Spin crossover in polymeric and heterometallic FeII species containing polytopic dipyridylamino-substituted-triazine ligands†
Tamsyn M.
Ross
,
Boujemaa
Moubaraki
,
Stuart R.
Batten
and
Keith S.
Murray
*
School of Chemistry, Building 23, Monash University, Clayton, Victoria 3800, Australia. E-mail: keith.murray@monash.edu; Fax: +61-3-99054597
First published on 22nd August 2011
Abstract
We report the synthesis and characterisation of the new polytopic ligands, ddta (N,N-di(pyridin-2-yl)-4,6-di(1,4,7,10-tetraoxa-13-azacyclopentadecan-13-yl)-1,3,5-triazin-2-amine) and tptd (N2,N2,N4,N4-tetra(pyridin-2-yl)-6-(1,4,7,10-tetraoxa-13-azacyclopentadecan-13-yl)-1,3,5-triazine-2,4-diamine). Each contains N-donor dipyridylamino binding sites as well as separate and distinct mono-aza-15-crown-5 binding sites. The ligand ddta has been used to synthesise the polymeric heterometallic SCO compound trans-[FeII(NCS)2(ddta)2Na2](ClO4)2·4CH3CH2CH2OH, 1, and tptd has been used to synthesise the polymeric SCO compound trans-[FeII(NCS)2(tptd)]·CH3OH, 2, and the dinuclear compound cis-[(FeII)2(NCS)4(tptd)2], 3. Magnetic susceptibility measurements show that 1 and a desolvated sample of 2 each undergo a gradual, one-step spin transition with T½ values of ∼240 K and ∼110 K, respectively. The paucity of inter-chain intermolecular interactions, as well as the flexible, covalent bridges between FeII spin crossover sites, are likely to contribute to the gradual nature of the spin transition observed in each case. Variable temperature powder X-ray diffraction studies on 1 show the anisotropic behaviour of the unit cell parameters, where c and the b-c plane are most affected by structural changes occurring as the temperature is lowered.
Introduction
Spin crossover (SCO) compounds are those with a labile electronic configuration, switchable between high spin (HS) and low spin (LS) states via some external perturbation, most commonly a change in temperature or pressure, or irradiation at a particular wavelength.1 It has long been known that lattice solvent,2 anions,3 ligand conformation4 and intermolecular interactions within the crystal lattice5 can influence both the T½ (the temperature at which half of the SCO centres have undergone a spin transition) and abruptness of a spin transition (i.e. the degree of cooperativity). Within the last decade, focus in the field of SCO research has centred on the study of polymeric species due largely to the possibility of technological application,6 and interest in this area is ongoing.7 Currently there is emphasis on multifunctional SCO materials, especially those displaying synergy between the spin transition and other properties such as liquid crystalline phase transitions,8 or sorption of gas or solvent molecules into porous networks.9Several examples of heterometallic SCO species appear in the literature. Perhaps most notably these include the 2D and 3D Hoffman-type phases which incorporated bridging M(CN)42− (where M = e.g. NiII, PtII or PdII) species between FeII centres, and displayed different temperature dependent magnetic behaviour depending on the nature of the tetracyanometallate ligand.10 Further, a 1D cyano-bridged FeII/MnII species displayed a low temperature spin transition as well as anti-ferromagnetic interactions between FeII and MnII centres.11 In addition to this, a series of SCO compounds of type [FeIIIM(salten)]pac]X (where salten = 4-aza-heptamethylene-1,7-bis(salicylideneiminate), pac = N-(4-picolyl)-aza-15-crown-5-ether, M = Li+, Na+, K+ or Rb+, X = ClO4− or BPh4−) were reported, where the nature of the spin transition occurring at FeIII centres was influenced by both the presence or absence of a guest within the mono-aza-15-crown-5 ring of pac, and by the nature of the guest.12 No structures were reported.
Judicious ligand design is an important approach to targeted syntheses in the field of metallosupramolecular chemistry,13 and the use of polytopic ligands incorporating crown moieties provides an effective means of developing new heterometallic species, as exemplified by the [FeIIIM(salten)]pac]X series,12 as well as exciting metallosupramolecular constructs. This second point is highlighted by a recent study of the versatile, polytopic ligand N,N′-bis(4-pyridyl-methyl)diaza-18-crown-6, which may function as a variable length ligand, giving different separation distances of the 2-connecting bis-pyridine moieties as dependent on the guest species bound within the di-aza-crown.14
The new polytopic ligands presented here, ddta (N,N-di(pyridin-2-yl)-4,6-di(1,4,7,10-tetraoxa-13-azacyclopentadecan-13-yl)-1,3,5-triazin-2-amine) and tptd (N2,N2,N4,N4-tetra(pyridin-2-yl)-6-(1,4,7,10-tetraoxa-13-azacyclopentadecan-13-yl)-1,3,5-triazine-2,4-diamine), shown in Fig. 1, have been designed for incorporation into heterometallic FeII SCO compounds, and each contain several distinct coordination sites which are geared specifically for the binding of particular, different metallic species. The dipyridylamino chelators in each ligand are intended here as bidentate N,N-donors for FeII centres such that an FeII(NCX)2(py)4-type (where NCX = NCS−, NCSe− or NCBH3−etc., and py = a pyridyl donor) coordination sphere may be obtained, as this combination has been shown to impart a ligand field appropriate for SCO in FeII species.15 The mono-aza-15-crown-5 moieties in ddta and tptd may separately bind guest species such as, for example, some group 1 and 2 metals of varying sizes.
 | ||
| Fig. 1 (a) The ligand ddta (N,N-di(pyridin-2-yl)-4,6-di(1,4,7,10-tetraoxa-13-azacyclopentadecan-13-yl)-1,3,5-triazin-2-amine); and (b) the ligand tptd (N2,N2,N4,N4-tetra(pyridin-2-yl)-6-(1,4,7,10-tetraoxa-13-azacyclopentadecan-13-yl)-1,3,5-triazine-2,4-diamine). | ||
The current study proceeds from previous work within our group on FeII SCO compounds containing dipyridylamino-ligands which have displayed intriguing magnetic properties, such as the 1D trans-[FeII(NCX)2(L)]·Solvent series (where L is an s-triazine species with two dipyridylamino substituents). Here, some compounds were shown to undergo a ‘half’ spin transition with ordered, alternating –HS–LS–HS–LS– FeII centres within 1D chains in the low temperature plateau region, as well as interesting solvent dependence of T½, cooperativity and the mechanism of the spin transition, and some LIESST (that is light induced excited spin state trapping)16 activity.17–19 Reedjik et al. have also reported FeII complexes, containing ligands similar to L, which display novel magnetic properties.20–22 The use of polytopic ligands comprising crown ether moieties is a relatively unexplored means by which to obtain heterometallic SCO species, with the [FeIIIM(salten)]pac]X series,12 to our knowledge, the only example of such a study to appear in the literature. The primary route to heterometallic SCO species involves the use of metalloligands, as in the Hoffman-type phases mentioned above.10
Initially, here, we sought to determine whether functionalisation of L-type ligands with mono-aza-crowns would afford heterometallic FeII SCO species, with a view to determining to what extent targeted syntheses were possible, and to conduct magnetostructural studies on any resulting compounds. While we have determined that ddta may be used to afford heterometallic FeII SCO species, targeted syntheses in this area have proven challenging, and we present our results from initial work with the ligands ddta and tptd, including magnetic susceptibility measurements on the novel 1D heterometallic SCO compound trans-[FeII(NCS)2(ddta)2Na2](ClO4)2·4CH3CH2CH2OH, 1, as well as on a desolvated sample of the 1D SCO species trans-[FeII(NCS)2(tptd)]·CH3OH, 2. Structural data on 1 are presented at both LS and HS temperatures, and are accompanied by variable temperature powder X-ray crystallographic data mapping the unit cell parameters over the course of the spin transition. Structural data are presented on 2 and also on the dinuclear compound cis-[(FeII)2(NCS)4(tptd)2], 3. Briefly, a qualitative discussion of factors influencing the nature of the spin transition for 1 and the desolvated sample of 2 is given, and future directions for this research are mentioned in the Conclusions.
Results and discussion
Syntheses
The ligands ddta and tptd (Fig. 1) were synthesised via the reaction of an s-triazine containing one or two dipyridylamino substituent(s) with, respectively, two or one equivalent(s) of mono-aza-15-crown-5. The 1H NMR and 13C NMR spectra for ddta and tptd appear in the ESI.†Mono-aza-15-crown-5, as contained in the ligands ddta and tptd, may play host to a range of different guests, such as the s-block group 1 and 2 metals, and some lanthanides, main group and transition metals.23 The structures of these crown-5-guest crystals23 generally show the cation to be out of the plane of the four O atoms even for the small Li+ and the larger Ba2+. Thus, the new ligands ddta and tptd present the possibility for a substantial number of novel heterometallic FeII SCO compounds, with a range of possible dimensionalities and different non-coordinating anions. However, as described below, significant difficulties have been met in obtaining the intended heterometallic products.
The coordination polymer 1 was synthesised via the reaction of two equivalents of the ddta ligand with one equivalent of Fe(NCS)2, giving a yellow solution in 1-propanol. Upon addition of a solution of excess NaClO4 in 1-propanol, a yellow precipitate formed immediately, which was subsequently dissolved by the addition of a small amount of methanol. The resultant yellow solution was allowed to slowly evaporate, giving crystals of 1. Although many attempts were made to incorporate other crown guest species into the mono-aza-15-crown-5 moieties of ddta using this method, including the smaller cation Li+, and also to isolate an ‘empty’ crown analogue of 1 in crystalline form, none were successful here.
The synthesis of 2 was carried out using an H-shaped tube; here Fe(NCS)2 was placed in one arm, while in the other was placed a mixture of tptd (in molar amounts equivalent to a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio for Fe(NCS)2:tptd), along with excess Ba(ClO4)2. Despite conducting the synthesis of 2 in the presence of Ba(ClO4)2, with Ba2+ known to bind to mono-aza-15-crown-5 ligands,23 none was found in the structure of 2. The distinctive, needle-shaped crystals of 2 were hand separated from a small amount of foreign matter present in the H-shaped tube prior to bulk measurements. Hand-sorting took place over the course of one week, during which time crystals of 2 were open to the atmosphere and lost all lattice methanol, as indicated by microanalysis. Due to the very small size of the crystals, hand-sorting under a blanket of methanol in order to preserve lattice solvation was extremely difficult, and was not carried out. The synthesis of the dinuclear compound 3 was carried out in an analogous fashion to that of 2, although no Ba(ClO4)2 was introduced into the reaction mixture, and the concentration of reactants in solution was lower than that used for 2. Compound 3 was formed in almost negligible quantities. Although the synthesis of 3 was repeated a number of times in the hope of gathering sufficient sample for bulk measurements, significant difficulty was encountered in isolating the small quantities of tiny crystallites that formed in all cases, and bulk measurements could not be carried out. It is to be noted that, for each of 2 and 3, crystals were of insufficient size for single crystal X-ray experiments using laboratory sources, and synchrotron radiation was used for data collection.
:1 ratio for Fe(NCS)2:tptd), along with excess Ba(ClO4)2. Despite conducting the synthesis of 2 in the presence of Ba(ClO4)2, with Ba2+ known to bind to mono-aza-15-crown-5 ligands,23 none was found in the structure of 2. The distinctive, needle-shaped crystals of 2 were hand separated from a small amount of foreign matter present in the H-shaped tube prior to bulk measurements. Hand-sorting took place over the course of one week, during which time crystals of 2 were open to the atmosphere and lost all lattice methanol, as indicated by microanalysis. Due to the very small size of the crystals, hand-sorting under a blanket of methanol in order to preserve lattice solvation was extremely difficult, and was not carried out. The synthesis of the dinuclear compound 3 was carried out in an analogous fashion to that of 2, although no Ba(ClO4)2 was introduced into the reaction mixture, and the concentration of reactants in solution was lower than that used for 2. Compound 3 was formed in almost negligible quantities. Although the synthesis of 3 was repeated a number of times in the hope of gathering sufficient sample for bulk measurements, significant difficulty was encountered in isolating the small quantities of tiny crystallites that formed in all cases, and bulk measurements could not be carried out. It is to be noted that, for each of 2 and 3, crystals were of insufficient size for single crystal X-ray experiments using laboratory sources, and synchrotron radiation was used for data collection.
It is clear that, for the polyfunctional ligands shown in Fig. 1, the nature of the final product is governed by subtle factors, and targeted syntheses remain a challenge for ongoing work in this area. Determination of intermediate solution-phase species present during formation of the final, crystalline products may be aided by such techniques as those employed by the L. Cronin group in the detection of self-assembled nano-structures in the solution phase.24
Magnetic susceptibility data for trans-[FeII(NCS)2(ddta)2Na2](ClO4)2·4CH3CH2CH2OH, 1, and a desolvated sample of trans-[FeII(NCS)2(tptd)]·CH3OH, 2
Variable temperature magnetic susceptibility measurements were carried out on a sample of 1 that was maintained in 1-propanol both prior to and during measurements, to ensure that there was no loss of the 1-propanol molecules which comprise the lattice solvent in 1. As seen in Fig. 2(a), 1 undergoes a gradual spin transition. At 270 K, per FeII, χMT = 3.1 cm3 mol−1 K, which indicates that 1 is almost, but not completely, HS at this temperature. As the temperature is lowered from 270 K, χMT values begin to drop immediately, and continue to decrease in a smooth, gradual fashion until ∼175 K, at which point a low of χMT = 0.1 cm3 mol−1 K is reached and a plateau follows, typical of the LS state for FeII. A T½ value of ∼240 K is observed for 1.The χMT vs. T plot for a desolvated sample of 2 is shown in Fig. 2(b). As seen for 1, the desolvated complex 2 undergoes a complete, continuous spin transition. At ∼300 K a χMT = 3.4 cm3 mol−1 K is observed for 2, which is indicative of HS FeII. This χMT = 3.4 cm3 mol−1 K is maintained as the temperature is lowered until ∼175 K, at which point χMT begins to drop gradually. Values of χMT decrease from ∼175 K until ∼60 K, whereupon a low value of χMT = ∼0.3 cm3 mol−1 K is reached. The small increase in χMT values below ∼60 K is due to some thermal HS trapping (this effect has been caused by quench cooling of the sample of 2, indicating that magnetic susceptibility measurements on 2 have been made upon heating from low temperatures to high temperatures, rather than cooling from room temperature). A T½ of ∼110 K is observed for the desolvated complex 2.
Structural data on trans-[FeII(NCS)2(ddta)2Na2](ClO4)2·4CH3CH2CH2OH, 1, at 123 K and 270 K
The asymmetric units of 1a (1 at 123 K) and 1b (1 at 270 K) are identical, and each consists of one half occupancy FeII atom, lying on a centre of inversion, that is coordinated to the chelating dipyridylamino moiety of a single unique ddta ligand. The dipyridylamino moiety of ddta chelates to this FeII centre in equatorial positions, and a single unique NCS− ligand is located in an axial position (see Fig. 3). Thus, the first coordination sphere of each FeII centre, when completed by symmetry, consists of (NCS)2(py)4. A single, unique Na+ cation is bound at one mono-aza-15-crown-5 moiety of the single unique ddta ligand, and one perchlorate anion and two molecules of 1-propanol are present as separate entities located within interstitial space in the crystal lattice (see Fig. 3).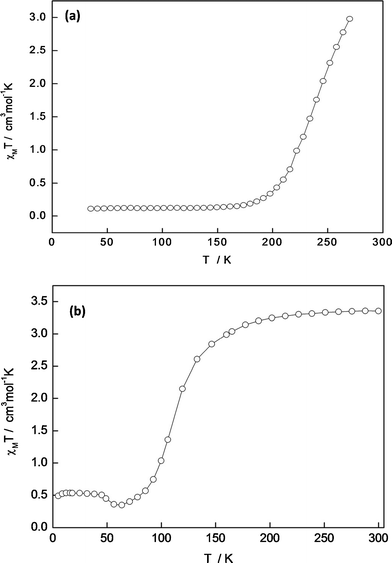 | ||
| Fig. 2 (a) Plot of χMT vs. T for 1; and, (b) plot of χMT vs. T for a desolvated sample of 2. | ||
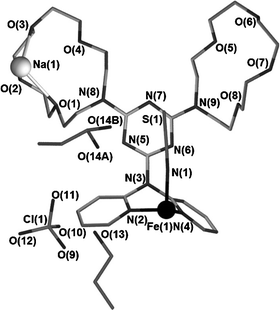 | ||
| Fig. 3 Asymmetric unit for 1a. The FeII atom is shown as a black sphere, while the Na+ atom is shown as a white sphere. Hydrogen atoms and labels for carbon atoms have been omitted for clarity. | ||
The extended structure of 1 consists of infinite 1D polymeric chains, in which each [FeII(NCS)2(ddta)2] monomeric unit is connected to the next via four bridging Na+ cations which are held within the mono-aza-15-crown-5 moieties of ddta (see Fig. 4(a)). Na+ cations are sandwiched between the mono-aza-15-crown-5 moieties on adjacent ddta ligands within the 1D chains. The sandwiched Na+ cations are bound asymmetrically, where all four oxygen donors of one mono-aza-15-crown-5 moiety coordinate to Na+ cations, while only three oxygen donor atoms of a second mono-aza-15-crown-5 moiety coordinate to Na+ cations, affording an O7 first coordination sphere to each Na+ cation (Fig. 4(b)). Such asymmetric binding of bridging Na+ cations to the oxygen donor atoms of sandwiching mono-aza-15-crown-5 species has been seen previously.25
![(a) A {[FeII(NCS)2(ddta)2Na2]2+}∞ chain in 1; (b) sandwiched Na+ atoms within 1; (c) packing of 1 with view down the a-axis, and; (d) packing of 1 with view down the c-axis. Hydrogen atoms, lattice solvent and perchlorate anions have been omitted for clarity. FeII atoms are shown as black spheres, while Na+ atoms are shown as white spheres.](/image/article/2012/DT/c1dt10818b/c1dt10818b-f4.gif) | ||
| Fig. 4 (a) A {[FeII(NCS)2(ddta)2Na2]2+}∞ chain in 1; (b) sandwiched Na+ atoms within 1; (c) packing of 1 with view down the a-axis, and; (d) packing of 1 with view down the c-axis. Hydrogen atoms, lattice solvent and perchlorate anions have been omitted for clarity. FeII atoms are shown as black spheres, while Na+ atoms are shown as white spheres. | ||
The parallel-packed 1D chains of {[FeII(NCS)2(ddta)2Na2]2+}∞ propagate in the direction of the a-axis (Fig. 4(a)). Fig. 4(c) and (d) show packing diagrams for 1. As can be seen in Fig. 4(c), the Na+ complexed mono-aza-15-crown-5 substituents of ddta ligands have considerable steric bulk and result in somewhat larger interchain Fe⋯Fe separations than have been noted for 1D polymeric species which incorporate similar, but less bulky ligands (see Fe⋯Fe separations in Table 1: in 1a, the closest inter-chain Fe⋯Fe separation is 13.2322(8) Å). In comparison, for the 1D trans-[FeII(NCX)2(L)]·solvent series, an inter-chain Fe⋯Fe separation of ca. 10 Å is observed.17,18 Accommodation of the non-coordinating perchlorate anions and the 1-propanol molecules into interstitial lattice space also contributes to the large inter-chain Fe⋯Fe separations observed for 1. The closest inter-chain Na+⋯Na+ separation is 7.946(3) Å in 1a (8.065(4) Å in 1b). Within the {[FeII(NCS)2(ddta)2Na2]2+}∞polymers an intra-chain Fe⋯Fe separation of 12.2574(7) Å for 1a is observed.
| 1a | 1b | 2 | 3 | |
|---|---|---|---|---|
| a Σ° = the sum of |90−θ| for the twelve N–Fe–N angles in the octahedron.15 b The intra-molecular Fe⋯Fe separation. c The closest intermolecular Fe⋯Fe distance. | ||||
| T/K | 123 | 270 | 100 | 100 |
| Fe1–NNCS1(Å) | 1.945(3) | 2.068(5) | 2.071(3) | 1.987(7) |
| Fe1–NNCS2(Å) | — | — | 2.034(3) | 1.968(7) |
| Fe1–Npy(Å) | 1.995(3) | 2.168(4) | 2.130(4) | 1.999(7) |
| 1.985(3) | 2.145(4) | 2.133(4) | 2.011(6) | |
| 2.126(3) | 2.030(6) | |||
| 2.141(3) | 1.999(7) | |||
| Fe1–N–CNCS1° | 174.5(3) | 174.4(4) | 148.4(3) | 167.8(6) |
| Fe1–N–CNCS2° | — | — | 169.2(4) | 164.9(8) |
| ΣFe1° a | 27 | 41 | 45 | 27 |
| Fe1⋯Fe1intrab | 12.2574(7) | 12.304(2) | 9.250(1) | 9.244(5) |
| Fe1⋯Fe1interc | 13.2322(8) | 13.400(2) | 9.553(1) | 8.406(5) |
Parameters describing the first coordination sphere of FeII centres in 1 are located in Table 1. Both Fe–Npy and Fe–NNCS bond lengths in 1a are indicative of LS FeII, in agreement with magnetic susceptibility measurements which suggest that the spin transition for 1 is complete by 123 K. Within 1b, Fe–N bond lengths are indicative of an admixed HS/LS character, with more HS than LS contribution, which also agrees with magnetic susceptibility measurements on the bulk, indicating an almost, but not entirely, HS state exists in 1 at 270 K. The octahedral distortion of the first coordination sphere of the FeII site, as measured by Σ° (see Table 1), is smaller in 1a than in 1b, in line with previous studies finding that octahedral distortion decreases as the spin transition takes place upon lowering of the temperature.15 Generally, for 1, the Na–O bond lengths do not change over the course of the spin transition, although an almost negligible contraction of Na–O5 is noted between 1a and 1b (in 1a Na–O5 is 2.766(3) Å and in 1b Na–O5 is 2.798(4) Å).
No prominent intermolecular inter-chain interactions, such as hydrogen bonding or aromatic interactions, are seen within the structure of 1, suggesting that dispersion forces are important for crystal cohesion here. Intra-molecular S⋯π interactions are noted to occur between the electron-rich S atoms of NCS− ligands and the electron deficient central s-triazine ring of ddta ligands,26 and in 1a the S⋯centroidtriazine separation is 3.40 Å, while in 1b this distance is 3.50 Å. A hydrogen bonding interaction occurs between NCS− ligands and one orientation of the hydroxyl group of the 1-propanol molecule in which the hydroxyl group is disordered over two positions (see Fig. 3); for this interaction in 1a the S⋯O distance is 3.266(8) Å, while in 1b the S⋯O distance is 3.50(2) Å. A hydrogen bonding interaction is also noted to occur between the second unique 1-propanol molecule (i.e. the 1-propanol molecule that does not participate in a hydrogen bonding interaction with NCS− ligands) and the ClO4− anion (here, the H⋯O distance is 2.08 Å for 1a, and a H⋯O contact of 2.16 Å for 1b).
Structural data on trans-[FeII(NCS)2(tptd)]·CH3OH, 2, and cis-[(FeII)2(NCS)4(tptd)2], 3, at 100 K
The asymmetric units of both 2 and 3 consist of one unique tptd ligand which is coordinated via only one of its chelating dipyridylamino groups to one unique FeII centre at full occupancy. The single unique FeII centre in both 2 and 3 is also ligated to two unique NCS− ligands, NCS1− and NCS2− – in 2 these NCS− ligands occupy axial positions, whereas in 3 they are arranged to give a cis-conformation. For each of 2 and 3, a FeII(NCS)2(py)4-type coordination sphere is observed, when completed by symmetry, as seen for 1. The asymmetric unit of 2 also includes one methanol molecule which engages in a hydrogen bonding interaction with the NCS1− ligand (here the S⋯H contact is 2.59 Å), whereas 3 is unsolvated. Minor positional disorder within the mono-aza-15-crown-5 moiety of tptd is noted in both 2 and 3. Full, labelled asymmetric unit diagrams are located in the ESI.†In an analogous fashion to what has been seen for the 1D trans-[FeII(NCX)2(L)]·solvent series,17,18 the extended structure of 2 consists of 1D polymeric chains (Fig. 5(a), (b) and (c)) which propagate parallel to the c-axis, for which the asymmetric unit may be regarded as the monomeric unit. Within the 1D chains, FeII atoms lie close to, but not on, the c-glide plane (Fig. 5(b)). In contrast, the extended structure of 3 consists of dinuclear molecules (Fig. 6(a)) in which two tptd ligands bridge between two FeII sites. Each tptd ligand in 3 shares two common FeII sites with its symmetry generated equivalent – this arrangement is in contrast to that seen in 2, where each tptd ligand shares only one common FeII site with its symmetry generated equivalents.
 | ||
| Fig. 5 (a) The 1D chain structure of 2; (b) packing of 2 with view down the c-axis; and (c) packing of 2 with view down the b-axis. Hydrogen atoms and lattice solvent have been omitted for clarity. FeII atoms are shown as black spheres. | ||
As seen in Table 1, the Fe–Npy and Fe–NNCS bond lengths in 2 suggest an admixed HS/LS character at FeII sites, with more HS than LS contribution. Within 3, both the Fe–NNCS and Fe–Npy bond lengths indicate that FeII is in a predominantly LS state. The smallest inter-dinuclear Fe⋯Fe separation in 3 is 8.406(5) Å (Table 1), which is somewhat smaller than the inter-chain Fe⋯Fe separation noted for 2 (9.553(1) Å, which occurs between diagonally opposed 1D chains (Fig. 5(b)). The intra-dinuclear Fe⋯Fe separation in 3 is 9.244(5) Å which is, predictably, not significantly different to the intra-chain Fe⋯Fe separation for 2.
It may be notable that, in 2, the angles Fe1–N–CNCS° and Fe2–N–CNCS° differ significantly (Table 1). Within 2, the S atom of NCS2− is disordered over two orientations, modelled at 30% and 70% occupancy for S2A and S2B, respectively. The S atoms of NCS1− and NCS2− each participate in an intra-molecular S⋯π interaction with the central s-triazine core of tptd, where for parts S2A and S2B the S⋯centroidtriazine separation is 3.56 Å and 3.76 Å, respectively. In comparison to the position of the NCS2− ligands, the NCS1− ligands are somewhat off-centre with respect to the s-triazine core of tptd ligands, with a S⋯centroidtriazine separation of 4.11 Å. Within 3, no intra-molecular S⋯π interactions are noted, although they are noted between the S atoms of NCS1− ligands and the central s-triazine rings of tptd ligands in adjacent dinuclear molecules (see Fig. 6(b)), with a corresponding S⋯centroidtriazine separation of 3.24 Å.
 | ||
| Fig. 6 (a) The dinuclear structure of 3, and (b) packing of 3 with view down the b-axis, showing inter-dinuclear S⋯π interactions as black dotted lines. Hydrogen atoms have been omitted for clarity, and FeII atoms are shown as black spheres. | ||
Within the extended structure of 2, an edge-to-face C–H⋯π interaction occurs between one unique pyridyl ring of the tptd ligands within a particular 1D chain and the central s-triazine cores of tptd ligands within a diagonally opposed 1D chain (Fig. 5(b)), where these are the diagonally opposed chains between which the closest inter-chain Fe⋯Fe distance is noted. Here, the closest H⋯Ctriazine contact is at a distance of 2.67 Å (the H⋯centroidtriazine distance is 3.04 Å). Another close C⋯H contact, between pyridyl rings on adjacent 1D chains, is also observed (here the closest C⋯H contact is 2.91 Å), suggesting another inter-chain edge-to-face C–H⋯π interaction. Within 3, a close C⋯H contact of 3.19 Å between pyridyl rings on adjacent dinuclear molecules may suggest an edge-to-face C–H⋯π interaction. Close inter-chain and inter-dinuclear S⋯H contacts of ∼2.8–2.9 Å, occurring between NCS− ligands and pyridyl rings or aliphatic C–H within mono-aza-15-crown-5 moieties of tptd, are noted within both 2 and 3.
Variable temperature powder X-ray diffraction studies on trans-[FeII(NCS)2(ddta)2Na2](ClO4)2·4CH3CH2CH2OH, 1
Variable temperature powder X-ray diffraction measurements on 1 were taken at the Australian Synchrotron. A sample of 1, damp with 1-propanol to preserve the lattice solvent, was loaded into a glass capillary. The sample was quench cooled to 150 K, and powder X-ray diffraction images were collected at 5 or 10 K intervals as the temperature was raised to 300 K. The lattice parameters at each temperature were determined from a single Le Bail fit to the data based on unit cell data determined using single crystal X-ray diffraction techniques, and are listed in the ESI.† Further details are contained within the Experimental section. Compound 1 crystallises in the triclinic space groupP![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and no crystallographic phase transitions were observed over the temperature range investigated, in agreement with single crystal data.
and no crystallographic phase transitions were observed over the temperature range investigated, in agreement with single crystal data.
For 1, a total unit cell volume contraction of ca. 4.3% of the HS volume (taken here as the unit cell volume at 300 K) is observed. It has been determined in earlier studies comparing SCO compounds with non-SCO analogues that a unit cell volume contraction of ca. 2% may be expected to occur due to the spin transition,27 indicating that a significant portion of the unit cell volume contraction seen for 1 is due solely to general thermal contraction effects.
Fig. 7 (a) gives the change in the unit cell parameters a, b and c as a proportion of their highest value over the temperature range 150–300 K, while Fig. 7(b) shows the behaviour of the angles α, β and γ, given as a proportion of their highest value over the temperature range 150–300 K. It may be seen that, with falling temperature, a and b decrease in an almost linear fashion, giving little indication of the completion of the spin transition below ∼200 K.
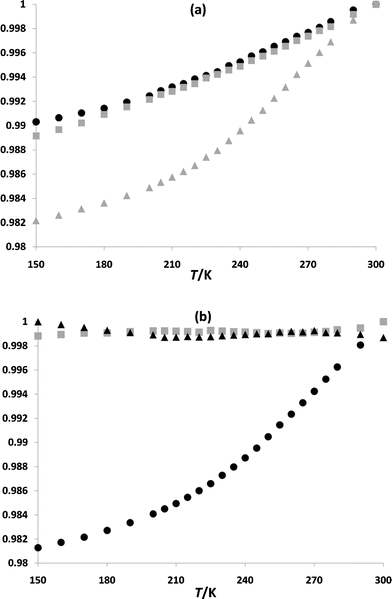 | ||
| Fig. 7 (a) The temperature dependent behaviour of a (shown as black circles), b (shown as gray squares) and c (shown as gray triangles) for 1, and (b) the temperature dependent behaviour of the angle α (shown as black circles), the angle β (shown as gray squares) and the angle γ (shown as black triangles). Each parameter is given as a proportion of its highest value (the vertical axes) over the temperature range investigated. | ||
It has previously been noted that the curve described by the temperature dependent behaviour of the unit cell volume reflects the shape of the χMT vs. T susceptibility plot for a particular compound.27 For 1, the curve described by the temperature dependent behaviour of the unit cell volume is very similar to that of c (Fig. 7 (a)) reflecting the gradual nature of the spin transition for 1 (see magnetic discussion above and the ESI† for further details).
From Fig. 7(a) and (b) it may be seen that c and the b–c plane are most affected by structural changes occurring as the temperature is lowered. Previous studies have shown that the anisotropic behaviour of the lattice parameters are determined by the crystal packing, which dictates how the changes due to the spin transition are distributed in different directions in space.15,28 It is likely that the contraction of c is less inhibited by steric interactions between 1D chains than that of b – several close contacts are found, for example, between 1D chains related by symmetry by a one unit cell length translation in the direction of the b-axis, while, largely, 1D chains generated by a one unit cell-length translation in the direction of the c-axis are separated by lattice solvent and anions (see Fig. 4(c)). 1D chains run parallel to a, and it is likely that inhibited contraction within the 1D chains may be the cause of the comparatively small contraction in this direction (similarly, the 1D chains run across the a–c and a–b planes, which is a likely to have influenced the smaller deformations of these planes in comparison to the b–c plane, see Fig. 4(a), (c) and (d)).
Discussion of factors influencing the nature of the spin transition observed for 1 and 2
While a full understanding of cooperativity lies in the area of solid state physics and the complex areas of phonon effects and elastic/mechanical effects,29 we present here a brief discussion of chemical/structural influences on magnetic character at a qualitative level. It has been established in previous work that the 4,6-bis(dipyridylamine)-1,3,5-triazine}-type covalent bridges between FeII centres, afforded by ligands of type L such as tptd, are ineffective in the communication of structural changes occurring due to the spin transition.17,18 This is in agreement with previous studies where flexible covalent bridges between SCO centres in polymeric species have been shown to result in poorly cooperative systems.30 More effective cooperative pathways within this family of compounds may involve intermolecular interactions between 1D chains, such as in the compound trans-[FeII(NCS)2(DPT)]·2CH3OH·H2O (where DPT = 6-phenoxy-N2,N2,N4,N4-tetra-2-pyridinyl-1,3,5-triazine-2,4-diamine) which contains a localised hydrogen bonding network between NCS− ligands on SCO centres,18 or in the related monomeric compound trans-[FeII(NCS)2(dpyatriz)2] (where dpyatriz = N2,N2,N4,N4,N6,N6-hexa(pyridin-2-yl)-1,3,5-triazine-2,4,6-triamine) where S⋯S interactions between NCS− ligands on adjacent monomers, as well as aromatic interactions, were thought to improve the cooperativity of the system.21 This is in line with previous findings that intermolecular interactions such as strong aromatic31 or hydrogen bonding32 interactions may improve the cooperativity of SCO systems.Compound 1 contains no significant, strong, directional intermolecular contacts (e.g.hydrogen bonding or aryl interactions) directly linking FeII centres on adjacent 1D chains, and this, in combination with the long, flexible bridges between FeII centres within 1D chains, is likely to contribute to the gradual nature of the spin transition observed viamagnetic susceptibility measurements. It is likely that the gradual nature of the spin transition observed for the desolvated sample of 2 is attributable, at least partly, to similar structural aspects, although a limited number of aromatic interactions were identified in the structure of solvate 2. Further, magnetic susceptibility measurements on the desolvated sample of 2 suggest that at 100 K (the temperature at which structural data for 2 were collected) the spin transition is approximately two-thirds complete, which is at odds with the Fe–N bond lengths (Table 1), suggesting more HS than LS contribution at this temperature. As previously mentioned, magnetic susceptibility measurements on 2 were carried out on a desolvated sample which had lost lattice methanol, and it is likely that the loss of lattice methanol has caused a shift in T½ – in this case to a higher temperature than that which would have been observed for the parent solvate. Quench cooling of the crystal of 2 used for single crystal studies may also have contributed to the longer than expected Fe–N bond lengths.
Compound 2 constitutes a comparatively rare example, within the trans-[FeII(NCX)2(L)]·solvent series,17,18 of a 1D polymer with a single unique FeII centre; the only other known example is the HS phase trans-[FeII(NCS)2(cddt)2]·0.5CHCl3·H2O (where cddt = 6-chloro-N2,N2,N4,N4-tetra(pyridin-2-yl)-1,3,5-triazine-2,4-diamine), where all other known trans-[FeII(NCX)2(L)]·solvent species contain two unique FeII centres.17,18 That only one unique FeII centre is found in 2 may contribute to the fact that a complete, one-step spin transition is observed, rather than the ‘half’ SCO observed elsewhere within the trans-[FeII(NCX)2(L)]·solvent series, although it has to be mentioned that compounds containing a single unique SCO site have been observed previously to undergo two-step or incomplete spin transitions.19,33
It was anticipated that the binding of different cation guest species within, or close to, aza-crown moieties of tptd and ddta within heterometallic SCO compounds would affect the magnetic behaviour of this species, as for the [FeIIIM(salten)]pac]X series.12 It is likely that differences in magnetic behaviour within such an analogous series would be qualitatively attributable to factors such as packing differences, or differences in the electronic influence of guest species. Difficulty in synthesising analogues of 1 containing different guest species within the aza-crown moiety renders it difficult to comment here on the precise nature of the influence of Na+ atoms on the magnetic behaviour of this compound.
Conclusions
Compound 1 shows that the synthesis of heterometallic FeII SCO compounds that incorporate the type of mono-aza-crown-functionalised, dipyridylamino-substituted s-triazine ligands presented here is possible. Difficulties in obtaining targeted heterometallic products with these polyfunctional ligands relate primarily to the failure to capture the intended guest within the mono-aza-crown moiety (as demonstrated by 2 and 3). Magnetic susceptibility measurements on 1 and on a desolvated sample of 2 show that each undergoes a gradual, one-step spin transition, with T½ values of ∼240 K and ∼110 K, respectively. The paucity of strong, directional intermolecular interactions between 1D chains in 1 and 2, as well as the flexible covalent bridges between FeII centres, are likely to have contributed to the gradual nature of the spin transition observed for both species. The gradual nature of the spin transition observed for 1 is also reflected in the temperature dependent behaviour of the unit cell parameters.For the previously mentioned [FeIIIM(salten)]pac]X series, no structural data were obtained, and, similarly, for the heterometallic SCO Hoffmann-type phases, structural data are rather limited.10,12 Although some difficulties have been met in the present study in obtaining the desired heterometallic species, it has been found, here and in previous studies, that FeII systems comprising dipyridylamino-substituted s-triazine ligands have a good ability to crystallise.17,18,20–22 Polytopic dipyridylamino-substituted s-triazine ligands, such as ddta and tptd, are thus good candidates for the investigation of heterometallic SCO species using single crystal techniques, a situation found not to be previously possible. Within our group work is proceeding using polytopic ligands similar to ddta and tptd with aza-crown moieties of different sizes.
Experimental
General
All reagents and solvents were purchased from Sigma-Aldrich Pty. Ltd. and used as received. The syntheses of the ligands ddta and tptd were carried out under a flow of N2 gas. 1H NMR and 13C NMR spectra were recorded on a Bruker DPX 200 MHz, Bruker DPX 300 MHz or Bruker Avance 400 MHz NMR spectrometer at ambient temperatures. Low resolution ESI mass spectra were recorded using a Micromass (now Waters) Platform II QMS connected to a Waters Alliance 2690 LC system for automatic flow injections. IR Spectra were recorded on a Bruker Equinox 55 FT-IR fitted with a Judson MCT detector and Specac ‘Golden Gate’ diamond ATR. Microanalyses were performed by Campbell Microanalytical Laboratory, Department of Chemistry, University of Otago, Dunedin, New Zealand. The compounds 4,6-dichloro-N,N-di-2-pyridinyl-1,3,5-triazin-2-amine,34cddt,35 and 1,4,7,10-tetraoxa-13-azacyclopentadecane,36 as used in the synthesis of the ligands ddta and tptd, were prepared as previously described.Magnetic susceptibility measurements
Magnetic susceptibility data were collected using a Quantum Design MPMS 5 SQUID magnetometer under an applied field of 1 T. A solvated sample of 1 was placed in a quartz tube along with 1-propanol, and care was taken to avoid solvent loss. An unsolvated sample of 2 was dispersed in a small amount of grease and placed in a gel capsule held within a plastic straw.Crystallographic data collection and refinement
Single crystal diffraction data for the structures 1a and 1b were collected on a Bruker APEX X8 diffractometer using Mo-Kα (λ = 0.71073 Å) radiation and equipped with an Oxford Instruments N2 gas cryostream. Crystals were mounted on a glass fibre in a small amount of oil. For 1a, a single crystal was quench-cooled to 273(2) K for data collection, and then the sample crystal was cooled slowly to 123(2) K for 1b. Initial diffraction data analysis was performed using the APEXII software package.37 The structures were solved by direct methods using SHELXS-97, and least-squares refinements against F2 were carried out using SHELXL-97, using the program X-Seed as a graphical interface.38 All non–hydrogen atoms in the structures were refined anisotropically (unless specified) and hydrogen atoms were generated using the riding model.Data for 2 were collected at the Australian Synchrotron using the MX1 beamline operating at ∼16 KeV (λ = 0.71079 Å). Data for 3 were collected at the Australian Synchrotron using the MX2 beamline operating at ∼16 KeV (λ = 0.71073 Å). The collection temperature was maintained at specified temperatures using an open-flow N2 cryostream. In each case single crystals of 2 and 3 were quench cooled to 100(2) K.
Data were collected using the Blue Ice software.39 Initial data processing was carried out using the XDS package.40 Both structures were solved by direct methods using SHELXS-97, and least-squares refinements against F2 were carried out using SHELXL-97, using the program X-Seed as a graphical interface.38Hydrogen atoms were generated using the riding model. Special refinement details are located in the ESI.†
CIFs of 1a, 1b, 2 and 3 are available in the ESI, reference numbers: 823862–823865.†Crystallographic data and refinement details are given in Table 2.
| 1a | 1b | 2 | 3 | |
|---|---|---|---|---|
| a R1 = Σ∥Fo| − |Fc∥/Σ|Fo|, b wR2 = [Σw(Fo2 − Fc2)2/Σ(Fo2)2]½ | ||||
| Formula | C80H128Cl2FeN18Na2O28S2 | C80H128Cl2FeN18Na2O28S2 | C36H40FeN12O5S2 | C70H72Fe2N24O8S4 |
| FW (g mol−1) | 2026.85 | 2026.85 | 840.77 | 1617.46 |
| T (K) | 123 | 270 | 100 | 100 |
| Crystal aystem | Triclinic | Triclinic | Monoclinic | Monoclinic |
| Space group |
P
|
P
|
P21/c | P21/n |
| Z | 1 | 1 | 4 | 2 |
| a (Å) | 12.2574(5) | 12.304(2) | 17.495(2) | 14.83(1) |
| b (Å) | 13.2322(8) | 13.400(2) | 12.707(2) | 14.78(1) |
| c (Å) | 16.4992(10) | 16.754(3) | 18.477(2) | 16.483(8) |
| α (°) | 76.909(2) | 78.495(7) | — | — |
| β (°) | 87.406(2) | 88.175(7) | 104.548(3) | 94.52(4) |
| γ (°) | 65.031(2) | 65.049(7) | — | — |
| V (Å3) | 2359.0(2) | 2449.9(7) | 3974.3(9) | 3602(4) |
| ρcalc (gm cm−3) | 1.427 | 1.374 | 1.405 | 1.491 |
| μ (mm−1) | 0.356 | 0.343 | 0.543 | 0.594 |
| Collected data | 15330 | 20028 | 56749 | 54682 |
| Unique data (Rint) | 8052 (0.0496) | 8449 (0.0546) | 8543 (0.0785) | 8046 (0.1964) |
| Observed data (I > 2σ(I)) | 5387 | 4797 | 7441 | 4243 |
| R 1(I > 2σ(I), all)a | 0.0540, 0.0967 | 0.0714, 0.1360 | 0.0807, 0.0899 | 0.1102, 0.1862 |
| wR2(I > 2σ(I), all)b | 0.1066, 0.1240 | 0.1938, 0.2541 | 0.2059, 0.2129 | 0.2933, 0.3425 |
| GOF | 1.014 | 1.043 | 1.047 | 1.043 |
Powder synchrotron X-ray diffraction
A microcrystalline sample of 1 which was dampened with 1-propanol was loaded into a soda-glass Mark-tube of 0.5 mm diameter and sealed with wax. Diffraction data were collected at the Powder Diffraction beamline at the Australian Synchrotron. The glass capillary was mounted and aligned concentric to the rotation of the three-axis diffractometer, and data were collected using a MYTHEN strip detector. The wavelength used was determined to be 0.77370 Å using NIST standard 660a, LaB6. The sample temperature was controlled using an open-flow N2 cryostream. The sample of 1 was quench-cooled to 150 K and data were collected at a 90 s exposure time at five or ten degree intervals from 150 K to 300 K. Le Bail analyses of the variable-temperature diffraction data were performed within RIETICA.41 Details of the refinements are included in Table S1, ESI.†Syntheses
:1 acetonitrile:toluene (20 ml, v/v). To this solution, N,N-diisopropylethylamine (5 ml, 28.7 mmol) was added. The solution was refluxed for four hours, and then the resultant golden yellow solution was reduced in vacuo to give a yellow oil. Deionised water (50 ml) was added, and a pale yellow solid precipitate was formed. This pale yellow solid was isolated by filtration, washed with deionised water, and then recrystallised from acetone–hexane to give a white microcrystalline powder (1.45 g, yield 37.8%). M.P.: 159.4–160.3 °C; IR(cm−1): 2861 (br), 2051(w), 1547 (s), 1495 (s), 1387 (s), 1353 (s), 1244 (s), 1119 (br), 984 (w), 804 (w); ESI-MS (m/z): 685 [M+H]+, 707 [M+Na]+; 1H NMR (DMSO-d6, 300 MHz, δ, TMS): 8.28 (ddd, 2H, J = 4.8 Hz, 1.8 Hz, 0.9 Hz), 7.77 (ddd, 2H, J = 8.1 Hz, 7.2 Hz, 1.8 Hz), 7.57 (dt, 2H, J = 8.1 Hz, 0.9 Hz), 7.15 (ddd, 2H, J = 7.2 Hz, 4.8 Hz, 1.2 Hz), 3.51 (m, 40H); 13C NMR (DMSO-d6, 200 MHz, δ, TMS): 165.18, 164.14, 155.78, 147.82, 136.83, 122.46, 120.32, 70.22, 70.18, 69.54, 69.43, 69.05, 68.51, 68.47, 49.42, 49.30; Anal. Calcd. For C33H48N8O8(684.78): C 57.88, H 7.07, N 16.36; found C 57.41, H 7.14; N 15.87.
:1 acetonitrile:toluene (20 ml, v/v). To this solution, the compound 1,4,7,10-tetraoxa-13-azacyclopentadecane (0.6 g, 2.7 mmol) was added, along with N,N-diisopropylethylamine (5 ml, 28.7 mmol), and the mixture was refluxed for two hours. The resultant golden yellow solution was reduced in vacuo to give a yellow oil, and to this yellow oil methanol (5 ml) and deionised water (20 ml) were added. The solution was filtered, and the filter cake washed with ethanol (20 ml) and hexane (20 ml). The filtrate was then reduced in volume, in vacuo, and the resultant yellow oil was purified viacolumn chromatography using a multiple phase solvent system (SiO2, 1:1 diethylether:tetrahydrofuran (v/v), Rf 0.14; 1:1 tetrahydrofuran:dichloromethane (v/v), Rf 0.36) and a yellow solid obtained. The yellow solid was dissolved in minimum hot ethanol, and hexane added until a white precipitate was obtained. The white precipitate was isolated by filtration and washed with diethylether and water (0.385 g, yield 34.2%). M.P.: 172.2–172.8 °C; IR (cm−1): 3007 (w), 2876 (w), 2111 (w), 1587 (m), 1524 (s), 1460 (s), 1363 (sh), 1291 (s), 1247 (s), 1122 (s), 990 (w), 928 (w), 768 (w); ESI-MS (m/z): 637 [M+H]+, 659 [M+Na]+; 1H NMR (DMSO-d6, 200 MHz, δ, TMS): 8.27 (ddd, 4H, J = 4.8 Hz, 1.8 Hz, 0.8 Hz), 7.75 (ddd, 4H, J = 8.0 Hz, 7.2 Hz, 1.8 Hz), 7.50 (dm, 4H, J = 8.0 Hz), 7.14 (ddd, 4H, J = 7.2 Hz, 4.8 Hz, 0.8 Hz), 3.43 (m, 20H); 13C NMR (DMSO-d6, 400 MHz, δ, TMS): 165.32, 164.06, 155.32, 147.97, 137.15, 122.23, 120.70, 70.17, 69.47, 68.99, 68.17, 49.56; Anal. Cald. for C33H36N10O4 (636.70): C 62.25, H 5.70, N 22.00; found C 62.41, H 5.86, N 21.82.
Acknowledgements
This work was supported by ARC Discovery Grants to KSM and SRB. Part of this research was undertaken on the MX1, MX2 and Powder Diffraction beamlines at the Australian Synchrotron, Victoria, Australia. The staff at the Powder Diffraction, MX1 and MX2 beamlines at the Australian Synchrotron are thanked for their generous help. Professor J.-F. Létard is thanked most kindly for very constructive discussions about solution-phase SCO with these compounds. Emily J. Mensforth kindly assisted with the crystal data collection.References
- P. Gütlich and H. A. Goodwin, Top. Curr. Chem., 2004, 233, 1–47 Search PubMed.
- See for example: M. Hostettler, K. W. Tornroos, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Angew. Chem., Int. Ed., 2004, 43, 4589–4594 CrossRef CAS; D. Chernyshov, M. Hostettler, K. W. Tornroos and H.-B. Bürgi, Angew. Chem., Int. Ed., 2003, 42, 3825–3930 CrossRef; K. W. Tornroos, M. Hostettler, D. Chernyshov, B. Vangdal and H.-B. Bürgi, Chem.–Eur. J., 2006, 12, 6207–6215 CrossRef; D. Chernyshov, B. Vangdal, K. W. Tornroos and H.-B. Bürgi, New J. Chem., 2009, 33, 1277–1282 RSC.
- See for example: M. Quesada, F. Prins, E. Bill, H. Kooijman, P. Gamez, O. Roubeau, A. L. Spek, J. G. Haasnoot and J. Reedjik, Chem.–Eur. J., 2008, 14, 8486–8499 CrossRef CAS; O. Roubeau, J. M. A. Gomez, E. Balskus, J. J. A. Kolnaar, J. G. Haasnoot and J. Reedjik, New J. Chem., 2001, 25, 144–150 RSC; K. Nishi, A. Shinobu, N. Matsumoto, S. Iijima, Y. Sunatsuki, H. Ishida and M. Kojima, Inorg. Chem., 2010, 49, 1517–1523 CrossRef; M. M. Dortu, A. Rotaru, D. Gillard, J. Linares, E. Codjovi, B. Tinant and Y. Garcia, Inorg. Chem., 2009, 48, 7838–7852 CrossRef.
- See for example: R. Pritchard, S. A. Barrett, C. A. Kilner and M. A. Halcrow, Dalton Trans., 2008, 3159–3168 RSC; T. M. Ross, S. M. Neville, D. S. Innes, D. R. Turner, B. Moubaraki and K. S. Murray, Dalton Trans., 2010, 39, 149–159 RSC; B. J. Kennedy, A. C. McGrath, K. S. Murray, B. W. Skelton and A. H. White, Inorg. Chem., 1987, 26, 483–495 CrossRef CAS.
- See for example: C. Carbonera, C. A. Kilner, J.-F. Létard and M. A. Halcrow, Dalton Trans., 2007, 1284–1292 RSC; R. Pritchard, C. A. Kilner and M. A. Halcrow, Chem. Commun., 2007, 577–579 RSC; K. Takahashi, H. Mori, H. Kobayashi and O. Sato, Polyhedron, 2009, 28, 1776–1781 CrossRef CAS; G. Dupouy, M. Marchivie, S. Triki, J. Sala-Pala, J.-Y. Salaün, C. J. Gómez-Garcia and P. Guionneau, Inorg. Chem., 2008, 47, 8921–8931 CrossRef.
- J. Kröber, J-. P. Audière, R. Claude, E. Codjovi, O. Kahn, J. G. Haasnoot, F. Grolière, C. Jay, A. Bousseksou, J. Linarès, F. Varret and A. Gonthier-Vassal, Chem. Mater., 1994, 6, 1404–1412 CrossRef CAS; O. Kahn and J. Martinez, Science, 1998, 279, 44–48 CrossRef.
- For recent reviews, see: K. S. Murray, Aust. J. Chem., 2009, 62, 1081–1101 CrossRef CAS; P. Gamez, J. S. Costa, M. Quesada and G. Aromí, Dalton Trans., 2009, 7845–7853 RSC.
- See: A. B. Gaspar, M. Seredyuk and P. Gütlich, Coord. Chem. Rev., 2009, 253, 2399–2413 CrossRef CAS , and references therein.
- S. M. Neville, G. J. Halder, K. W. Chapman, M. B. Duriska, B. Moubaraki, K. S. Murray and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 12106–12108 CrossRef CAS; P. D. Southon, L. Liu, E. A. Fellows, D. J. Price, G. J. Halder, K. W. Chapman, B. Moubaraki, K. S. Murray, J.-F. Létard and C. J. Kepert, J. Am. Chem. Soc., 2009, 131, 10998–11009 CrossRef.
- Recent publications in this area include: V. Martinez, A. B. Gaspar, C. M. Munoz, G. V. Bukin, G. Levchenko and J. A. Real, Chem.–Eur. J., 2009, 15, 10960–10971 CrossRef CAS; M. Seredyuk, A. B. Gaspar, V. Ksenofontov, Y. Galyametdinov, M. Verdaguer, F. Villain and P. Gütlich, Inorg. Chem., 2010, 49, 10022–10031 CrossRef.
- S. Hayami, G. Juhász, Y. Maeda, T. Yokoyama and O. Sato, Inorg. Chem., 2005, 44, 7289–7291 CrossRef CAS.
- Y. Maeda, M. Suzuki, S. Hirose, S. Hayami, T. Oniki and S. Sugihara, Bull. Chem. Soc. Jpn., 1998, 71, 2837–2843 CrossRef CAS.
- See for example: H. Fenton, I. S. Tidmarsh and M. D. Ward, Dalton Trans., 2010, 39, 3805–3815 RSC.
- M. B. Duriska, S. M. Neville and S. R. Batten, Chem. Commun., 2009, 5579–5581 RSC.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and D. Chasseau, Top. Curr. Chem., 2004, 234, 97–128 CAS.
- A. Hauser, Top. Curr. Chem., 2004, 234, 155–198 Search PubMed; J.-F. Létard, J. Mater. Chem., 2006, 16, 2550–2559 RSC.
- S. M. Neville, B. A. Leita, D. A. Offerman, M. B. Duriska, B. Moubaraki, K. W. Chapman, G. J. Halder and K. S. Murray, Eur. J. Inorg. Chem., 2007, 1073–1085 CrossRef CAS.
- T. M. Ross, B. Moubaraki, D. R. Turner, G. J. Halder, G. Chastanet, S. M. Neville, J. D. Cashion, J.-F. Létard, S. R. Batten and K. S. Murray, Eur. J. Inorg. Chem., 2011, 9, 1395–1417 CrossRef.
- S. M. Neville, B. A. Leita, G. J. Halder, C. J. Kepert, B. Moubaraki, J-. F Létard and K. S. Murray,, Chem.–Eur. J., 2008, 14, 10123–10133 CrossRef CAS.
- M. Quesada, P. de Hoog, P. Gamez, O. Roubeau, G. Aromí, B. Donnadieu, C. Massera, M. Lutz, A. L. Spek and J. Reedjik, Eur. J. Inorg. Chem., 2006, 1353–1361 CrossRef CAS.
- M. Quesada, M. Monrabal, G. Aromí, V. de la Peña-O'Shea, M. Gich, E. Molins, O. Roubeau, S. J. Teat, E. J. MacLean, P. Gamez and J. Reedjik, J. Mater. Chem., 2006, 16, 2669–2676 RSC.
- M. Quesada, V. de la Peña-O'Shea, G. Aromí, S. Geremia, C. Massera, O. Roubeau, P. Gamez and J. Reedjik, Adv. Mater., 2007, 19, 1397–1402 CrossRef CAS.
- B. Witulski, M. Weber, U. Bergstrasser, J.-P. Desvergne, D. M. Bassani and H. Bouas-Laurent, Org. Lett., 2001, 3, 1467–1470 CrossRef CAS; K. Kubo, E. Yamamoto, N. Kato and A. Mori, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 1999, 55, 1819 Search PubMed; Y. Habata, T. Saeki, S. Akabori, X. X. Zhang and J. S. Bradshaw, J. Heterocycl. Chem., 2001, 38, 253–258 CrossRef; Y. Habata and S. Akabori, J. Heterocycl. Chem., 2001, 38, 471–474 CrossRef; Y. Habata, C. Okazaki, K. Ogura, S. Akabori, X. X. Zhang and J. S. Bradshaw, Inorg. Chem., 2007, 46, 8264–8270 CrossRef; P. D. Prince, P. J. Cragg and J. W. Steed, Chem. Commun., 1999, 1179–1180 RSC; E. Yamamoto, K. Kubo, N. Kato and A. Mori, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2000, 56, e329 CrossRef; L. Luo, M.-L. Wei, S.-P. Yan, G.-L. Wang, H.-G. Wang and X.-K. Yao, Chem. J. Chin. Univ. (Gaodeng Xuexiao Huaxue Xuebao), 1992, 13, 1202 Search PubMed; K. A. Byriel, K. R. Dunster, L. R. Gahan, C. H. L. Kennard and J. L. Latten, Inorg. Chim. Acta, 1992, 196, 35 CrossRef; K. Byriel, K. R. Dunster, L. R. Gahan, C. H. L. Kennard, J. L. Latten, I. L. Swann and P. A. Duckworth, Polyhedron, 1992, 11, 1205–1212 CrossRef; S. Itoh, H. Kumei, S. Nagatomo, T. Kitagawa and S. Fukuzumi, J. Am. Chem. Soc., 2001, 123, 2165–2175 CrossRef.
- See for example: H. N. Miras, D. J. Stone, E. J. L. McInnes, R. G. Raptis, P. Baran, G. I. Chilas, M. P. Sigalas, T. K. Kabanos and L. Cronin, Chem. Commun., 2008, 4703 RSC.
- E. S. Meadows, L. J. Barbour, R. Ferdani and G. W. Gokel, J. Supramol. Chem., 2001, 1, 111–115 CrossRef CAS.
- T. J. Mooibroek and P. Gamez, Inorg. Chim. Acta, 2007, 360, 381–404 CrossRef CAS.
- P. Guionneau, M. Marchivie, G. Bravic, J.-F. Létard and Chasseau, J. Mater. Chem., 2002, 12, 2546–2551 RSC; L. Wiehl, H. Spiering, P. Gütlich and K. Knorr, J. Appl. Crystallogr., 1990, 23, 151 CrossRef CAS; L. Kusz, J. Spiering and P. Gütlich,, J. Appl. Crystallogr., 2000, 33, 201 CrossRef; Kusz, J. Spiering and P. Gütlich, J. Appl. Crystallogr., 2001, 34, 229 CrossRef; M. Seredyuk, A. B. Gaspar, J. Kusz, G. Bednarek and P. Gülich, J. Appl. Crystallogr., 2007, 40, 1135–1145 CrossRef.
- J. A. Real, B. Gallois, T. Granier, F. Suez-Panamá and J. Zarembowitch, Inorg. Chem., 1992, 31, 4972–4979 CrossRef CAS.
- See for example: H. Spiering, Top. Curr. Chem., 2004, 235, 171–195 CrossRef CAS , and references thereinRecent work in this area includes: M. Kepenekian, J. A. Costa, B. Le Guennic, P. Maldivi, S. Bonnet, J. Reedjik, P. Gamez and V. Robert, Inorg. Chem., 2010, 49, 11057–11061 CrossRef.
- P. J. van Koningsbruggen, Y. Garcia, O. Kahn, L. Fournés, H. Kooijman, A. L. Spek, J. G. Haasnoot, J. Moscovici, K. Provost, A. Michalowicz, F. Renz and P. Gütlich, Inorg. Chem., 2000, 39, 1891–1900 CrossRef CAS; A. Białońska and R. Bronisz, Inorg. Chem., 2010, 49, 4534–4542 CrossRef.
- S. Hayami, Z. Z. Gu, M. Shiro, Y. Einaga, A. Fujishima and O. Sato, J. Am. Chem. Soc., 2000, 122, 7126 CrossRef CAS.
- K. H. Sugiyarto, D. C. Craig, A. D. Rae and H. A. Goodwin, Aust. J. Chem., 1994, 47, 869–890 CrossRef CAS; K. H. Sugiyarto, K. Weitzner, D. C. Craig and H. A. Goodwin, Aust. J. Chem., 1997, 50, 869–873 CrossRef; K. H. Sugiyarto, M. L. Scudder, D. C. Craig and H. A. Goodwin, Aust. J. Chem., 2000, 53, 755–765 CrossRef; M. M. Dîrtu, C. Neuhausen, A. D. Naik, A. Rotaru, L. Spinu and Y. Garcia, Inorg. Chem., 2010, 49, 5723–5736 CrossRef.
- See for example: S. Bonnet, G. Molnár, J. S. Costa, M. A. Siegler, A. L. Spek, A. Bousseksou, W.-T. Fu, P. Gamez and J. Reedjik, Chem. Mater., 2009, 21, 1123–1136 CrossRef CAS.
- T. Kudo, Y. Oishi, J. Oravec and K. Mori, J. Photopolym. Sci. Technol., 2004, 17, 259–262 CrossRef CAS.
- P. de Hoog, P. Gamez, W. L. Driesson and J. Reedjik, Tetrahedron Lett., 2002, 43, 6783–6786 CrossRef CAS.
- H. Maeda, S. Furuyoshi, Y. Nakatsuji and M. Okahara, Bull. Chem. Soc. Jpn., 1983, 56, 212–218 CrossRef CAS.
- APEXII Software Package v 2.0, Bruker AXS Inc., Madison, WI, 2005 Search PubMed.
- SHELXL97. Program for crystal structural solution and refinement, Bruker Analytical Instruments Inc., Madison, Wisconsin, USA, 1997 Search PubMed; L. J. Barbour, X-SEED, University of Stellenbosch, South Africa, 1999 Search PubMed.
- T. M. McPhillips, S. E. McPhillips, H. J. Chiu, A. E. Cohen, A. M. Deacon, P. J. Ellis, E. Garman, A. Gonzalez, N. K. Sauter, R. P. Phizackerley, S. M. Soltis and P. Kuhn, J. Synchrotron Radiat., 2002, 9, 401–406 CrossRef CAS.
- W. J. Kabsch, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- B. A. Hunter, C. J. Howard, In A Computer Program for Rietveld Analysis of X-Ray and Neutron Powder Diffraction Patterns, LucasHeights Research Laboratories, Sydney, Australia, 1998, p 1 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 1H NMR and 13C NMR data for the ligands ddta and tptd. Labelled asymmetric unit diagrams for the structures 1a, 1b, 2 and 3, with ellipsoids at 50% probability. Special refinement details for single crystal and powder X-ray diffraction data. CCDC reference numbers 823862–823865. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c1dt10818b |
| This journal is © The Royal Society of Chemistry 2012 |