Alternative approaches for the aqueous–organic biphasic hydroformylation of higher alkenes
Lorenz
Obrecht
,
Paul C. J.
Kamer
* and
Wouter
Laan
*
School of Chemistry, University of St Andrews, North Haugh, St Andrews, UK. E-mail: wwl1@st-andrews.ac.uk; pcjk@st-andrews.ac.uk
First published on 14th September 2012
Abstract
The biphasic hydroformylation of linear alkenes using the Rh–TPPTS catalyst system is one of the cornerstones of aqueous biphasic catalysis, but due to mass-transfer limitations its application is restricted to short alkenes. This perspective provides an overview of various alternative approaches which have been developed to extend the aqueous biphasic methodology to the hydroformylation of higher alkenes.
Introduction
Hydroformylation (also known as oxo synthesis or oxo process) is one of the largest industrial applications of homogeneous catalysis, with a current world-wide production of approximately 9 million tons per year.1 The process entails the transition-metal catalysed reaction of alkenes with carbon-monoxide and hydrogen gas affording aldehydes. The aldehydes are easily hydrogenated to alcohols, which can be converted to important products such as detergents and plasticizers. Both branched and linear aldehydes can be formed, but their more prevalent use in downstream processes makes linear aldehydes the more desirable products.2Catalyst separation is crucial for industrial processes. Catalyst recovery allows for the implementation of catalyst recycling strategies, thus reducing the overall process costs. Also contamination of the product with the catalyst or metal species can pose problems for downstream processes or applications. This issue has been very effectively tackled for the hydroformylation of propene and butene by the development of the aqueous biphasic Ruhrchemie/Rhône-Poulenc (RCH/RP) process, commercially used since 1984.3 Water is a cheap, environmentally friendly and safe solvent, which renders it very attractive for green and sustainable production processes. The RCH/RP process uses the highly water-soluble ligand triphenylphosphine-3,3′,3′′-trisulfonate (TPPTS) to immobilize a rhodium catalyst in the aqueous phase, while the substrate and products form a second phase. The catalyst can be completely and quickly recovered by phase-separation. The process produces almost exclusively aldehydes with over 96% selectivity for the linear products under relatively mild conditions. This technology is currently in operation at five plants worldwide, furnishing an annual production of 800![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tons aldehydes.4 The operational simplicity, robustness and the excellent economics of the process (loss of rhodium by leaching into the organic phase lies in the ppb range) make the RCH/RP a benchmark process in the field of aqueous biphasic transition-metal catalysis.
000 tons aldehydes.4 The operational simplicity, robustness and the excellent economics of the process (loss of rhodium by leaching into the organic phase lies in the ppb range) make the RCH/RP a benchmark process in the field of aqueous biphasic transition-metal catalysis.
The solubilities of propene and C4 alkenes in the aqueous phase are sufficient to allow chemical reactions to occur at an acceptable rate without phase transfer limitations. However, for longer alkenes, the reaction rate is too low to be economically viable. For example, hydroformylation of 1-octene under standard conditions (125 °C, 30 bar, [Rh] = 300 ppm) proceeds with a rate constant of 5.3 × 10−4 min−1.2
For the hydroformylation of higher alkenes (>C5) homogenous processes are used. For alkenes up to C8 commercial processes using phosphine-modified rhodium catalysts are known. For longer alkenes the distillation conditions required to separate the high-boiling products may lead to catalyst decomposition and concomitant loss of metal. Therefore cheaper cobalt catalysts are used, ensuring the processes are still economically viable. However, cobalt catalysts are less active than rhodium-catalysts, requiring higher reaction temperatures and pressures.5
For these reasons the development of a process for the hydroformylation of higher alkenes using rhodium catalysts under biphasic conditions, which allows for catalyst recycling by phase separation, is highly desirable. Using the unmodified Rh–TPPTS system, only minor improvements can be achieved by varying the reaction conditions such as the syngas pressure or the ligand–metal ratio.6 Many approaches have been developed to tackle the issue of low space-time yield in biphasic aqueous–organic reaction systems involving poorly water-soluble substrates. In this perspective, we will discuss various alternative approaches which have been developed to overcome the mass-transfer limitations in the biphasic hydroformylation of higher 1-alkenes. Unless stated otherwise, the catalyst system applied in the studies discussed below is based on the Rh–TPPTS system.
Additives
Various additives are available to improve the solubility or the transport of hydrophobic substrates into the aqueous phase. To be suitable, an additive should (i) be inert with respect to the catalyst, substrate and reaction products; (ii) not increase the solubility of the catalyst in the product phase; (iii) exert a minimal influence on phase separation and (iv) show minimal leaching into the product phase.Co-solvents
One of the first strategies employed to improve mass transfer in biphasic catalysis is the use of co-solvents. Co-solvents increase the lipophilicity of the aqueous phase, thereby increasing the solubility of alkenes in the catalyst phase. For example, the solubility of 1-octene has been estimated to be 104 times higher in 50% ethanol than in pure water.7 Indeed, the use of ethanol, acetone, acetonitrile or methanol as co-solvent in the hydroformylation of 1-octene leads to a significant increase of the reaction rate.7,8 However, the addition of co-solvents results in a decrease in the linear selectivity, i.e. from 98 to 92% upon addition of 20% methanol to the alkene phase.6 Transfer of the co-solvent from the aqueous to the organic phase due to the formation of nonanal has been reported.7 This might affect the partition coefficient of the catalyst between the two phases. Also, ethanol was found to react with the product nonanal to yield acetals. A buffer solution of sodium carbonate and bicarbonate in the aqueous phase (pH ∼ 10) was found to be effective in suppressing acetal formation.7The erosion of the linear selectivity, the fact that they react with the product, their leaching into the product phase as well as the potential leaching of catalyst make co-solvents an unattractive additive for commercial applications.
Surfactants
Above the critical micelle concentration surfactants form micelles, which can solubilize hydrophobic compounds within their hydrophobic cores. The formation of micelles increases the interfacial area in biphasic systems, thus increasing the phase transfer. The first reports of rate-enhancements in biphasic hydroformylation by using surfactants came from Johnson Matthey.9 Despite their ability to encapsulate substrates, neutral and anionic surfactants show no activating effect on reaction rates.6,10 In contrast, cationic surfactants greatly enhance reaction rates. For example, no aldehydes are formed during the hydroformylation of 1-dodecene in the presence of sodium dodecyl sulfate (SDS), while the use of cetyltrimethylammonium bromide (CTAB 1, Fig. 1) afforded 61% conversion.10 High reaction rates can be obtained using CTAB: turnover frequency (TOF) values up to 900 h−1 have been reported.11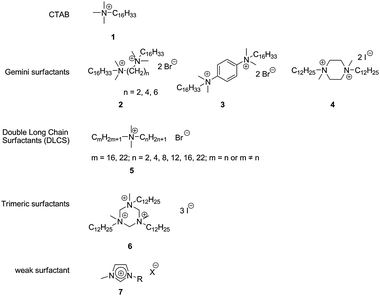 | ||
| Fig. 1 Cationic surfactants applied in aqueous biphasic hydroformylation. | ||
The difference in the ability of anionic and cationic surfactants to enhance the reaction rate has been ascribed to electrostatic interaction between the surface of the micelle and the catalyst. Electrostatic repulsion between the negatively charged catalyst and the surface of micelles formed by anionic surfactants prevents reaction of the catalyst with the substrate. In the case of cationic surfactants, however, electrostatic attraction between the negatively charged sulfonate group of the TPPTS ligand and the cationic headgroup of the surfactant leads to sequestering of the catalyst on the micelle surface, providing a high local concentration of catalyst in the vicinity of a high local concentration of substrate and thus causing an increase in reaction rate. Indeed, 31P-NMR data suggest a strong interaction between the catalyst and CTAB micelles.12 Light-scattering experiments confirmed that the rate increase of the hydroformylation of 1-dodecene in the presence of CTAB is due to solubilisation of the substrate inside the micelle and the binding of [HRh(CO)[TPPTS3] to the micelle surface, leading to a 90 fold increase of the rhodium concentration in the micelle interface compared to the bulk water phase.13
The reported effects of surfactants on the linear selectivity are inconsistent. For instance, for the use of CTAB, both an increase10 and a decrease6 in the linear selectivity have been reported. Interestingly, Chen et al. observed a synergistic effect of Na-TPPTS and Na-TPPDS (disodium triphenylphosphine-3,3′-disulfonate) on the regioselectivity of 1-dodecene hydroformylation in the presence of CTAB, increasing the ratio of linear to branched products (l/b) from 6.5 to 22.3 for TPPTS/TTPDS = 2:1.14
Motivated by the high reaction rates obtained with CTAB, several other types of cationic surfactants have been explored. Li, Chen and co-workers found that gemini surfactants 2 (Fig. 1) containing alkyl bridges afford slightly higher rates and selectivities than CTAB in the hydroformylation of 1-dodecene.15 Notably, shorter alkyl bridges lead to increased selectivities. The surfactants with shorter bridges were suggested to form more compact micelles, favouring the formation of the less crowded linear aldehyde. Also gemini-surfactants 3 and piperazine-based 4, as well as trimeric triazine-based surfactant 6 (Fig. 1) were explored, with 4 performing the best, affording a TOF of 1845 h−1 in the hydroformylation of 1-decene.16
Nonetheless, the highest rate enhancements to date have been achieved by using double long-chain surfactants (DLCS, 5). Using 5 (Fig. 1), Fu et al. achieved a TOF of 7472 h−1 for the biphasic hydroformylation of 1-dodecene, with an aldehyde selectivity of 94%; values which are comparable to those of the equivalent homogeneous reaction.17 Threshold values for the length of both chains were determined: high reaction rates are obtained when n ≥ 8 if m = 22 and n ≥ 12 if m = 16 (see Fig. 1).17
Although the addition of surfactants provides high reaction rates and the leaching of metal into the product phase is generally low, it often makes reaction mixtures prone to emulsification, particularly at high stirring rates and conversions, rendering phase separation difficult.18
Desset et al. found that the weak surfactant 1-octyl-3-methylimidazolium bromide 7 ([Octmim]Br, Fig. 1; X = Br−, R = C8H17) provides high reaction rates while at the same time allows rapid and complete phase separation and good catalyst retention, albeit with a slight decrease in the linear selectivity. While imidazolium and triethylammonium salts with longer alkyl “tails” (R>C8) also provide large rate enhancements, these amphiphiles lead to stable emulsions.19 It appears that [Octmim]Br and related molecules are promising surfactants for increasing the rate of aqueous biphasic hydroformylation without compromising the inherent strength characteristic of the RCH/RP process.
Polyethylene glycol
Polyethylene glycol (PEG) of medium molecular mass (400–1000) is an effective additive for increasing the reaction rate.6 It significantly enhances the reaction rate, although it leads to a slight decrease in the l/b ratio. Leaching of the PEG and rhodium into the organic phase is very low, and phase separation is fast and straightforward.Inverse phase transfer catalysts
Another approach to facilitate the migration of hydrophobic molecules from an organic to an aqueous phase is the use of inverse phase transfer catalysts. The two most widely studied classes of compounds are cyclodextrins20 and calixarenes,21 which are widely applied in aqueous biphasic catalysis. Cyclodextrins 8 (CD, Fig. 2) are cyclic oligosaccharides, and the three main types (α, β or γ) are composed of 6, 7 or 8 α-D-glucopyranoside units respectively. CDs are shaped like conical cylinders with a hydrophobic inner surface and a hydrophilic outer surface; the wider rim contains secondary hydroxyl-groups while primary hydroxyl groups occupy the narrower rim (R1 = R2 = R3 = OH). | ||
| Fig. 2 Inverse phase transfer catalysts applied in aqueous biphasic hydroformylation. | ||
Classical α- and γ-CD show no effect while native β-CD only provides a moderate 2-fold increase in reaction rate.22 However, the effectiveness of CDs can be greatly improved by chemical modification of the hydroxyl-groups with hydrophobic or hydrophilic groups such as methyl, acetyl or 2-hydroxypropyl. Randomly methylated β-CD (RAME-β-CD; on average 12.6 methyl groups added on positions 2, 3 and 6) is particularly effective.22 This was initially ascribed to the solubility of the modified CD in both the aqueous and organic phase. Indeed, CDs which are little soluble in either water or the organic phase have little influence on the reaction rate. Complementary experiments have demonstrated that modified CDs concentrate at the liquid–liquid interface, where substrate recognition leads to the formation of inclusion complexes which facilitate the reaction between the substrate and the water-soluble organometallic catalyst.23 Interestingly, positive synergistic effects of binary mixtures of RAME-α-,β-, and γ-CDs have been observed, suggesting the formation of 2:1 ternary complexes.24
Unfortunately, the use of RAME-β-CD leads to a significant reduction of the linear selectivity. This decrease in selectivity stems from the formation of inclusion complexes between TPPTS and the CD. The formation of such complexes facilitates ligand dissociation, leading to catalyst species which afford lower linear selectivity.25 To prevent this, various combinations of CD and ligands have been explored.
Due to their smaller cavity size, chemically modified α-CDs do not interact with the catalyst, and RAME-α-CD enhances the reaction rate while not affecting the l/b ratio.26 Also α- and β-CDs bearing alkyl chains on the secondary face and sulfoalkyl- or poly(ethyleneoxide) chains on the primary face afford higher rates and regioselectivity than randomly methylated β-CD.27
Noteworthy is the use of the sulphonated xantphos ligand (9, Fig. 2) in combination with RAME-α and β-CDs. Although the CDs bind a ligand phenyl ring, ligand dissociation does not occur and improved chemo- and regioselectivities in the hydroformylation of 1-octene and 1-decene are obtained.28 The ligand 1,3,5-triaza-7-phosphaadamantane 10 (PTA) and its N-benzylated derivative 11 (Fig. 2) were recently found not to interact with RAME-β-CD during the biphasic hydroformylation of 1-decene. Although this system affords poor linear selectivity, PTA may provide a suitable framework for ligand optimization.29
In general, upon addition of CDs to biphasic hydroformylation reactions the phase-separation is still excellent, and leaching of the catalyst is minimal. Also catalyst recycling can be done without significant loss of activity. Even an increase in activity upon recycling has been reported; this was attributed to a gradual supramolecular organization of the rhodium complex, its solvation sphere and cyclodextrin in the interphase.30
Supramolecular organization has also been suggested to explain the performance of recently introduced β-CD functionalized polymers in biphasic hydroformylation.31 Acrylamide polymers 12 with degrees of β-CD substitution up to 50% were synthesized (Fig. 2). Whereas highly substituted polymers were found to be nearly as effective as RAME-β-CD as mass-transfer promoters in the hydroformylation of 1-decene, the ineffectiveness of less substituted polymers was interpreted to stem from unfavourable organisation of the main-chains of the polymers at the phase-boundary, leading to shielding of the substrate from the CDs. With 1-hexadecene as substrate, highly substituted polymers were much more effective than RAME-β-CD. Whereas the substrate is too long to be efficiently transferred by a single β-CD cavity, cooperative multivalent substrate recognition by close-in-space polymer-bound CDs leads to efficient recognition and higher reaction rates.
Calixarenes are cyclic oligomers of substituted benzene units, synthesised from phenols and aldehydes. While calixarenes have been applied as phase-transfer agents in a number of biphasic catalysis reactions, to the best of our knowledge their use as additive in biphasic hydroformylation of linear alkenes has not been reported.
The group of Vogt recently applied polystyrene-based latices as phase transfer agents in the biphasic hydroformylation of 1-octene.32 The latices 17, obtained from the polymerization of PEGylated styrene 13, divinylbenzene 14, styrene 15 and styryl-salts 16 (Fig. 2; R = H, SO3Na or CH2NMe3BF4), consist of a lipophilic core and a hydrophilic shell. It was found that the rate-enhancement critically depends on the nature of the styryl-salt: the highest rate (TOF of 150 h−1) was achieved using ammonium-modified styrene, while latices containing unmodified or sulfonated styrene had little effect. In analogy to the situation with cationic surfactants, this was explained by the association of the anionic catalyst with the cationic ammonium-group on the outer shell.
Activated carbon
Recently Monflier introduced activated carbon (e.g. Nuchar® WV-B) as a novel mass-transfer additive for aqueous biphasic catalysis.33 Considerable rate-enhancements are achieved in biphasic hydroformylation using either cobalt/trisulfonated tris(biphenyl)phosphine (BiphTS) or the Rh–TPPTS system, with little effect on the l/b ratio. Also the aqueous phase catalyst and activated carbon can be recovered easily and recycled multiple times without significant loss of activity.34 It was proposed that the activated carbon facilitates the mixing of the aqueous and organic phases, leading to a more effective interfacial area concomitant with confinement of the reactants and the catalyst.33Ligand variation
TPPDS, TPPMS, TPP
Substituting TPPTS by the less water-soluble triphenylphosphine-3,3′disulfonate (TTPDS) or triphenylphosphine-3-sulfonate (TPPMS) as ligand leads to a slight increase in reaction rate, but also leads to significant leaching of the ligand into the organic phase.6,35Chaudhari et al. proposed the use of promoter ligands which are exclusively soluble in the organic phase. This was suggested to lead to the formation of mixed ligand complexes which would concentrate at the phase boundary, affording interfacial catalysis. Although inclusion of triphenylphosphine (TPP) in the hydroformylation of 1-octene with HRh(CO)(TPPTS)3 leads to a 10–50 fold increase in the reaction rate,36 it has been argued that TPP simply increases the solubility of the catalytic species in the organic phase, which may affect downstream processing.6
Amphiphilic ligands
Another approach that has been explored is the use of ligands which combine surface-activity with water solubility. Analogous to surfactants, the aggregation of amphiphilic ligands may lead to the formation of micelles, which are capable of encapsulating substrates in their hydrophobic core. This increases the substrate solubility in the aqueous phase and also brings the substrate and the catalyst in close proximity, thus increasing the reaction rate.Fell and Papadogianakis were the first to use a specifically designed amphiphilic ligand in biphasic hydroformylation.37 Application of the zwitterionic trisulfoalkylated tris(2-pyridyl)phosphine (18, Fig. 3) for the conversion of 1-tetradecene resulted in TOFs up to 340 h−1. The best yields were obtained with the ligand where n = 5; longer chains afforded lower yields while stable emulsions were formed with n = 9, 11. The catalysts with n = 0–7 could be quantitatively recovered by a simple phase separation. Various other amphiphilic monodentate ligands superior to TPPTS in biphasic hydroformylation have been reported. Most of these ligands bear the hydrophilic group (either sulfonate or phosphate) and the phosphorous atom on the two ends of the alkyl chain, including phosphonate-phosphines developed by Bischoff and the Hanson group’s sulfonated tris(ω-phenylalkyl)phosphines.8a,38
 | ||
| Fig. 3 Amphiphilic ligands used in the aqueous biphasic hydroformylation. | ||
Also ligands containing the hydrophilic group and the phosphorous atom on the same end of the hydrocarbon have been successfully applied. For example the sodium salt of sulfonated n-C12H25O·C6H4P(C6H4)-p(CH3O)2 (DMOPPS, 19, Fig. 3) affords significant rate enhancements in the biphasic hydroformylation of long chain alkenes, not only providing TOFs up to 673 h−1 for 1-dodecene but even a TOF of 100 h−1 for 1-hexadecene, with l/b ratios around 2.4.39
Surface active ligands based on diphosphine frameworks such as 2,2′-bis(diphenylphosphino)-1,1′-biphenyl (BISBI) and 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl (BINAP) have also been developed.40 Sulfonated xantphos-derivatives containing long apolar chains (20, Fig. 3) spontaneously form large and thermally stable vesicles in water.41 These vesicles were proven to increase the solubility of 1-octene in aqueous solution, and the activity of the vesicle-forming ligands in the biphasic hydroformylation was found to be 12–14 times higher than that of xantphos. It was shown that the aggregates stay intact during recycling, the rhodium is quantitatively retained in the aqueous phase and the TOF and linear selectivity remained the same in four consecutive runs.
Monflier and co-workers found that the performance of the surface-active ligands (21, 22, Fig. 3) in the biphasic hydroformylation of 1-decene could be improved, by the addition of stoichiometric amounts of ionic beta-cyclodextrins, leading to a 4-fold increase in reaction rate and an excellent phase separation. In contrast, the addition of neutral β-cyclodextrins led to a decrease in performance, which was shown to be due to destruction of the micelles formed by the ligand.42 This shows that for amphiphilic ligands, addition of CD with the right structure may provide an elegant way to further improve their performance in biphasic catalysis.
Ligands containing cyclodextrins or calixarenes
In addition to their use as additives in biphasic catalysis, cyclodextrins and calixarenes have also been incorporated as phase-transfer agents in ligand structures to develop dual-function catalysts.Reetz reported the evaluation of a series of diphosphine-ligands modified with β-CD in the rhodium catalyzed biphasic hydroformylation of 1-octene (23, Fig. 4). Very high activities were achieved with l/b ratios of 3.2. Disappointingly, the activity dropped 50% upon reuse.43 Also methylated α-cyclodextrins capped with a diphosphine/rhodium moiety were active catalysts in the biphasic hydroformylation of 1-octene, but significant leaching of the catalyst to the organic phase occurred over the course of the reaction.44
 | ||
| Fig. 4 Ligands containing cyclodextrins or calixarenes. | ||
Using rhodium complexes of water-soluble calix[4]arene based ligands modified with two phosphines (24, Fig. 4) on the upper rim provided considerable rate-enhancements but slightly reduced l/b ratios in the biphasic hydroformylation of 1-octene. Remarkably, the system showed almost complete retention of activity and selectivity over two recycling runs.45
The use of hemispherical 1,3-calix[4]arene-diphosphites 25 (Fig. 4) as ligands in combination with upper rim sulfonated calix[4]arenes as surfactants in the biphasic hydroformylation of 1-octene and 1-hexene was recently reported by Matt and co-workers. In the absence of surfactant, catalysis takes place in the organic phase. In the presence of surfactant, the activity increased, and TOFs of 1160 h−1 and 750 h−1 were obtained for 1-octene and 1-hexene, respectively, with high l/b ratios (up to 61.8 for 1-octene using a 10-fold excess of ligand). However, stable emulsions were recovered after each reaction including surfactant. Although no recycling experiments were reported, this will probably hamper efficient catalyst recovery.46
Polymer supported ligands
Water soluble polymers as support
In this approach the catalytically active metal is complexed to a water soluble polymer modified with donor groups. In most cases the coordinating groups are phosphorous based: mono-47 or bidentate48 phosphines, phosphine oxides,49 phosphinites,47g phosphonites47g or bidentate P–N50 ligands. There are also examples where bidentate N–N-ligands,50 carbenes,51 acetylacetonate52 or glycolate53 groups act as donor ligands for a rhodium metal, although these ligands afford less active hydroformylation catalysts.These polymer bound catalysts are believed to act as phase transfer agents47e and are successfully used in the aqueous biphasic hydroformylation of water insoluble alkenes. In general rhodium leaching to the organic phase is low and the catalyst can easily be reused. TOFs obtained are low (<100 h−1) for these systems, while the l/b ratio varies from moderate to excellent (1.3–8.5) depending on the conditions, the polymer and the donor atoms used.47b,d–g,48,52
There are two groups who obtained remarkably high TOF (>500 h−1) for the same reaction when using water soluble polymers as support.
The group of Ritter used a polyethylene glycolate complex which was synthesised from rhodium trichloride and polyethylene glycol. They assumed that the reaction takes place at the aqueous/organic interphase as the TOF is almost unaffected by the alkene chain length – and therefore their water solubility though they did not neglect the possibility of the catalyst being transferred into the organic phase.53 For the hydroformylation of 1-dodecene the rhodium content in the organic phase after five catalytic cycles was measured as 1.9 ppm. The high TOF cannot be attributed to leached rhodium as upon addition of 4 equivalents of TPPTS to the reaction mixture the amount of leached rhodium drops below the detection limit of 0.1 ppm while the TOF even increased. Unfortunately their system is rather unselective, yielding a l/b ratio of ≈0.9.
Weberskirch and his group postulated that their tailor made amphiphilic block copolymers form micelles encapsulating the olefin and the catalytic centre (see Fig. 5).47a,51,54 All their catalysts were evaluated using 1-octene as substrate, resulting in TOFs as high as 3700 h−1 and a moderate selectivity (l/b ≈ 3) in most cases. When rhodium leaching and catalyst recycling were investigated for one of their systems the former was found to be low (0.4 ppm). Reusing this catalyst four times resulted in an increasing TOF in the first three cycles (from 1100 to 2185 h−1), after which it remained constant in the next cycle. On the other hand the l/b ratio of the aldehydes formed dropped from 2.57 to 1.33 during the first three cycles, indicating an increase in free rhodium, and remained constant in the last run.51 Phase separation after the reaction is highly dependent on the polymer used. For some polymers a simple decantation can be applied while for others the separation was poor.
 | ||
| Fig. 5 Example of an amphiphilic block copolymer forming micelles. | ||
While the use of most water soluble polymers is unattractive due to low TOF the use of polyethylene glycol as support gives high TOFs but low selectivities. The use of tailor made amphiphilic block copolymers is probably the most interesting approach as the TOFs are high, the l/b ratio can be tuned, rhodium leaching is small and recycling is possible.
Proteins as support
A promising approach was invented by the group of Marchetti. Instead of using rather poorly defined man-made polymers they use proteins as support. Although the protein structure itself is well defined the protein–rhodium-complexes synthesised are undefined as the proteins contain numerous donor atoms.They investigated the proteins human serum albumin,55 as well as papain and egg albumin56 in combination with Rh(I) in the hydroformylation of 1-octene. The latter complexes are not stable under hydroformylation conditions but precipitate and release rhodium to the organic phase while the rhodium–human serum albumin complex can be reused several times. Unfortunately rhodium leaching was quite significant in the range of 8 ppm.55
Supported aqueous phase catalysis
One approach to increase the surface area of the interphase between aqueous and organic phase and therefore the reaction rate is Supported Aqueous Phase Catalysis (SAPC), which was developed in 1989.57In SAPC a thin aqueous layer containing a water soluble catalyst complex is adsorbed onto hydrophilic solids. The solid should have a large surface area and should be inert to thermal and mechanical stress and all chemicals present in the reaction mixture. For hydroformylation most supports are based on silica, but other supports like cation exchange resin,58 glass59 and apatitic tricalcium phosphates60 have been investigated as support as well. In most cases the catalyst is based on rhodium but some examples for cobalt-58a,59,61 and platinum-based catalysts61 exist. The reaction itself takes place at the interphase of the organic phase and the water layer absorbed to the particles (see Fig. 6), therefore the reaction rate is almost unaffected by the water solubility of the substrate. At the end of the reaction the organic phase containing the products can be separated from the support and its catalyst containing water layer by filtration and the latter can be reused. SAPC not only depends on the nature of the metal-complex, it is also highly dependent on the inherent properties of the support such as particle size and surface area. Additionally the hydration of the support is important.62 The influence of the hydration on the hydroformylation of long chain 1-alkenes was shown very clearly by Kalck et al. They varied the water content near the optimal hydration degree of their system in steps of 0.1%. Changing their optimal hydration by 0.2% in either ways reduces the conversion by about 13% while the selectivity remained unchanged.63
 | ||
| Fig. 6 SAPC concept. | ||
In SAPC, the activity is low (TOF <100 h−1) in most cases for both rhodium61,64 and cobalt based58a,59 catalysts. The group of Hanson showed that in SAPC the reaction takes place at the phase-boundary: conversion of 1-octene, 1-decene and 1-dodecene occurred with similar TOFs of ≈45 h−1 affording the corresponding aldehydes with a l/b ratio of ≈2.3.61 Ligand tuning is a powerful tool to influence the selectivity in SAPC. Using a xantphos-derivative as ligand results in a linear to branched selectivity of >3065 which is almost as high as in biphasic catalysis.66 Using TPPTS as ligand under almost the same conditions gives a much lower selectivity (l/b = 3).64 There are some examples with high TOFs (>100 h−1),61,64,65,67 two of which use TPPTS as ligand and silica as support, the high TOFs are most likely due to optimised conditions.61,64
In one of the other successful approaches a mixture of polyethyleneglycol and water instead of pure water is adsorbed onto the particle.67a Both the TOF (≈1000 h−1) as well as the selectivity (l/b ≈ 16) are remarkably high in this case but it should to be mentioned that the TOF was measured by the initial gas uptake of the reaction. This approach is prone to errors and the TOF calculated this way is higher than conventionally measured TOFs. No products were formed when reusing the organic phase, indicating that no rhodium had leached.
Another approach was published by Zhu and co-workers.65 Their system is a combination of SAPC and heterogeneous catalysis as there are rhodium particles on the support in addition to the adsorbed water-soluble catalyst (Fig. 7). Both TOF (>300 h−1) and selectivity (l/b ≈ 6) are good and rhodium leaching is low (<0.1 ppm). The catalyst could be reused three times without significant erosion of performance.
 | ||
| Fig. 7 Combining SAPC with heterogeneous catalysis. | ||
Yuan et al. reported high TOF (up to >500 h−1) and good selectivities (up to l/b ≈ 6) when using fumed silica as support.67b
Smart systems
In smart systems the aqueous biphasic system can be triggered to turn into a monophasic system during the reaction. Upon completion of the reaction it can be turned back into a biphasic system to allow for the separation of catalyst and products. There are small variations of this concept (Fig. 8) and a variety of different triggers exist.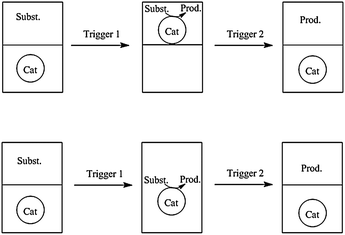 | ||
| Fig. 8 Principle of smart systems. | ||
Smart systems for biphasic hydroformylation were already reported in 1973. Using cobalt, rhodium or iridium complexes with basic aminophosphine ligands such as P(CH2CH2CH2NEt2)3, the catalyst could be extracted either by aqueous solution of carbon dioxide under pressure or by using “standard” acids/bases like sulphuric acid and sodium hydroxide. Reusing the catalyst afforded very similar conversions and selectivities.68
The group of van Leeuwen and Kamer reported several rhodium complexes with pH dependent solubility.69 In neutral solutions these complexes are soluble in the organic layer while upon acidification they become water soluble. Acidic extraction into the water layer and/or backwash upon neutralisation into a new organic layer proved to be good for two ligands (26, 27, Fig. 9) of a series of mono- and bidentate ligands. In the case of these ligands 97–98% of rhodium could be transferred to a new organic layer. When the xantphos based ligand 26 was tested in the rhodium catalysed hydroformylation of 1-hexene, 1-octene and 1-dodecene a remarkably high selectivity (l/b = 48–52) and good TOF (≈200 h−1) were obtained. They investigated the hydroformylation of 1-octene at a higher temperature (100 °C instead of 80 °C) and obtained a slightly lower selectivity (l/b ≈ 45) and slightly more isomerisation but a much higher activity (≈900 h−1). Most likely the results will be similar for different 1-alkenes. Unfortunately, for both ligands the activity in a new catalytic run went down to ≈86%.69b,c It was shown that this drop in activity was due to catalyst decomposition during the acidic extraction.69c In addition to the significant rhodium leaching and catalyst decomposition, salt is formed in the acid–base reaction as undesired byproduct.
 | ||
| Fig. 9 pH dependent smart ligands. | ||
The group of Cole-Hamilton uses the pH change caused by bubbling carbon dioxide through the solution to obtain a water soluble ligand, e.g.29. Upon bubbling nitrogen through the solution carbon dioxide is released and the ligand becomes soluble in organic solvents again (Fig. 10).70 The system has a remarkably high initial TOF (>10000 h−1 measured by the initial gas uptake) and the linear to branched selectivity is highly dependent on the type of ligand used: when using ligand 28 the l/b ratio is ≈3. When using a xantphos based bidentate ligand (30, Fig. 11) the l/b ratio is ≈20 for the hydroformylation of 1-octene. Rhodium leaching of 1.9 ppm was measured for 28. Although the level of rhodium leaching when using 30 was not determined it was visually deemed to be very high. When 28 was reused the activity dropped to 92%. Upon reusing it for three times an activity of 91% was observed. The l/b ratio remains almost unchanged during these recycle experiments. In contrast to the approach of van Leeuwen, the use of gases to tune catalyst solubility does not suffer from the build-up of byproducts and the catalyst does not have to withstand harsh recycling conditions.
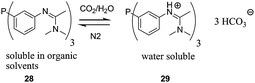 | ||
| Fig. 10 A smart system using gases to change catalyst solubility. | ||
Although not strictly aqueous–organic biphasic reactions, some alternative approaches are also worth mentioning. In the Union Carbide process for the hydroformylation of higher alkenes, the reaction is carried out as a homogenous process using the TPPMS ligand in N-methylpyrrolidone (NMP) as solvent. The addition of water after the reaction brings about a phase separation, resulting in complete transfer of the catalyst to the aqueous phase, which is then recovered.71 In another approach a homogeneous mixture of THF and water (7:3 (v:v)) was used as homogeneous reaction medium yielding a good TOF (≈400 h−1) and moderate selectivity (l/b = 2.4).72 At the end of the reaction carbon dioxide pressure is applied to induce phase separation. Different ligands were tested and it was shown that the aqueous layer containing the catalyst could be reused when using TPPMS as ligand for three times without changing the activity. Furthermore the rhodium content in the organic layer was below the detection limit of 1 ppm.
Using temperature as trigger to change the solubility was investigated by various groups.
The group of Monflier73 used a thermo controlled cyclodextrin based system to obtain really high TOF (6000–7000 h−1). At low temperatures an amphiphilic phosphine ligand is totally or partly located within the hydrophobic cavity of a modified cyclodextrin. Upon increasing the temperature the proportion of ligand within the cyclodextrin is reduced, consequently raising the ligand concentration at the aqueous/organic interphase. When the temperature is reduced this process is reversed, which gives rise to an easy phase separation. They investigated this concept for various 1-alkenes and the TOFs obtained were independent of the alkene chain length. Unfortunately the linear selectivity was rather poor (l/b = 1.7–2.0). They investigated the possibility to recycle the aqueous layer and found it could be recycled at least twice with little effect on conversion or selectivity. Unfortunately no data on the amount of rhodium or ligand leaching were published.
Another ligand class of thermo regulated ligands used for the hydroformylation of long chain 1-alkenes was intensively investigated by the group of Jin.74 The ligands are based on triphenylphosphines modified with a polyethylene chain 31–33 (Fig. 12) and have an inverse temperature dependent solubility in water. Good to excellent activities were found for the biphasic hydroformylation of various 1-alkenes (300–4000 h−1), while the l/b ratio tends to be poor (l/b = 0.4–2.1). It was shown that the l/b ratio is highly temperature dependent: increasing the temperature from 80 to 120 °C caused the ratio to drop from 2.06 to 0.64. Though the selectivity is unsatisfying the reusability of this type of ligands is almost perfect as they could reuse one of their catalysts twenty times without significantly affecting its activity.74g
 | ||
| Fig. 12 Temperature dependent smart ligands. | ||
Outlook
Transferring the effectiveness and elegance of the RCH/RP process to the biphasic hydroformylation of higher alkenes has been a long-standing goal in transition-metal catalysis research. Despite intensive and creative research leading to a variety of approaches to overcome the mass-transfer limitations, most of these have drawbacks inhibiting their practical application, like reduced linear selectivity or poor catalyst recovery/recyclability. Nonetheless, some promising approaches like those based on the use of certain additives or smart ligands have been established, which provide considerable rate-enhancements while preserving some of the inherent strength of the original process. To date, none of the concepts outlined above has been applied to the industrial hydroformylation of higher olefins. There is still a need for an approach that meets all of the strict requirements of a technical two-phase process, such as complete catalyst retention, high activity and stability, high aldehyde selectivity, simple phase separation, and low ligand costs in order to be economically competitive with the currently used processes.Acknowledgements
We thank the European Union (Marie Curie excellence grant (MEXT-2004-014320), NEST Adventure STREP Project (FP6-2003-NEST-B3 15471)) and Sasol Technology UK Ltd. for funding.References
- S. K. Sharma, P. A. Parikh and R. V. Jasra, J. Mol. Catal. A: Chem., 2010, 316, 153 CrossRef CAS.
- B. Cornils and W. A. Herrmann, Aqueous-Phase Organometallic Catalysis, Wiley-VCH, Weinheim, 2004 Search PubMed.
- C. W. Kohlpaintner, R. W. Fischer and B. Cornils, Appl. Catal., A, 2001, 221, 219 CrossRef CAS.
- E. Wiebus and B. Cornils, in Catalyst Separation, Recovery and Recycling, ed. D. J. Cole-Hamilton and R. P. Tooze, Springer, Dordrecht, 2006, p. 105 Search PubMed.
- P. W. N. M. Van Leeuwen, Homogeneous Catalysis, Kluwer Academic Publishers, Dordrecht, 2004 Search PubMed.
- H. Bahrmann, S. Bogdanovic and P. W. N. M. v. Leeuwen, in Aqueous Phase Organometallic Catalysis, ed. B. Cornils and W. A. Hermanns, Wiley-VCH, Weinheim, 2004 Search PubMed.
- P. Purwanto and H. Delmas, Catal. Today, 1995, 24, 135 CrossRef CAS.
- (a) H. Ding, B. E. Hanson, T. Bartik and B. Bartik, Organometallics, 1994, 13, 3761 CrossRef CAS; (b) F. Monteil, R. Queau and P. Kalck, J. Organomet. Chem., 1994, 480, 177 CrossRef CAS.
- (a) M. J. H. Russell, Platinum Met. Rev., 1988, 32, 179 CAS; (b) M. J. Hayling and B. A. Murrer, GB2085874A, Johnson Matthey, 1982.
- H. Chen, Y. Z. Li, J. R. Chen, P. M. Cheng, Y. E. He and H. J. Li, J. Mol. Catal. A: Chem., 1999, 149, 1 CrossRef CAS.
- Y. Q. Zhang, Z. S. Mao and T. Y. Chen, Catal. Today, 2002, 74, 23 CrossRef CAS.
- A. Riisager and B. E. Hanson, J. Mol. Catal. A: Chem., 2002, 189, 195 CrossRef CAS.
- L. B. Wang, H. Chen, Y. A. He, Y. Z. Li, M. Li and X. J. Li, Appl. Catal., A, 2003, 242, 85 CrossRef CAS.
- H. Chen, Y. Z. Li, J. R. Chen, P. M. Cheng and X. J. Li, Catal. Today, 2002, 74, 131 CrossRef CAS.
- M. Li, H. Y. Fu, M. Yang, H. J. Zheng, Y. E. He, H. Chen and X. J. Li, J. Mol. Catal. A: Chem., 2005, 235, 130 CrossRef CAS.
- H. Fu, M. Li, H. Mao, Q. Lin, M. Yuan, X. Li and H. Chen, Catal. Commun., 2008, 9, 1539 CrossRef CAS.
- H. Fu, M. Li, H. Chen and X. Li, J. Mol. Catal. A: Chem., 2006, 259, 156 CrossRef CAS.
- C. Yang, X. Y. Bi and Z. S. Mao, J. Mol. Catal. A: Chem., 2002, 187, 35 CrossRef CAS.
- (a) S. L. Desset, D. J. Cole-Hamilton and D. F. Foster, Chem. Commun., 2007, 1933 RSC; (b) S. L. Desset, S. W. Reader and D. J. Cole-Hamilton, Green Chem., 2009, 11, 630 RSC.
- (a) H. Bricout, F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, Curr. Org. Chem., 2010, 14, 1296 CrossRef CAS; (b) F. Hapiot, A. Ponchel, S. Tilloy and E. Monflier, C. R. Chim., 2011, 14, 149 CrossRef CAS.
- J. B. Simoes, D. L. da Silva, A. de Fatima and S. A. Fernandes, Curr. Org. Chem., 2012, 16, 949 CrossRef CAS.
- (a) J. R. Anderson, E. M. Campi and W. R. Jackson, Catal. Lett., 1991, 9, 55 CrossRef CAS; (b) E. Monflier, G. Fremy, Y. Castanet and A. Mortreux, Angew. Chem., Int. Ed. Engl., 1995, 34, 2269 CrossRef CAS.
- (a) F. Hapiot, L. Leclercq, N. Azaroual, S. Fourmentin, S. Tilloy and E. Monflier, Curr. Org. Synth., 2008, 5, 162 CrossRef CAS; (b) L. Leclercq, H. Bricout, S. Tilloy and E. Monflier, J. Colloid Interface Sci., 2007, 307, 481 CrossRef CAS.
- M. Ferreira, F. X. Legrand, C. Machut, H. Bricout, S. Tilloy and E. Monflier, Dalton Trans., 2012, 41, 8643 RSC.
- (a) E. Monflier, H. Bricout, F. Hapiot, S. Tilloy, A. Aghmiz and A. M. Masdeu-Bulto, Adv. Synth. Catal., 2004, 346, 425 CrossRef CAS; (b) S. Tilloy, G. Crowyn, E. Monflier, P. van Leeuwen and J. N. H. Reek, New J. Chem., 2006, 30, 377 RSC.
- L. Leclercq, M. Sauthier, Y. Castanet, A. Mortreux, H. Bricout and E. Monflier, Adv. Synth. Catal., 2005, 347, 55 CrossRef CAS.
- (a) N. Badi, P. Guegan, F.-X. Legrand, L. Leclercq, S. Tilloy and E. Monflier, J. Mol. Catal. A: Chem., 2010, 318, 8 CrossRef CAS; (b) P. Blach, D. Landy, S. Fourmentin, G. Surpateanu, H. Bricout, A. Ponchel, F. Hapiot and E. Monflier, Adv. Synth. Catal., 2005, 347, 1301 CrossRef CAS; (c) D. Kirschner, T. Green, E. Hapiot, S. Tilloy, L. Leclercq, H. Bricout and E. Monflier, Adv. Synth. Catal., 2006, 348, 379 CrossRef CAS; (d) D. Kirschner, M. Jaramillo, T. Green, F. Hapiot, L. Leclercq, H. Bricout and E. Monflier, J. Mol. Catal. A: Chem., 2008, 286, 11 CrossRef CAS.
- L. Leclercq, F. Hapiot, S. Tilloy, K. Ramkisoensing, J. N. H. Reek, P. van Leeuwen and E. Monflier, Organometallics, 2005, 24, 2070 CrossRef CAS.
- F.-X. Legrand, F. Hapiot, S. Tilloy, A. Guerriero, M. Peruzzini, L. Gonsalvi and E. Monflier, Appl. Catal., A, 2009, 362, 62 CrossRef CAS.
- M. Dessoudeix, M. Urrutigoity and P. Kalck, Eur. J. Inorg. Chem., 2001, 1797 CrossRef CAS.
- J. Potier, S. Menuel, D. Fournier, S. Fourmentin, P. Woisel, E. Monflier and F. Hapiot, ACS Catal., 2012, 1417 CrossRef CAS.
- K. Kunna, C. Muller, J. Loos and D. Vogt, Angew. Chem., Int. Ed., 2006, 45, 7289 CrossRef CAS.
- N. Kania, B. Leger, S. Fourmentin, E. Monflier and A. Ponchel, Chem.–Eur. J., 2010, 16, 6138 CrossRef CAS.
- (a) A. A. Dabbawala, H. C. Bajaj, H. Bricout and E. Monflier, Appl. Catal., A, 2012, 413, 273 CrossRef; (b) J. Boulanger, A. Ponchel, H. Bricout, F. Hapiot and E. Monflier, Eur. J. Lipid Sci. Technol., 2012 DOI:10.1002/ejlt.201200146.
- P. J. Baricelli, E. Lujano, M. Modrono, A. C. Marrero, Y. M. Garcia, A. Fuentes and R. A. Sanchez-Delgado, J. Organomet. Chem., 2004, 689, 3782 CrossRef CAS.
- R. V. Chaudhari, B. M. Bhanage, R. M. Deshpande and H. Delmas, Nature, 1995, 373, 501 CrossRef CAS.
- B. Fell and G. Papadogianakis, J. Mol. Catal., 1991, 66, 143 CrossRef CAS.
- (a) S. Bischoff and M. Kant, Ind. Eng. Chem. Res., 2000, 39, 4908 CrossRef CAS; (b) E. Paetzold, G. Oehme, C. Fischer and M. Frank, J. Mol. Catal. A: Chem., 2003, 200, 95 CrossRef CAS; (c) Q. R. Peng, X. L. Liao and Y. Z. Yuan, Catal. Commun., 2004, 5, 447 CrossRef CAS; (d) B. E. Hanson, H. Ding and C. W. Kohlpaintner, Catal. Today, 1998, 42, 421 CrossRef CAS.
- H. Fu, M. Li, J. Chen, R. Zhang, W. Jiang, M. Yuan, H. Chen and X. Li, J. Mol. Catal. A: Chem., 2008, 292, 21 CrossRef CAS.
- H. Ding, J. X. Kang, B. E. Hanson and C. W. Kohlpaintner, J. Mol. Catal. A: Chem., 1997, 124, 21 CrossRef CAS.
- M. S. Goedheijt, B. E. Hanson, J. N. H. Reek, P. C. J. Kamer and P. van Leeuwen, J. Am. Chem. Soc., 2000, 122, 1650 CrossRef CAS.
- M. Ferreira, H. Bricout, N. Azaroual, D. Landy, S. Tilloy, F. Hapiot and E. Monflier, Adv. Synth. Catal., 2012, 354, 1337 CrossRef CAS.
- M. T. Reetz and S. R. Waldvogel, Angew. Chem., Int. Ed. Engl., 1997, 36, 865 CrossRef CAS.
- D. Armspach and D. Matt, Chem. Commun., 1999, 1073 RSC.
- S. Shimizu, S. Shirakawa, Y. Sasaki and C. Hirai, Angew. Chem., Int. Ed., 2000, 39, 1256 CrossRef CAS.
- L. Monnereau, D. Semeril, D. Matt and L. Toupet, Adv. Synth. Catal., 2009, 351, 1629 CrossRef CAS.
- (a) O. Nuyken, P. Persigehl and R. Weberskirch, Macromol. Symp., 2002, 177, 163 CrossRef CAS; (b) Y. Uozumi and M. Nakazono, Adv. Synth. Catal., 2002, 344, 274 CrossRef CAS; (c) E. A. Karakhanov, Y. S. Kardasheva, A. L. Maximov, E. A. Runova, T. A. Buchneva, M. A. Gaevskiy, A. Y. Zhuchkova and T. Y. Filippova, Polym. Adv. Technol., 2001, 12, 161 CrossRef CAS; (d) T. Malmström, C. Andersson and J. Hjortkjaer, J. Mol. Catal. A: Chem., 1999, 139, 139 CrossRef; (e) E. A. Karakhanov, Y. S. Kardasheva, A. L. Maksimov, V. V. Predeina, E. A. Runova and A. M. Utukin, J. Mol. Catal. A: Chem., 1996, 107, 235 CrossRef CAS; (f) M. Ahlmann, O. Walter, M. Frank and W. Habicht, J. Mol. Catal. A: Chem., 2006, 249, 80 CrossRef CAS; (g) E. A. Karakhanov, Y. S. Kardasheva, E. A. Runova and V. A. Semernina, J. Mol. Catal. A: Chem., 1999, 142, 339 CrossRef CAS.
- A. N. Ajjou and H. Alper, J. Am. Chem. Soc., 1998, 120, 1466 CrossRef CAS.
- L. Xiaozhong, L. Hongmei and K. Fanzhi, J. Organomet. Chem., 2002, 664, 1 CrossRef.
- B. C. E. Makhubela, A. Jardine and G. S. Smith, Green Chem., 2012, 14, 338 RSC.
- M. T. Zarka, M. Bortenschlager, K. Wurst, O. Nuyken and R. Weberskirch, Organometallics, 2004, 23, 4817 CrossRef CAS.
- J. Chen and H. Alper, J. Am. Chem. Soc., 1997, 119, 893 CrossRef CAS.
- T. Borrmann, H. W. Roesky and U. Ritter, J. Mol. Catal. A: Chem., 2000, 153, 31 CrossRef CAS.
- (a) B. Gall, M. Bortenschlager, O. Nuyken and R. Weberskirch, Macromol. Chem. Phys., 2008, 209, 1152 CrossRef CAS; (b) M. Bortenschlager, N. Schöllhorn, A. Wittmann and R. Weberskirch, Chem.–Eur. J., 2007, 13, 520 CrossRef CAS.
- M. Marchetti, G. Mangano, S. Paganelli and C. Botteghi, Tetrahedron Lett., 2000, 41, 3717 CrossRef CAS.
- C. Bertucci, C. Botteghi, D. Giunta, M. Marchetti and S. Paganelli, Adv. Synth. Catal., 2002, 344, 556 CrossRef CAS.
- J. P. Arhancet, M. E. Davis, J. S. Merola and B. E. Hanson, Nature, 1989, 339, 454 CrossRef CAS.
- (a) M. K. Markiewicz and M. C. Baird, Inorg. Chim. Acta, 1986, 113, 95 CrossRef CAS; (b) R. T. Smith, R. K. Ungar, L. J. Sanderson and M. C. Baird, Organometallics, 1983, 2, 1138 CrossRef CAS.
- I. Guo, B. E. Hanson, I. Tóth and M. E. Davis, J. Organomet. Chem., 1991, 403, 221 CrossRef CAS.
- M. Dessoudeix, U. J. Jáuregui-Haza, M. Heughebaert, A. M. Wilhelm, H. Delmas, A. Lebugle and P. Kalck, Adv. Synth. Catal., 2002, 344, 406 CrossRef CAS.
- I. Tóth, I. Guo and B. E. Hanson, J. Mol. Catal. A: Chem., 1997, 116, 217 CrossRef.
- G. Fremy, E. Monflier, J. F. Carpentier, Y. Castanet and A. Mortreux, J. Catal., 1996, 162, 339 CrossRef CAS.
- P. Kalck, L. Miquel and M. Dessoudeix, Catal. Today, 1998, 42, 431 CrossRef CAS.
- A. J. Sandee, V. F. Slagt, J. N. H. Reek, P. C. J. Kamer and P. W. N. M. van Leeuwen, Chem. Commun., 1999, 1633 RSC.
- H. Zhu, Y. Ding, H. Yin, L. Yan, J. Xiong, Y. Lu, H. Luo and L. Lin, Appl. Catal., A, 2003, 245, 111 CrossRef CAS.
- M. Schreuder Goedheijt, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Mol. Catal. A: Chem., 1998, 134, 243 CrossRef CAS.
- (a) M. J. Naughton and R. S. Drago, J. Catal., 1995, 155, 383 CrossRef CAS; (b) Z. Li, Q. Peng and Y. Yuan, Appl. Catal., A, 2003, 239, 79 CrossRef CAS.
- (a) G. Gregorio and A. Andreeta, Ger. Offen., 1973, 231, 302 Search PubMed , Montecatini Edison Spa; (b) A. Andreeta, G. Barberis and G. Gregorio, Chim. Ind. (Milan, Italy), 1978, 60, 887 Search PubMed.
- (a) A. Buhling, J. W. Elgersma, S. Nkrumah, P. C. J. Kamer and P. W. N. M. van Leeuwen, J. Chem. Soc., Dalton Trans., 1996 Search PubMed; (b) A. Buhling, P. C. J. Kamer, P. W. N. M. van Leeuwen and J. W. Elgersma, J. Mol. Catal. A: Chem., 1997, 116, 297 CrossRef CAS; (c) A. Buhling, P. C. J. Kamer, P. W. N. M. van Leeuwen, J. W. Elgersma, K. Goubitz and J. Fraanje, Organometallics, 1997, 16, 3027 CrossRef CAS.
- (a) S. L. Desset and D. J. Cole-Hamilton, Angew. Chem., Int. Ed., 2009, 48, 1472 CrossRef CAS; (b) P. Kalck, Polyhedron, 1988, 7, 2441 CrossRef CAS.
- A. G. Abatjoglou and D. R. Bryant, US 4.731.486, Union Carbide Corporation, 1988.
- J. P. Hallett, J. W. Ford, R. S. Jones, P. Pollet, C. A. Thomas, C. L. Liotta and C. A. Eckert, Ind. Eng. Chem. Res., 2008, 47, 2585 CrossRef CAS.
- N. Six, A. Guerriero, D. Landy, M. Peruzzini, L. Gonsalvi, F. Hapiot and E. Monflier, Catal. Sci. Technol., 2011, 1, 1347 CAS.
- (a) Z. Jin, X. Zheng and B. Fell, J. Mol. Catal. A: Chem., 1997, 116, 55 CrossRef CAS; (b) X. Zheng, J. Jiang, X. Liu and Z. Jin, Catal. Today, 1998, 44, 175 CrossRef CAS; (c) J. Jiang, Y. Wang, C. Liu, F. Han and Z. Jin, J. Mol. Catal. A: Chem., 1999, 147, 131 CrossRef CAS; (d) Y. Wang, J. Jiang, R. Zhang, X. Liu and Z. Jin, J. Mol. Catal. A: Chem., 2000, 157, 111 CrossRef CAS; (e) Y. Wang, J. Jiang, Q. Miao, X. Wu and Z. Jin, Catal. Today, 2002, 74, 85 CrossRef CAS; (f) J. Jiang, Y. Wang, C. Liu, Q. Xiao and Z. Jin, J. Mol. Catal. A: Chem., 2001, 171, 85 CrossRef CAS; (g) C. Liu, J. Jiang, Y. Wang, F. Cheng and Z. Jin, J. Mol. Catal. A: Chem., 2003, 198, 23 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2013 |