
Layered inorganic–organic frameworks based on the 2,2-dimethylsuccinate ligand: structural diversity and its effect on nanosheet exfoliation and magnetic properties†
Abstract
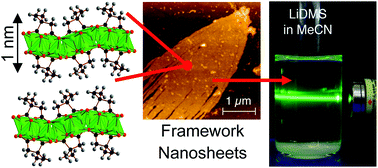
The structures of four new 2,2-dimethylsuccinate frameworks suitable for exfoliation into
- This article is part of the themed collections: Spotlight Collection: 2D Materials Chemistry and In celebration of Tony Cheetham’s 70th birthday
 Please wait while we load your content...
Please wait while we load your content...

