Heteropoly acid catalysts for the synthesis of fragrance compounds from biorenewables: isomerization of limonene oxide†
Vinícius V.
Costa
a,
Kelly A.
da Silva Rocha
b,
Ivan V.
Kozhevnikov
c,
Elena F.
Kozhevnikova
c and
Elena V.
Gusevskaya
*a
aDepartamento de Química, Universidade Federal de Minas Gerais 31270-901, Belo Horizonte, MG, Brazil. E-mail: elena@ufmg.br; Fax: (+)55 31 34095700; Tel: (+)55 31 34095741
bDepartamento de Química, Universidade Federal de Ouro Preto, 35400-000, Ouro Preto, MG, Brazil
cDepartment of Chemistry, University of Liverpool, Liverpool L69 7ZD, UK
First published on 4th September 2012
Abstract
The liquid-phase isomerization of limonene oxide was studied in the presence of heteropoly acid catalysts in aprotic solvents in homogeneous and heterogeneous systems. Among the catalysts were bulk and silica-supported tungstophosphoric acid H3PW12O40 and its acidic Cs salt Cs0.5H0.5PW12O40 (CsPW). The reaction gave dihydrocarvone, a valuable fragrance intermediate, as the main product with turnover numbers of up to 8000. The nature of the solvent had a strong effect on reaction rate and selectivity. CsPW (0.1 mol%) was found to be a highly efficient and truly heterogeneous catalyst for this reaction, providing 82% yield of dihydrocarvone in 1,4-dioxane as a solvent under ambient conditions. This simple catalytic method represents economically attractive route to industrially important compounds starting from bio-renewable substrates easily available from essential oils.
1. Introduction
Monoterpenic compounds are the main components of various essential oils and represent an important renewable hydrocarbon feedstock for the flavor and fragrance industry.1,2 Acid-catalyzed transformations are used for the commercial production of various terpenic fragrance compounds from more abundant terpenoids. Many of these processes still employ mineral acids as homogeneous catalysts, which results in serious technological and environmental problems. The development of efficient solid acid catalysts for the reactions involving terpenic compounds is an important task for the valorization of original essential oils.For example, limonene oxide, obtained from limonene epoxidation or extracted from citric essential oils, can be converted by acid-catalyzed transformations to various valuable synthetic intermediates and ingredients for the flavor industry, such as carveol, exo-carveol, dihydrocarvone and carvenone.3 This reaction, however, usually gives a complex mixture of products resulting from its isomerization, which can be accompanied by addition of solvent or residual water. For this reason, the development of selective syntheses of particular products from limonene oxide is a challenge. In most reported procedures, the isomerization of limonene oxide gives mainly para-menthenic allylic alcohols: carveol and exo-carveol.4–10 For example, gold nanoparticles supported on TiO2 are efficient catalysts for selective isomerization of limonene oxide into exo-carveol.9 On the other hand, there are only a few reports describing selective isomerization of limonene oxide into carbonyl compounds, such as carvenone and dihydrocarvone.3,11–13 Dihydrocarvone has been synthesized from limonene oxide in 50–70% yield using ZnBr2 as a homogeneous catalyst in benzene3 or solid LiClO4,11 BF3-etherate,11 or alumina6 in toluene, with all these catalysts used in nearly stoichiometric amounts.
Heteropoly acids (HPAs) of the Keggin series have attracted interest as promising acid catalysts for the clean synthesis of fine and specialty chemicals.14,15 Due to their stronger acidity, HPAs are usually more efficient catalysts compared to conventional acid catalysts, such as mineral acids, zeolites, mixed oxides, and ion-exchange resins. HPAs are less corrosive than mineral acids and do not effect undesirable side reactions, such as sulfonation and chlorination. HPAs are insoluble in non-polar solvents and scarcely soluble in dry carboxylic acids, allowing truly heterogeneous acid catalysis in such media. On the other hand, in many polar solvents, where HPAs are highly soluble, homogeneous catalysis is the only option. HPA salts with large monovalent cations are insoluble in water and polar solvents and can be prepared with large surface areas.16–18 In particular, the hydrophobic acidic salt, Cs2.5H0.5PW12O40, which has strong Brønsted acid sites and large surface area (100–150 m2 g–1), is a very efficient solid-acid catalyst for a variety of liquid-phase organic reactions.19–22
In our previous studies, HPAs have been used as catalysts for isomerization,23–26 hydration and esterification27–30 of terpenic compounds in liquid phase. In α-pinene oxide isomerization23,24 remarkable effect of solvent polarity and basicity on reaction chemoselectivity has been found. This allows selective synthesis of various valuable fragrance compounds through the choice of appropriate solvent. Further using cerium and tin catalysts we have shown that the balance between different reaction pathways is mostly determined by the nature of solvent rather than by the catalyst or other reaction variables.31
Within our program aimed at adding value to natural ingredients of renewable essential oils, we now investigate the application of HPA catalysts for the isomerization of limonene oxide, the reaction which has attracted much less attention as compared to the widely studied isomerization of α-pinene oxide. We explore heterogeneous HPA catalysis for limonene oxide isomerization using Cs2.5H0.5[PW12O40] (CsPW) and silica-supported H3PW12O40 as solid acid catalysts in liquid phase in a range of aprotic solvents. Our primary goal is to improve the selectivity to dihydrocarvone through catalyst and solvent optimization. To our knowledge, no attempt to use HPA catalysts for this reaction has been made so far.
2. Experimental methods
2.1. Chemicals
All chemicals were purchased from commercial sources and used as received, unless otherwise stated. (+)-Limonene oxide (a mixture of cis and trans isomers) was from Aldrich, H3PW12O40 hydrate from Aldrich and Aerosil 300 silica from Degussa.2.2. Characterization techniques
31P MAS NMR spectra were recorded at room temperature and 4 kHz spinning rate on a Bruker Avance DSX 400 NMR spectrometer using 85% H3PO4 as a reference. Powder X-ray diffraction (XRD) of the catalysts was performed on a Rigaku Geigerflex-3034 diffractometer with CuKα radiation. The textural characteristics were determined from nitrogen physisorption measured on a Micromeritics ASAP 2010 instrument at 77 K. Tungsten and phosphorus content in HPA catalysts was determined by inductively coupled plasma (ICP atomic emission spectroscopy) on a Spectro Ciros CCD spectrometer.2.3. Catalyst preparation and characterization
The 20 wt% H3PW12O40/SiO2 catalyst (PW/SiO2) was prepared by impregnating Aerosil 300 (SBET, 300 m2 g−1) with an aqueous PW solution followed by calcination at 130 °C/0.2–0.3 Torr for 1.5 h, as described elsewhere.32 The PW content determined by ICP was 20 wt%. The specific surface area determined by the BET method was 200 m2 g−1. The average pore diameter and the single point total pore volume were 144 Å and 0.53 cm3 g−1, respectively. The 31P MAS NMR spectrum of the catalyst (Fig. S1, ESI†) displayed only a single peak at ca. −15 ppm, characteristic of PW confirming the integrity of the Keggin structure.33 From XRD (Fig. S2, ESI†), the catalyst included finely dispersed PW on the silica surface, as practically no PW crystal phase can be seen.The acidic heteropoly salt CsPW was prepared according to the literature procedure34 by adding dropwise the required amount of aqueous solution of cesium carbonate (0.47 M) to aqueous solution of H3PW12O40 (0.75 M) at room temperature with stirring. The precipitate obtained was aged in aqueous mixture for 48 h at room temperature and dried in a rotary evaporator at 45 °C per 3 kPa and after that in an oven at 150 °C per 0.1 kPa for 1.5 h. CsPW thus prepared had a surface area of 111 m2 g−1, pore volume 0.07 cm3 g−1, and pore diameter 24 Å. The acid strength of CsPW and silica-supported PW was characterized calorimetrically using ammonia and pyridine adsorption and discussed previously.35,36
2.4. Catalytic reactions
The reactions were carried out in a glass reactor equipped with a magnetic stirrer and a condenser. In a typical run, a mixture (total volume of 10.0 mL) of limonene oxide (1.5–3.0 mmol), dodecane (1.0 mmol, GC internal standard) and the catalyst (PW (3.5–17.5 μmol), PW/SiO2 (5 mg, 0.35 μmol of PW), or CsPW (5 mg, 1.5 μmol)) in a specified solvent was intensely stirred under air at a specified temperature (25–140 °C). The reactions were followed by gas chromatography (GC) using a Shimadzu 17 instrument fitted with a Carbowax 20 M capillary column and a flame ionization detector. At indicated reaction times, stirring was stopped and after quick catalyst settling down aliquots were taken and analyzed by CG. The mass balance, product selectivity and yield were determined using dodecane as an internal standard.To control catalyst leaching and the possibility of a homogeneous reaction, the catalyst was removed by centrifugation of the reaction mixture at the reaction temperature to avoid re-adsorption of active components onto silica, then the supernatant was added with a fresh portion of substrate, if necessary, and allowed to react on. The absence of further reaction in such experiments indicated the absence of PW leaching and vice versa.
Products 2, 3 and 4 were separated by a column chromatography (silica gel 60) using mixtures of hexane and CH2Cl2 as eluents and identified by GC-MS, 1H, and 13C-NMR. The 1H and 13C-NMR signals were assigned using bidimensional techniques. NMR spectra were recorded in CDCl3 using a Bruker 400 MHz spectrometer, with TMS as an internal standard. Mass spectra were obtained on a Shimadzu QP2010-PLUS instrument operating at 70 eV. Products 5 and 6 were identified by GC/MS (Shimadzu QP2010-PLUS instrument, 70 eV) by comparison with an authentic sample.
3. Results and discussion
The results of limonene oxide (1) isomerization in the presence of PW catalysts in various solvents are presented in Table 1. In all solvents, limonene oxide was stable in the absence of catalysts. In blank reactions (not shown in Table 1), only negligible conversions were observed in 6 h. Detected in reaction were the following products (Scheme 1): dihydrocarvone (2), 1-methyl-3-isopropenyl-cyclopentyl-1-carboxaldehyde (3), limonene-1,2-diol (4), exo-carveol (5) and carveol (6). Our efforts were directed to achieve high selectivity to any of these products, especially 2, through the choice of catalyst and solvent and to perform the process under truly heterogeneous conditions, i.e., in the absence of leaching of HPA from the catalyst. As PW is insoluble in cyclohexane, dichloromethane and dichloroethane, we started with PW/SiO2 catalyst using these solvents.| Run | Solvent | Catalyst (mg) | PW/μmol | T/°C | Time/min | Conversion (%) | Product selectivity (%) | TONb | TOFb (min−1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | |||||||||
| a Substrate 1.5 mmol, total volume 10 mL. Conversion and selectivity were determined by GC; the difference in mass balance was due to the formation of high-boiling products; tr. – trace amounts; DMA – dimethylacetamide. b TON (turnover number) in moles of substrate converted per mole of a total amount of PW in catalyst. TOF – the average turnover frequency. c After runs 1, 2 and 3, the catalyst was removed, the solution was recharged with fresh substrate (1.5 mmol) and the reactions were allowed to proceed further, with no further conversion observed thereupon. d Substrate 3.0 mmol. | |||||||||||||
| 1c | Cyclohexane | PW/SiO2 (5) | 0.35 | 25 | 10 | 90 | 42 | 7 | 19 | tr. | tr. | 385 | |
| 60 | 97 | 47 | 5 | 14 | tr. | tr. | 4156 | ||||||
| 2c | Dichloromethane | PW/SiO2 (5) | 0.35 | 25 | 10 | 100 | 44 | 12 | 15 | tr. | tr. | 4285 | 428 |
| 3c | Dichloroethane | PW/SiO2 (5) | 0.35 | 25 | 10 | 100 | 48 | 23 | 17 | tr. | tr. | 4285 | 428 |
| 4d | Dichloromethane | PW/SiO2 (5) | 0.35 | 25 | 90 | 96 | 58 | 22 | 9 | tr. | tr. | 8228 | 91 |
| 5 | 1,4-Dioxane | PW (10) | 3.5 | 25 | 10 | 62 | 65 | 5 | 14 | 1 | 2 | 7 | |
| 60 | 100 | 65 | 8 | 12 | 4 | 7 | 428 | ||||||
| 6 | PhNO2 | PW (10) | 3.5 | 25 | 15 | 100 | 36 | 12 | 22 | 2 | 7 | 428 | 28 |
| 7 | DMA | PW (10) | 3.5 | 25 | 480 | <5 | tr. | ||||||
| 8 | DMA | PW (10) | 3.5 | 100 | 240 | 36 | 62 | tr. | tr. | 19 | 15 | 154 | 0.6 |
| 9 | DMA | PW (10) | 3.5 | 130 | 240 | 70 | 63 | tr. | tr. | 16 | 15 | 300 | 1.3 |
| 10 | DMA | PW (10) | 3.5 | 140 | 240 | 85 | 56 | 4 | 4 | 18 | 10 | ||
| 480 | 93 | 56 | 4 | 5 | 18 | 12 | 400 | 0.8 | |||||
| 11 | DMA | PW (50) | 17.5 | 100 | 60 | 93 | 50 | 4 | 9 | 18 | 10 | 80 | 1.3 |
| 12 | DMA | PW (50) | 17.5 | 140 | 60 | 96 | 53 | 4 | 5 | 20 | 15 | 82 | 1.5 |
 | ||
| Scheme 1 Main products of acid-catalyzed transformations of limonene oxide (1). | ||
It should be mentioned that the total selectivity for the detected products in some runs did not reach 100% mainly due to the formation of high-boiling products, which were not observable by GC. In the presence of only 0.1 wt% of PW/SiO2 in cyclohexane (relative to the whole reaction mixture), a 90% conversion of limonene oxide was observed in 10 min at 25 °C, with the reaction nearly completed in 1 h (Table 1, run 1). Dihydrocarvone was detected as the main product formed with 47% selectivity as a mixture of exo and endo isomers in comparable amounts. Along with dihydrocarvone, diol 4 and ring contraction product 3 were formed in small amounts. High turnover number and turnover frequency per mol of the total amount of PW were achieved in this run (TON = 4156 and initial TOF = 385 min−1). But the total yield of isomerization products 2 and 3 in cyclohexane was rather low (ca. 50%). The formation of diol 4 (14%) was probably due to epoxide ring opening by adventitious water present in the porous PW/SiO2 catalyst, commercial solvent and/or limonene oxide.
Further it has been found that the nature of solvent exerts remarkable effect on acid-catalyzed transformations of limonene oxide. In more polar solvents, such as dichloroethane and dichloromethane, much better reaction selectivities were obtained than in cyclohexane. Isomerization products 2 and 3 were formed in ca. 70% total yield along with ca. 15% of diol 4 (Table 1, runs 2 and 3). The reactions were very fast at 25 °C and showed complete conversion at first sampling in 10 min, with average TOFs of ca. 430 min−1. At a higher substrate concentration, the selectivity to diol 4 decreased probably due to a smaller water/substrate ratio, with the total yield of 2 and 3 as high as 80% (Table 1, run 4).
The possibility of HPA leaching from the PW/SiO2 catalyst and any contribution of homogeneous catalysis in reaction in cyclohexane, dichloromethane and dichloroethane was verified in special experiments. After runs 1, 2, and 3 (Table 1), the catalyst was separated by centrifugation, and the filtrates were recharged with fresh substrate and allowed to react. No further reaction was observed after catalyst removal, which shows the absence of any contribution of homogeneous catalysis, implying that there was no significant HPA leaching in these reaction systems.
To examine the effect of solvent on limonene oxide isomerization, we have tested several aprotic solvents which readily dissolved PW and could affect homogeneous catalysis (Table 1, runs 5–12). These solvents included 1,4-dioxane, nitrobenzene and dimethylacetamide (DMA), which differ in their polarity and basicity (Table 2). The polarity is represented by dielectric constants and the basicity by pKa values for the corresponding conjugate acids. In 1,4-dioxane, a non-polar basic solvent, dihydrocarvone, was obtained in 65% yield along with small amounts of 3, 5, and 6 at 25 °C (Table 1, run 5). It is evident that the reaction in rather basic dioxane is much slower than in neutral cyclohexane, dichloromethane and dichloroethane, regardless of the solvent polarity (Table 1, compare runs 1, 2 and 3 with run 5 with 10 times more PW). On the other hand, in highly polar but less basic nitrobenzene, the reaction was faster than in 1,4-dioxane, although less selective to dihydrocarvone (Table 1, run 6 vs. run 5).
The effect of solvent on reaction selectivity was especially pronounced when the reaction was performed in DMA, a highly polar and basic solvent (Table 1, runs 7–12). In DMA, allylic alcohols 5 and 6 were formed in 35% total selectivity together with 50–65% of dihydrocarvone. 90% total yield of 2, 5 and 6 was obtained under optimized conditions in DMA (Table 1, runs 11 and 12). Aldehyde 3 and diol 4 were detected in DMA only in small amounts.
At room temperature in the presence of PW, limonene oxide was nonreactive in DMA (Table 1, run 7). But at 100 °C, fast conversion of limonene oxide into products 2, 5 and 6 with a total selectivity of 96% was observed (Table 1, run 8). The reaction, however, stopped at about 35% conversion in 60 min (Fig. 1). At higher temperature (140 °C), the reaction went almost to completion (Fig. 1) to give dihydrocarvone with 60% selectivity and allylic alcohols 5 and 6 with 30% combined selectivity (Table 1, run 10). This behavior was not the result of thermodynamic control since addition of larger amounts of catalyst increased the conversion, as illustrated by runs 8 and 11 (Table 1). Pre-treatment of DMA with Amberlyst-15 acidic resin to remove possible adventitious basic impurities had no effect on the reaction. Therefore a likely explanation is that the catalyst was partially deactivated by interaction with the solvent. At 140 °C, even small amounts of PW converted all substrate in 8 h with a TON of 400 (Table 1, run 10). Thus, the deactivation is probably reversible since the catalyst can be re-activated by increasing the temperature. Similar effect has been observed previously in isomerization of α-pinene oxide in DMA.7 In this regard, high boiling point of DMA (166 °C) is an important advantage for these reactions.
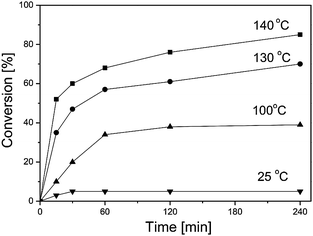 | ||
| Fig. 1 Isomerization of limonene oxide (0.15 M) catalyzed by H3PW12O40 (0.35 mM) in DMA at different temperatures. | ||
In Table 1 one can see that TON and TOF values are about one order of magnitude lower for PW in homogeneous systems than for the supported PW catalyst in heterogeneous systems. This can be explained by the solvent effect on the strength of active proton sites. Protons in the homogeneous systems (1,4-dioxane, DMA and PhNO2) are strongly solvated hence weaker and less active than those much less solvated in the heterogeneous systems (cyclohexane, dichloromethane and dichloroethane). The TON and TOF values were calculated per total number of PW molecules in the catalysts, assuming that all the protons were active. This is obviously true for homogeneous PW systems and also appears to be true for PW/SiO2 at moderate PW loadings (≤20%).36
The proposed mechanism for the acid-catalyzed transformations of limonene oxide 1 into products 2–6 is shown in Scheme 2. The opening of epoxy ring in 1 is induced by protonation of the oxygen atom to give carbenium ion A. The latter can undergo several competing transformations. It can form dihydrocarvone 2 through C2–C1 hydride shift followed by proton abstraction from carbenium ion B. Attack of water on A can result in the formation of limonene diol 4. Deprotonation of A at C7 or C6 can give directly exo-carveol 5 or carveol 6. Deprotonation could be assisted by basic solvents, such as DMA and dioxane, in which products 5 and 6 were formed in appreciable amounts. Finally, A can rearrange into carbenium ion C through electron pair transfer from C2–C3 σ-bond to C3. Then, C can lose a proton to give the ring contraction product, aldehyde 3.
 | ||
| Scheme 2 Schematic representation of acid-catalyzed transformations of limonene oxide (1). | ||
Thus, the application of PW catalysts for the isomerization of limonene oxide allows obtaining dihydrocarvone in a 50–60% yield along with only one or two minor products: aldehyde 3 or carveol and exo-carveol, which are also valuable fragrance compounds. The best selectivities were obtained under homogeneous rather than heterogeneous conditions in oxygenated solvents (1,4-dioxane and DMA) which dissolve PW. The use of solid PW/SiO2 catalyst in these solvents is not possible due to PW leaching.
Further, we tested the CsPW salt as the catalyst for limonene oxide isomerization. Importantly, the lack of solubility of CsPW as compared to PW allowed the development of truly heterogeneous synthesis of dihydrocarvone in appropriate solvents. Representative results for the reaction at 25 °C are given in Table 3. The reaction in cyclohexane was very fast in the presence of small amounts of CsPW (0.1 mol%); it reached 90% in the first aliquot taken for analysis in 10 min. Dihydrocarvone was obtained in higher yield (60%), than with PW/SiO2 under similar conditions (Table 1, run 1 vs.Table 3, run 1). The advantage of CsPW over PW in terms of reaction selectivity was even greater in other solvents. The reactions in dichloromethane, dichloroethane and nitrobenzene gave dihydrocarvone and aldehyde 3 in nearly 70 and 20% yields, respectively (Table 3, runs 2, 3, 4, and 7). These results correspond to TONs of 1000–2000 per mol of the total amount of CsPW. Considering that a part of acid sites may be located in the bulk of the solid phase and hence not accessible to the substrate, the real efficiency of the surface active sites could be even higher.
| Run | Solvent | Time (min) | Conversion (%) | Product selectivity (%) | TONb | TOFb (min−1) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 2 | 3 | 4 | 5 | 6 | ||||||
| a Substrate 1.5 mmol, catalyst 5 mg (1.5 μmol); total volume 10 mL, 25 °C. Conversion and selectivity were determined by GC; the difference in mass balance was due to the formation of high-boiling products; tr. – trace amounts; DMA – dimethylacetamide. b TON in moles of substrate converted per mole of a total amount of CsPW, TOF – the average turnover frequency. c After runs 1, 2, 5 and 7, the catalyst was removed, the solution was recharged with fresh substrate (1.5 mmol) and the reactions were allowed to proceed further, with no further conversion observed thereupon. d Substrate 3.0 mmol. | ||||||||||
| 1c | Cyclohexane | 10 | 90 | 50 | 12 | 25 | 5 | 3 | ||
| 60 | 98 | 63 | 15 | 10 | 4 | 2 | 980 | 16.3 | ||
| 2c | Dichloromethane | 240 | 100 | 69 | 20 | 8 | tr. | tr. | 1000 | 4.2 |
| 3b | Dichloroethane | 240 | 100 | 68 | 16 | 10 | tr. | tr. | 1000 | 4.2 |
| 4d | Dichloromethane | 360 | 96 | 72 | 20 | 6 | tr. | tr. | 1920 | 5.3 |
| 5c | 1,4-Dioxane | 10 | 90 | 81 | 6 | 4 | 1 | 1 | ||
| 60 | 100 | 82 | 7 | 4 | 2 | 2 | 1000 | 16.7 | ||
| 6d | 1,4-Dioxane | 10 | 91 | 80 | 9 | 5 | 3 | 3 | ||
| 60 | 98 | 80 | 6 | 6 | 2 | 3 | 1960 | 32.7 | ||
| 7c | PhNO2 | 15 | 98 | 71 | 17 | 6 | tr. | tr. | 980 | 65.3 |
Although dihydrocarvone was the major reaction product in all solvents tested, the highest dihydrocarvone yield (82%) was obtained in 1,4-dioxane (Table 3, runs 5 and 6). This is probably the best result ever reported for this reaction. CsPW is a highly efficient catalyst, operating under ambient conditions. It is truly heterogeneous, as confirmed by the filtration test described above, and can be separated from the reaction mixture by simple centrifugation and reused.
The nature of aprotic solvent used was found to affect significantly both the reaction rate and selectivity. The effect of solvent was similar for both catalysts studied and can be summarized as follows. The solvent polarity (dielectric constant) does not seem to have strong effect on the reaction rate (conversion). Non-polar solvents, such as cyclohexane, dichloromethane and dichloroethane, and polar nitrobenzene showed high reaction rates with PW and CsPW catalysts. In contrast, the solvent basicity had profound influence on both the rate and selectivity of limonene oxide isomerization. The more basic solvents, DMA and 1,4-dioxane, tend to predictably decrease the reaction rate due to decreasing catalyst acid strength. At the same time, these solvents show higher selectivities to dihydrocarvone. This may be explained assuming that in the basic “slow” solvents the reaction occurs under kinetic control, whereas in the non-basic “fast” solvents the reaction is equilibrium controlled. Therefore by choosing the appropriate solvent the reaction can be directed towards the desired product(s).
4. Conclusions
Silica-supported PW and its acidic Cs salt are efficient, environmentally benign and versatile heterogeneous catalysts for the liquid-phase isomerization of limonene oxide. The reaction can be performed under ambient conditions at low catalyst loadings and gives dihydrocarvone as the main product. High yields of this valuable fragrance intermediate were achieved through the choice of the solvent, whose polarity and basicity strongly affected the reaction rate and selectivity. This simple catalytic method represents economically attractive route to industrially valuable compounds starting from bio-renewable substrates easily available from essential oils.Acknowledgements
Financial support and scholarships from CNPq, CAPES, FAPEMIG, and INCT-Catálise (Brazil) are acknowledged.Notes and references
- W. E. Erman, Chemistry of Monoterpenes. An Encyclopedic Handbook, Marcel Dekker, New York, 1985 Search PubMed.
- C. Sell, in The Chemistry of Fragrances: from Perfumer to Consumer, ed. C. Sell, RSC Publishing, Dorset, UK, 2nd edn, 2006, vol. 2, pp. 52–88 Search PubMed.
- R. L. Settine, G. L. Parks and G. L. K. Hunter, J. Org. Chem., 1964, 29, 616–618 CrossRef CAS.
- I. C. Nigam and L. Levi, Can. J. Chem., 1968, 46, 1944–1947 CrossRef CAS.
- E. H. Eschinasi, Isr. J. Chem., 1968, 6, 713–721 CAS.
- K. Arata and K. Tanabe, Chem. Lett., 1976, 321–322 CrossRef CAS.
- J. Jayasree and C. S. Narayanan, Res. Chem. Intermed., 1997, 23, 169–178 CrossRef CAS.
- EP Pat., 1 404 635, 2003 Search PubMed.
- C. Raptis, H. Garcia and M. Stratakis, Angew. Chem., Int. Ed., 2009, 48, 3133–3136 CrossRef CAS.
- US Pat., 7 884 252, 2011 Search PubMed.
- K. Arata, S. Akutagatva and K. Tanabe, J. Catal., 1976, 41, 173–179 CrossRef CAS.
- JP Pat., 62114926, 1987 Search PubMed.
- T. Kurata, T. Koshiyama and H. Kawashima, Yukagaku, 1985, 34, 1032–1034 CAS.
- I. V. Kozhevnikov, Catalysts for Fine Chemicals, Catalysis by Polyoxometalates, Wiley, Chichester, 2002, vol. 2 Search PubMed.
- T. Okuhara, N. Mizuno and M. Misono, Appl. Catal., A, 2001, 222, 63–77 CrossRef CAS.
- I. V. Kozhevnikov, Appl. Catal., A, 2003, 256, 3–18 CrossRef CAS.
- J. B. Moffat, Metal-Oxygen Clusters. The Surface and Catalytic Properties of Heteropoly Oxometalates, Kluwer Academic Publishers, New York, 2001 Search PubMed.
- H. Niiyama, Y. Saito, S. Yoshida and E. Echigoya, Nippon Kagaku Kaishi, 1982, 4, 569–573 CrossRef.
- J. B. Moffat, J. Mol. Catal., 1989, 52, 169–191 CrossRef CAS.
- T. Okuhara, N. Mizuno and M. Misono, Adv. Catal., 1996, 41, 113–252 CrossRef CAS.
- M. Misono and T. Okuhara, CHEMTECH, 1993, 23, 23–29 CAS.
- T. Okuhara, T. Nishimura, H. Watanabe and M. Misono, Stud. Surf. Sci. Catal., 1994, 90, 419–428 CrossRef CAS.
- K. A. da Silva Rocha, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2005, 294, 106–110 CrossRef CAS.
- K. A. da Silva Rocha, J. L. Hoehne and E. V. Gusevskaya, Chem.–Eur. J., 2008, 14, 6166–6172 CrossRef CAS.
- K. A. da Silva Rocha, P. A. Robles-Dutenhefner, E. M. B. Sousa, E. F. Kozhevnikova, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2007, 317, 171–174 CrossRef CAS.
- K. A. da Silva Rocha, P. A. Robles-Dutenhefner, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2009, 352, 188–192 CrossRef.
- P. A. Robles-Dutenhefner, K. A. da Silva, M. R. H. Siddiqui, I. V. Kozhevnikov and E. V. Gusevskaya, J. Mol. Catal. A: Chem., 2001, 175, 33–42 CrossRef CAS.
- K. A. da Silva, I. V. Kozhevnikov and E. V. Gusevskaya, J. Mol. Catal. A: Chem., 2003, 192, 129–134 CrossRef CAS.
- K. A. da Silva Rocha, N. V. S. Rodrigues, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2010, 374, 87–94 CrossRef.
- A. L. P. de Meireles, K. A. da Silva Rocha, I. V. Kozhevnikov and E. V. Gusevskaya, Appl. Catal., A, 2011, 409–410, 82–86 CrossRef CAS.
- V. V. Costa, K. A. da Silva Rocha, L. F. de Sousa, P. A. Robles-Dutenhefner and E. V. Gusevskaya, J. Mol. Catal. A: Chem., 2011, 345, 69–74 CrossRef CAS.
- I. V. Kozhevnikov, A. Sinnema, A. J. A. van der Weerdt and H. van Bekkum, J. Mol. Catal. A: Chem., 1997, 120, 63–70 CrossRef CAS.
- I. V. Kozhevnikov, Chem. Rev., 1998, 98, 171–198 CrossRef CAS.
- Y. Izumi, M. Ono, M. Kitagawa, M. Yoshida and K. Urabe, Microporous Mater., 1995, 5, 255–262 CrossRef CAS.
- E. F. Kozhevnikova and I. V. Kozhevnikov, J. Catal., 2004, 224, 164–169 CrossRef CAS.
- A. M. Alsalme, P. V. Wiper, Y. Z. Khimyak, E. F. Kozhevnikova and I. V. Kozhevnikov, J. Catal., 2010, 276, 181–189 CrossRef CAS.
- G. Erős, H. Mehdi, I. Pápai, T. A. Rokob, P. Király, G. Tárkányi and T. Soós, Angew. Chem., Int. Ed., 2010, 49, 6559–6563 CrossRef.
- J. March, Advanced Organic Chemistry: Reactions, Mechanisms, and Structures, Wiley, NY, 4th edn, 1992, p. 250 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20526b |
| This journal is © The Royal Society of Chemistry 2013 |