 Open Access Article
Open Access ArticleSynthesis, Cu(II) complexation, 64Cu-labeling and biological evaluation of cross-bridged cyclam chelators with phosphonate pendant arms†
Riccardo
Ferdani
a,
Dannon J.
Stigers
b,
Ashley L.
Fiamengo
a,
Lihui
Wei
a,
Barbara T. Y.
Li
b,
James A.
Golen
c,
Arnold L.
Rheingold
d,
Gary R.
Weisman
*be,
Edward H.
Wong
*bf and
Carolyn J.
Anderson
*agh
aMallinckrodt Institute of Radiology, Washington University School of Medicine, St. Louis, Missouri, 63110, USA
bDepartment of Chemistry, University of New Hampshire, Durham, New Hampshire, 03824, USA
cDepartment of Chemistry and Biochemistry, University of Massachusetts, Dartmouth, North Dartmouth, Massachusetts, 02747, USA
dDepartment of Chemistry and Biochemistry, University of California, San Diego, La Jolla, California, 92093, USA
eE-mail: gary.weisman@unh.edu
fE-mail: ehw@unh.edu
gDepartment of Biochemistry and Molecular Biophysics; Department of Chemistry, Washington University, St. Louis, Missouri, 63110, USA
hCurrent address: Department of Radiology, University of Pittsburgh, Pittsburgh, PA 15219, USA. E-mail: andersoncj@upmc.edu
First published on 15th December 2011
Abstract
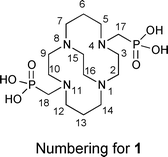
A new class of cross-bridged cyclam-based macrocycles featuring phosphonate pendant groups has been developed. 1,4,8,11-tetraazacyclotetradecane-1,8-di(methanephosphonic acid) (CB-TE2P, 1) and 1,4,8,11-tetraazacyclotetradecane-1-(methanephosphonic acid)-8-(methanecarboxylic acid) (CB-TE1A1P, 2) have been synthesized and have been shown to readily form neutral copper(II) complexes at room temperature as the corresponding dianions. Both complexes showed high kinetic inertness to demetallation and crystal structures confirmed complete encapsulation of copper(II) ion within each macrocycle's cleft-like structure. Unprecedented for cross-bridged cyclam derivatives, both CB-TE2P (1) and CB-TE1A1P (2) can be radiolabeled with 64Cu at room temperature in less than 1 h with specific activities >1 mCi μg−1. The in vivo behavior of both 64Cu-CB-TE2P and 64Cu-CB-TE1A1P were investigated through biodistribution studies using healthy male Lewis rats. Both new compounds showed rapid clearance with similar or lower accumulation in non-target organs/tissues when compared to other copperchelators including CB-TE2A, NOTA and Diamsar.
Introduction
The quest for an optimal bifunctional chelator (BFC) continues to be one major goal in the research and development of metal-based radiopharmaceuticals for clinical imaging and therapeutic applications. An ideal BFC should bind the targeted radiometal with high specificity, yield, and with alacrity. Further, the resulting complex should be both thermodynamically stable and, more importantly, kinetically inert to in vivo transchelation or transmetallation processes. Copper-64 (T1/2 = 12.7 h; β+: 0.656 MeV, 17.8%; β−: 0.573 MeV, 38.4%) is a promising radiometal with potential for applications in diagnostic imaging (positron emission tomography (PET)) and targeted radiotherapy.1–3 It can also be produced in high yield and specific activity from a medical cyclotron.4 Targeting 64Cu(II), a variety of acyclic, macrocyclic, as well as macrobicyclic polyamine BFC's have been studied and reported.2,5–10 Kinetic stability of Cu(II) complexes has been shown to be more predictive of in vivo stability than thermodynamic stability.11 Macrocyclic chelators of Cu(II) such as TETA (1,4,8,11-tetraazacyclotetradecane-1,4,8,11-tetraacetic acid) demonstrated higher kinetic inertness and thus in vivo stability relative to acyclic chelators such as EDTA (ethylenediamine tetraacetic acid).12,13 However, biodistribution and metabolism studies of 64Cu-labeled TETA and TETA-biological molecule conjugates have revealed significant transchelation of 64Cu to superoxide dismutase and metallothionein in liver and albumin in blood resulting in high background radioactivity.14,15We have previously developed a dicarboxymethyl pendant-armed cyclam macrocycle featuring an ethylene cross-bridge, CB-TE2A (4,11-bis(carboxymethyl)-1,4,8,11-tetraazabicyclo[6.6.2]hexadecane; Fig. 1),16,17 and shown that it possesses many of the desirable attributes to be an effective BFC for 64Cu(II).‡ The resulting radiolabeled complex18 has been found to exhibit significantly improved in vivo behavior compared to non-cross-bridged analogs like TETA.15,17,19–21 We hypothesize that both its kinetic inertness to 64Cu(II) loss and resistance to biological reduction contribute to these enhanced properties. CB-TE2A9,17,20,22–25 and derivatives26–29 have been successfully bioconjugated to targeting peptides and investigated by us and other research groups.
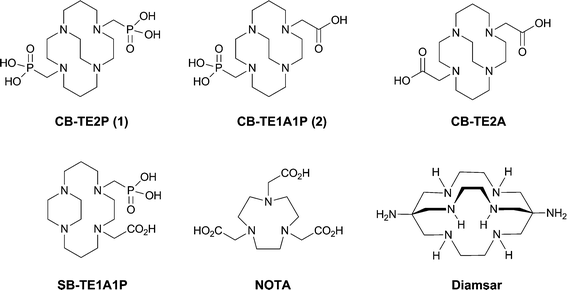 | ||
| Fig. 1 Structures of ligands studied or discussed. | ||
A less-than-optimal aspect of the useful BFCCB-TE2A lies in the rather sluggish radio-copper binding kinetics necessitating relatively harsh labeling conditions (95 °C heating for 1–1.5 h) which can handicap its bioconjugation to more fragile targeting moieties. To overcome this limitation, we have developed a second generation of cross-bridged chelators incorporating methanephosphonic acid pendant arms. A number of phosphonate pendant-armed tetraazamacrocyclic chelators have been studied previously.30–46 Substitution of methanephosphonic acid pendant arms for acetic acid functional groups has resulted in complexes having higher selectivity for Cu(II) and an increased thermodynamic and kinetic stability.47 Radiochemistry, in vitro and in vivo stability, and biodistribution studies were reported for 64Cu-labeled cyclen derivatives having 2, 3 or 4 methanephosphonate pendant arms (DO2P, DO3P and DOTP, respectively).48Cu(II)-DO2P was found to have a very high formation constant (logK = 28.7), while its 64Cu-complex exhibited high in vitro stability in rat serum and a very rapid disappearance from the blood, liver and kidney. A higher bone uptake for 64Cu-DO3P and 64Cu-DOTP was observed, most likely due to the fact that methanephosphonate sidearms are known to have great affinity for the hydroxyapatite in the bone.49–52 A side-bridged cyclam derivative with mixed phosphonate/acetate pendant arms, SB-TE1A1P (Fig. 1), has also been reported, though heating was still required for high 64Cu-labeling yields.53 A related side-bridged phosphonate BFC precursor has also been reported.54
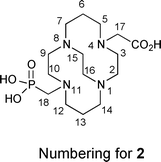
Recently, we communicated in this journal the synthesis and characterization of the first methanephosphonic acid derivative of cross-bridged cyclam, CB-TE2P (1,4,8,11-tetraazacyclotetradecane-1,8-di(methanephosphonic acid), Fig. 1), its Cu(II) complex Cu-CB-TE2P, and 64Cu(II)labeling and biodistribution studies.55 Pleasingly, the anticipated acceleration of Cu(II) complexation was realized. However, the actual use of CB-TE2P as a BFC using conventional bioconjugation protocols is less straightforward. We have now prepared a mixed-armed variation, CB-TE1A1P (1,4,8,11-tetraazacyclotetradecane-1-(methanephosphonic acid)-8-(methanecarboxylic acid), Fig. 1), featuring both a methanephosphonic acid as well as a carboxymethyl pendant arm. We surmise that the former pendant arm will enhance the Cu(II) binding rate while the latter provides a scaffold to facilitate bioconjugation. In this report, we detail the synthesis and characterization of both CB-TE2P and CB-TE1A1P, and their respective copper(II) complexes, Cu-CB-TE2P and Cu-CB-TE1A1P.56 Their 64Cu-labeling along with biodistribution studies have also been carried out for comparison with other widely used BFC's including CB-TE2A, NOTA (1,4,7-triazacyclononane-1,4,7-triacetic acid), and Diamsar (1,8-diamino-3,6,10,13,16,19-hexaazabicyclo[6.6.6]icosane) (Fig. 1).
Results and discussion
Ligand synthesis and characterization
As previously communicated,55CB-TE2P (1) was synthesized in two steps from cross-bridged cyclam 316 (Scheme 1(a)). 3 was first converted to bis-diethylphosphonate4 by a variant of the Kabachnik–Fields three-component reaction57–61 and this synthetic intermediate was then hydrolyzed to CB-TE2P. Thus, reaction of 3 with triethylphosphite and paraformaldehyde in anhydrous THF under nitrogen at room temperature for 4 days gave pure 4 in 94% yield after extractive workup. 4 was hydrolyzed in 6 M HCl under reflux for 24 h to give 1 in 64% yield as a hydrochloride after purification by ion exchange chromatography.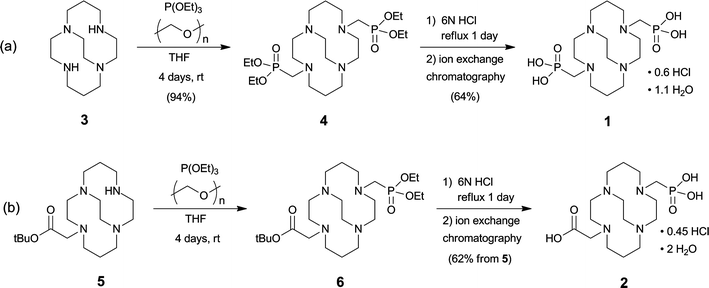 | ||
| Scheme 1 | ||
NMR spectroscopic results for this salt are similar to NMR spectra of previously studied di-inside-protonated salts of cross-bridged ligands62 and are consistent with a C2-symmetric diamond-lattice [2323]/[2323] conformation16,63,64 for the bicyclic backbone in solution, which is the conformation that has been observed in the solid state for such cross-bridged salts.62 In the 1H NMR spectrum, every geminal pair of hydrogens is diastereotopic and anisochronous and exhibits the coupling patterns and coupling constants expected of an equatorial/axial pair of hydrogens in a diamond-lattice arrangement (see the Experimental for detailed interpretation and the ESI for spectra†). However, neither a related non-diamond-lattice [2233]/[2233] conformation, which differs from the former by a change in sign of the cross-bridge N–C–C–N torsion angle, nor a time-averaged dynamic mixture of these two conformations in solution can be rigorously excluded as possibilities. Nevertheless, we were surprised when an X-ray crystal structure of 1·HCl·4H2O (Fig. 2), reported in our preliminary communication, showed the ligand adopting a [2233]/[2233] conformation rather than the expected [2323]/[2323] diamond-lattice conformation. A possible explanation for this observation lies in the fact that net-positively-charged di-inside-protonated ligand possesses one phosphonic acid arm and one phosphonate arm and these are shown to be intramolecularly O–H⋯O hydrogen bonded (disordered) in the solid state. This conformation of the ligand backbone may be necessary in order to facilitate the appropriate arm-arm distance for the intramolecular H-bond. In aqueous solution, solvation of phosphonate arms would likely dominate over arm-arm intramolecular H-bonding, so the diamond-lattice backbone conformation may be favored in solution.
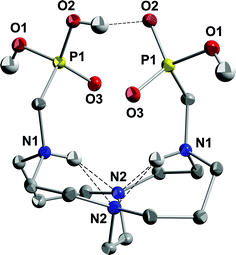 | ||
| Fig. 2 X-ray structure of 1·HCl·4H2O (50% thermal ellipsoids for all non-hydrogen atoms). Intramolecular hydrogen bonds are shown as dashed lines. One of the two half-occupancy symmetry-related hydrogens on O2 has been removed for clarity so that the H-bond can be shown. | ||
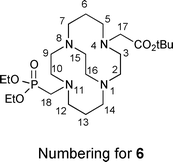
CB-TE1A1P (2) was synthesized similarly in two steps from the previously reported unsymmetrical synthetic intermediate CB-TE1A tert-butyl ester 5 (Scheme 1(b)).195 was reacted with triethylphosphite and paraformaldehyde in anhydrous THF for 4 days at room temperature to give, after an extractive workup, the corresponding phosphonate ester 6, which was taken on to the next step without further purification. Hydrolysis of both the diethylphosphonate and tert-butyl carboxylic ester moieties of 6 was accomplished in refluxing 6 M HCl to give 2 in 62% overall (two step) yield from 5 as a hydrochloride salt after purification by ion exchange chromatography (Scheme 1(b)). This salt can be further purified if necessary by reversed-phase HPLC (C18; 0.1% TFA in H2O/0.1% TFA in MeCN) to give 2 as a di-TFA salt. The detailed NMR characterization reported in the Experimental section is of 2·2.5TFA. As was the case for the salt of 1, the NMR spectroscopic results for 2·2.5TFA are indicative of a di-inside-protonated species having a diamond-lattice [2323]/[2323] conformation in solution (see the Experimental for detailed interpretation and the ESI for spectra†). In the 1H spectrum, each geminal pair of hydrogens is diastereotopic and anisochronous, exhibiting the characteristic coupling patterns consistent with the diamond-lattice [2323]/[2323] geometry, although neither the [2233]/[2233] nor a time average of the two can be excluded. Both the 1H and 13C{1H} NMR spectra exhibit the expected 31P couplings and point to the expected lack of symmetry. The 31P{1H} spectrum exhibits one signal.
Synthesis and characterization of copper(II) complexes of CB-TE2P and CB-TE1A1P
The Cu(II) complex of CB-TE2P was initially prepared from CuCl2 and the ligand in refluxing methanol with the pH adjusted to 8 using aqueous NaOH. Subsequently, it was found that complexation in methanol was already complete in less than 5 min at ambient temperature. The blue complex has a d-d absorption maximum at λmax (MeOH)/nm 639 (ε/dm3 mol−1 cm−1 35). Its cyclic voltammogram in 0.1 M aq. NaOAc exhibited a quasi-reversible reduction at −0.96 V (Ag/AgCl), typical of Cu(II) cross-bridged cyclam complexes. Its acid inertness was assayed under pseudo first-order conditions in 5 M HCl at 90 °C and found to have a half life of 3.8(1) h. While this is much less impressive than the 154 h previously reported for Cu-CB-TE2A, it is nonetheless significantly more inert than both Cu-DOTA and Cu-TETA which completely decomplexed within minutes.As desired, the complexation of copper(II) by CB-TE1A1P also proceeded readily at room temperature even in acidic aqueous solution. The isolated dark-blue complex exhibited a carboxylate stretching band at 1619 cm−1 in its IR spectrum and a d–d band maximum at 613 nm (ε/dm3 mol−1 cm−1 24) in its electronic spectrum. In contrast to previously reported cross-bridged cyclam copper(II) complexes including Cu-CB-TE2P, the electrochemical reduction of Cu-CB-TE1A1P has been found to be irreversible with a peak potential at around −1.0 V (Ag/AgCl) accompanied by a large copper stripping peak in the reverse scan. Acid inertness studies of the complex in 5 M HCl at 90 °C gave a half-life of 6.8(1) h which is comparable to Cu-CB-TE2P though both are significantly less acid-resistant than Cu-CB-TE2A (half-life = 154(6) h).
X-ray structures of Cu-CB-TE2P and Cu-CB-TE1A1P
The X-ray crystal structure of Cu-CB-TE2P55 is shown in Fig. 3. Full envelopment of the cation within the ligand's N4O2 donor set results in a distorted octahedral coordination mode. Observed elongated bond lengths of 2.20 Å for N(4)–Cu(1) and 2.45 Å for Cu(1)–O(6) designate the expected Jahn–Teller distortion axis. Further, an N(1)–Cu(1)–N(3) bond angle of 174.8° confirms a good fit of the cation inside the chelator cavity. This can be compared to the 177.5° for the analogous angle in the Cu-CB-TE2A structure. Interestingly, here the ligand adopts a [2233]/[2233] conformation rather than the distorted diamond-lattice [2323]/[2323] conformation more commonly seen in reported Cu(II) complexes of cross-bridged cyclams, including Cu-CB-TE2A.16,19,65–68 However, the [2233]/[2233] conformation has also been found in the structure of monoprotonated Cu-CB-TE2A (as a perchlorate salt)11 and in a Cu(II) complex of the diamide analog of CB-TE2A.69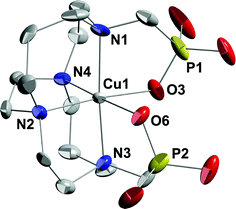 | ||
| Fig. 3 X-ray structure of Cu-CB-TE2P (50% thermal ellipsoids for all non-hydrogen atoms). Only one of two independent complexes is shown. Both exhibit distorted octahedral coordination at Cu and have the same ligand conformation. Hydrogens, sodium and chloride ions, and solvent molecules have been removed for clarity. | ||
The X-ray structure of Cu-CB-TE1A1P also has the expected full six-coordinate N4O2 envelopment of the cation with the methylphosphonate arm monoprotonated to give a charge-neutral complex overall (Fig. 4). The Jahn–Teller elongation here is found along the O(1)–Cu(1)–N(2) axis with the phosphonate Cu–O 2.544(3) Å and Cu–N 2.220(3) Å. In comparison, the Cu(1)–O(4) bond to the carboxylate oxygen is much shorter at 1.993(2) Å. It should be noted that each phosphonate P(O)OH moiety is hydrogen bonded to the P(O)OH of another complex in a dimeric structure, so it may be that the weaker phosphonate coordination and apparent Jahn–Teller elongation is partly a consequence of this competing hydrogen bonding. An observed exo N(1)–Cu(1)–N(3) angle of 175.0(1)° indicates that the cation is slightly sunken into the ligand cleft. Again, the ligand here adopts the [2233]/[2233] conformation suggesting that methanephosphonate pendant arms, with their longer P–C and P–O bonds compared to the C–C and C–O bonds of acetate arms, may dictate this optimum cross-bridged cyclam conformation for Cu(II) binding. Alternatively, the conformations may be similar enough in energy that solvation or packing effects could determine the preference.
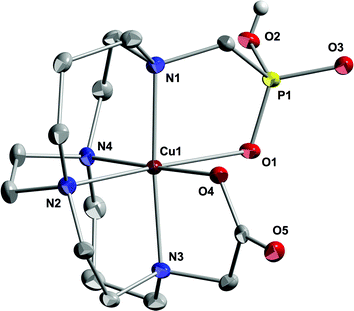 | ||
| Fig. 4 X-ray structure of Cu-CB-TE1A1P (50% thermal ellipsoids for all non-hydrogen atoms). Hydrogens other than the phosphonatehydrogen and solvent molecules have been removed for clarity. | ||
Significant bonding data for both complexes are listed for comparison in Table 1. To sum up the structural results, both CB-TE2P and CB-TE1A1P have been shown to readily form six-coordinate copper(II) complexes at ambient temperature resulting in full envelopment of the cation by the respective chelator. Each of these complexes adopts the more unusual [2233]/[2233] conformation in the solid state. Our assays of these products further confirmed that both should be sufficiently inert to acid decomplexation and resistant to reduction by common biological reductants.
| Cu-CB-TE2P | Cu-CB-TE1A1P | ||
|---|---|---|---|
| Bond distances in Å and bond angles in (°) | |||
| (Numbers in parentheses refer to estimated standard deviations) | |||
| Cu(1)–O(6) | 2.450(5) | Cu(1)–O(1) | 2.544(3) |
| Cu(1)–O(3) | 1.998(5) | Cu(1)–O(4) | 1.993(2) |
| Cu(1)–N(1) | 2.087(6) | Cu(1)–N(1) | 2.053(3) |
| Cu(1)–N(2) | 2.035(7) | Cu(1)–N(2) | 2.220 (3) |
| Cu(1)–N(3) | 2.041(6) | Cu(1)–N(3) | 2.078(3) |
| Cu(1)–N(4) | 2.205(7) | Cu(1)–N(4) | 2.032(3) |
| O(3)–Cu(1)–N(2) | 174.4(2) | O(4)–Cu(1)–N(4) | 178.5(1) |
| O(3)–Cu(1)–N(2) | 88.8(2) | O(4)–Cu(1)–N(1) | 92.4(1) |
| N(2)–Cu(1)–N(3) | 85.6(3) | N(4)–Cu(1)–N(1) | 86.3(1) |
| O(3)–Cu(1)–N(1) | 87.5(2) | O(4)–Cu(1)–N(3) | 83.5(1) |
| N(2)–Cu(1)–N(1) | 98.1(3) | N(4)–Cu(1)–N(3) | 97.9(1) |
| N(3)–Cu(1)–N(1) | 174.8(3) | N(1)–Cu(1)–N(3) | 175.0(1) |
| O(3)–Cu(1)–N(4) | 94.5(2) | O(4)–Cu(1)–N(2) | 93.1(1) |
| N(4)–Cu(1)–N(2) | 86.2(3) | N(4)–Cu(1)–N(2) | 86.3(1) |
| N(3)–Cu(1)–N(4) | 99.7(2) | N(1)–Cu(1)–N(2) | 99.1(1) |
| N(1)–Cu(1)–N(4) | 84.3(2) | N(3)–Cu(1)–N(2) | 83.9(1) |
Density Functional Theory (DFT) MO calculations on Cu-CB-TE2A, Cu-CB-TE2P and Cu-CB-TE1A1P
The structural results (vide supra) led us to investigate whether the preference of Cu-CB-TE2P and Cu-CB-TE1A1P for the [2233]/[2233] conformation over the more commonly observed [2323]/[2323] conformation observed in Cu-CB-TE2A and most other cross-bridged cyclam metal complexes is a result of fundamental geometric and electronic factors within each coordination complex, or whether the energies of the conformations are so similar that differences in solvation and packing might logically be determining solid-state conformation. While the conformational change is largely associated with the organic portion of the coordination complex, we felt that a molecular mechanics (force field) approach would not properly account for differences in copper coordination and so proceeded to carry out density functional theory (DFT) calculations (gas phase) on the [2233]/[2233] and [2323]/[2323] conformations of Cu-CB-TE2P, Cu-CB-TE1A1P and Cu-CB-TE2A using basis sets as large as practical considering the size of these coordination complexes. Two density functionals were employed, the very commonly used B3LYP70–74 and the M06 functional of Truhlar,75,76 which has been shown to give better results for metal complexes.77 In each case, the all-electron 6-31G* basis set was employed and geometry optimization was carried out (E, kcal mol−1). Furthermore, in each case a frequency calculation was carried out so that zero-point energy (ZPE) corrections could be made (Ecorr, kcal mol−1) and free energies (G, kcal mol−1 at 298 K) could be calculated. In the case of the M06 functional, single-point calculations at the M06/6-31+G**//M06/6-31G* level were also carried out for both conformations of all three complexes. The results (energies in kcal mol−1) are summarized in Table 2.| a ZPE-corrected | |||
|---|---|---|---|
| Cu-CB-TE2P | |||
| B3LYP/6-31G*//B3LYP/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 0 | 0 | 0.18 |
| [2323]/[2323] | 0.49 | 0.01 | 0 |
| M06/6-31G*//M06/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 0 | 0 | 0 |
| [2323]/[2323] | 0.92 | 0.70 | 0.50 |
| M06/6-31+G**//M06/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 0 | — | — |
| [2323]/[2323] | 0.86 | — | — |
| Cu-CB-TE1A1P | |||
| B3LYP/6-31G*//B3LYP/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G(298 K) |
| [2233]/[2233] | 0.31 | 0.07 | 0.06 |
| [2323]/[2323] | 0 | 0 | 0 |
| M06/6-31G*//M06/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 0 | 0 | 0 |
| [2323]/[2323] | 0.53 | 0.70 | 0.69 |
| M06/6-31+G**//M06/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 0 | — | — |
| [2323]/[2323] | 0.67 | — | — |
| Cu-CB-TE2A | |||
| B3LYP/6-31G*//B3LYP/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 1.36 | 1.55 | 1.68 |
| [2323]/[2323] | 0 | 0 | 0 |
| M06/6-31G*//M06/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 1.16 | 0.58 | 0.60 |
| [2323]/[2323] | 0 | 0 | 0 |
| M06/6-31+G**//M06/6-31G*: | Relative energies (kcal mol−1) | ||
| Conformer | E | E corr a | G (298 K) |
| [2233]/[2233] | 0.68 | — | — |
| [2323]/[2323] | 0 | — | — |
Inspection of the relative (gas phase) ZPE-corrected energies and free energies in Table 2 show that the calculations using the more reliable M06 functional qualitatively reproduce the relative conformational energies implied by the crystallographic results. The M06/6-31G*//M06/6-31G* calculations show the [2233]/[2233] conformer to be more stable for CB-TE2P and CB-TE1A1P and the [2323]/[2323] conformer to be favored for CB-TE2A. The single point M06/6-31+G**//M06/6-31G* energy differences are not significantly different from the lower level basis E differences for Cu-CB-TE2P and Cu-CB-TE1A1P. The gap is decreased somewhat for Cu-CB-TE2A but the [2323]/[2323] conformation is still predicted to be favored as observed in the crystal structure.16 The B3LYP/6-31G*//B3LYP/6-31G* calculations have the two conformations essentially equienergetic for Cu-CB-TE2P and Cu-CB-TE1A1P and favor the [2323]/[2323] conformation strongly for Cu-CB-TE2A, so the trend is similar but shifted toward favoring the [2323]/[2323] conformation. Practical computational limitations prevented us from investigating higher level (triple-zeta) basis sets and our calculations take no account of solvation. Based on benchmarking that has been done in the literature for transition metal complexes and the fact that M06 has been shown to be superior to B3LYP in terms of medium-range correlation energies,75,77 we are inclined to trust the M06 results more than the B3LYP results. On the basis of the results (Table 2), we tentatively conclude that fundamental differences related to the pendant arm influence the conformation and that the result is not simply a matter of solvation and packing. It is clear from this work that the [2233]/[2233] non-diamond-lattice conformation is energetically viable and must be considered as well as the diamond-lattice [2323]/[2323] conformation as a possible global minimum for cross-bridged metal complexes.
Radiolabeling of CB-TE2P and CB-TE1A1P
Even though cold chemistry experiments showed that the copper(II) complexes of CB-TE1A1P and CB-TE2P formed quickly and under mild conditions in organic solvents (e.g., methanol), radiochemistry experiments were performed in aqueous solvents for translation to in vivo investigations. Radiolabeling reactions of 64Cu to CB-TE2P and CB-TE1A1P were performed under aqueous conditions with various buffers, pH values (4.5–8.1), and temperatures (25–95 °C). The radiolabeling yield was determined by radio-HPLC, with the free 64Cu having a retention time of ∼4 min and the 64Cu complexes having a retention time of ∼20 min. The HPLC peaks corresponding to 64Cu-CB-TE1A1P and 64Cu-CB-TE2P were assigned by LC-MS of a carrier-added sample. Note that under no carrier-added conditions the concentration of the 64Cu complexes was too low to be detected by either UV or MS.Using mM concentrations of CB-TE2P or CB-TE1A1P, radiolabeling was complete under 1 h, but only at higher temperatures or with carrier-added Cu(II). Using μM concentrations, the CB-TE2P and CB-TE1A1P were labeled with 64Cu at room temperature in shorter time periods. The optimized conditions for 64Cu-labeled CB-TE1A1P and CB-TE2P were as follows: 100 μL of a 26.5 μM solution of chelator in 0.1 M NH4OAc (pH = 8.1) was incubated with 1 mCi of 64CuCl2 at room temperature. Radiochemical yields of >95% were achieved after 30 min. This was the first successful attempt to efficiently radiolabel cross-bridged cyclam derivatives at room temperature. Copper-64-labeling of CB-TE2A under the same conditions resulted in negligible radiochemical yields. As previously reported, CB-TE2A requires temperatures >90 °C in order to achieve complete radiolabeling within 1 h.18
Biodistribution studies of 64Cu-CB-TE2P and 64Cu-CB-TE1A1P
In order to gain insight into the pharmacokinetics of the 64Cu-labeled complexes, biodistribution studies were performed. 64Cu-CB-TE2P and 64Cu-CB-TE1A1P (50 μCi (∼1 μCi μg−1) in 150 μL of saline) were injected through the tail vein in male, Lewis rats (26 d). The animals were sacrificed at 1, 2, 4 and 24 h after injection, the organs were harvested, and their activity measured with a γ counter. The radiolabeled compounds that were injected for biodistribution studies were positively identified as 64Cu-CB-TE2P and 64Cu-CB-TE1A1P by LC-MS and radio-LC-MS, and were compared to an authentic standard under carrier-added conditions.As discussed in our communication,5564Cu-CB-TE2P clears rapidly from the blood (from 0.15%ID g−1 at 1 h to 0.002%ID/g at 24 h) and has only minor residual uptake in non-target organs such as liver, spleen, lung, heart and marrow (not higher than 0.03%ID g−1 at 24 h). Rapid clearance of 64Cu-CB-TE2P from the liver was indicative of its high in vivo stability, as it is known that dissociated 64Cu is rapidly coordinated by liver proteins such as superoxide dismutase (SOD), ceruloplasmin and metallothionein and is retained in liver, kidneys and the blood circulation.
A biodistribution study of 64Cu-CB-TE1A1P in Lewis rats (Fig. 5) demonstrated rapid blood clearance (0.08 ± 0.02%ID g−1 at 1 h postinjection) and low liver accumulation (0.11 ± 0.01%ID g−1 at 1 h postinjection), suggesting that 64Cu-CB-TE1A1P had comparable stability to 64Cu-CB-TE2A and 64Cu-CB-TE2Pin vivo. Higher uptake was observed in the kidney (1.20 ± 0.43%ID g−1 at 1 h postinjection which decreased to 0.30 ± 0.03%ID g−1 at 24 h), while no significant uptake was detected in lung, spleen, heart or stomach. These data suggest that 64Cu-CB-TE1A1P clears quickly through the kidneys, with a marginal amount clearing through the liver. Bone uptake cleared significantly by 24 h (0.19 ± 0.04%ID g−1 and 0.044 ± 0.003%ID g−1 at 1 and 24 h postinjection, respectively). Overall, the biodistribution of CB-TE1A1P was similar to that of CB-TE2P, except for a lower accumulation in the bone at all time points (p < 0.0001). Note that unlike the recently-reported 4,11-dimethyl-cyclam-1,8-bis(methylphosphonic acid),41 both CB-TE2P and CB-TE1A1P form Cu(II) complexes that are suitable for radiopharmaceutical applications because of their inertness in vivo, a direct consequence of the cross-bridged bicyclic structure.
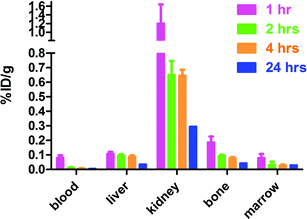 | ||
| Fig. 5 Biodistribution of 64Cu-CB-TE1A1P (%ID g−1) in male, Lewis rats in selected organs/tissues. | ||
Fig. 6 shows a comparison of the biodistributions of the methanephosphonic acid derivatives of CB-cyclam, 64Cu-labeled CB-TE2P and CB-TE1A1P, compared to CB-TE2A, Diamsar and NOTA. All complexes rapidly clear from the blood, with 64Cu-NOTA showing the highest remaining accumulation at 24 h (0.017 ± 0.001%ID g−1). 64Cu-CB-TE2A has the lowest retention in the marrow, liver and kidney at 24 h postinjection (p < 0.05). 64Cu-NOTA showed significantly higher liver retention (0.086 ± 0.01%ID g−1), while 64Cu-Diamsar has higher kidney retention (1.75 ± 0.50%ID g−1) compared to the other complexes (24 h postinjection; p < 0.0005). Overall, 64Cu-CB-TE2P and 64Cu-CB-TE1P demonstrated significantly, but not dramatically, higher accumulations in the marrow, liver and kidneys than 64Cu-CB-TE2A (p < 0.05). However, 64Cu-CB-TE2P and 64Cu-CB-TE1P had significantly lower accumulations in the marrow than 64Cu-Diamsar and 64Cu-NOTA, and significantly more clearance from the blood compared to 64Cu-NOTA (p < 0.05).
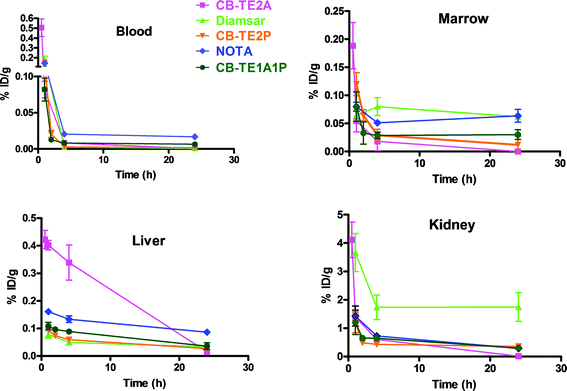 | ||
| Fig. 6 Comparison of the clearance (%ID g−1) in male Lewis rats of 64Cu-labeled CB-TE2A (□), Diamsar (△), CB-TE2P (▽), NOTA (◊) and CB-TE1A1P (○) from blood, marrow, liver and kidney. | ||
These results are important since CB-TE2P and CB-TE1A1P maintain most of the positive attributes of CB-TE2A including high in vivo stability of the 64Cu-complexes; however, a major advantage is that they can be quantitatively labeled at room temperature. 64Cu-CB-TE2A has been shown to have unparalleled in vivo stability that is much higher than the commonly used DOTA and TETAchelators. However, the high temperatures required for labelingCB-TE2A with 64Cu have limited its conjugation to only robust peptides and small molecules. CB-TE1A1P will likely eliminate this restriction and allow for its broader use in 64Culabeling of all types of targeting molecules, including temperature-sensitive proteins such as monoclonal antibodies.
Along with in vivo stability, charge and lipophilicity likely play a role in the difference in liver and kidney uptake. For example, the neutral 64Cu-CB-TE2A complex has the highest liver uptake at 1 h post-injection, but has the most extensive clearance of the five complexes, likely due to its superior in vivo stability. 64Cu-diamsar, which has a 2+ charge, has the highest retention in the kidneys, and this is consistent with positively charged metal complexes being retained in the kidneys.78 The macrocycles with the phosphonate pendant arms as well as the NOTA complex of 64Cu(II) are all negatively charged, and thus clear rapidly from the liver and kidneys. To our knowledge, charge does not have a significant impact on blood clearance or retention in the bone marrow.
It is important to emphasize that these data with the 64Cu-chelates may not predict with certainty the in vivo stability with 64Cu-labeled chelator-biomolecule conjugates, as the linkage between the chelator and the biomolecule, as well as the nature of the biomolecule have a significant impact on biodistribution. Conjugation of CB-TE1A1P to a biomolecule will result in the conversion of the carboxylate group to an amide moiety. We previously demonstrated that the amideoxygen decreases in vivo stability compared to the carboxylate group, and this may negatively impact stability of a CB-TE1A1P-biomolecule conjugate. However, preliminary data from the Anderson lab79 has demonstrated that a 64Cu-labeled CB-TE1A1P conjugate of the somatostatin analog, Y3-TATE, has a similar biodistribution to 64Cu-CB-TE2A-Y3-TATE,17 including blood and liver clearance. Maecke et al. compared 64Cu-labeled NOTA and CB-TE2A analogs of a somatostatin antagonist, LM3, and a αvβ3 integrin-targeted RGDpeptide in tumor-bearing mouse models.80,81 Interestingly, the 64Cu-labeled NOTA (NODAGA) LM3 conjugate showed significantly higher tumor![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :non-target tissue ratios compared to 64Cu-CB-TE2A-LM3;80 however, for RGD, the CB-TE2A conjugates showed higher tumor:blood and tumor:liver ratios at 18 h postinjection.81 This underscores the likelihood that there is not one single Cu(II)chelator that is optimal for all biomolecules.
:non-target tissue ratios compared to 64Cu-CB-TE2A-LM3;80 however, for RGD, the CB-TE2A conjugates showed higher tumor:blood and tumor:liver ratios at 18 h postinjection.81 This underscores the likelihood that there is not one single Cu(II)chelator that is optimal for all biomolecules.
Summary and conclusions
A new class of cross-bridged cyclammacrocycles having methanephosphonic acid pendant arms has been developed. These compounds readily formed copper(II) complexes and the 64Cu-labeled chelates demonstrate promising kinetic inertness in vivo. Both the crystal structures of Cu(II)-CB-TE2P and Cu(II)-CB-TE1A1P showed the copper(II) ion within the bicyclic tetraamine cleft, with the pendant arms completing the coordination sphere for complete encapsulation of copper. Both crystal structures exhibit the [2233]/[2233] ligand conformation rather than the more common distorted diamond-lattice [2323]/[2323] cross-bridged cyclam conformation found in Cu-CB-TE2A. DFT calculations reproduce this conformational change, which may be a consequence of the longer C–P and P–O bonds in the phosphonate arms as compared to the C–C and C–O bonds of the acetate arms. Acid inertness and electrochemical assays support the viability of these cross-bridged complexes in vitro. Biodistribution of 64Cu-CB-TE2P and CB-TE1A1P in normal rats show comparable in vivo stability to the current “gold standard”, 64Cu-CB-TE2A, but they both have the major advantage of being amenable to 64Culabeling at room temperature with high specific activity. CB-TE2P and CB-TE1A1P are the first examples of cross-bridged cyclam derivatives that can be radiolabeled with 64Cu at room temperature under no-carrier-added conditions. CB-TE1A1P has the potential for enabling the design of a new generation of imaging agents that can be quickly labeled at room temperature and with high stability in vivo, expanding the portfolio of potential agents beyond heat-stable small molecules and peptides into heat-sensitive proteins such as monoclonal antibodies. Bioconjugation of CB-TE1A1P and associated radiolabeling and biological studies toward cancer diagnosis will be reported in future publications.Experimental
Materials and methods
Reagents: Cupric chloride dihydrate was obtained from Aldrich Chemical Company. Paraformaldehyde and triethyl phosphite were obtained from Acros Organics. 1,1,1,3,3,3-hexafluoro-2-propanol was purchased from Alfa Aesar. Trace Metal™ Grade Hydrochloric acid was obtained from Fischer Scientific. Amberlite CG50 was obtained from Sigma. Solvents: Methanol (MeOH, ACS grade) was obtained from Pharmco Products Inc. Diethyl ether (Et2O) and toluene (PhCH3, ACS grade) were obtained from EMD Chemicals Inc. The solvents were stored in an Innovative Technology Inc. Pure-Solv SolventPurification System. Prior to use, each solvent was passed through the system's alumina column under low pressure to remove trace impurities.Melting points (mp) were obtained on a Thomas Hoover capillary melting point apparatus and were uncorrected. Elemental analyses were obtained at Atlantic Microlab Inc., Norcross, GA and Schwartzkopf Microanalytical Laboratory, Woodside, NY. Infrared (IR) spectra were run on a Nicolet MX-1 FT-IR spectrometer and absorptions are reported in wavenumbers (cm−1). High-resolution mass spectra were obtained at the Mass Spectrometry Facility at the University of Notre Dame on a JEOL AX505HA high-resolution mass spectrometer. 1H NMR, 13C{1H} NMR and 31P{1H} NMR spectra were acquired on Varian Mercury-400BB and/or INOVA-500NMR spectrometers. 1H and 13C chemical shifts are reported in parts per million (ppm) relative to internal Me4Si (TMS) unless otherwise noted and coupling constants (J) are reported in Hertz (Hz). 31P chemical shifts are reported in parts per million (ppm) relative to an external 85% phosphoric acid standard.
UV-Vis spectra were obtained on a Varian Cary 50 spectrophotometer. Electrochemical studies were performed on a BAS 100B electrochemical analyzer in 0.1 M aq. NaOAc solution using a glassy carbonworking electrode, a platinum wire auxiliary electrode, and Ag/AgCl reference electrode. Acid inertness studies were carried out under pseudo first-order conditions in 5 M HCl solutions at 90 °C by monitoring the time-dependent decay of the electronic absorption maximum at 630 nm.
Ligand synthesis
CB-TE2A was synthesized as previously published.16,17 Diamsar82–84 and NOTA85 were synthesized as described in the literature.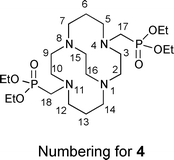
The reaction was also scaled up and the product carried directly to the next step: Triethyl phosphite (0.18 mL, 1.0 mmol) and paraformaldehyde (32.5 mg, 2.5 molar equivalents of CH2O) were added to a solution of 5 (200 mg, 0.587 mmol) in dry THF (4 mL) and stirred under N2 for 4 days. Solvent was removed, the residue was dissolved in H2O (20 mL), and the solution was made strongly basic (pH = 14, slow addition of KOH pellets). The resulting solution was extracted with toluene (5 × 20 mL), the combined organic phases were dried over anhyd. Na2SO4, and solvent was removed to give crude product (0.29 g, oil) that was used for the next reaction without further purification.
Copper(II) complex synthesis
Cu-CB-TE2P . A solution of CuCl2·2H2O (0.0132 g, 0.077 mmol) in 2 mL MeOH was added to a 2 mL solution of 1·0.6HCl·1.1H2O (0.035 g, 0.077 mmol) in MeOH. Aq. NaOH (0.3 mL of a 0.4 M solution) was added to raise the pH to 5. The resulting blue solution was evaporated to dryness and extracted with 1 mL of 1,1,1,3,3,3-hexafluoro-2-propanol. The NaCl precipitate was separated from the blue supernatant after centrifugation. Diethyl ether diffusion into the supernatant yielded blue microcrystals (0.047 g, 86%) (Found: C, 29.48; H, 4.96; N, 7.91; Cl, 1.78; P, 8.66. Calc. for C14H30N4O6P2Cu·H2O·0.3NaCl·1.2C3H2F6O: C, 29.64; H, 4.86; N, 7.86; Cl, 1.49; P 8.69%); UV-Vis: λmax (MeOH)/nm 639 (ε/dm3 mol−1 cm−1 35); HRFABMSm/z(M+H)+ exact mass for C14H31N4P2Cu: 476.1015; Found: 476.1009 (error −0.6 mmu/−1.2 ppm). Cyclic voltammogram (0.1 M aq. NaOAc): −0.96 V (Ag/AgCl) quasi-reversible. X-ray crystals were obtained by diethyl ether diffusion into a hexafluoro-2-propanol solution of the complex inside a 2 mm inner diameter glass tube.Cu-CB-TE1A1P . An amount of CB-TE1A1P·0.45HCl·2H2O (84 mg, 0.19 mmol) was dissolved in 2 mL water along with Cu(ClO4)2·6H2O (75 mg, 0.20 mmol). The solution pH was adjusted to 2 using 0.1 N NaOH and stirred for 1 h. After removal of solvent by rotary evaporation, the blue residue was dissolved in 1 mL of methanol. After a small amount of insoluble material was removed by centrifugation, the supernatant was placed in a vial inside a chamber and diethyl ether allowed to diffuse into it. After 2 days, a dark-blue solid deposited which was separated and dried. About 1 mL of hexafluoro-isopropanol was added to this solid and the resulting suspension centrifuged to remove a small amount of insoluble matter. The supernatant was returned to the ether diffusion chamber yielding a dark-blue solid product (120 mg, 95% yield) upon standing. Elemental analysis: found: C, 31.71; H, 4.72; N, 8.04; Cl, 3.12. Calc. for CuC15H29N4O5P·C3F6OH2·0.60(NaClO4): C, 31.73; H, 4.58; N, 8.22; Cl, 3.12%. UV-Vis: λmax (aq)/nm 613 (ε/dm3 mol−1 cm−1 24); FT-IR (KBr): 1619, 1262, 1218, 1100 cm−1. Cyclic voltammetry (0.1 M NaOAc, 40 mV s−1): reduction peak potential −1.03 V (irrev, Ag/AgCl), copper stripping peak +0.10 V. Crystals suitable for X-ray diffraction study were obtained by slow evaporation of a 6 M aqueous ammonia sample solution.
X-ray structural studies
A summary of crystal data for the two X-ray structures can be found in Table 3.| Cu-CB-TE2P | ||
|---|---|---|
| Empirical formula | C14 H34Cl0.50 CuN4Na0.50O9.50P2 | |
| Formula weight | 565.15 | |
| Temperature | 100(2) K | |
| Wavelength | 1.54178 Å | |
| Crystal system | Triclinic | |
| Space group |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif)
|
|
| Unit cell dimensions | a = 9.5428(5) Å | α = 80.039(4)° |
| b = 15.5173(7) Å | β = 81.081(4)° | |
| c = 16.9240(10) Å | γ = 89.874(3)° | |
| Volume | 2437.7(2) Å3 | |
| Z | 4 | |
| Density (calculated) | 1.540 g cm−3 | |
| Absorption coefficient | 3.574 mm−1 | |
| F(000) | 1180 | |
| Crystal size | 0.31 × 0.21 × 0.05 mm3 | |
| Theta range for data collection | 4.28 to 59.37° | |
| Index ranges | −10 ≤ h ≤ 10, −17 ≤ k ≤ 17, −16 ≤ l ≤ 18 | |
| Reflections collected | 16637 | |
| Independent reflections | 6611 [R(int) = 0.0970] | |
| Completeness to theta = 59.37° | 92.9% | |
| Absorption correction | None | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data/restraints/parameters | 6611/0/593 | |
| Goodness-of-fit on F2 | 1.020 | |
| Final R indices [I > 2σ(I)] | R 1 = 0.0741, wR2 = 0.1968 | |
| R indices (all data) | R 1 = 0.1213, wR2 = 0.2186 | |
| Largest diff. peak and hole | 0.941 and −0.730 e Å−3 | |
| Cu-CB-TE1A1P | ||
|---|---|---|
| Empirical formula | C30H80Cu2N8O21P2 | |
| Formula weight | 1078.04 | |
| Temperature | 100(2) K | |
| Wavelength | 0.71073 Å | |
| Crystal system | Orthorhombic | |
| Space group | Pccn | |
| Unit cell dimensions | a = 10.1989(19) Å | α = 90°. |
| b = 35.067(7) Å | β = 90°. | |
| c = 12.943(3) Å | γ = 90°. | |
| Volume | 4629.2(15) Å3 | |
| Z | 4 | |
| Density (calculated) | 1.547 Mg m−3 | |
| Absorption coefficient | 1.073 mm−1 | |
| F(000) | 2288 | |
| Crystal size | 0.20 × 0.20 × 0.10 mm3 | |
| Crystal color/habit | blue/plate | |
| Theta range for data collection | 2.08 to 28.29°. | |
| Index ranges | −13 ≤ h < = 13, −46 ≤ k ≤ 45, −16 ≤ l ≤ 17 | |
| Reflections collected | 57822 | |
| Independent reflections | 5510 [R(int) = 0.0564] | |
| Completeness to theta = 25.00° | 99.9% | |
| Absorption correction | multi-scan/sadabs | |
| Max. and min. transmission | 0.9003 and 0.8141 | |
| Refinement method | Full-matrix least-squares on F2 | |
| Data/restraints/parameters | 5510/17/322 | |
| Goodness-of-fit on F2 | 1.157 | |
| Final R indices [I > 2σ(I)] | R 1 = 0.0484, wR2 = 0.1350 | |
| R indices (all data) | R 1 = 0.0568, wR2 = 0.1392 | |
| Largest diff. peak and hole | 0.585 and −0.520 e.Å−3 | |
Cu-CB-TE2P . A crystal was mounted on a Cryoloop with Paratone-N oil and cooled to −173 °C under a stream of dry nitrogen gas. Data were collected on a BRUKER APEX2 CCD X-ray system using Cu K alpha radiation at −173 °C and integrated using APEX2 software (SHELXL) corrected for adsorption using SADABS. Structure was solved by direct methods and all non-hydrogen atoms were refined as being anisotropic. Two molecules were found in the asymmetric unit. One of the molecules was disordered and this was treated by assuming a two-position disorder model. Although all the hydrogen atoms were not found, they were included in the chemical formula to account for F000, density and molecular weight.
Disorder associated with Cu-CB-TE2P was treated by using a 50% occupancy model for atoms C15, C16, C17, C18, C23, C24, C25, C26, C27, N5, N6, O7, O8, O9, P3, O2SA, O2SB, O6SA, O6SB, C15′, C16′ C17′, C18′, C23′, C24′, C25′, C26′ C27′, N5′, N6′, O7′, O8′, O9′, P3′ and their associated hydrogen atoms. Occupancies were set for the following atoms at values indicated: Atoms O5SA 40%, O5SB 60%, O7SA and O7SB both at 30% occupancy while O7SC was at 40% occupancy; atoms Na1A and Na1B 75% and 25%, respectively. The hydrogen atoms on O8 and O8′ were fixed in position with appropriate riding models.
Cu-CB-TE1A1P . A blue crystal was mounted on a Cryoloop with Paratone-N oil. Data were collected on a Bruker APEX II CCD system using Mo K alpha radiation in a nitrogen gas stream at 100(2) K using phi and omega scans. Crystal-to-detector distance was 60 mm and exposure time was 10 s per frame using a scan width of 0.5°. Indexing and unit cell refinement indicated a primitive, orthorhombic lattice. The space group was found to be Pccn. The data were integrated using the Bruker SHELXTL software program and scaled using the SADABS software program. Solution by direct methods (SHELXS) and all non-hydrogen atoms were refined anisotropically by full-matrix least-squares (SHELXL-97). All hydrogen atoms except H2P on O2 were placed on their appropriate parent atom using a riding model. Hydrogen H2P was determined from a Fourier difference map and was allowed to refine. Hydrogen atoms on water molecules were restrained using DFIX and DANG commands.
Radiochemistry
64Cu(OAc)2 was obtained by adding 200 μL of 0.4 M NH4OAc (pH = 6.5) to 5 mCi of 64CuCl2 in 5 μL of 2 M HCl and shaking the reaction for 10 min at room temperature.:methanol/10% ammonium acetate 1:4). The 64Cu-CB-TE2P was diluted with 2.7 mL of saline and divided into 150 μL aliquots (42 μg chelator; 45 μCi 64Cu per dose).
:4). The 64Cu-CB-TE1A1P was diluted with 3.15 mL of saline and divided into 150 μL aliquots (48 μg chelator; 40 μCi each).
64Cu-Diamsar was prepared by incubating the chelator (1 mg) with 64Cu in 0.5 M NH4OAcbuffer (pH = 10) at room temperature in 30 min. A single peak (Rf ∼ 0.2) corresponding to 64Cu-Diamsar was verified by radio-TLC using a mobile phase of ethanol on ITLC-SG plates (Rf (64Cu-acetate) = 0). Radiochemical purity of 64Cu-Diamsar was > 95% as confirmed by radio-TLC and radio-HPLC (retention time (64Cu-Diamsar) = 32 min). HPLC analysis of the radiolabeled complex was performed using an EPS column with the following gradient: solvent A: 10 mM NH4OAc, pH 5.5; solvent B: 0.1% TFA in acetonitrile; 0% B to 50% B in 40 min; 0.5 mL min−1 flow rate.
The Cu(II) complex of Diamsar (natCu-Diamsar) was prepared with high-purity natural copper chloride (CuCl2) using the procedure described above except both non-radioactive CuCl2 and radioactive 64CuCl2 (∼50 μCi) were added to the reaction mixture. Complex formation was determined by radio-TLC and confirmed by radio-HPLC. After sufficient time for radio-decay, formation of natCu-Diamsar was demonstrated by ESI-MS (calc. m/z (M–H)+, 376.23; found, 376.20).
Biodistribution studies
Animal experiments were carried out in compliance with the Guidelines for the Care and Use of Research Animals established by the Animal Studies Committee of Washington University. Male Lewis rats (34–35 day old) were injected with 64Cu-NOTA, 64Cu-Diamsar, 64Cu-CB-TE2P or 64Cu-CB-TE1A1P (40–60 μCi; 0.5–130 nmol ligand) intravenously via the tail vein. Tissue biodistribution data were obtained at 1, 4, and 24 h postinjection. Animals were sacrificed at 1, 4 and 24 h postinjection, organs of interest were removed, weighed, and counted on a gamma counter (Beckman). The percent injected dose per gram (%ID g−1) and percent injected dose per organ (%ID organ−1) were measured by comparison against a weighed, counted standard.Computational chemistry
All DFT calculations were carried out in parallel mode using Spartan'10 version 1.1.0 (Wavefunction, Inc., Irvine, CA) running the Q-Chem computational engine87 on a Mac Pro dual Quad-Core Intel Xeon (2.93 GHz) computer with 16 GB of RAM.Acknowledgements
This work was supported by Award Number R01CA093375 from the National Cancer Institute (USA). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. The authors thank Elizabeth Garcia (University of New Hampshire) for synthesis of NOTA and Christopher D. Sherman (Washington University) for performing the biodistribution studies.Notes and references
- S. Bhattacharyya and M. Dixit, Dalton Trans., 2011, 40, 6112–6128 RSC.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Chem. Rev., 2010, 110, 2858–2902 CrossRef CAS.
- C. J. Anderson and M. J. Welch, Chem. Rev., 1999, 99, 2219–2234 CrossRef CAS.
- D. W. McCarthy, R. E. Shefer, R. E. Klinkowstein, L. A. Bass, W. H. Margenau, C. S. Cutler, C. J. Anderson and M. J. Welch, Nucl. Med. Biol., 1997, 24, 35–43 CrossRef CAS.
- P. S. Donnelly, Dalton Trans., 2011, 40, 999–1010 RSC.
- B. M. Zeglis and J. S. Lewis, Dalton Trans., 2011, 40, 6168 RSC.
- R. E. Mewis and S. J. Archibald, Coord. Chem. Rev., 2010, 254, 1686–1712 CrossRef CAS.
- M. Shokeen and C. J. Anderson, Acc. Chem. Res., 2009, 42, 832–841 CrossRef CAS.
- T. J. Wadas, E. H. Wong, G. R. Weisman and C. J. Anderson, Curr. Pharm. Des., 2007, 13, 3–16 CrossRef CAS.
- S. V. Smith, J. Inorg. Biochem., 2004, 98, 1874–1901 CrossRef CAS.
- K. S. Woodin, K. J. Heroux, C. A. Boswell, E. H. Wong, G. R. Weisman, W. J. Niu, S. A. Tomellini, C. J. Anderson, L. N. Zakharov and A. L. Rheingold, Eur. J. Inorg. Chem., 2005, 4829–4833 CrossRef CAS.
- M. K. Moi, C. F. Meares, M. J. Mccall, W. C. Cole and S. J. Denardo, Anal. Biochem., 1985, 148, 249–253 CrossRef CAS.
- W. C. Cole, S. J. Denardo, C. F. Meares, M. J. Mccall, G. L. Denardo, A. L. Epstein, H. A. Obrien and M. K. Moi, J. Nucl. Med., 1987, 28, 83–90 CAS.
- L. A. Bass, M. Wang, M. J. Welch and C. J. Anderson, Bioconjugate Chem., 2000, 11, 527–532 CrossRef CAS.
- C. A. Boswell, X. K. Sun, W. J. Niu, G. R. Weisman, E. H. Wong, A. L. Rheingold and C. J. Anderson, J. Med. Chem., 2004, 47, 1465–1474 CrossRef CAS.
- E. H. Wong, G. R. Weisman, D. C. Hill, D. P. Reed, M. E. Rogers, J. S. Condon, M. A. Fagan, J. C. Calabrese, K. C. Lam, I. A. Guzei and A. L. Rheingold, J. Am. Chem. Soc., 2000, 122, 10561–10572 CrossRef CAS.
- J. E. Sprague, Y. J. Peng, X. K. Sun, G. R. Weisman, E. H. Wong, S. Achilefu and C. J. Anderson, Clin. Cancer Res., 2004, 10, 8674–8682 CrossRef CAS.
- X. K. Sun, M. Wuest, G. R. Weisman, E. H. Wong, D. P. Reed, C. A. Boswell, R. Motekaitis, A. E. Martell, M. J. Welch and C. J. Anderson, J. Med. Chem., 2002, 45, 469–477 CrossRef CAS.
- J. E. Sprague, Y. Peng, A. L. Fiamengo, K. S. Woodin, E. A. Southwick, G. R. Weisman, E. H. Wong, J. A. Golen, A. L. Rheingold and C. J. Anderson, J. Med. Chem., 2007, 50, 2527–2535 CrossRef CAS.
- T. J. Wadas, M. Eiblmaier, A. Zheleznyak, C. D. Sherman, R. Ferdani, K. Liang, S. Achilefu and C. J. Anderson, J. Nucl. Med., 2008, 49, 1819–1827 CrossRef CAS.
- T. J. Wadas and C. J. Anderson, Nat. Protoc., 2006, 1, 3062–3068 CrossRef CAS.
- J. E. Sprague, H. Kitaura, W. Zou, Y. Ye, S. Achilefu, K. N. Weilbaecher, S. L. Teitelbaum and C. J. Anderson, J. Nucl. Med., 2007, 48, 311–318 CAS.
- L. Wei, Y. Ye, T. Wadas, J. S. Lewis, M. J. Welch, S. Achilefu and C. J. Anderson, Nucl. Med. Biol., 2009, 36, 277–285 CrossRef CAS.
- L. Wei, C. Butcher, Y. Miao, F. Gallazzi, T. P. Quinn, M. J. Welch and J. S. Lewis, J. Nucl. Med., 2007, 48, 64–72 CAS.
- J. C. Garrison, T. L. Rold, G. L. Sieckman, S. D. Figueroa, W. A. Volkert, S. S. Jurisson and T. J. Hoffman, J. Nucl. Med., 2007, 48, 1327–1337 CrossRef CAS.
- W. Liu, G. Hao, M. A. Long, T. Anthony, J.-T. Hsieh and X. Sun, Angew. Chem., Int. Ed., 2009, 48, 7346–7349 CrossRef CAS.
- E. Lewis, R. Boyle and S. Archibald, Chem. Commun., 2004, 2212–2213 RSC.
- C. A. Boswell, C. A. S. Regino, K. E. Baidoo, K. J. Wong, A. Bumb, H. Xu, D. E. Milenic, J. A. Kelley, C. C. Lai and M. W. Brechbiel, Bioconjugate Chem., 2008, 19, 1476–1484 CrossRef CAS.
- A. Y. Lebedev, J. P. Holland and J. S. Lewis, Chem. Commun., 2010, 1706–1708 RSC.
- I. Svobodová, P. Lubal, J. Plutnar, J. Havlícková, J. Kotek, P. Hermann and I. Lukes, Dalton Trans., 2006, 5184–5197 RSC.
- F. K. Kalman, Z. Baranyai, I. Toth, I. Banyai, R. Kiraly, E. Brucher, S. Aime, X. K. Sun, A. D. Sherry and Z. Kovacs, Inorg. Chem., 2008, 47, 3851–3862 CrossRef CAS.
- C. Bianchini, G. Giambastiani, F. Laschi, P. Mariani, A. Vacca, F. Vizza and P. Zanello, Org. Biomol. Chem., 2003, 1, 879–886 CAS.
- C. F. G. C. Geraldes, M. P. M. Marques, B. de Castro and E. Pereira, Eur. J. Inorg. Chem., 2000, 559–565 CrossRef CAS.
- J. Kotek, P. Lubal, P. Hermann, I. Cisarova, I. Lukes, T. Godula, I. Svobodova, P. Taborsky and J. Havel, Chem.–Eur. J., 2003, 9, 233–248 CrossRef CAS.
- S. Füzerová, J. Kotek, I. Císařová, P. Hermann, K. Binnemans and I. Lukeš, Dalton Trans., 2005, 2005, 2908–2915 RSC.
- K. P. Guerra, R. Delgado, L. S. M. P. Lima, M. G. B. Drew and V. T. Felix, Dalton Trans., 2004, 1812–1822 RSC.
- P. Lubal, M. Kyvala, P. Hermann, J. Holubova, J. Rohovec, J. Havel and I. Lukes, Polyhedron, 2001, 20, 47–55 CrossRef CAS.
- J. Kotek, F. K. Kalman, P. Hermann, E. Brucher, K. Binnemans and I. Lukes, Eur. J. Inorg. Chem., 2006, 1976–1986 CrossRef CAS.
- L. M. P. Lima, R. Delgado, M. G. B. Drew, P. Brandao and V. Felix, Dalton Trans., 2008, 6593–6608 RSC.
- L. Gano, F. Marques, M. P. Campello, M. Balbina, S. Lacerda and I. Santos, Q. J. Nucl. Med. Mol. Imaging, 2007, 51, 6–15 CAS.
- I. Svobodová, J. Havlíćková, J. Plutnar, P. Lubal, J. Kotek and P. Hermann, Eur. J. Inorg. Chem., 2009, 2009, 3577–3592 CrossRef.
- J. Havlíčková, H. Medová, T. Vitha, J. Kotek, I. Císařová and P. Hermann, Dalton Trans., 2008, 5378–5386 RSC.
- G. Giambastiani, W. Oberhauser, C. Bianchini, F. Laschi, L. Sorace, P. Brueggeller, R. Gutmann, A. Orlandini and F. Vizza, Eur. J. Inorg. Chem., 2005, 2027–2031 CrossRef CAS.
- A. D. Sherry, J. Alloys Compd., 1997, 249, 153–157 CrossRef CAS.
- I. Svobodová, P. Lubal, P. Hermann, J. Kotek and J. Havel, Microchim. Acta, 2004, 148, 21–26 Search PubMed.
- I. Svobodová, P. Lubal, P. Hermann, J. Kotek and J. Havel, J. Inclusion Phenom. Macrocyclic Chem., 2004, 49, 11–15 CrossRef.
- I. Lukeš, J. Kotek, P. Vojtíšek and P. Hermann, Coord. Chem. Rev., 2001, 216, 287–312 CrossRef.
- X. K. Sun, M. Wuest, Z. Kovacs, A. D. Sherry, R. Motekaitis, Z. Wang, A. E. Martell, M. J. Welch and C. J. Anderson, JBIC, J. Biol. Inorg. Chem., 2003, 8, 217–225 CrossRef CAS.
- H. A. Fleisch, R. G. G. Russell, S. Bisaz, R. C. Mühlbauer and D. A. Williams, Eur. J. Clin. Invest., 1970, 1, 12–18 CrossRef CAS.
- R. G. G. Russell, R. C. Mühlbauer, S. Bisaz, D. A. Williams and H. Fleisch, Calcif. Tissue Res., 1970, 6, 183–196 CrossRef CAS.
- A. Zieba, G. Sethuraman, F. Perez, G. H. Nancollas and D. Cameron, Langmuir, 1996, 12, 2853–2858 CrossRef CAS.
- Z. Amjad, Langmuir, 1987, 3, 1063–1069 CrossRef CAS.
- C. A. Boswell, C. A. S. Regino, K. E. Baidoo, K. J. Wong, D. E. Milenic, J. A. Kelley, C. C. Lai and M. W. Brechbiel, Bioorg. Med. Chem., 2009, 17, 548–552 CrossRef CAS.
- J. Plutnar, J. Havlickova, J. Kotek, P. Hermann and I. Lukes, New J. Chem., 2008, 32, 496–504 RSC.
- D. J. Stigers, R. Ferdani, G. R. Weisman, E. H. Wong, C. J. Anderson, J. A. Golen, C. Moore and A. L. Rheingold, Dalton Trans., 2010, 39, 1699–1701 RSC.
- C. J. Anderson and R. Ferdani, Cancer Biother. Radiopharm., 2009, 24, 379–393 CrossRef CAS.
- P. Kafarski and J. Zon, in Aminophosphonic Aminophosphinic Acids, ed. V. P. Kukhar and H. R. Hudson, John Wiley & Sons, Ltd, Chichester, 2000, ch. 2, pp. 33–74 Search PubMed.
- M. Syamala, Org. Prep. Proced. Int., 2005, 37, 103–171 CrossRef CAS.
- S. Bhagat and A. K. Chakraborti, J. Org. Chem., 2007, 72, 1263–1270 CrossRef CAS.
- N. S. Zefirov and E. D. Matveeva, ARKIVOC, 2008, 1, 1–17 Search PubMed.
- R. A. Cherkasov and V. I. Galkin, Russ. Chem. Rev., 1998, 67, 857–882 CrossRef.
- G. Weisman, M. Rogers, E. Wong, J. Jasinski and E. Paight, J. Am. Chem. Soc., 1990, 112, 8604–8605 CrossRef CAS.
- J. Dale, Acta Chem. Scand., 1973, 27, 1115–1129 CrossRef CAS.
- J. Dale, in Topics in Stereochemistry, ed. N. L. Allinger and E. L. Eliel, John Wiley & Sons, New York, 1976, vol. 9, pp. 199–270 Search PubMed.
- K. J. Heroux, K. S. Woodin, D. J. Tranchemontagne, P. C. B. Widger, E. Southwick, E. H. Wong, G. R. Weisman, S. A. Tomellini, T. J. Wadas, C. J. Anderson, S. Kassel, J. A. Golen and A. L. Rheingold, Dalton Trans., 2007, 2150–2162 RSC.
- T. Hubin, N. Alcock and D. Busch, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, C56, 37–39 CrossRef CAS.
- T. Hubin, J. McCormick, N. Alcock, H. Clase and D. Busch, Inorg. Chem., 1999, 38, 4435–4446 CrossRef CAS.
- G. R. Weisman, E. H. Wong, D. C. Hill, M. E. Rogers, D. P. Reed and J. C. Calabrese, Chem. Commun., 1996, 947–948 RSC.
- W. Niu, E. Wong, G. Weisman, L. Zakharov, C. Incarvito and A. Rheingold, Polyhedron, 2004, 23, 1019–1025 CrossRef CAS.
- A. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- S. H. Vosko, L. Wilk and M. Nusair, Can. J. Phys., 1980, 58, 1200–1211 CrossRef CAS.
- J. Tirado-Rives and W. L. Jorgensen, J. Chem. Theory Comput., 2008, 4, 297–306 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Acc. Chem. Res., 2008, 41, 157–167 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- C. J. Cramer and D. G. Truhlar, Phys. Chem. Chem. Phys., 2009, 11, 10757 RSC.
- T. M. Jones-Wilson, K. A. Deal, C. J. Anderson, D. W. McCarthy, Z. Kovacs, R. J. Motekaitis, A. D. Sherry, A. E. Martell and M. J. Welch, Nucl. Med. Biol., 1998, 25, 523–530 CrossRef CAS.
- Y. Guo, R. Ferdani and C. J. Anderson, J. Lab. Comp. Radiopharm., 2011, 54 suppl 1, S368 Search PubMed.
- M. Fani, L. Del Pozzo, K. Abiraj, R. Mansi, M. Tamma, R. Cescato, B. Waser, W. Weber, J. Reubi and H. Maecke, J. Nucl. Med., 2011, 52, 1110–1118 CrossRef CAS.
- R. A. Dumont, F. Deininger, R. Haubner, H. R. Maecke, W. A. Weber and M. Fani, J. Nucl. Med., 2011, 52, 1276–1284 CrossRef CAS.
- N. M. Di Bartolo, A. M. Sargeson, T. M. Donlevy and S. V. Smith, J. Chem. Soc., Dalton Trans., 2001, 2303–2309 RSC.
- G. A. Bottomley, I. J. Clark, I. I. Creaser, L. M. Engelhardt, R. J. Geue, K. S. Hagen, J. M. Harrowfield, G. A. Lawrance, P. A. Lay, A. M. Sargeson, A. J. See, B. W. Skelton, A. H. White and F. R. Wilner, Aust. J. Chem., 1994, 47, 143–179 CrossRef CAS.
- R. J. Geue, T. W. Hambley, J. M. Harrowfield, A. M. Sargeson and M. R. Snow, J. Am. Chem. Soc., 1984, 106, 5478–5488 CrossRef CAS.
- K. Wieghardt, U. Bossek, P. Chaudhuri, W. Herrmann, B. C. Menke and J. Weiss, Inorg. Chem., 1982, 21, 4308–4314 CrossRef CAS.
- C. A. Boswell, P. McQuade, G. R. Weisman, E. H. Wong and C. J. Anderson, Nucl. Med. Biol., 2005, 32, 29–38 CrossRef CAS.
- Y. Shao, L. F. Molnar, Y. Jung, J. Kussmann, C. Ochsenfeld, S. T. Brown, A. T. B. Gilbert, L. V. Slipchenko, S. V. Levchenko, D. P. O'Neill, R. A. DiStasio Jr, R. C. Lochan, T. Wang, G. J. O. Beran, N. A. Besley, J. M. Herbert, C. Yeh Lin, T. Van Voorhis, S. Hung Chien, A. Sodt, R. P. Steele, V. A. Rassolov, P. E. Maslen, P. P. Korambath, R. D. Adamson, B. Austin, J. Baker, E. F. C. Byrd, H. Dachsel, R. J. Doerksen, A. Dreuw, B. D. Dunietz, A. D. Dutoi, T. R. Furlani, S. R. Gwaltney, A. Heyden, S. Hirata, C.-P. Hsu, G. Kedziora, R. Z. Khalliulin, P. Klunzinger, A. M. Lee, M. S. Lee, W. Liang, I. Lotan, N. Nair, B. Peters, E. I. Proynov, P. A. Pieniazek, Y. Min Rhee, J. Ritchie, E. Rosta, C. David Sherrill, A. C. Simmonett, J. E. Subotnik, H. Lee, Woodcock Iii, W. Zhang, A. T. Bell, A. K. Chakraborty, D. M. Chipman, F. J. Keil, A. Warshel, W. J. Hehre, H. F. Schaefer Iii, J. Kong, A. I. Krylov, P. M. W. Gill and M. Head-Gordon, Phys. Chem. Chem. Phys., 2006, 8, 3172–3191 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of HPLCpurification of CB-TE1A1P (2), X-ray software employed; detailed energetic results for DFT calculations; and NMR spectra of ligand 2 and precursor 6. CCDC reference number 843734. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1dt11743b |
| ‡ In this paper ligand acronyms and numbers refer not only to the neutral species shown in Fig. 1 and elsewhere, but also to the various deprotonated forms that may complex metal cation, with net charge indicating ionization state of the ligand or ligand portion of any complex. |
| This journal is © The Royal Society of Chemistry 2012 |