz;a ±0.2 MHz;b ±0.1 MHz;c ±0.2 MHz;d ±0.1. | ||||||||
| [Cu(Salen)] | 50.5 | 37.4 | 38.5 | −1.15 | 0.70 | 0.45 | −2.3 | 0.2 |
| [CuPc] | 56.4 | 44.8 | 45.7 | −0.79 | 0.82 | 0.03 | ||
| [Cu(1a)] | 45.6 | 35.9 | 36.7 | −0.87 | 0.97 | −0.10 | −2.3 | 0.2 |
| [Cu(1b)] | 39.8 | 33.1 | 32.9 | −0.57 | 0.52 | 0.05 | ||
| [Cu(1c)] | 41.9 | 32.5 | 32.8 | −0.87 | 0.97 | −0.10 | −2.5 | 0.15 |
The 14N couplings are in fact extremely well resolved at Q-band (Fig. 4B) enabling the angular selective data to be simulated more accurately (Table 2). The hyperfine and quadrupolar coupling from the 14N (I = 1) nuclei in the [Cu(1a)] complex was found to deviate slightly from axial symmetry and the largest principal axes was approximately directed to the copper ion. The observed hyperfine (Ai) and quadrupolar (Pi) parameters are very similar to those reported for other CuII centres bearing strongly coupled N4 or N2O2 donor ligand sets (Table 2).
Whilst the quadrupolar 14N parameters are similar for the two heteroleptic complexes, [Cu(1a)] and [Cu(1c)], the hyperfine parameters are slightly different in each case. These differences in NAi are consistent with the earlier variations noted in the g/CuA values by EPR (vide supra) and again suggest a slightly different degree of distortion in the CuII–N2 plane. An even larger difference in hyperfine (Ai) and quadrupolar (Pi) parameters is observed between the homoleptic ([Cu(1b)]) and heteroleptic ([Cu(1a)]) complexes (Table 2). In particular, the NAi and NPi parameters are smaller in the homoleptic complex (Table 2), and this is entirely consistent with the redistribution of the unpaired spin density in the CuII–N4 complex compared to the CuII–N2 complex.
1H and 19F ENDOR
The hyperfine couplings to the proton and fluorine nuclei of the complexes were well resolved by ENDOR at Q-band. The spectra recorded at the principal turning points (g = g|| and g = g⊥) for [CuII(1a–c)] are shown in Fig. 5. The presence of a weakly coupled 19F nucleus in [CuII(1a)] is evident in Fig. 5a,d, which must arise from coordinated TfO− groups. By comparison, in the homoleptic complex [CuII(1b)], only a matrix 19F peak centred on νn for fluorine, is observed (Fig. 5b,e) and this emanates from remote (non-coordinated) TfO− ions in the surrounding solvent. The small 19F couplings in [CuII(1a)] produced a well resolved spectrum which was simulated at multiple field positions in order to extract the 19F hyperfine parameters (Fig. 6). The resulting FA hyperfine parameters are given in Table 3. Analysis of the FA data using a simple point dipole approximation suggests a Cu⋯19F distance of ca. 7.78 Å based on a dipolar coupling of 0.327 MHz. Furthermore the largest contribution to this coupling was observed along the g = g|| direction with an angle of θH = 0° (angle between g3 and Br), consistent with the TfO− groups coordinating along the axial position of the CuII complex (orthogonal to the Cu–N2 plane). This picture is in fact consistent with the crystal structure of [CuII(1)](OTf)2(H2O)2 reported by Evans et al.,34 where the triflate groups were also oriented along the axial position. However the reported Cu⋯F distances varied from 4.834–6.049 Å in the crystal structure,34 compared to 7.78 Å estimated by ENDOR. This large discrepancy must arise from the differences in counterion positioning in the solid state single crystal compared to the solvated complex in frozen solution, as measured by ENDOR; the presence of solvent molecules may then cause the Cu⋯TfO− distance to increase.![Q-band CW 1H ENDOR spectra (10 K) of (a,d) [Cu(1a)] (1 : 1 THF–DCM), (b,e) [Cu(1b)] (1 : 1 AcN–DCM) and (c,f) [Cu(1c)] (1 : 1 AcN–DCM). The spectra were recorded at the field positions corresponding to: (a,b,c) g = g⊥ and (d,e,f) g = g||.](/image/article/2012/DT/c2dt31273e/c2dt31273e-f5.gif) | ||
| Fig. 5 Q-band CW 1H ENDOR spectra (10 K) of (a,d) [Cu(1a)] (1:1 THF–DCM), (b,e) [Cu(1b)] (1:1 AcN–DCM) and (c,f) [Cu(1c)] (1:1 AcN–DCM). The spectra were recorded at the field positions corresponding to: (a,b,c) g = g⊥ and (d,e,f) g = g||. | ||
![Q-band CW 19F ENDOR (10 K) of [CuII(1a)] dissolved in THF–DCM recorded at the field positions (a) 1180.4, (b) 1176.9, (c) 1159.7, (d) 1124.4, (e) 1080.0 and (f) 1030.8 mT. The corresponding simulations are given in a′–f′. Solvent 1 : 1 THF–DCM.](/image/article/2012/DT/c2dt31273e/c2dt31273e-f6.gif) | ||
| Fig. 6 Q-band CW 19F ENDOR (10 K) of [CuII(1a)] dissolved in THF–DCM recorded at the field positions (a) 1180.4, (b) 1176.9, (c) 1159.7, (d) 1124.4, (e) 1080.0 and (f) 1030.8 mT. The corresponding simulations are given in a′–f′. Solvent 1:1 THF–DCM. | ||
|
A
1a |
A
2a |
A
3b |
a iso | α′ | β′ | γ c | A || | R/Å | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| ENDOR | X-ray | ||||||||||
| All values are given in MHz;a ±0.2 MHz;b ±0.1 MHz;c ±10°;d ±0.5 MHz. | |||||||||||
| 19F | 0.49 | 0.49 | 0.98 | 0.65 | 0 | 0.32 | 7.78 | 4.834 | |||
| 1H2O | Expt | −10.8 | −5.02 | 8.6 | −2.41 | 20 | 38 | 0 | 11.01 | 2.52 | 2.410 |
| DFT | −12.01 | +9.11 | −5.82 | −2.90 | 20 | 30 | 20 | ||||
| α-1H | Expt | −2.1 | −1.7 | 5.9 | 0.7 | 11 | 63 | 34 | 5.20 | 3.18 | 3.185 |
| DFT | −1.98 | −0.97 | 5.57 | 0.87 | 11 | 63 | 34 | ||||
| o-1HPhen | Expt | −3.0 | −1.20d | 1.20d | −1.0 | 0 | 11 | 0 | 2.20 | 4.36 | 4.001 |
| DFT | 2.47 | −1.31 | −1.19 | −0.01 | −21.0 | 38 | 24 | ||||
Furthermore the crystal structure of [CuII(1)](OTf)2(H2O)2 notably contains two coordinated water molecules in the equatorial position.34 This is also consistent with the current ENDOR data for [Cu(1a)] which reveals the presence of strongly coupled protons which are too large to arise from the ligand, and must therefore originate from bound water molecules. A large coupling of ca. 10 MHz is observed in the experimental spectrum (Fig. 5a; g = g⊥ position) which was not observed in either the homoleptic complex [Cu(1b)] or in the heteroleptic complex formed from the CuIICl2 salt, [Cu(1c)] (Fig. 5b,c; g = g⊥ position). We therefore tried to prepare the [Cu(1a)] complex under rigorous anhydrous and anaerobic conditions, in order to suppress or eliminate the H2O derived peaks from the ENDOR spectra. Although a small suppression was observed, we could not completely eliminate the H2O peaks. This indicates that [CuII(1)](OTf)2 prepared on the bench using commercially available CuII(OTf)2, is always likely to contain some coordinated water in solution.
The large couplings assigned to the bound water molecules in Fig. 5a, were simulated at multiple field positions and the resulting angular selective simulations are given in Fig. 7. Owing to the close proximity of the H2O to the CuII centre (Cu⋯HH2O distance of 2.410 Å from the crystal structure), a large aiso contribution is expected (Table 3). Furthermore, analysis of the experimental hyperfine tensor suggests a Cu⋯HH2O distance of 2.52 Å (Adipolar = 11 MHz), which is in reasonable agreement with the crystal structure.
![Q-band CW 1H ENDOR (10 K) of [CuII(1a)] dissolved in THF–DCM recorded at the field positions (a) 1180.4, (b) 1176.9, (c) 1159.7, (d) 1124.4, (e) 1080.0 and (f) 1030.8 mT. The corresponding simulations are given in a′–h′. Solvent 1 : 1 THF–DCM.](/image/article/2012/DT/c2dt31273e/c2dt31273e-f7.gif) | ||
| Fig. 7 Q-band CW 1H ENDOR (10 K) of [CuII(1a)] dissolved in THF–DCM recorded at the field positions (a) 1180.4, (b) 1176.9, (c) 1159.7, (d) 1124.4, (e) 1080.0 and (f) 1030.8 mT. The corresponding simulations are given in a′–h′. Solvent 1:1 THF–DCM. | ||
The remaining proton couplings observed in the ENDOR spectra (Fig. 5 and 7) arise from the BOX ligand nuclei. In particular the nearest neighbour protons which interact with CuII arise from the α-H at the asymmetric carbon of the BOX ring (labelled * in Scheme 1), with a Cu⋯α-HBOX distance of 3.185 Å, and from the ortho-1H of the phenyl ring, with a Cu⋯o-Hphenyl distance of 4.001 Å. These two protons are most likely responsible for the observed couplings at A1 = 3.0 MHz and 5.9 MHz in Fig. 5a–c. The principal hyperfine values for the α-H are given in Table 3, with an estimated Cu⋯α-HBOX distance of 3.18 Å, in good agreement with the X-ray data. Unfortunately, owing to the overlapping features with the more remote protons from the BOX ligand, a reliable estimation of the A2,3 components of the o-Hphenyl is not possible, hence the large difference in Cu⋯o-Hphenyl distances between the ENDOR data versus the X-ray data (Table 3).
DFT calculations
The spin Hamiltonian parameters were also calculated for the [CuII(1a)] and [CuII(1c)] complex in order to compare to the experimental data. The calculations were performed using the ORCA package37–40 and based on the reported crystal structures of [CuII(1)](OTf)2(H2O)234 and [CuII(1)](Cl)2.14 The relevant EPR parameters are listed in Tables 1 and 3. Current state-of-the-art DFT methods still struggle to reproduce accurately the g and metal hyperfine values for the transition metal ions,47 hence the discrepancy between the experimental and calculated g/CuA values in Table 1. Nevertheless, the general trends are in good agreement with each other. In particular the decrease in g3 and A3 observed experimentally upon complex formation is satisfactorily reproduced in the computations. Indeed the structure of the [CuII(1)](Cl)2 complex used in the calculation had a slight twisted arrangement around the Cu-N2Cl2 plane, in agreement with the earlier EPR observations.Ligand hyperfine parameters are more reliably determined by DFT, particularly for weakly coupled protons (Table 3). As expected the coordinated H2O molecules are predicted to produce the largest couplings, and these values are in good agreement with the experimental ENDOR data. The α-H and ortho-phenyl protons of the BOX ligand also produce appreciable hyperfine couplings (Table 3). Although the A1,2 couplings of these protons could not be confidently extracted from the powder ENDOR spectra, the largest calculated A1 component agrees well with the experimental values (Table 3).
Role of the counterion in CuIIBOX complexes
As stated earlier, the CuIIbis(oxazoline) complexes are used in a variety of different asymmetric reactions, including the Diels–Alder reaction,7–16 cyclopropanation,17–20 and aziridination.21–23 In most cases, the metal based catalysts are prepared in situ by mixing the metal salt and BOX ligands prior to catalysis. Depending on the type and amount of BOX ligand used, and the nature of the counterion, this may result in the formation of a heteroleptic or homoleptic complex, such as [CuII(1a)] or [CuII(1b)] respectively, and this has important implications in catalysis. An excess of the BOX ligand clearly increases the likelihood of the formation of a homoleptic complex. However, as the current results show, this is heavily dependent on the choice of counterion. The more labile TfO− facilitates the formation of [CuII(1b)] when BOX is present in excess (Cu–BOX ratio 1:6), whereas the Cl− prevents this from occurring; only the heteroleptic complex [CuII(1c)] is observed under analogous preparative conditions.
The change in catalytic activity between the hetero- and homoleptic CuIIBOX complexes have been explored by Hager et al.48 Competitive experiments were performed to monitor the yields and ee's, which demonstrated that the homoleptic complex was catalytically inactive. These complexes therefore required prolonged reaction times, and significantly lower yields were observed in these cases. Indeed Le Roux et al.,49 highlighted the necessity for controlled synthesis conditions (slow addition of ligand to metal, under dilute metal concentrations) to prevent the formation of homoleptic complexes. Attempts have therefore been made to increase the steric bulk of the bis(oxazoline) ligand in order to prevent formation of the homoleptic species,49 whereas the current work reveals a change in counterion may also achieve a similar result.
Currently most of the catalysis work involving Lewis metal based bis(oxazoline) complexes have utilised OTf− (OTf = CF3SO3) or SbF6 counterions. For enantioselective aziridination using CuIIbis(oxazoline), Evans et al.,21 reported that the CuIICl2 and CuIIBr2 salts were prohibitively slow with poor enantioselectivities, so that a highly electronegative counterion is a design prerequisite for efficient asymmetric catalysis. Fraile et al.,26 postulated that the choice of counterion can affect the nature of the reaction mechanism, leading to undesired side-reactions that are non-asymmetric resulting in lower ee's. Furthermore, it is well known that the geometry of the CuIIBOX complex, which is heavily dependent on the counterion, affects the catalysis.50 For example, when triflate is utilized as the counterion the X-ray crystal structure of the resulting CuIIBOX complex reveals a Jahn–Teller distorted octahedral complex with TfO− coordinated in axial positions and water coordinated in the equatorial plane. By contrast, when CuIICl2 is employed as the starting salt, the resulting complex exhibits a distorted square-planar geometry with two chloride counterions coordinated to the metal center at ∼33° out of the copper-ligand plane. Furthermore, the presence of the coordinating water molecules affects the direction of approach of substrates to the metal center, resulting in differences in enantioselectivity. As the current results reveal, these changes to the CuIIBOX complexes induced by the different counterions can be examined by EPR and ENDOR techniques.
Bolm et al.,32,33 previously reported the influence of the counterion and choice of starting metal salt for Diels–Alder reactions using the structurally similar CuIIbis(sulfoximine) complexes. The EPR spectra of the copper-bissulfoximine complexes showed significant differences depending on the starting CuII-salt (CuCl2, CuBr2, Cu(OTf)2 and CuCl2–AgSbF6). Upon subsequent addition of a substrate molecule, (N-(1-oxoprop-2-en-1-yl)oxazolidin-2-one, changes were observed in the spectra, with distinct behaviour noted for the different counterions showing again how EPR can be successfully used to monitor such reactions. We are currently studying the mechanistic details involving these homo- and heteroleptic BOX complexes with CuCl2, Cu(OTf)2 and CuCl2–AgSbF6 for asymmetric aziridination and Diels–Alder reactions.
Conclusions
In the current study we have presented a detailed EPR and ENDOR investigation of a series of heteroleptic and homoleptic copper bis(oxazoline) complexes, [CuII(1a–c)]. The geometry of the hetereoleptic complexes [CuII(1a)] and [CuII(1c)] is dependent on the choice of counterion used in the synthesis, since different g/CuA parameters are observed by EPR. Since the geometry is closely linked to the resulting catalytic activity, this work reveals the potential of EPR to study such complexes in solution. The homoleptic complex [CuII(1b)] was only formed using an excess of the BOX ligand (1) in the presence of the CuII(OTf)2 salt; the CuIICl2 salt prevented this from occurring. The hyperfine technique of ENDOR enabled the hyperfine and quadrupole parameters of the surrounding nuclei to be determined. Significant differences were observed in the NA values for [CuII(1a)] and [CuII(1c)], consistent with the more distorted arrangement in the latter complex, whereas smaller NA and NP values were detected for [Cu(1b)] attributed to the redistributed spin density in the homoleptic complex. Well resolved 19F couplings in [CuII(1a)] confirmed the presence of coordinated TfO− counterions along the axial direction, while strong 1H couplings from bound water molecules along the equatorial direction were also observed for this complex. These results reveal how the inner and indeed outersphere coordination environment of CuIIBOX complexes, of relevance to catalysis, can be studied by EPR and ENDOR in the ‘solvated’ environment where counterion effects are still manifested.Acknowledgements
Funding from EPSRC (EP/H023879) is gratefully acknowledged.References
- T. Katsuki and K. B. Sharpless, J. Am. Chem. Soc., 1980, 102, 5974–5976 CrossRef CAS
.
- M. Movassaghi and E. N. Jacobsen, Science, 2002, 298, 1904–1905 CrossRef CAS
- T. P. Yoon and E. N. Jacobsen, Science, 2003, 299, 1691–1693 CrossRef CAS
-
P. J. Walsh and M. C. Kozlowsk, Fundamentals of Asymmetric Catalysis, University Science Books, 2009 Search PubMed
- A. K. Ghosh, P. Mathivanan and J. Cappiello, Tetrahedron: Asymmetry, 1998, 9, 1–45 CrossRef CAS
- J. M. Fraile, J. I. García and J. A. Mayoral, Coord. Chem. Rev., 2008, 252, 624–646 CrossRef CAS
- D. A. Evans, S. J. Miller and T. Lectka, J. Am. Chem. Soc., 1993, 115, 6460–6461 CrossRef CAS
- M. Johannsen and K. Anker Jørgensen, J. Chem. Soc., Perkin Trans. 2, 1997, 1183–1186 RSC
- D. A. Evans, S. J. Miller, T. Lectka and P. von Matt, J. Am. Chem. Soc., 1999, 121, 7559–7573 CrossRef CAS
- D. A. Evans, D. M. Barnes, J. S. Johnson, T. Lectka, P. von Matt, S. J. Miller, J. A. Murry, R. D. Norcross, E. A. Shaughnessy and K. R. Campos, J. Am. Chem. Soc., 1999, 121, 7582–7594 CrossRef CAS
- D. A. Evans, J. S. Johnson, C. S. Burgey and K. R. Campos, Tetrahedron Lett., 1999, 40, 2879–2882 CrossRef CAS
- D. A. Evans, J. S. Johnson and E. J. Olhava, J. Am. Chem. Soc., 2000, 122, 1635–1649 CrossRef CAS
- V. K. Aggarwal, D. E. Jones and A. M. Martin-Castro, Eur. J. Org. Chem., 2000, 2939–2945 CrossRef CAS
- J. Thornhauge, M. Roberson, R. G. Hazell and K. A. Jorgensen, Chem.–Eur. J., 2002, 8, 1888–1898 CrossRef
- A. Landa, B. Richter, R. L. Johansen, A. Minkkilä and K. A. Jørgensen, J. Org. Chem., 2007, 72, 240–245 CrossRef CAS
- S. Tanaka, M. Tada and Y. Iwasawa, J. Catal., 2007, 245, 173–183 CrossRef CAS
- R. E. Lowenthal, A. Abiko and S. Masamune, Tetrahedron Lett., 1990, 31, 6005–6008 CrossRef CAS
- D. A. Evans, K. A. Woerpel, M. M. Hinman and M. M. Faul, J. Am. Chem. Soc., 1991, 113, 726–728 CrossRef CAS
- A. M. Harm, J. G. Knight and G. Stemp, Tetrahedron Lett., 1996, 37, 6189–6192 CrossRef CAS
- M. J. Fernández, J. M. Fraile, J. I. García, J. A. Mayoral, M. I. Burguete, E. García-Verdugo, S. V. Luis and M. A. Harmer, Top. Catal., 2000, 13, 303–309 CrossRef
- D. A. Evans, M. M. Faul, M. T. Bilodeau, B. A. Anderson and D. M. Barnes, J. Am. Chem. Soc., 1993, 115, 5328–5329 CrossRef CAS
- C. Langham, P. Piaggio, P. McMorn, D. J. Willock, G. J. Hutchings, D. Bethell, D. F. Lee, P. C. Bulman Page, C. Sly, F. E. Hancock and F. King, Chem. Commun., 1998, 1601–1602 RSC
- C. Langham, D. Bethell, D. F. Lee, P. McMorn, P. C. Bulman Page, D. J. Willock, C. Sly, F. E. Hancock, F. King and G. J. Hutchings, Appl. Catal., A, 1999, 182, 85–89 CrossRef
- G. Desimoni, G. Faita and K. A. Jørgensen, Chem. Rev., 2006, 106, 3561–3651 CrossRef CAS
- H. A. McManus and P. J. Guiry, Chem. Rev., 2004, 104, 4151–4202 CrossRef CAS
- J. M. Fraile, J. I. García, J. A. Mayoral and T. Tarnai, J. Mol. Catal. A: Chem., 1999, 144, 85–89 CrossRef CAS
- D. A. Evans, J. A. Murry, P. von Matt, R. D. Norcross and S. J. Miller, Angew. Chem., Int. Ed. Engl., 1995, 34, 798–800 CrossRef CAS
- D. A. Evans, M. C. Kozlowski, C. S. Burgey and D. W. C. MacMillan, J. Am. Chem. Soc., 1997, 119, 7893–7894 CrossRef CAS
- P. J. Alonso, J. M. Fraile, J. García, J. I. García, J. I. Martínez, J. A. Mayoral and M. C. Sánchez, Langmuir, 2000, 16, 5607–5612 CrossRef CAS
- Y. Traa, D. M. Murphy, R. D. Farley and G. J. Hutchings, Phys. Chem. Chem. Phys., 2001, 3, 1073–1080 RSC
- V. Caplar, Z. Raza, M. Roje, V. Tomisic, G. Horvat, J. Pozar, I. Piantanida and M. Zinic, Tetrahedron, 2004, 60, 8079–8087 CrossRef CAS
- C. Bolm, M. Martin, G. Gescheidt, C. Palivan, D. Neshchadin, H. Bertagnolli, M. Feth, A. Schweiger, G. Mitrikas and J. Harmer, J. Am. Chem. Soc., 2003, 125, 6222–6227 CrossRef CAS
- C. Bolm, M. Martin, G. Gescheidt, C. Palivan, T. Stanoeva, H. Bertagnolli, M. Feth, A. Schweiger, G. Mitrikas and J. Harmer, Chem.–Eur. J., 2007, 13, 1842–1850 CrossRef CAS
- D. A. Evans, C. S. Burgey, N. A. Paras, T. Vojkovsky and S. W. Tregay, J. Am. Chem. Soc., 1998, 120, 5824–5825 CrossRef CAS
- T. Spalek, P. Pietrzyk and Z. Sojka, J. Chem. Inf. Model., 2005, 45, 18–29 CrossRef CAS
- S. Stoll and A. Schweiger, J. Magn. Reson., 2006, 178, 42–55 CrossRef CAS
- F. Neese, J. Chem. Phys., 2001, 115, 11080–11096 CrossRef CAS
- F. Neese, J. Phys. Chem. A, 2001, 105, 4290–4299 CrossRef CAS
- F. Neese, J. Chem. Phys., 2003, 118, 3939–3948 CrossRef CAS
- F. Neese, J. Chem. Phys., 2005, 122, 034107–034113 CrossRef
- This basis is based on the TurboMole DZ basis developed by Ahlrichs and co-workers and obtained from the basis set library under ftp.chemie.uni-karlsruhe.de/pub/basen, R. Ahlrichs and co-workers (unpublished).
-
V. Barone, Recent Advances in Density Functional Methods, World Scientific Publ. Co., Singapore, 1996 Search PubMed
- H. R. Gersmann and J. D. Swalen, J. Chem. Phys., 1962, 36, 3221–3233 CrossRef CAS
- N. Niklas and R. Alsfasser, Dalton Trans., 2006, 3188–3199 RSC
- W. E. B. J. Peisach, Arch. Biochem. Biophys., 1974, 165, 691–708 CrossRef
-
J. R. Pilbrow, Transition Ion Electron Paramagnetic Resonance, Oxford Science Publications, Oxford University Press, 1st edn, 1990 Search PubMed
- F. Neese, JBIC, J. Biol. Inorg. Chem., 2006, 11, 702–711 CrossRef CAS
- M. Hager, S. Wittmann, A. Schätz, F. Pein, P. Kreitmeier and O. Reiser, Tetrahedron: Asymmetry, 2010, 21, 1194–1198 CrossRef CAS
- E. Le Roux, N. Merle and K. W. Tornroos, Dalton Trans., 2011, 40, 1768–1777 RSC
- K. A. Jørgensen, M. Johannsen, S. Yao, H. Audrain and J. Thorhauge, Acc. Chem. Res., 1999, 32, 605–613 CrossRef
- D. M. Murphy, I. Caretti, E. Carter, I. A. Fallis, M. C. Göbel, J. Landon, S. V. Doorslaer and D. J. Willock, Inorg. Chem., 2011, 50, 6944–6955 CrossRef CAS
- C. Finazzo, C. Calle, S. Stoll, S. Van Doorslaer and A. Schweiger, Phys. Chem. Chem. Phys., 2006, 8, 1942–1953 RSC
Footnote |
| † Electronic supplementary information (ESI) available: X-band EPR of CuCl2:BOX, Q-band 14N ENDOR of [Cu(1b)], Q-band 14N ENDOR of [Cu(1c)]. See DOI: 10.1039/c2dt31273e |
| This journal is © The Royal Society of Chemistry 2012 |