Challenges in the catalytic synthesis of cyclic and polymeric carbonates from epoxides and CO2
Paolo P.
Pescarmona
* and
Masoumeh
Taherimehr
Centre for Surface Chemistry and Catalysis, KU Leuven, Kasteelpark Arenberg 23, 3001 Heverlee, Belgium. E-mail: paolo.pescarmona@biw.kuleuven.be
First published on 29th June 2012
Abstract
The addition of carbon dioxide to epoxides to produce either cyclic carbonates or polycarbonates is an important reaction allowing the conversion of a renewable, inexpensive and non-toxic feedstock such as CO2 into useful products with many potential applications. In this perspective article, an overview of the type of catalysts used for this reaction, the mechanisms with which they operate and the parameters influencing their activity and selectivity are presented and discussed critically. In this context, the main challenges to be tackled in this vibrant area of research are highlighted.
 Paolo P. Pescarmona | Paolo Pescarmona was born in 1973 in Torino, Italy. After receiving his Master Degree in Chemistry from the University of Torino, he moved to the Netherlands where he obtained his Ph.D. from the Delft University of Technology in 2003. Since 2005 he works at the University of Leuven (KU Leuven), Belgium, first as post-doc researcher and since 2008 as professor. Presently, his group consists of six Ph.D. students and two post-doc researchers. His research interests are steadily growing and embrace many aspects of catalysis and of materials chemistry. A common thread in his research is the attention towards sustainability, as exemplified by the topic of this perspective article. His curiosity is not limited to chemistry and he is keen on travelling and learning about different cultures around the world. |
 Masoumeh Taherimehr | Masoumeh Taherimehr was born in 1983 in Ahvaz, Iran. In 2005 she received the Degree in Chemistry from the Chamran University of Ahvaz and in 2007 she obtained her Master Degree from the Lorestan University, Iran. In 2010 she started her Ph.D. at the KU Leuven under the supervision of Prof. Pescarmona. Her research interests include homogeneous and heterogeneous catalysis for the synthesis of organic compounds from renewable sources, with a focus on the conversion of carbon dioxide to valuable products. |
1. Introduction
The growing concerns about the environmental impact of carbon dioxide emissions produced by fuel combustion and other human activities prompted the scientific community to research ways to store or, preferably, reuse this molecule.1 In this context, the chemical fixation of CO2 to produce useful compounds has attracted increasing interest.2,3 Carbon dioxide is an inexpensive, widely available, non-toxic and renewable carbon source, but its use as feedstock for the production of valuable chemicals faces the challenge of its high thermodynamic stability.4 This implies that the conversion of carbon dioxide may require a high energy input, as in the case of the photocatalytic reaction with water to generate oxygen and methanol or methane.5 An alternative approach consists in the reaction of CO2 with compounds with a relatively high free energy, thus providing a thermodynamically favourable process.4 Among the reactions employing this last strategy, the addition of carbon dioxide to epoxides is a relevant topic of research since it is an atom-efficient reaction that can generate useful classes of compounds as cyclic carbonates and polycarbonates (Scheme 1).4,6–9 This perspective article focuses on this reaction and on the catalytic systems used to promote it, with the purpose of providing a systematic analysis of the topic and of underlining the challenges that still need to be tackled. Both cyclic carbonates and polycarbonates are valuable compounds with many established and potential applications. Cyclic carbonates are the thermodynamically favoured products of the reaction of epoxides with carbon dioxide.4,6,8 Their application range is broad, the most employed compounds being ethylene carbonate and propylene carbonate.4,6,7 Cyclic carbonates can be used as intermediates in the synthesis of fine or bulk chemicals,1,6 as exemplified by the Asahi Kasei industrial process for the production of polycarbonates from bisphenol A.10 This multi-step process involves the use of ethylene carbonate as reagent and represents a greener alternative to the currently main industrial process, which is based on the highly toxic phosgene as reagent. Other applications of cyclic carbonates are as environmentally friendly polar aprotic solvents, with low odour and toxicity,7,11,12 and as electrolyte solvents for lithium-ion batteries.13 The second class of organic carbonates that can be obtained by the reaction of carbon dioxide with epoxides are the polycarbonates. At this stage, it is important to note that the term polycarbonate is used to refer to various types of polymers, which can present rather different physicochemical properties from each other. A polycarbonate is defined as a polymer containing carbonate groups (–O−(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)–O–) in its backbone chain: this description includes both the commercial polycarbonates that are synthesised using bisphenol A as building block, and the polymers obtained by the alternating copolymerisation of CO2 with an epoxide, which are discussed in this article. The bisphenol-based polycarbonates are thermoplastic polymers with excellent mechanical and physical properties, including high strength, rigidity, toughness, durability, transparency to light, heat resistance and good electrical insulation. Owing to these properties, bisphenol-based polycarbonates can substitute glass or metal in many uses and find widespread application as materials for data storage (e.g. CDs and DVDs), for electronic components, for lenses, as construction materials and as automotive and aircraft components.6,10 On the other hand, polycarbonates obtained by copolymerising carbon dioxide and epoxides display less suitable properties including low rigidity (glass transition temperature of 35–40 °C for poly(propylene carbonate) compared to about 150 °C for bisphenol-based polycarbonates) and moderate thermal stability.8,14 So far these features limited the application of these polymers,14 despite their biodegradability and the fact that they can be prepared in a green process from renewable carbon dioxide. These observations lead us to the identification of a first challenge (1): the properties of alternating copolymers obtained from the coupling reaction of carbon dioxide and epoxide should be improved in order to attain practical applications for these materials. This might be achieved by investigating different types of epoxides as substrates for the copolymerisation reaction with CO2; by optimising the catalysts in order to increase the molecular weight and crystallinity of the polymers; by cross-linking the polymer chains; by copolymerisation involving two different epoxides or an epoxide and another type of monomer; through the use of fillers during the synthesis to form (nano)composites; through blending with other polymers.14,15 The improved polymeric materials could find a vast range of applications as engineering thermoplastics, elastomers, packaging materials, binders, adhesives and coatings.8,14
O)–O–) in its backbone chain: this description includes both the commercial polycarbonates that are synthesised using bisphenol A as building block, and the polymers obtained by the alternating copolymerisation of CO2 with an epoxide, which are discussed in this article. The bisphenol-based polycarbonates are thermoplastic polymers with excellent mechanical and physical properties, including high strength, rigidity, toughness, durability, transparency to light, heat resistance and good electrical insulation. Owing to these properties, bisphenol-based polycarbonates can substitute glass or metal in many uses and find widespread application as materials for data storage (e.g. CDs and DVDs), for electronic components, for lenses, as construction materials and as automotive and aircraft components.6,10 On the other hand, polycarbonates obtained by copolymerising carbon dioxide and epoxides display less suitable properties including low rigidity (glass transition temperature of 35–40 °C for poly(propylene carbonate) compared to about 150 °C for bisphenol-based polycarbonates) and moderate thermal stability.8,14 So far these features limited the application of these polymers,14 despite their biodegradability and the fact that they can be prepared in a green process from renewable carbon dioxide. These observations lead us to the identification of a first challenge (1): the properties of alternating copolymers obtained from the coupling reaction of carbon dioxide and epoxide should be improved in order to attain practical applications for these materials. This might be achieved by investigating different types of epoxides as substrates for the copolymerisation reaction with CO2; by optimising the catalysts in order to increase the molecular weight and crystallinity of the polymers; by cross-linking the polymer chains; by copolymerisation involving two different epoxides or an epoxide and another type of monomer; through the use of fillers during the synthesis to form (nano)composites; through blending with other polymers.14,15 The improved polymeric materials could find a vast range of applications as engineering thermoplastics, elastomers, packaging materials, binders, adhesives and coatings.8,14
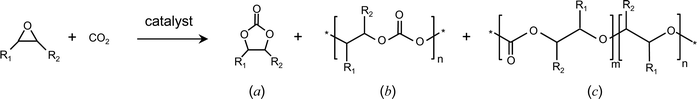 | ||
| Scheme 1 Possible products of the reaction of CO2 with epoxides: cyclic carbonate (a), polycarbonate (b) and polycarbonate containing ether linkages (c). | ||
From the point of view of green chemistry and sustainability, the relevance of the synthesis of organic carbonates through the reaction of carbon dioxide with epoxides lies in the low cost, availability and renewable character of CO2 as carbon source and in the features and potential applications of the products: cyclic carbonates can be used as green solvents and provide an alternative to the use of the highly toxic phosgene as reagent,7,10,11 while polycarbonates could substitute synthetic polymers prepared from non-renewable petroleum feedstock.8 On the other hand, it should be noted that the benefits on the mitigation of the impact of CO2 emissions through this reaction are expected to be limited.16 Presently, only ∼0.5% of the CO2 generated by human activities is reused by the industry, and of this amount only a small fraction is employed for the synthesis of organic carbonates (i.e. acyclic carbonates, cyclic carbonates and polycarbonates).1 The processes for the synthesis of organic carbonates have the potential to reach a larger scale, particularly if the applicability of the products and the efficiency of their synthesis will be improved (see next sections for a more detailed discussion). However, the yearly need for these and other compounds that could be produced by CO2 fixation is likely to remain low compared to the yearly amount of CO2 generated worldwide by fuel combustion. The minimisation of carbon dioxide emissions will require most probably a combination of approaches including the use of greener energy sources as alternative to fossil fuels,1 the improvement of the efficiency of existing processes generating CO2 as a waste (e.g. transportation, heating, industrial processes) and worldwide policies to avoid an unsustainable growth of the human population.
In this perspective article, the different mechanisms leading to the catalytic synthesis of either cyclic or polycarbonates from the reaction of carbon dioxide with epoxides are reviewed and rationalised, and the parameters governing the activity and the selectivity of the catalysts are critically discussed. The definition of this general picture provides the context to introduce the various open challenges in this field of research. The target of this article is to help understanding the catalytic systems used for this reaction and to suggest relevant research directions, and not to provide a comprehensive overview of all homogeneous and heterogeneous catalysts reported for the reaction of CO2 with epoxides, for which purpose excellent reviews are available.4,6–9,14,17–19
2. Mechanism of the addition of carbon dioxide to epoxides
The reaction of CO2 with epoxides can generate two types of products: cyclic carbonates and polycarbonates (a and b in Scheme 1). Moreover, the consecutive insertion of two epoxides in the polymer chain can occur instead of the alternating copolymerisation of carbon dioxide and epoxide.8,14 This leads to the presence of ether linkages in the polycarbonate (c in Scheme 1), or to the formation of polyethers in the extreme case of polymerisation occurring exclusively between epoxide molecules (m = 0 in structure c).The addition of CO2 to epoxides is generally carried out in the presence of a catalytic system that can initiate the reaction by activating either the epoxide or carbon dioxide, or both at the same time. The epoxide is typically activated by interaction of the oxygen atom with a Lewis acid, followed by a nucleophilic attack that causes the opening of the epoxide ring. CO2 is a linear, apolar molecule, though the two CO bonds are polar, implying that the carbon atom has a partial positive charge and the oxygen atoms have a partial negative charge. Therefore, the carbon atom can act as an electrophile and the oxygen atoms as nucleophiles: the activation of CO2 can thus occur both through a nucleophilic and an electrophilic attack (vide infra). It follows from these considerations that most catalytic systems that have been investigated for this reaction contain Lewis acid sites for the electrophilic activation of epoxide and/or carbon dioxide and Lewis base sites acting as nucleophiles. The two sites can belong to two different compounds – for example the metal of a complex as Lewis acid and the anion of a salt as Lewis base – or be parts of a single compound, as in a complex containing a cationic metal centre and a labile anionic ligand.
Different mechanisms for the reaction of carbon dioxide with epoxides have been proposed, and in many cases demonstrated by means of characterisation with various spectroscopic and analytical techniques. The predominance of a specific reaction path depends on the nature of the catalytic system, but also on the substrate and on the reaction conditions. In all cases the activation of the epoxide requires a nucleophile, which is the common feature of all catalytic systems employed for the reaction of carbon dioxide with epoxides. Regardless of the nature of the first step, all mechanisms involve an intermediate containing a carbonate precursor, which can either react further through consecutive additions of other epoxide and CO2 molecules (propagation) to generate a polycarbonate, or undergo a back-biting reaction leading to ring closure with formation of the cyclic carbonate (Scheme 2). If polycarbonates are the products of the reaction, the nucleophilic species that serves as the initiating group will also be the end group of the polymer chain.
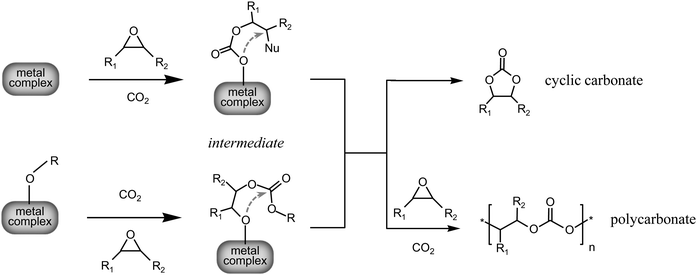 | ||
| Scheme 2 Possible intermediates of the reaction of epoxides with CO2, exemplified in the case of the reaction catalysed by a metal complex. Nu is a nucleophile that can originate from the metal complex or from a co-catalyst. RO- is an alkoxide or an aryloxide that can act as a nucleophile. The dotted arrows indicate the possible back-biting reactions that would lead to the formation of cyclic carbonate. | ||
The most widely studied type of catalysts for the reaction of carbon dioxide with epoxides are homogeneous metal complexes, which can be employed alone if they contain a ligand that can act as a nucleophile, or in combination with a co-catalyst providing the nucleophilic species. A variety of metal centres acting as Lewis acid sites has been studied for these complexes: Al, Cr, Mn, Co, Fe, Cu, Zn, Cd, Mg, Li and lanthanoids.8,14 Both monometallic and bimetallic complexes have been employed as catalysts for this reaction. The most common ligands used for the synthesis of these complexes are: salen and related ligands, β-diiminates (BDI), phenolates and porphyrins (Fig. 1, 2, 4 and 7).19 Non-metallic homogeneous catalysts based on salts of organic cations have been reported as well (see Section 2.3). Heterogeneous catalysts are also known: the most widely applied is zinc glutarate, but heterogeneous systems based on the immobilisation of homogeneous catalysts on a support are receiving increasing attention (see Section 4).14,18,19
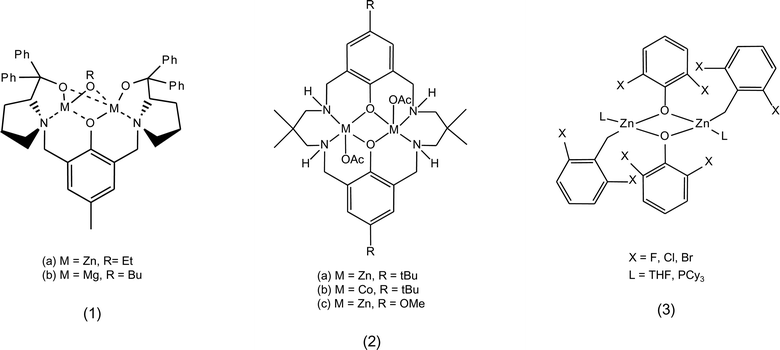 | ||
| Fig. 1 Phenolate metal complexes. | ||
 | ||
| Fig. 2 Metal complexes with salen and related ligands. | ||
Most of the mechanisms proposed for the reaction of CO2 with epoxides have been derived from studies of homogeneous catalytic systems, but heterogeneous catalysts are considered to follow similar pathways.20 In order to facilitate the systematic discussion of the various reaction mechanisms, we divided them into three categories based on the number of metal atoms involved in the catalytic cycle: monometallic pathways, bimetallic pathways and non-metallic pathways.
2.1 Monometallic pathways
This type of mechanisms is exemplified by the wide group of homogeneous catalytic systems based on metal complexes. These metal complexes can include a ligand that can act as nucleophile. Alternatively the metal complex, usually referred to as the catalyst, is used in combination with an added nucleophile generally denoted as the co-catalyst.(a) A common monometallic mechanism for the addition of CO2 to epoxide catalysed by a metal complex proceeds through the steps summarised in Scheme 3. First, the epoxide coordinates to a Lewis acidic metal centre and gets activated towards attack by a nucleophile. The nucleophilic attack leads to opening of the epoxide ring and formation of a metal-bound alkoxide. This alkoxide can in turn act as a nucleophile leading to CO2 insertion into the metal alkoxide bond, generating a metal carbonate intermediate. This carbonate species can either evolve towards the cyclic carbonate or propagate by further addition of epoxide and CO2 with formation of a polycarbonate (vide supra). The occurrence of this mechanism has been supported by an isotope-labelling experiment.21
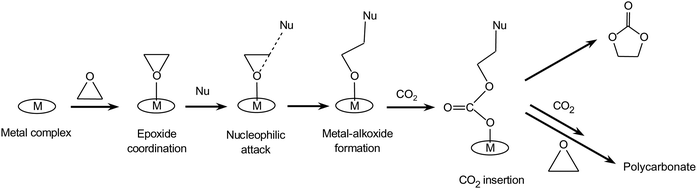 | ||
| Scheme 3 Reaction of CO2 with epoxides: monometallic pathway involving one nucleophile. The nucleophile (Nu) may originate from the metal complex or from a co-catalyst. | ||
(b) A monometallic mechanism involving two nucleophiles has been proposed for metal salen complexes (e.g. Cr-salen), which are characterised by a planar configuration with a tetradentate coordination of the salen ligand (Fig. 2). The axial positions of the metal are available for coordination with nucleophiles. Of the two nucleophiles involved in the mechanism, one is part of the metal complex as axial ligand (Nua) and the second one (Nub) is added to the system and coordinates to the metal in the remaining trans axial position, thus generating a six-coordinated intermediate (II in Scheme 4).9,14,22 The coordination of the second nucleophile serves to labilise the other metal-nucleophile bond, favouring coordination and nucleophilic attack of the epoxide (III). The formed alkoxide species (IV) acts in turn as a nucleophile that attacks CO2 to form a metal carbonate species. The presence of a nucleophile in the trans position to this carbonate (V) has been reported to favour the further nucleophilic attack and ring opening of a new epoxide molecule (similarly to what occurs in step III). Therefore, the alternating copolymerisation of epoxide and carbon dioxide leading to the formation of polycarbonates is the preferential path with this mechanism.23 In general, the nucleophiles can be neutral or ionic species. Kinetic studies indicated that anionic six-coordinated intermediates (II in Scheme 4) are more effective both in the opening of the epoxide ring and in the CO2 insertion compared to their neutral six-coordinated analogues.9 It is has been proposed, though not unequivocally established, that both Nua and Nub can serve as initiator groups leading to the growth of two polymer chains from one metal centre.9,14,24
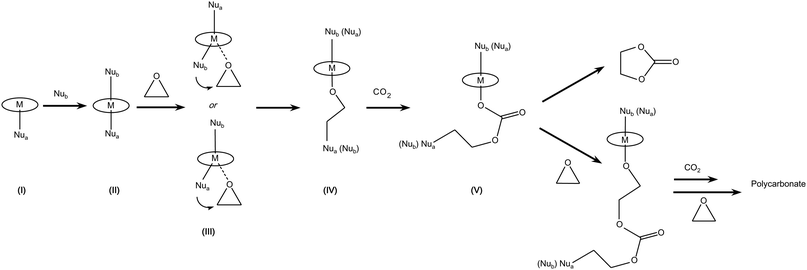 | ||
| Scheme 4 Reaction of CO2 with epoxides: monometallic pathway involving two nucleophiles. Note that Nua and Nub may be the same or different type of nucleophile. | ||
2.2 Bimetallic pathways
The addition of CO2 to epoxides can follow a bimetallic mechanism if the catalyst contains two metal centres or if two monometallic catalysts act in collaboration.(a) A bimetallic pathway involving two separate metal complexes has been inspired by a mechanism proposed for the asymmetric ring opening of epoxides.25 This intermolecular mechanism is similar to the monometallic pathway (2.1.a), with the difference that the nucleophile that attacks the epoxide activated by the metal centre is coordinated to a second metal centre (Scheme 5).8 The bimetallic initiation is followed by a monometallic pathway leading to cyclisation and formation of cyclic carbonate or to propagation of the alternating polycarbonate. Kinetic studies by means of in situ infrared spectroscopy supported this mechanism for a Cr-salen complex.26 This pathway is expected to occur when low epoxide/catalyst ratios are employed.19 It has been suggested that this mechanism tends to be followed when a weaker Lewis base is used as co-catalyst, while mechanism 2.1.b is more likely to occur in the presence of a stronger Lewis base that is able to weaken the metal-nucleophile bond and allow concerted opening of the epoxide ring.27 If this mechanism is followed, the reaction rate has a second-order dependence on the metal concentration, while a first-order dependence is observed in the case of a monometallic pathway.
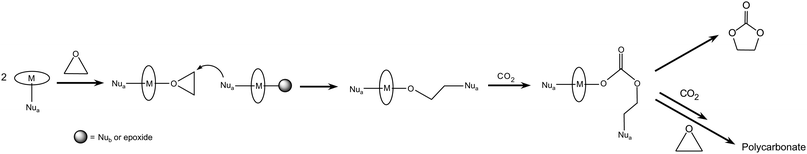 | ||
| Scheme 5 Reaction of CO2 with epoxides: bimetallic pathway involving two monometallic complexes. | ||
(b) Metal complexes containing two neighbouring metal centres may activate epoxide and CO2 simultaneously (Scheme 6). This promotes the intramolecular nucleophilic attack of the alkoxide to the carbon atom of the activated CO2 molecule. This mechanism has been proposed to explain the improved catalytic activity of bimetallic [Al(salen)]2O complexes (Fig. 8) compared to monometallic salen complexes.28 It has been suggested that in this mechanism the co-catalyst, tetratbuylammonium bromide (Bu4NBr), plays a second role besides the ring opening of the epoxide by the bromide. Tetrabuylammonium bromide can decompose to tributylamine, which can form a carbamate salt with CO2 (Bu3N+–CO2−) and this salt can coordinate more easily to the second Al-centre compared to CO2 alone.29
 | ||
| Scheme 6 Reaction of CO2 with epoxides: bimetallic pathway with simultaneous activation of epoxide and CO2 on a complex with two adjacent metal centres. | ||
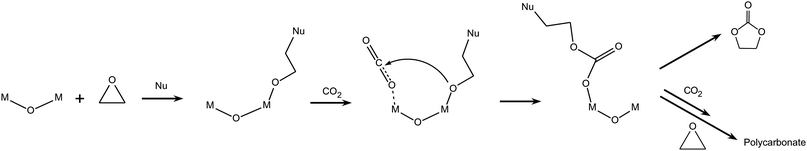
(c) The first step of the catalytic addition of carbon dioxide to epoxides is not always the activation of the epoxide. Some metal complexes containing an alkoxide or aryloxide group (–OR) are able to undergo CO2 insertion to form a bidentate carbonate as the first step (Scheme 7).8,14 This is not unexpected, because the –OR groups are analogous to the alkoxide intermediates formed upon epoxide insertion in the monometallic mechanisms described above, and such metal-bound alkoxides are able to react with CO2 (see Section 2.1). The reaction continues through the coordination of the epoxide followed by ring opening and insertion into the metal carbonate bond, which was proposed to be the rate-determining step of the process.30 This type of mechanism has been proposed and investigated in detail for bulky β-diiminate zinc complexes containing two adjacent metal centres bridged by alkoxide (or carboxylate) groups (Fig. 4).8,30 β-Diiminate zinc complexes can exist both in dimeric (bimetallic) or monomeric (monometallic) form. The most likely pathway is bimetallic, as sketched in Scheme 7, but a contribution from a monometallic mechanism cannot be excluded.30 The prevalence of the monomeric or dimeric form of the complex in solution strongly depends on the steric hindrance of the substituents.8 Catalysts for which the first step of the mechanism is CO2 insertion with formation of a bidentate species generally display high selectivity towards the copolymerisation reaction.8,9 In this context, it has been suggested that the presence of two adjacent metal centres can promote the selectivity of the reaction towards the growth of a polycarbonate chain by simultaneously coordinating two species (see Scheme 7), thus favouring the interaction of the growing chain with new incoming reagents.19 Nevertheless, the presence of two neighbouring metal centres is not a sufficient condition for selectively generating polycarbonates, as proven by the high selectivity towards cyclic carbonate displayed by Al-salen bimetallic complexes following mechanism 2.2.b.28
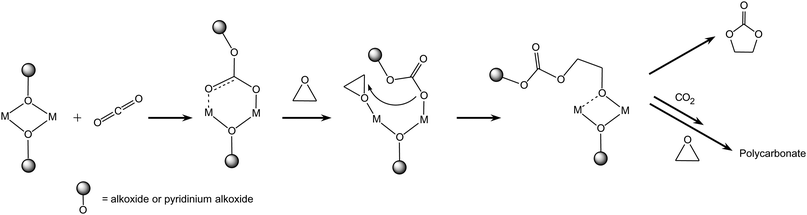 | ||
| Scheme 7 Reaction of CO2 with epoxides: bimetallic pathway with complexes containing two adjacent metal centres and –OR groups. | ||
2.3 Non-metallic pathways
Salts of organic cations have been employed alone as catalysts for the reaction of carbon dioxide with epoxides. Particularly, ionic liquids have been studied either as homogeneous or heterogeneous catalysts, in the latter case in the form of supported species.18 Carbon dioxide can dissolve in considerable amounts in ionic liquids, implying that the ionic liquids can be employed with the double role of solvents and catalysts. In general, reactions with ionic liquids as catalysts without the assistance of metal centres require relatively high temperature (≥80 °C) in order to achieve good product yields, thus favouring the formation of the thermodynamically more stable cyclic carbonate as the main product. The mechanism followed by these non-metallic catalysts depends on the nature of the anion and cation (X− Y+) constituting the salt. Two possible pathways may lead to formation of the carbonate product (Scheme 8):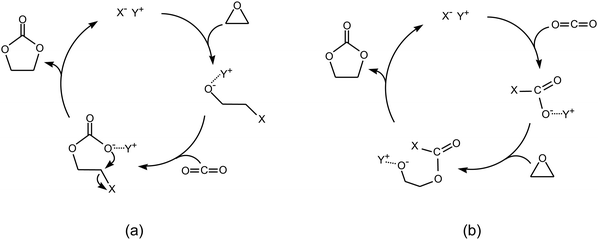 | ||
| Scheme 8 Reaction of CO2 with epoxides: non-metallic pathways with epoxide activation as the first step (a) or with CO2 activation as the first step (b). | ||
(a) The most common mechanism involves a nucleophilic attack of the epoxide by the anion, which causes opening of the epoxide ring, followed by the insertion of carbon dioxide and finally by ring closure to generate the cyclic carbonate. This mechanism is very similar to the monometallic pathway described in 2.1.a.
(b) If the anion is bulky (e.g. BF4−, PF6−), the interaction between the epoxide and the anion is very weak and the anion rather interacts with CO2 to generate a new ionic species [X−CO2]−, that is more basic than the original anion X− and thus able to attack the epoxide to generate a carbonate intermediate and finally the cyclic product.18 This pathway was proposed on the basis of a mechanistic study by means of in situ ATR infrared spectroscopy.31
The heterogenisation of ionic liquids has been employed as a strategy to develop recyclable catalysts for the synthesis of cyclic carbonate from CO2 and epoxide.32–36 Among these supported ionic liquids, the imidazolium-based ionic liquids containing an –OH or a –COOH group in the cation are particularly interesting from a mechanistic point of view.34,35 It has been proposed that the –OH of the alcoholic or carboxylic group can interact with the oxygen atom of the epoxide, thus helping the nucleophilic attack of the epoxide by the anion (Scheme 9). The role of the –OH is similar to that of the Lewis acidic metal centre in the metallic mechanisms (vide supra).
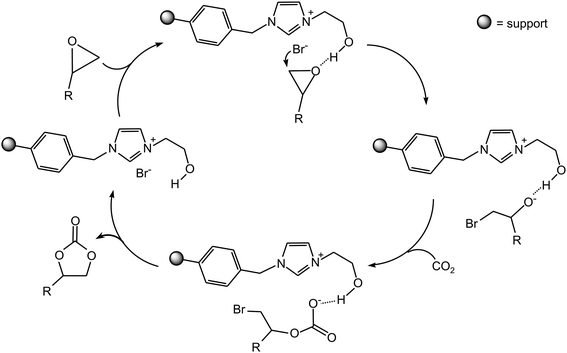 | ||
| Scheme 9 Synthesis of cyclic carbonate catalysed by an immobilised ionic liquid with a side chain terminating with an –OH group. | ||
3. Catalytic activity and selectivity in the synthesis of cyclic carbonates and polycarbonates
As discussed in Section 1, cyclic carbonates and polycarbonates are both relevant compounds. However, if we want to design a catalyst that is able to produce any of them efficiently it is crucial to understand and control the parameters determining the selectivity of the reaction of CO2 and epoxides towards each of these two families of carbonates (challenge 2). The formation of polycarbonates is thermodynamically less favourable than the synthesis of cyclic carbonates, but it generally requires lower activation energy.9,37 This means that by selecting a suitable catalytic system and by tuning the reaction conditions it should be possible to selectively produce either one or the other type of carbonate. In the case in which the polycarbonate is the desired product it is also important to avoid the formation of polyether linkages (see Section 2) and to achieve polymers with high molecular weight and low polydispersity. These parameters can play a relevant role in obtaining polycarbonates with suitable properties for application (see challenge 1). The control of the selectivity is not the only challenge connected to the design of improved catalysts for this reaction. The catalytic activity can still be improved compared to the already known, promising catalysts, in order to reach higher turnover frequencies under mild temperature and pressure conditions with different types of substrates, including sterically hindered and internal epoxides and oxetanes (challenge 3).A rationalisation of the factors determining the activity and selectivity of known catalysts for the reaction of CO2 with epoxides can be very useful for designing novel catalysts with improved performance. For this purpose, the influence of the nature of the catalyst, of the co-catalyst (if present), of the substrate and the role of the solvent and of the reaction conditions will be analysed and discussed in this section for selected catalysts reported in the literature.
3.1 Metal-containing catalysts
Metal complexes are the most widely studied class of metal-containing catalysts for the reaction of carbon dioxide with epoxides (see Section 2). The knowledge gained by studying the parameters that influence the catalytic behaviour of these complexes can be rather general and apply to other catalytic systems, including heterogeneous ones. Both the metal and the ligands play a role in defining the properties of metal complexes as homogeneous catalysts for the reaction between CO2 and epoxides.(a) The nature of the metal centre in metal complexes plays a crucial role in their catalytic performance. The monometallic and bimetallic pathways discussed in Section 2 are characterised by alkoxide and carbonate intermediates containing a metal–oxygen bond. The strength of this bond is an important factor in determining the activity and selectivity of the catalyst. A too strong metal–oxygen bond makes the intermediate inactive both towards another insertion, which would lead to propagation and copolymer production, and towards ring closure, which would yield a cyclic carbonate. On the other hand, a weak metal–oxygen bond can be displaced by a nucleophile or a solvent molecule, leading to lower activity but also favouring the back-biting reaction and, thus, increasing the selectivity towards the cyclic carbonate. It can be concluded that intermediate bond strength is needed for a good catalytic performance and that the properties of the catalyst can be tuned by the choice of the metal centre.38 The relevance of the nature of the metal centre can be recognised by comparing the catalytic behaviour of complexes with the same ligand scaffold but with different metal centres.
• Replacing the two zinc atoms in the bimetallic phenolate complex 1 in Fig. 1 with magnesium atoms increased the catalytic activity if the reaction was carried out at low CO2 pressure (1 atm, see Table 1).14,37,39 It was hypothesised that the harder Lewis acid nature and higher oxophilicity of Mg2+ in comparison with Zn2+ might make the former more effective for epoxides activation under these reaction conditions.37
| Catalyst | Epoxidea | Catalyst loading/mol% | T/°C | p(CO2)/bar | Reaction time/h | TONb | TOFc/h−1 | Major carbonate product [selectivity (%)] | M n (kg mol−1) | PDId (Mw/Mn) | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| a CHO = cyclohexene oxide; PO = propylene oxide; SO = styrene oxide; b TON = mol of epoxide converted per mol of metal; c TOF = mol of epoxide converted per mol of metal per hour; d PDI = polydispersity index; Mn = number average molar mass; Mw = weight average molar mass; e Based on the monomeric complex. | |||||||||||
| 1a | CHO | 5 | 60 | 1 | 6 | 10 | 2 | Polycarbonate | 19.2 | 1.56 | 39 |
| 1b | CHO | 0.67 | 60 | 1 | 2 | 43 | 22 | Polycarbonate | 12.6 | 1.29 | 37 |
| 2a | CHO | 0.1 | 80 | 1 | 24 | 219 | 9 | Polycarbonate [96] | 6.2 | 1.19 | 42 |
| 2b | CHO | 0.1 | 80 | 1 | 2 | 170 | 86 | Polycarbonate [>99] | 5.1 | 1.26 | 43 |
| 3a [X = F, L = THF] | CHO | 0.1 | 80 | 55 | 48 | 364 | 8 | Polycarbonate | 42 | 6 | 56 |
| 4b/PPNCl | PO | 0.05 | 25 | 15 | 2 | — | 328 | Polycarbonate [>99] | 25.4 | 1.14 | 45 |
| 5b [X = OCCCl3]/PPNCl | PO | 0.05 | 25 | 15 | 1.5 | — | 568 | Polycarbonate [>99] | 28.1 | 1.14 | 45 |
| 5c/PPNCl | PO | 0.05 | 25 | 1.5 | 2 | — | 235 | Polycarbonate [>99] | 15.5 | 1.18 | 45 |
| 6b/Bu4NI | SO | 2.5 | 45 | 10 | 3 | 39 | 13 | Cyclic carbonate [>99] | — | — | 69 |
| 7a [X = NO3]/DMAP | PO | 0.1 | 25 | 15 | 3 | — | 86 | Polycarbonate [>99] | 13.2 | 1.29 | 22 |
| 8/PPNCl | CHO | 0.1 | 70 | 34 | 2 | — | 230 | Polycarbonate [>99] | 91 | 1.15 | 50 |
| 9 | PO | 0.05 | 25 | 14 | 12 | 1540 | 128 | Polycarbonate [99] | 23.9 | 1.14 | 51 |
| 10 | PO | 0.001 | 70 | 20 | 1 | 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 000 |
16000 |
Polycarbonate [>99] | 300 | 1.31 | 54 |
| 11 [R = iPr, R′ = Me] | CHO | 0.1e | 50 | 7 | 2 | 449 | 224 | Polycarbonate [95] | 19.1 | 1.07 | 59 |
| 13d | CHO | 0.1 | 50 | 7 | 0.5 | 364 | 729 | Polycarbonate | 23.3 | 1.15 | 30 |
| 14c | CHO | 0.009 | 80 | 12 | 10 | 2720 | 272 | Polycarbonate | 261 | 1.6 | 61 |
| 15b/DMAP | CHO | 0.2 | 80 | 50 | 24 | 500 | 21 | Polycarbonate [>99] | 14.5 | 1.13 | 67 |
| 16/Bu4NBr | SO | 2.5 | 25 | 1 | 3 | 12 | 4 | Cyclic carbonate [>99] | — | — | 68 |
• Compared with their organozinc counterparts, many organoaluminium-based catalysts are less active and selective in the synthesis of polycarbonates and tend to promote cyclic carbonate production.40 An explanation for the preferential formation of cyclic carbonates with aluminium salen complexes has been provided by theoretical calculations.41 It was found that the dissociation of the growing polymer chain from aluminium salen complexes is more favourable than from chromium salen complexes. The dissociation can be followed by ring closure with formation of the cyclic carbonate, thus accounting for the higher selectivity towards this product.
• Dizinc macrocyclic complex 2a in Fig. 1 displays high activity in the copolymerisation of CO2 and cyclohexene oxide at low CO2 pressures.42 This catalytic behaviour was attributed to the coordinative flexibility of the ligand and to the vicinity of the two metal centres, which can facilitate bidentate binding of the growing carbonate chain (see mechanism 2.2.c), thus lowering the activation energy for CO2 insertion.14 Replacing zinc with cobalt (2b) in this type of bimetallic complexes caused a significant increase of the catalytic activity (Table 1). Considering that the rate determining step is likely to be the epoxide ring opening by the nucleophilic carbonate chain, the higher activity of the cobalt complex can be attributed to an increased nucleophilicity of the carbonate propagating species in the attack of the epoxide ring when the metal centre is Co(II) compared to Zn(II).43
• The electronic configuration of the metal centre can determine the type of coordination of the metal complex. In the case of metal salen catalysts (Fig. 2), it has been shown that a six-coordinated metal complex with a distorted octahedral geometry (see mechanism 2.1.b) is necessary for the alternating copolymerisation to proceed efficiently.44 For example, six-coordinated salen complexes with high catalytic activity are formed with chromium(III) and cobalt(III), which have half-filled t2g orbitals. On the other hand, Schiff base Mn(III) complexes are preferentially five-coordinated. These Mn-complexes have low tendency to bind the epoxide substrates and, therefore, are ineffective in catalysing the reaction between CO2 and epoxides.
(b) The ligands in a metal complex can be strongly coordinated to the metal centre or be labile. Both types play a role in determining the electronic and steric properties of the metal complex. The latter ones can also actively participate in the catalytic reaction. From these considerations, it becomes clear that a careful selection of the ligands is crucial for optimising the catalytic activity and selectivity of metal complexes.
The importance of the strength of the metal–oxygen bond in the alkoxide and carbonate intermediates was underlined in Section 3.1.a. The strength of this bond is not only influenced by the metal, but also by the nature of the ligand(s) coordinated to it. Electron-donating ligands and electron-donating groups on the ligands can lead to a decrease in the metal–oxygen bond strength, thus favouring the back-biting reaction and increasing the selectively towards the production of cyclic carbonate. Conversely, electron-withdrawing groups will decrease the electron density of the metal centre, thus increasing its Lewis acid character. The role of the ligand is intimately related to that of the metal centre, of the co-catalyst and of the reaction conditions. Therefore, the optimum features of the ligand can be extremely different for different systems. Many studies demonstrated the impact of changes in the structural and chemical features of the ligand and in the nature of its substituents on the catalytic activity and selectivity of metal complexes.
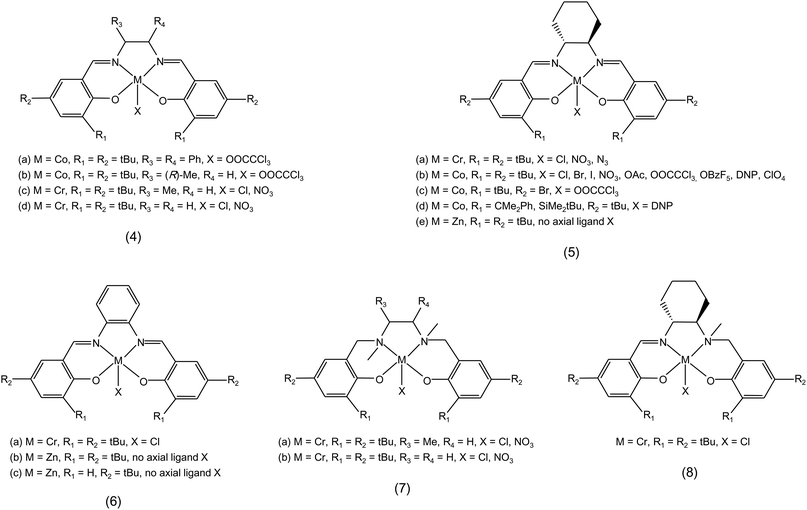
• Various studies on the effect of changes in the structure and in the substituents of the ligand have been carried out with metal salen complexes. In the case of cobalt salen complexes (X = OOCCCl3, complexes 4a, 4b and 5b in Fig. 2), ligands with a non-cyclic diimine backbone showed lower catalytic activity for copolymerisation of propylene oxide and CO2 compared to their counterparts with the cyclic diimine backbone (Table 1). Moreover, it was found that electron-withdrawing substituents in the para-position of the phenolate are detrimental to the copolymerisation activity (compare 5b with X = OOCCCl3 and 5c in Table 1).45
• The substituents on the aromatic ring of the salen ligand can also have a steric effect: in the reaction of styrene oxide with CO2, a significant decrease in the activity and polymer selectivity of a Co-salen complex was observed when changing the groups in the ortho-position of the phenolate from tert-butyl to very bulky groups (i.e. −CMe2Ph, −SiMe2tBu, see 5b with X = DNP and 5d in Fig. 2).46
• Changing the cyclohexane moiety of the diimine backbone of a salen ligand to a phenyl group (salphen ligand, 6 in Fig. 2) was reported to affect the selectivity of the chromium complex in the reaction of CO2 with propylene oxide. The Cr-salphen complex (6a) has a fully conjugated ligand structure and is more selective towards poly(propylene carbonate) (if using no more than one equivalent of co-catalyst, see Section 3.2.a). The Cr-salen complex (5a with X = Cl) has a non-conjugated fragment and is more selective towards cyclic propylene carbonate. This result was explained by the more electron-donating nature of the salen ligand, which weakens the metal–oxygen bond, thus favouring the dissociation of the intermediate species and leading to the cyclic product (see Section 3.1.a).47
• The complexity of the catalytic reaction of carbon dioxide and epoxides is underlined by the different effects that a same change in the ligand can have with different metals. Switching from a salen to a salphen ligand had a major effect on the catalytic activity of Zn complexes employed with Bu4NI as co-catalyst but did not affect the selectivity, which was directed towards cyclic carbonates with a variety of epoxides (5e and 6b in Fig. 2 and Table 1).48 The higher activity of the Zn-salphen complexes stems from the higher Lewis acidity of the zinc centre, which is ascribed not only to the electronic effects of the ligand but also to the rigid, planar geometry imposed by the ligand scaffold. The effect of the substituent in the para-position on the phenolate was also investigated (6b and 6c in Fig. 2): bulky groups in these positions are needed to prevent the dimerisation of the Zn-salphen complexes, which would cause a dramatic drop in catalytic activity.49
• The nature of the ligand determines whether the complex has a rigid or flexible structure. In different contexts, both features have been claimed to be advantageous. Chromium salan complexes (7 in Fig. 2 and Table 1) contain nitrogen atoms with sp3 hybridisation, which makes them more flexible than salen complexes and decreases the electrophilicity of the metal centre. These features were proposed to favour the dissociation and re-association of the co-catalyst and of the growing polymer chain, thus explaining the much higher activity in the reaction of propylene oxide and CO2 with the salan complexes compared to their salen counterparts (7a and 7b vs. 4c and 4d).22 Similarly, the high activity of the chromium salalen complex 8 (where the salalen ligand contains a nitrogen atom with sp2 hybridisation and the other with sp3 hybridisation, see Fig. 2 and Table 1) in the copolymerisation of cyclohexene oxide and CO2 under mild conditions was ascribed to its coordinative flexibility, which can allow bidentate binding of the growing polymer chain, thereby decreasing the energy barrier for CO2 insertion.50 More in general, it has been suggested that the ability of a metal complex to afford bidentate coordination or coordination of two species (i.e. epoxide and carbonate or CO2 and ring-opened epoxide) at the same time, tends to favour the propagation reaction, making the catalyst selective for polycarbonate synthesis.19
• Functionalisation of the ligand with tailored groups can positively affect the catalytic performance of a metal complex. The presence of a side arm bearing a protonated piperidinium group on a salen ligand was proposed to prevent cyclic carbonate formation by protonating the polymer chain in case it dissociated from the metal centre, thus avoiding the back-biting reaction that would lead to the cyclisation reaction (9 in Fig. 3 and Table 1).51
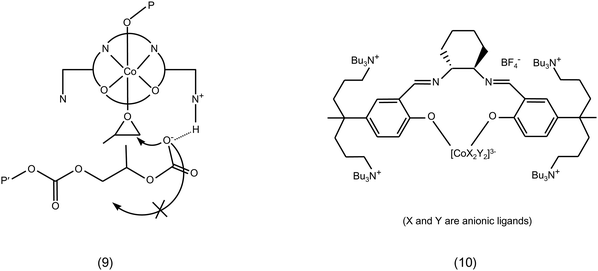 | ||
| Fig. 3 Cobalt salen complexes with functionalised side arms. | ||
Another approach based on the functionalisation of the ligand aimed at keeping dissociated anionic polymer chains in the proximity of the metal centre by binding the co-catalyst to the ligand: cobalt salen complexes with bound quaternary ammonium units (10 in Fig. 3) displayed improved activity and selectivity for copolymerisation and generated polycarbonates with very high molecular weight (Table 1).52–55
• Introducing halide substituents in the ortho-position of the aromatic rings of the ligands of the bimetallic phenolate zinc complex 3 (Fig. 1 and Table 1) caused an increase in the activity of the catalyst in the copolymerisation of cyclohexene oxide with CO2. The increase was proportional to the electronegativity of the halide (Br < Cl < F), which causes a decrease in electron density at the metal centre and, thus, an increase in the binding ability of the metal to the epoxide.56,57 This is in line with the rate-determining step for bimetallic zinc complexes being the activation of the epoxide rather than the insertion of CO2 (see mechanism 2.2.c). The same trend is followed by the bimetallic zinc complexes 2 in Fig. 1, which showed a decrease in catalytic activity upon variation of the substituent in the para-position on the aromatic ring from an alkyl group to an electron-donating methoxy group.58
• Modifying a ligand towards optimum catalytic performance of the metal complex often requires a fine tuning. This is exemplified by the β-diiminate zinc complexes (Fig. 4 and Table 1), for which the substituents at both the N-aryl ring and the diimine backbone can influence strongly the catalytic behaviour of the complex.8,59 For the β-diiminate zinc acetate complexes, the N-aryl groups should provide sufficient steric bulk to prevent the formation of tightly-bound, inactive dimeric species, but should also not be so bulky to generate encumbered, inactive monomeric species. The most active species were shown to have intermediate features, allowing the formation of highly-active loosely-bound dimers (12 in Fig. 4).30 The introduction of electron-withdrawing groups (e.g. −CN, −CF3, 13e–g in Fig. 4) on the diimine backbone leads to a relevant enhancement of the catalytic activity in the copolymerisation reaction.60 However, the presence of more electron-withdrawing groups is detrimental to the activity (13h in Fig. 4), probably because the consequently increased Lewis acidity of the metal centre results in too strong metal–oxygen bonds (see Section 3.1.a).19
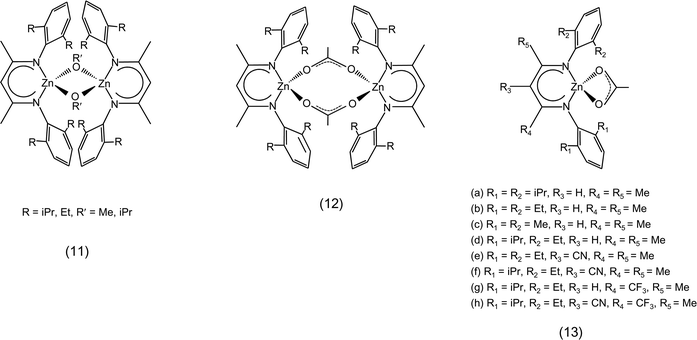 | ||
| Fig. 4 β-Diiminate zinc complexes. | ||
• Another example of the electronic and steric effect of the substituents is provided by bimetallic zinc anilido-aldimine complexes as catalysts for the copolymerisation of cyclohexene oxide and CO2 (Fig. 5 and Table 1). Bulky groups on the aromatic rings can have either a positive or negative effect on the activity according to their position within the complex. An increase in activity was also observed using electron-withdrawing fluorine substituents, whose proposed double role is to decrease the electron density at the metal centres, increasing the ability to bind CO2 and epoxide, and to decrease the basicity of the ligand and, thus, the sensitivity of the complex to protic impurities.61,62
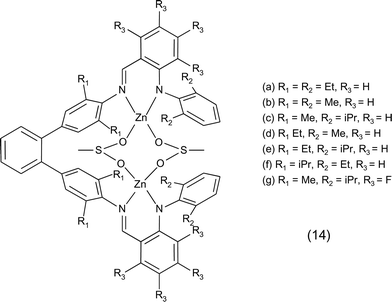 | ||
| Fig. 5 Bimetallic zinc anilido-aldimine complex. | ||
Two of the most widely studied types of ligands in metal complexes employed as catalysts for the reaction of carbon dioxide with epoxides are the porphyrin and the salen ligands (see Section 2). Both provide complexes with a planar structure and tetradentate coordination of the metal centre. Depending on the nature of the metal, these complexes may accommodate also ligands in the two axial positions. These axial ligands can be neutral or anionic, they are generally labile and, therefore, can be displaced during the reaction.
Generally, the rate-determining step of the coupling reaction between epoxides and CO2 is the epoxide ring-opening by a nucleophile. This can be an external nucleophile or a nucleophile belonging to the catalyst. In the case of salen or porphyrin complexes, the nucleophiles occupy the axial sites in the complex (see mechanisms 2.1.b and 2.2.a). Nucleophile ligands in axial position may have two roles: as a nucleophile that dissociates from the metal centre to attack the epoxide substrate and open its ring; or as a nucleophile that decreases the strength of the bond between the metal and the nucleophile in the trans axial position (see mechanisms in Section 2). The features of the axial ligand can affect both the degree of epoxide conversion and the selectivity of the reaction. To achieve good activity, the axial ligand should not be too strongly bound to the metal, so that is can dissociate from it, while still being a sufficiently good nucleophile to attack the epoxide. If the axial ligand is able to dissociate easily from the complex, it might also be a good leaving group from the metal-bound carbonate intermediates, thus facilitating the back-biting reaction and favouring cyclic carbonate production (see Scheme 2). On the other hand, a nucleophile with poor leaving ability can suppress the formation of cyclic carbonates and increase the selectivity towards polycarbonates. If a complex with a nucleophile in the axial position is used in combination with a co-catalyst providing a second nucleophilic species to the reaction, the effect of the axial ligand is tightly related to the nature of the co-catalyst (see mechanism 2.1.b). These considerations indicate that a fine tuning is generally required for finding the optimum axial ligand for the desired reaction (cycloaddition or copolymerisation).
• For example, the selectivity between polymeric and cyclic carbonate in the reaction of propylene oxide with CO2 catalysed by a cobalt salen complex in combination with Bu4NBr as co-catalyst was dramatically increased (from 3 to 78%) by changing the axial ligand (X) of the complex from acetate to 2,4-dinitrophenolate (DNP) (5b in Fig. 2).63 When the same complex was used as catalyst with bis(triphenylphosphine)iminium chloride [(PPN)Cl] as co-catalyst, a high selectivity towards polycarbonate was observed also with axial groups that gave low selectivity with Bu4NBr.45 The reaction rate was only slightly affected by changes in the axial group, apart from the case in which this was the non-nucleophilic ClO4−, which caused a complete loss of activity. In another example with the same type of cobalt salen complex but without any co-catalyst, it was found that changes in the axial ligand resulted in substantial differences in the rate of reaction between propylene oxide and CO2. The activity decreased in the order X = Br > OBzF5 ∼ OAc > Cl > I, with a complete selectivity towards the polycarbonate in all cases.64 This trend was not maintained if the reaction was carried out in the presence of (PPN)Cl as co-catalyst: in such a case, the rates were significantly higher with all axial ligands, but the highest turnover was now observed with X = OBzF5 (pentafluorobenzoate), while the selectivity towards the polymer slightly decreased with X = OAc or Br.45,64
3.2 Co-catalysts and non-metallic catalysts
(a) With a variety of metal complex catalysts for the coupling reaction between epoxides and CO2, the catalytic performance can be greatly improved by the addition of a Lewis base as co-catalyst.14,45 If the metal complex does not contain a group that can act as a nucleophile (e.g. in Zn-salphen, see Section 3.1.b), the co-catalyst is actually essential to observe any conversion in the reaction between epoxide and CO2.48 Similarly to the role of the nucleophilic axial groups in salen complexes (see Section 3.1.b), the co-catalyst can play two different roles: as a nucleophile that attacks and opens the epoxide coordinated to the metal centre; or as a nucleophile that coordinates to the metal centre increasing its electron density and thus weakening the bond with other nucleophilic species (e.g. the growing polymer chain) and providing a favourable electronic environment for the insertion of epoxide and CO2 molecules (see mechanisms in Section 2).A specific balance between nucleophilicity (towards attack of the epoxide), coordination ability (towards the metal centre), and leaving ability (from the carbonate intermediate species) of the co-catalyst is required for enhancing the activity and for tuning the selectivity of each different metal complex. The selectivity is often controlled by the leaving ability of the nucleophile: a poor leaving group will favour the growth of the polymer chain while a good leaving group will promote the back-biting reaction with cyclic carbonate production. Co-catalysts can be neutral (e.g. an organic base) or ionic (e.g. an ammonium salt), see Fig. 6. With ionic co-catalysts, both the anion and the cation may influence the reaction between CO2 and epoxide.45 For a given anion, the nucleophilicity towards the reagents will be higher if its cation is bulkier and thus exerts lower ion-pairing electrostatic attraction towards the anion.18
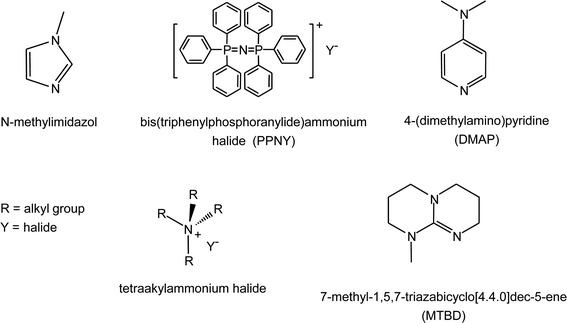 | ||
| Fig. 6 Co-catalysts employed in the reaction of CO2 with epoxides. | ||
• The ratio between co-catalyst and catalyst has been reported to play a decisive role in determining the selectivity towards cyclic or polymeric carbonates. A higher ratio of nucleophile to metal centres can favour the selectivity towards cyclic carbonate over polycarbonate, because the excess of nucleophile tends to displace the metal-bound carbonate intermediate, thereby favouring the back-biting reaction leading to the formation of the cyclic product.9,14,65 As an example, it has been found that a co-catalyst/catalyst ratio higher than 2 tends to favour the selectivity towards the cyclic carbonate with chromium salen complexes.41,47,66
• The importance of the use of a co-catalyst is exemplified by the catalytic behaviour of tetraphenylporphyrin aluminium complexes (15a in Fig. 7). These aluminium complexes without co-catalyst promoted the homopolymerisation of propylene oxide with very little incorporation of CO2 into the polymer chain. On the other hand, in the presence of 4-(dimethylamino)pyridine (DMAP) as co-catalyst, the alternating copolymerisation of propylene oxide and CO2 became the dominant reaction. The proposed role of DMAP is two-fold: it promotes the insertion of CO2 into the growing polymer chain and increases the rate of ring opening of the epoxide by the carbonate intermediate (mechanism 2.1.b).23
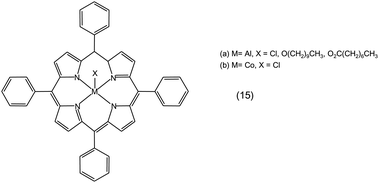 | ||
| Fig. 7 Tetraphenylporphyrin metal complexes. | ||
Similarly, the cobalt porphyrin complex 15b in Fig. 7 produced polycarbonate selectively from the reaction of cyclohexene oxide and CO2 in the presence of DMAP as co-catalyst in a 0.5–1 ratio to the complex (Table 1). Under the same reactions conditions, but without DMAP, poly(cyclohexene oxide) was the only product. If the DMAP/complex ratio was increased to 10, both polycarbonate and cyclic carbonate were produced, in agreement with what is observed with other types of complexes (vide supra).67
• An example of the effect of the leaving ability of the anion of the co-catalyst on the selectivity is provided by studies with the cobalt salen complex 5b (Fig. 2) as catalyst and a tetrabutylammonium salt (Bu4NY) as co-catalyst in the reaction of propylene oxide with CO2. Changing the anion (Y−) of the co-catalyst to poorer leaving groups, that is from I− and Br− to Cl−, F− or CH3COO−, suppressed the back-biting reaction and thus increased the selectivity towards the polycarbonate. In the case of F− and CH3COO− this was accompanied by a significant decrease in the reaction rate.45,63 The explanation of this decrease in activity is not straightforward as it requires considering the possible interactions of the anion with the ammonium cation, with the metal centre and with the reagents.
The same cobalt salen complex 5b (Fig. 2) was employed to study the role of the steric hindrance of the co-catalyst. It was proposed that the ideal ionic co-catalyst for achieving high activity and selectivity in the copolymerisation of carbon dioxide and epoxides consists of an anion with poor leaving ability (e.g. Cl−, see above) and of a bulky cation (e.g. bis(triphenylphosphoranylide)ammonium, PPN+). For neutral co-catalysts, sterically hindered strong organic Lewis bases (e.g. 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (MTBD)) are preferred over other strong but less bulky organic bases (e.g. N-methylimidazole), which can strongly coordinate to the cobalt centre, thus making it catalytically inactive.45
The positive features of bis(triphenylphosphoranylide)ammonium chloride as co-catalyst were also demonstrated by a study of the copolymerisation of cyclohexene oxide with CO2 with chromium salen complex 5a (X = Cl) in Fig. 2. The reaction rate was significantly increased using (PPN)Cl instead of N-methylimidazole as co-catalyst. The improvement was ascribed to the combination of an anionic nucleophile with a non-interacting cation as PPN+, in which the positive charge is delocalised over the large cation. With this Cr-salen complex and this co-catalyst, mechanism 2.1.b, which implies a shorter initiation time and thus a higher reaction rate, is expected to be favoured over mechanism 2.2.a.27
• A study of the effect of the anion of the co-catalyst used in combination with a bimetallic aluminium salen complex (16 in Fig. 8 and Table 1) was performed by carrying out the reaction of styrene oxide with CO2 in the presence of different tetrabutylammonium halides (Bu4NY, with Y = F, Cl, Br, I). In all cases the reaction was completely selective towards the cyclic carbonate, but the conversion of the epoxide varied between a maximum of 62% with Bu4NBr to almost no conversion with Bu4NF. The overall order of activity was Br > I > Cl > F.68 This somehow peculiar trend can be explained considering that the catalytic activity of the halide is determined by a balance between its nucleophilicity and leaving-group ability: a good nucleophile would favour the initial opening of the epoxide ring, but it would also be a worse leaving group in the last step of the reaction, i.e. in the back-biting reaction leading to the formation of cyclic carbonate. This balance is also influenced by the nature of the Lewis acid site, if present, and by the nature of the cation of the halide salt. Therefore, different catalytic systems can show different order of activity between the halides.18,68 For example, Zn-salphen complexes (6b in Fig. 2) display higher activity in the synthesis of various cyclic carbonates with Bu4NI as co-catalyst compared to Bu4NBr.69 Even for the same catalyst, the order of activity between halides can be different with different epoxides: with a bimetallic iron amine triphenolate complex as catalyst, Bu4NI led to higher rates of conversion of terminal epoxides compared to Bu4NBr, while with sterically hindered epoxides Bu4NBr was more active. This behaviour has been explained on the basis of the smaller size of Br−, which can be advantageous in the nucleophilic attack of sterically congested epoxides.70
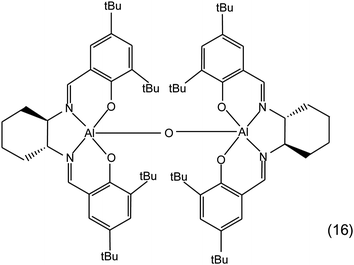 | ||
| Fig. 8 Bimetallic aluminium salen complex. | ||
Additionally, also the stability of the tetraalkylammonium salt during the reaction might play a role in the catalytic process (see Section 2.2.b).29
(b) Ionic or neutral nucleophilic compounds, similar to those used as co-catalysts together with metal catalysts, can also be employed alone as catalysts for the addition of carbon dioxide to epoxides (see Section 2.3). Examples of catalysts of this type include ionic liquids, ammonium, pyridinium and phosphonium salts, and strong organic bases.7,71 These non-metallic compounds can be used as homogeneous catalysts or be immobilised on a support to generate heterogeneous catalysts.18 These catalytic systems are intrinsically simpler compared to the binary systems consisting of a metal complex and a co-catalyst. This facilitates the correlation between their physicochemical features and their catalytic properties, particularly in the case of homogeneous systems. Still, more than one reaction mechanism is possible with non-metallic catalysts (see Section 2.3) and the reaction rate depends both on the nucleophilicity of the anion and on the features of the cation. For example when the anion is a halide, bulkier cations provide weaker electrostatic attraction to the halide, thus increasing their nucleophilicity towards the opening of the epoxide ring.18 This implies that the order of activity between halides of quaternary ammonium salts can change as a function of the nature of the ammonium cation (see previous paragraph).68,72,73 The absence of a metallic Lewis acid in these catalytic systems implies that generally higher reaction temperatures are needed to achieve a good degree of epoxide conversion, driving the reaction towards the thermodynamically favoured cyclic carbonates.7
3.3 Substrate
The rate of reaction and selectivity towards polymeric or cyclic carbonate formation does not depend only on the features of the catalytic system, but is also influenced by the nature of the epoxide substrate. Both steric and electronic effects play a role.• Steric congestion around the epoxide ring tends to cause a decrease in conversion rate in the reaction with carbon dioxide. This steric effect is more likely to hinder the nucleophilic attack of the epoxide rather than its coordination to the Lewis acid metal centre.48,69,70,74 This explains the lower conversion degree usually observed with internal epoxides (e.g. cyclohexene oxide) or α-disubstituted epoxides (e.g. 1,1-dimethyloxirane) compared to those of terminal epoxides with a linear alkyl chain (e.g. propylene oxide, 1,2-epoxyhexane).48,70
• Cyclohexene oxide has a higher tendency to form polycarbonate rather than cyclic carbonate compared to other epoxides such as propylene oxide or styrene oxide. This preference of cyclohexene oxide for polycarbonate formation is ascribed to the strain in forming the five-membered cyclic carbonate ring imposed by the adjacent cyclohexane ring.14,75 As a consequence, the synthesis of poly(propylene carbonate) is often accompanied by the formation of cyclic carbonate while the synthesis of poly(cyclohexene carbonate) tends to proceed more selectively.
• The nature of groups on the epoxide can affect its tendency to produce preferentially either the cyclic or the polymeric carbonate. For example, epoxides with electron-withdrawing groups, such as styrene oxide, have a higher tendency to produce cyclic carbonate compared to aliphatic terminal or internal epoxides.76 This behaviour has been explained on the basis of the most likely position at which the epoxide ring is attacked by a nucleophile, leading to ring opening (Fig. 9). For terminal epoxides such as propylene oxide, the nucleophilic attack is considered to occur predominantly at the less sterically hindered carbon atom of the epoxide ring (β-carbon),9 both due to its higher accessibility and to the electron-donating effect of the alkyl group. On the other hand, for epoxides such as styrene oxide the nucleophilic ring-opening occurs mainly at the α-carbon as a consequence of the electron-withdrawing inductive effect of the phenyl group. This can lead, after CO2 insertion, to an intermediate in which the electron-withdrawing nature of the aromatic ring can favour the back-biting reaction with formation of cyclic styrene carbonate (Scheme 10).46,76 Similarly, the electron-withdrawing nature of the chloromethyl group of epichlorohydrin tends to drive the reaction towards the cyclic carbonate product.77 In the case of propylene oxide, even if the nucleophilic attack at the β-carbon of the epoxide ring is favoured, the attack at the α-carbon can occur. This is demonstrated by the generally observed presence of head-to-head and tail-to-tail connections in the poly(propylene carbonate) chain, whereas if the nucleophilic attack occurred always at the same position the polymer would contain exclusively head-to-tail connections (Fig. 10).23,45 A high percentage of head-to-tail connections (regioregularity) is desirable as it can increase the crystallinity and improve properties of the polymer including thermal resistance, toughness and stiffness (see challenge 1).14 The nature of the catalytic system can strongly affect the regioregularity of the obtained polycarbonate.63,64 Particularly, it has been suggested that a chiral environment around the metal centre can promote the regioselectivity of the reaction.46
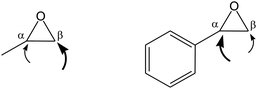 | ||
| Fig. 9 Most likely positions for a nucleophilic attack of the ring in different types of epoxides. | ||
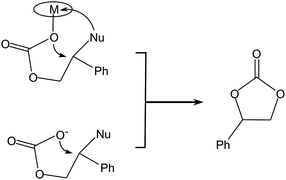 | ||
| Scheme 10 Possible back-biting reactions in the reaction of styrene oxide with CO2. | ||
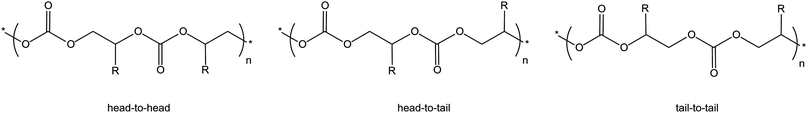 | ||
| Fig. 10 Possible epoxide/CO2 connections in polycarbonates. | ||
• The nature of the epoxide substrate has a strong influence on the properties of the alternating copolymer obtained upon reaction with CO2. As mentioned in challenge 1, the physicochemical properties of these polycarbonates should be enhanced in order to grant their applicability. This could be achieved by increasing the rigidity of the polymer backbone through the choice of suitable epoxides. Poly(propylene carbonate) has good mechanical properties but rather low glass transition temperature (∼40 °C), while poly(cyclohexene carbonate) has higher glass transition temperature (115 °C) but is brittle. Recently, indene oxide has been introduced as substrate for the synthesis of a promising polycarbonate, displaying the highest glass transition temperature (134 °C) reported so far for alternating copolymers of epoxides and carbon dioxide.14,78 This poly(indene carbonate) is thermally stable up to 249 °C, which is in the same range as the thermal decomposition temperature of poly(propylene carbonate) and lower than that of poly(cyclohexene carbonate) (∼300 °C).8,14
• Oxetanes represent another class of compounds that has been recently studied as substrates for the formation of cyclic and polymeric carbonates upon reaction with CO2.70,79,80 Compared to epoxides, oxetanes are characterised by lower ring strain, which implies that the ring opening step is more challenging. On the other hand, binding of the oxetanes to the Lewis acidic metal centre can be favoured by their higher basicity compared to epoxides.80 Another main difference between epoxides and oxetanes is that for the former the five-membered cyclic carbonates are the thermodynamically favoured products, while for the latter the polycarbonate is thermodynamically more stable than the six-membered cyclic carbonate.
3.4 Solvent and reaction conditions
(a) In order to achieve optimum performance of the catalytic system, the contact between all components taking part in the reaction should be maximised (challenge 4).69 In the case of a reaction with a binary homogeneous catalytic system, e.g. a metal complex and an ammonium salt, this implies trying to get a homogeneous solution containing CO2, the epoxide, the catalyst and the co-catalyst. This can be a challenging task, as these components can cover a rather broad range of polarity. To complicate the situation, the solubility of CO2 in liquids depends on its density,81 which increases with increasing pressure and with decreasing temperature, while the solubility of the catalyst and co-catalyst in an organic solution generally increases with temperature. This means that it is difficult to predict the effect of a change of temperature on the overall solubility between the components. Moreover, a high degree of dissolution of CO2 into the organic solution can cause a decrease in the solubility of the catalytic species and possibly induce their precipitation.82 In some cases, the catalyst and/or co-catalyst are not soluble in the epoxide substrate in the concentration chosen for the catalytic test, and a solvent is added to the reaction mixture to achieve full dissolution of the catalytic species. The presence of a solvent does not necessarily affect the solubility of CO2 into the reaction mixture, but would always cause a dilution of the solution, thus decreasing the contact between the reaction components. This can negatively affect the reaction rate, as demonstrated by the much higher activity of a Zn-salphen/Bu4NI catalytic system in a solvent-free, CO2-rich environment compared to that in CH2Cl2 or other solvents.49,69 This means that if the catalytic species dissolve in the epoxide at the temperature at which the reaction will be carried out, the use of any additional solvent should be generally avoided.Another example of the importance of the contact between the reaction components was provided by the synthesis of cyclic carbonates carried out in imidazolium-based ionic liquids with varying length of the alkyl chain of the cation (C2 to C8): choosing ionic liquids bearing a longer alkyl chain and working under conditions in which carbon dioxide is supercritical led to enhanced catalytic activity as a consequence of the higher solubility of epoxides and CO2 in the ionic liquid phase.83
If the desired product of the reaction of CO2 with epoxides is the polycarbonate, the presence of alcohols or water even in small amounts should be avoided, as these polar molecules can react with the growing polymer chain to yield a hydroxyl-terminated polycarbonate and a metal-alkoxide or metal-hydroxide species from which a new polymer chain can grow. Therefore, these chain transfer reactions limit the number of repeating units and, thus, the molecular weight of the polymer products.14 Moreover, it has been reported that the interaction of water with the metal centre of the catalyst can cause substantial loss in activity and polymer selectivity.45
(b) CO2 can be in its gas, liquid or supercritical state (i.e. above its critical point, 31.1 °C, 73.8 bar) under the reaction conditions. Performing the catalytic test in supercritical CO2 rather than with CO2 in the gas phase can be advantageous for maintaining a good contact between the components participating in the reaction, particularly if the epoxide substrate has a relatively low boiling point and would tend to shift to the gas phase at low CO2 pressure.69
Carbon dioxide dissolves into epoxides but in general the two species do not form a single phase. On the basis of the temperature and of the CO2 pressure, the amount of CO2 dissolved can vary significantly, and in some conditions the volume of the phase containing the epoxide can expand sensibly upon CO2 dissolution. Carrying out the reaction in a reactor with a viewing window can provide useful information on the phase behaviour of the system.69
The effect of CO2 pressure on the catalytic activity and selectivity was studied using cobalt and chromium salen complexes (Fig. 2) as catalysts for the copolymerisation of CO2 and epoxides.47,63,84 In general, it was found that the activity increases with CO2 pressure until a maximum, after which the insertion of CO2 is not anymore the rate-limiting step and further increase of the pressure causes dilution of catalyst and substrate, thus decreasing the reaction rate. On the other hand, the selectivity towards the cyclic carbonate side-product with the Cr-salen catalyst was lower at higher CO2 pressure, possibly because high CO2 concentrations favour the insertion of this molecule over the back-biting reaction leading to cyclic carbonate.84 A similar trend of activity as a function of CO2 pressure, with a maximum value at an intermediate pressure and a dilution effect causing a decrease of activity with increasing CO2 pressure, has been observed with heterogeneous catalysts based on supported ionic liquids.32,33,36
(c) Since cyclic carbonates are thermodynamically more stable than polycarbonates, carrying out the reaction of CO2 and epoxides at elevated temperatures can increase the selectivity towards the formation of cyclic carbonates. On the other hand, decreasing the reaction temperature will decrease the reaction rate but can enhance the selectivity towards the polycarbonates: since the activation energy for the synthesis of polycarbonates is usually lower than that for the thermodynamically more favoured cyclic carbonates, a lower reaction temperature can allow a kinetic control of the reaction, thus favouring the polycarbonates. This strategy has been employed to steer the reaction towards polycarbonates with epoxides that would otherwise tend to give higher selectivity towards the cyclic carbonate, as those bearing an electron-withdrawing group (see Section 3.3).76,77
4. Homogeneous vs. heterogeneous catalysts
The next generation of catalysts for the reaction of carbon dioxide with epoxides should not only display enhanced activity and selectivity towards the desired carbonate product (cyclic or polymeric), but should also fulfil the requirements for a green, sustainable process. Therefore, research efforts should be centred on catalytic systems that do not contain potentially toxic species (such as, e.g., chromium or 4-(dimethylamino)pyridine), are active in the absence of solvent (see Section 3.4) and can be easily separated from the reaction products and reused in consecutive runs without loss of activity (challenge 5). The last feature is essential in order to develop catalysts that could find industrial application in the production of cyclic and polymeric carbonates. On the basis of these considerations, a shift in the focus of the research from homogeneous towards heterogeneous catalysis is expected. So far, heterogeneous catalysts for the reaction of carbon dioxide with epoxides have been studied less extensively compared to homogeneous systems. The factors determining the activity and selectivity of homogeneous catalysts (see Section 3) can apply to their heterogeneous counterparts, but the design flexibility that is typical of metal complexes is restricted in the case of heterogeneous catalysts by the requirement of a material that can be readily recovered and reused. The most known and widely employed heterogeneous catalyst for the copolymerisation of CO2 and epoxides is zinc glutarate, [Zn(O2C(CH2)3CO2)]n.8,14 This zinc dicarboxylate catalyses the production of poly(propylene carbonate) with high molecular weight and with only minor contamination from ether linkages or cyclic carbonate.14 However, its turnover frequency is significantly inferior compared to that of the most active metal complex catalysts.19 The catalytic reaction catalysed by zinc glutarate has been proposed to follow the bimetallic mechanism described in Section 2.2.c.20 It has been suggested that the distance between two neighbouring zinc centres is crucial in determining the activity of this family of heterogeneous catalysts. Double metal cyanides (e.g. Zn3[M(CN)6]2 with M = Fe or Co) are another class of widely investigated heterogeneous catalysts for the copolymerisation of CO2 and epoxides.14 However, these catalysts can present relevant drawbacks, such as low CO2 incorporation in the polymer chain and the requirement for harsh reaction conditions. Metal oxides, zeolites and clays have also been studied as heterogeneous catalysts for the reaction of CO2 with epoxides, but all these systems suffer from one or more serious limitations such as the requirement for a homogeneous base as co-catalyst, low activities, need for harsh reaction conditions in terms of temperature and CO2 pressure, or leaching of the active species.85–87 A totally different strategy for preparing a heterogeneous catalyst for the coupling reaction of CO2 and epoxides consists in immobilising active homogeneous catalysts on the surface of a support.7,18,85 Typical materials that can be used as supports are polymers or silica-based materials, including mesoporous ordered materials with high specific surface area such as MCM-41 and SBA-15.32,36,88,89 The immobilisation of the catalytically active species should be performed through robust covalent grafting to the support, in order to avoid possible leaching of active species during the reaction.88,89 A general drawback of supported catalysts compared to their homogeneous counterparts is the lower activity due to diffusion limitations of reagents and products between the solution and the catalyst surface.90 This effect can be minimised by using supports with sufficiently large pores, and by tuning the loading of the supported species. On the other hand, in some cases the support can play a catalytic role and the activity of the heterogeneous catalyst can be higher than that of its homogeneous counterpart. For example, the silica-supported phosphonium salt SiO2-[PrBu3P]Br is more active than the homogeneous salt catalyst in the synthesis of cyclic carbonate from CO2 and propylene oxide.91 This result was ascribed to the presence of surface silanol groups in proximity of the immobilised salt (Scheme 11). These silanols can act as electrophiles and interact with the oxygen atom of the epoxide, increasing its reactivity towards the ring opening by the Br− nucleophile with a similar mechanism to that sketched in Scheme 9 (see Section 2.3). Among supported catalysts, relatively few examples of immobilisation of organometallic complexes have been reported, while supported non-metallic catalysts, and particularly ionic liquids, are receiving growing attention.7,18,85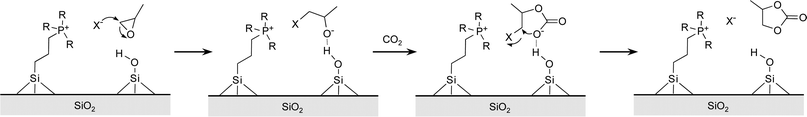 | ||
| Scheme 11 Proposed role of silanols in the reaction of CO2 with propylene oxide. | ||
Future research should aim at designing and preparing heterogeneous catalysts with improved activity and a good control of the product selectivity under milder reaction conditions compared to the high temperatures and CO2 pressures generally employed with the heterogeneous systems known so far. This will most probably imply developing bifunctional heterogeneous catalysts, containing both Lewis acidic metal centres and nucleophilic species. A promising example in this direction was provided recently by bimetallic Al-salen complexes with quaternary ammonium bromides covalently attached to the salen ligand, supported on amorphous silica or MCM-41 (Fig. 11).7,92 These materials were tested as heterogeneous catalysts for the reaction of different epoxides with CO2 both in batch and continuous flow reactors. The catalysts showed good activity, but tended to deactivate upon reuse in the batch mode, or after prolonged use in the flow mode. The deactivation was attributed to degradation through dequaternisation of the ammonium salt (see Section 2.2.b) and the catalysts could be reactivated by treating them with benzyl chloride to regenerate the quaternary ammonium species.
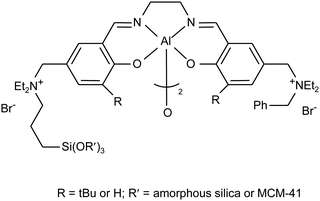 | ||
| Fig. 11 Supported bimetallic salen complex. | ||
In the perspective of an industrial application, it is also important to reduce the costs connected to catalysts production. This issue could be particularly relevant when developing heterogeneous catalysts based on metal complexes immobilised on a support, as the synthesis of many of the most commonly employed complexes (see Sections 2 and 3) is very expensive.93 Reduction of the costs could be achieved by using ligands that require less expensive synthesis procedures, while still leading to catalysts with a good performance.
5. Direct synthesis of cyclic carbonates and polycarbonates using alkenes as substrates
The epoxides used as substrates in the reaction with carbon dioxide are typically prepared by the catalytic oxidation of alkenes.94,95 The development of a process for the synthesis of cyclic carbonates and polycarbonates using alkenes as substrates would be attractive both from the point of view of sustainability and of industrial application (challenge 6). Performing the reaction with this single-step route would be intrinsically safer and involve lower costs, as it would not require the purification and handling of epoxides, which are much more toxic than alkenes and carbonates. This route would require a multifunctional catalyst that is able to promote the oxidation of alkenes to epoxides and the following reaction of the latter with CO2 to yield the carbonates (Scheme 12). Alternatively, separate, non-interfering catalysts could be employed. The process could be carried out as a one-pot reaction or by two consecutive steps, with the addition of CO2 being performed once the epoxidation is complete. On the basis of these considerations, it can be foreseen that developing an efficient process for the direct synthesis of organic carbonates from alkenes will involve challenges both in catalyst design and process technology. Few studies of catalysts for the direct synthesis of carbonates from alkenes have been reported, typically combining known catalysts for the two separate reactions (oxidation of alkenes and addition of CO2 to epoxides).7,96 However, these systems are still far from optimal and future research efforts should aim at achieving higher activity with truly heterogeneous catalysts, while minimising the formation of side-products of the epoxidation reaction, controlling the selectivity towards the desired carbonate, and using H2O2 or O2 as oxidant for the conversion of the alkene to the epoxide. | ||
| Scheme 12 Direct synthesis of cyclic carbonates and polycarbonates from alkenes. | ||
6. Conclusions
The use of carbon dioxide as a feedstock for the production of useful chemicals is a research topic that is receiving growing attention both in academia and industry. In this context, the reaction of CO2 with epoxides to produce either cyclic carbonates or polycarbonates shows a large potential for application. In this article, the effect of the various components of the catalytic system and of the reaction conditions on the reaction rate and selectivity were systematically discussed. This knowledge is important in the perspective of tackling the various open challenges that were outlined throughout the article:(1) Development of polycarbonates that have suitable properties for applications.
(2) Control of the selectivity of the reaction towards the desired carbonate (cyclic or polycarbonate).
(3) Improvement of the catalytic activity in order to reach high turnover frequencies under mild conditions for a broad range of substrates including sterically hindered and internal epoxides and oxetanes.
(4) Optimisation of the reaction conditions in order to maximise the contact between reagents and catalysts.
(5) Development of heterogeneous catalysts that can be easily separated and reused without loss of activity while being competitive with the best homogeneous catalysts.
(6) Direct synthesis of cyclic carbonates and polycarbonates from alkenes.
Achieving these targets will involve research efforts both from scientific and technological points of view.
The design of novel, enhanced catalysts for the reaction of CO2 with epoxides is a complex task: many parameters determine the performance of the catalysts, and these factors are often strongly interwoven. Therefore, changing the same parameter can have an opposite effect on different catalytic systems. This implies that the optimisation of the features of a given catalytic system will generally require a fine tuning of its different components (e.g. metal, ligands, co-catalyst). From these considerations it becomes clear that rational design of novel catalytic systems for the reaction of CO2 with epoxides should be supported by a rich experimental activity. For this purpose, the use of High-Throughput techniques can be very beneficial as they enable carrying out a large set of catalytic tests in parallel.97 This approach allows the rapid screening of libraries of catalysts, and at the same time grants a high degree of reliability in the comparison of different samples, as all tests can take place simultaneously under exactly the same conditions of temperature and pressure.36 Experimental work can be nicely complemented by theoretical calculations,20,41,98 which can provide useful insight into the interaction between the catalytic species and the reagents, thus helping the rationalisation of the experimental results and the design of new catalysts and processes.
References
- M. Mikkelsen, M. Jørgensen and F. C. Krebs, Energy Environ. Sci., 2010, 3, 43 CAS.
- M. Aresta and A. Dibenedetto, Dalton Trans., 2007, 2975 RSC.
- E. A. Quadrelli, G. Centi, J.-L. Duplan and S. Perathoner, ChemSusChem, 2011, 4, 1194 CrossRef CAS.
- T. Sakakura, J. C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi, Y. Fujii and M. Honda, J. Phys. Chem. B, 1997, 101, 2632 CrossRef CAS.
- T. Sakakura and K. Kohno, Chem. Commun., 2009, 1312 RSC.
- M. North, R. Pasquale and C. Young, Green Chem., 2010, 12, 1514 RSC.
- G. W. Coates and D. R. Moore, Angew. Chem., Int. Ed., 2004, 43, 6618 CrossRef CAS.
- D. J. Darensbourg, Chem. Rev., 2007, 107, 2388 CrossRef CAS.
- S. Fukuoka, M. Kawamura, K. Komiya, M. Tojo, H. Hachiya, K. Hasegawa, M. Aminaka, H. Okamoto, I. Fukawa and S. Konno, Green Chem., 2003, 5, 497 RSC.
- B. Schäffner, F. Schäffner, S. P. Verevkin and A. Börner, Chem. Rev., 2010, 110, 4554 CrossRef.
- P. Lenden, P. M. Ylioja, C. González-Rodríguez, D. A. Entwistle and M. C. Willis, Green Chem., 2011, 13, 1980 RSC.
- T. Ogasawara, A. Debart, M. Holzapfel, P. Novak and P. G. Bruce, J. Am. Chem. Soc., 2006, 128, 1390 CrossRef CAS.
- M. R. Kember, A. Buchard and C. K. Williams, Chem. Commun., 2011, 47, 141 RSC.
- G. A. Luinstra, Polym. Rev., 2008, 48, 192 CrossRef CAS.
- R. Zevenhoven, S. Eloneva and S. Teir, Catal. Today, 2006, 115, 73 CrossRef CAS.
- A. Decortes, A. M. Castilla and A. W. Kleij, Angew. Chem., Int. Ed., 2010, 49, 9822 CrossRef CAS.
- F. Jutz, J.-M. Andanson and A. Baiker, Chem. Rev., 2011, 111, 322 CrossRef CAS.
- S. Klaus, M. W. Lehenmeier, C. E. Anderson and B. Rieger, Coord. Chem. Rev., 2011, 255, 1460 CrossRef CAS.
- S. Klaus, M. W. Lehenmeier, E. Herdtweck, P. Deglmann, A. K. Ott and B. Rieger, J. Am. Chem. Soc., 2011, 133, 13151 CrossRef CAS.
- Y.-M. Shen, W.-L. Duan and M. Shi, J. Org. Chem., 2003, 68, 1559 CrossRef CAS.
- D.-Y. Rao, B. Li, R. Zhang, H. Wang and X.-B. Lu, Inorg. Chem., 2009, 48, 2830 CrossRef CAS.
- M. H. Chisholm and Z. Zhou, J. Am. Chem. Soc., 2004, 126, 11030 CrossRef CAS.
- D. J. Darensbourg, Inorg. Chem., 2010, 49, 10765 CrossRef CAS.
- E. N. Jacobsen, Acc. Chem. Res., 2000, 33, 421 CrossRef CAS.
- D. J. Darensbourg and J. C. Yarbrough, J. Am. Chem. Soc., 2002, 124, 6335 CrossRef CAS.
- D. J. Darensbourg, R. M. Mackiewicz, J. L. Rodgers and A. L. Phelps, Inorg. Chem., 2004, 43, 1831 CrossRef CAS.
- J. Meléndez, M. North and R. Pasquale, Eur. J. Inorg. Chem., 2007, 3323 CrossRef.
- M. North and R. Pasquale, Angew. Chem., Int. Ed., 2009, 48, 2946 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2003, 125, 11911 CrossRef CAS.
- T. Seki, J.-D. Grunwaldt and A. Baiker, J. Phys. Chem. B, 2009, 113, 114 CrossRef CAS.
- Y. Xie, Z. Zhang, T. Jiang, J. He, B. Han, T. Wu and K. Ding, Angew. Chem., Int. Ed., 2007, 46, 7255 CrossRef CAS.
- J.-Q. Wang, X.-D. Yue, F. Cai and L.-N. He, Catal. Commun., 2007, 8, 167 CrossRef CAS.
- J. Sun, W. Cheng, W. Fan, Y. Wang, Z. Meng and S. Zhang, Catal. Today, 2009, 148, 361 CrossRef CAS.
- L. Han, H.-J. Choi, S.-J. Choi, B. Liu and D.-W. Park, Green Chem., 2011, 13, 1023 RSC.
- C. Aprile, F. Giacalone, L. Liotta, J. A. Martens, P. P. Pescarmona and M. Gruttadauria, ChemSusChem, 2011, 4, 1830 CrossRef CAS.
- Y. L. Xiao, Z. Wang and K. L. Ding, Macromolecules, 2006, 39, 128 CrossRef CAS.
- R. Srivastava, T. Bennur and D. Srinivas, J. Mol. Catal. A: Chem., 2005, 226, 199 CrossRef CAS.
- Y. Xiao, Z. Wang and K. Ding, Chem.–Eur. J., 2005, 11, 3668 CrossRef CAS.
- H. Sugimoto and S. Inoue, Pure Appl. Chem., 2006, 78, 1823 CrossRef CAS.
- G. A. Luinstra, G. R. Haas, F. Molnar, V. Bernhart, R. Eberhardt and B. Rieger, Chem.–Eur. J., 2005, 11, 6298 CrossRef CAS.
- M. R. Kember, p. D. Knight, P. T. R. Reung and C. K. Williams, Angew. Chem., Int. Ed., 2009, 48, 931 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Macromolecules, 2010, 43, 2291 CrossRef CAS.
- D. J. Darensbourg and E. B. Frantz, Inorg. Chem., 2007, 46, 5967 CrossRef CAS.
- X.-B. Lu, L. Shi, Y.-M. Wang, R. Zhang, Y.-J. Zhang, X.-J. Peng, Z.-C. Zhang and B. Li, J. Am. Chem. Soc., 2006, 128, 1664 CrossRef CAS.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, B. Li, Y.-P. Zu and D. J. Darensbourg, Energy Environ. Sci., 2011, 4, 5084 CAS.
- R. Eberhardt, M. Allmendinger and B. Rieger, Macromol. Rapid Commun., 2003, 24, 194 CrossRef CAS.
- A. Decortes, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Chem. Commun., 2010, 46, 4580 RSC.
- A. Decortes and A. W. Kleij, ChemCatChem, 2011, 3, 831 CrossRef CAS.
- K. Nakano, M. Nakamura and K. Nozaki, Macromolecules, 2009, 42, 6972 CrossRef CAS.
- K. Nakano, T. Kamada and K. Nozaki, Angew. Chem., Int. Ed., 2006, 45, 7274 CrossRef CAS.
- E. K. Noh, S. J. Na, S. Sujith, S. W. Kim and B. Y. Lee, J. Am. Chem. Soc., 2007, 129, 8082 CrossRef CAS.
- S. Sujith, J. K. Min, J. E. Seong, S. J. Na and B. Y. Lee, Angew. Chem., Int. Ed., 2008, 47, 7306 CrossRef CAS.
- S. J. Na, S. Sujith, A. Cyriac, B. E. Kim, J. Yoo, Y. K. Kang, S. J. Han, C. Lee and B. Y. Lee, Inorg. Chem., 2009, 48, 10455 CrossRef CAS.
- J. Yoo, S. J. Na, H. C. Park, A. Cyriac and B. Y. Lee, Dalton Trans., 2010, 39, 2622 RSC.
- D. J. Darensbourg, J. R. Wildeson, J. C. Yarbrough and J. H. Reibenspies, J. Am. Chem. Soc., 2000, 122, 12487 CrossRef CAS.
- C. Koning, J. Wildeson, R. Parton, B. Plum, P. Steeman and D. J. Darensbourg, Polymer, 2001, 42, 3995 CrossRef CAS.
- M. R. Kember, A. J. P. White and C. K. Williams, Inorg. Chem., 2009, 48, 9535 CrossRef CAS.
- M. Cheng, D. R. Moore, J. J. Reczek, B. M. Chamberlain, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2001, 123, 8738 CrossRef CAS.
- D. R. Moore, M. Cheng, E. B. Lobkovsky and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2599 CrossRef CAS.
- B. Y. Lee, H. Y. Kwon, S. Y. Lee, S. J. Na, S. I. Han, H. S. Yun, H. Lee and Y. W. Park, J. Am. Chem. Soc., 2005, 127, 3031 CrossRef CAS.
- T. Bok, H. Yun and B. Y. Lee, Inorg. Chem., 2006, 45, 4228 CrossRef CAS.
- X.-B. Lu and Y. Wang, Angew. Chem., Int. Ed., 2004, 43, 3574 CrossRef CAS.
- C. T. Cohen, T. Chu and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 10869 CrossRef CAS.
- A. Buchard, M. R. Kember, K. G. Sandeman and C. K. Williams, Chem. Commun., 2011, 47, 212 RSC.
- D. J. Darensbourg, P. Bottarelli and J. R. Andreatta, Macromolecules, 2007, 40, 7727 CrossRef CAS.
- H. Sugimoto and K. Kuroda, Macromolecules, 2008, 41, 312 CrossRef CAS.
- W. Clegg, R. Harrington, M. North and R. Pasquale, Chem.–Eur. J., 2010, 16, 6828 CrossRef CAS.
- M. Taherimehr, A. Decortes, S. M. Al-Amsyar, W. Lueangchaichaweng, C. J. Whiteoak, E. C. Escudero-Adán, A. W. Kleij and P. P. Pescarmona, Catal. Sci. Technol., 2012 10.1039/c2cy20171b.
- C. J. Whiteoak, E. Martin, M. Martínez Belmonte, J. Benet-Buchholz and A. W. Kleij, Adv. Synth. Catal., 2012, 354, 469 CrossRef CAS.
- V. Caló, A. Nacci, A. Monopoli and A. Fanizzi, Org. Lett., 2002, 4, 2561 CrossRef.
- J.-Q. Wang, D.-L. Kong, J.-Y. Chen, F. Cai and L.-N. He, J. Mol. Catal. A: Chem., 2006, 249, 143 CrossRef CAS.
- Y. Zhao, J.-S. Tian, X.-H. Qi, Z.-N. Han, Y.-Y. Zhuang and L.-N. He, J. Mol. Catal. A: Chem., 2007, 271, 284 CrossRef CAS.
- D. J. Darensbourg, R. R. Poland and A. L. Strickland, J. Polym. Sci., Part A: Polym. Chem., 2012, 50, 127 CrossRef CAS.
- D. J. Darensbourg and A. L. Phelps, Inorg. Chem., 2005, 44, 4622 CrossRef CAS.
- G. P. Wu, S. H. Wei, X. B. Lu, W. M. Ren and D. Darensbourg, Macromolecules, 2010, 43, 9202 CrossRef CAS.
- G.-P. Wu, S.-H. Wei, W.-M. Ren, X.-B. Lu, T.-Q. Xu and D. J. Darensbourg, J. Am. Chem. Soc., 2011, 133, 15191 CAS.
- D. J. Darensbourg and S. J. Wilson, J. Am. Chem. Soc., 2011, 133, 18610 CrossRef CAS.
- D. J. Darensbourg and A. I. Moncada, Macromolecules, 2009, 42, 4063 CrossRef CAS.
- D. J. Darensbourg, A. Horn and A. I. Moncada, Macromolecules, 2010, 12, 1376 CAS.
- P. G. Jessop, T. Ikariya and R. Noyori, Chem. Rev., 1999, 99, 475 CrossRef CAS.
- P. G. Jessop and B. Subramaniam, Chem. Rev., 2007, 107, 2666 CrossRef CAS.
- H. Kawanami, A. Sasaki, K. Matsui and Y. Ikushima, Chem. Commun., 2003, 896 RSC.
- D. J. Darensbourg, R. M. Mackiewicz and D. R. Billodeaux, Organometallics, 2005, 24, 144 CrossRef CAS.
- W.-L. Dai, S.-L. Luo, S.-F. Yin and C.-T. Au, Appl. Catal., A, 2009, 366, 2 CrossRef CAS.
- M. Tu and R. J. Davis, J. Catal., 2001, 199, 85 CrossRef CAS.
- H. Yasuda, L. N. He, T. Takahashi and T. Sakakura, Appl. Catal., A, 2006, 298, 177 CrossRef CAS.
- C. Baleizão, B. Gigante, M. J. Sabater, H. García and A. Corma, Appl. Catal., A, 2002, 228, 279 CrossRef.
- M. Alvaro, C. Baleizão, D. Das, E. Carbonell and H. García, J. Catal., 2004, 228, 254 CrossRef CAS.
- K. Yu and C. W. Jones, Organometallics, 2003, 22, 2571 CrossRef CAS.
- T. Takahashi, T. Watahiki, S. Kitazume, H. Yasuda and T. Sakakura, Chem. Commun., 2006, 1664 RSC.
- M. North, P. Villuendas and C. Young, Chem.–Eur. J., 2009, 15, 11454 CrossRef CAS.
- M. North and C. Young, Catal. Sci. Technol., 2011, 1, 93 CAS.
- D. E. De Vos, B. F. Sels and P. A. Jacobs, Adv. Synth. Catal., 2003, 345, 457 CrossRef CAS.
- P. P. Pescarmona, K. P. F. Janssen and P. A. Jacobs, Chem.–Eur. J., 2007, 13, 6562 CrossRef CAS.
- J. Sun, L. Liang, J. Sun, Y. Jiang, K. Lin, X. Xu and R. Wang, Catal. Surv. Asia, 2010, 15, 49–54 CrossRef.
- P. P. Pescarmona, J. C. van der Waal, I. E. Maxwell and T. Maschmeyer, Catal. Lett., 1999, 63, 1 CrossRef CAS.
- Y. Ren, C.-H. Guo, J.-F. Jia and H.-S. Wu, J. Phys. Chem. A, 2011, 115, 2258 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |