Versatility of heterogeneous photocatalysis: synthetic methodologies epitomizing the role of silica support in TiO2 based mixed oxides
Harrison S.
Kibombo
*,
Rui
Peng
,
Shivatharsiny
Rasalingam
and
Ranjit T.
Koodali
*
University of South Dakota – Chemistry, 414 E. Clark Street, Vermillion South Dakota 57069, USA
First published on 13th June 2012
Abstract
Heterogeneous photocatalysis continues to be an active area of research with focus on developing catalytic systems that can degrade toxic pollutants in the gas and aqueous phase, and split water to generate hydrogen and oxygen. In this review, the incorporation of silica phases in titanium dioxide based photocatalysts is reviewed.
 Harrison S. Kibombo | Mr Harrison S. Kibombo is a PhD student pursuing research in the group of Dr Ranjit T. Koodali at the University of South Dakota (USD). His research interests are in sol–gel chemistry, photocatalysis, and environmental sciences. |
 Rui Peng | Mr Rui Peng is a PhD student in the group of Dr Ranjit T. Koodali at USD. His research work pertains to the development of mesoporous materials for photocatalytic splitting of water. |
 Shivatharsiny Rasalingam | Ms Shivatharsiny Rasalingam is a PhD student in the group of Dr Ranjit T. Koodali at USD. Her research interest is in the area of environmental remediation of organics and dyes. |
 Ranjit T. Koodali | Dr Ranjit T. Koodali is a faculty member at USD. His research interests include use of oxide materials for catalysis, environmental remediation, and solar energy conversion and storage. His research related to solar hydrogen production has been cited in the NSF budget for FY 13 to the Congress. |
1. Introduction
Sustainable development is one of the biggest global challenges of the 21st century. The world demand for energy is expected to reach 28 TW by 2050 and triple by the end of this century. A significant portion of the energy demand is currently met by fossil fuels.1 Increase in energy demand has led to consumption of fossil fuels at an alarming pace, with peak production anticipated in the next decade. A consequence of the continued use of fossil fuels is its impact on the environment. Global warming from greenhouse gases emitted by burning fossil fuels has been linked to disruptive climate changes and threatens human life. Thus, there is a need to find renewable sources of energy to power our planet. Solar energy provides the largest carbon neutral source of energy. Harnessing this abundant, renewable, and non-polluting source of energy is expected to usher in an era of global prosperity free from geopolitical tensions and unpredictable climate patterns.It is widely recognized that advances in the field of nanoscience and nanotechnology will afford us unprecedented opportunities to produce technological breakthroughs to solve challenges in energy and environment.2 A promising field to meet these challenges is photocatalysis.
The pioneering work of Fujishima and Honda in the 1970's has paved the path for the extensive application of titanium dioxide for solar energy conversion.3 Since then, a large number of competing photocatalysts have been developed. However, titanium dioxide (titania, TiO2) is still the popular choice since it is inexpensive, easy to synthesize, stable under irradiation conditions, and non-toxic.
Titanium dioxide has been used for the destruction of pollutants in the gas phase, aqueous phase, photosplitting of water, and destruction of bacteria.4–7 The wide band-gap of titania (3.2 eV) and the rapid recombination of the photoproduced electrons and holes are limitations that have precluded the wide spread use of the titania based materials for various applications. Efforts to shift the absorption of titania into the visible region include doping with transition metal ions, incorporation of noble metals, functionalization with electron acceptors, doping with non-metals, and coupling small-band gap semiconductors.8–17
In this review, we will first discuss the synthesis of titania–silica (TiO2–SiO2) mixed oxides. A variety of synthetic methods have been reported for the preparation of titania–silica catalysts and thus an extensive discussion is presented. Following this, the structural characterization of titania–silica is reviewed. The resulting (photo)catalytic activity is largely dependent on the physico-chemical properties and thus a variety of techniques that have been used to characterize the surface and textural properties are presented. In Section 4, the application of TiO2–SiO2 photocatalysts in aqueous phase, gas phase degradation reactions and photosplitting of water is discussed. Finally, other emerging areas of applications are explored. We first present an extensive discussion of the synthesis of TiO2–SiO2 by a sol–gel method.
2. Synthesis of TiO2–SiO2
Sol–gel processing is a versatile method that has demonstrated commercial success as desired properties of a material can be obtained through control over the preparation route. It provides inherent advantages such as molecular-scale mixing of reactants to produce highly homogeneous products.18 The synthesis usually involves the hydrolysis of titania and silica alkoxide precursors and their condensation processes that lead to branched polymeric gels. Optimizing the sequence of addition and other experimental parameters such as the choice and amounts of precursor, water, solvents, and the pH of the system allows for tailoring of the structural properties and the generation of highly porous materials with unique physico-chemical properties.The following section addresses the synthetic methods for the preparation of materials utilized for catalytic reactions, and will provide the reader with the rich and diverse preparative methods utilized for making titania–silica mixed oxides.
2.1. Hydrolytic methods
Various transition metal ions have been incorporated into the silicate network to form mixed oxides of variable surface and bulk properties. Uniform and homogeneous distribution of these metal ions in the silica matrix must be achieved in order to impart desirable properties that are responsible for enhanced photocatalytic activity. The focus of this review is on titania based materials, which have proven to produce highly active photocatalysts. Metal alkoxide precursors, Ti(OR)4, are preferred to inorganic salts such as TiCl4 due to the ease of solubility irrespective of the preparation route employed. An inherent challenge in the synthesis of mixed oxides by hydrolytic sol–gel processing is the different reaction rates of the alkoxide precursors that render the products vulnerable towards phase separation rather than the formation of homogeneous polymeric gel structures. This phenomenon is evidenced in the hydrolysis reactions that proceed via nucleophilic substitution mechanisms and depend on the electronic partial charge, δ+, on the metal atoms. The Ti atom (Ti4+) has a significantly large positive partial charge (δ+ ∼0.6) derived from its higher electrophilic character that imparts higher reactivity with water than the Si atom (Si4+) in tetra-alkoxysilanes. Thus, phase homogeneity has been improved by:1. the pre-hydrolysis of the SiO2 precursor
2. deceleration of the fast reacting electropositive Ti-alkoxide precursor via chemical modification involving the substitution of alkoxide groups with chelating multidentate ligands, and
3. the use of single-source precursors.19
Highly reproducible polymeric gel materials have been developed by extending these methodologies through the optimization of the sol–gel reaction. Attention can be drawn to the competing experimental parameters such as the alkoxide/water ratio, concentration of precursors, reaction temperature, reaction time, drying methods, choice and amount of catalyst, and solvent media utilized.20 Various conditions have been explored using the xerogel, hydrothermal, and aerogel synthetic procedures that have demonstrated competing structural properties and catalytic efficiencies as summarized in subsequent parts of this review.
| Si(OEt)4 + H2O → Si(OH)(OEt)3 + EtOH | (1) |
| 4Si(OH)(OEt)3 + Ti(OPri)4 → Ti[OSi(OEt)3]4 + 4PriOH | (2) |
| Ti[OSi(OEt)3]4 + 12H2O → Si–O–Ti–O–Si + 12EtOH | (3) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cosθ)/r, where θ = contact angle, and r = pore radius) will reduce the capillary pressure, Pc, during drying and will hence preserve the pore structure and maximize the surface area.27,39 Octadecyltrichlorosilane CH3(CH2)17SiCl3 (OTS) is a silica source with amphiphilic character that is well-documented for surface modification reactions with surface hydroxyl groups. It was demonstrated that the attachment of silica modifies the local environment of titanium surface species of titania in the presence of toluene as the solvent. The titanium ions bond with –Si(OH)2(OSi)2 to transform and stabilize a fraction of titanium ions in tetrahedral coordination.40
cosθ)/r, where θ = contact angle, and r = pore radius) will reduce the capillary pressure, Pc, during drying and will hence preserve the pore structure and maximize the surface area.27,39 Octadecyltrichlorosilane CH3(CH2)17SiCl3 (OTS) is a silica source with amphiphilic character that is well-documented for surface modification reactions with surface hydroxyl groups. It was demonstrated that the attachment of silica modifies the local environment of titanium surface species of titania in the presence of toluene as the solvent. The titanium ions bond with –Si(OH)2(OSi)2 to transform and stabilize a fraction of titanium ions in tetrahedral coordination.40
Simple organic solvents such as acetic acid (AcOH) have been utilized as modifying agents to decrease the reactivity of Ti(OPri)4 in aqueous solutions. The reaction occurs between the terminal OPri and bridging CH3COO− ligands, thus facilitating the combined steric effects of the ligands to decrease the efficiency of the hydrolysis reactions.41,42
| Ti(OPri)4 + CH3COOH → Ti(OPri)3OOCCH3 + PriOH | (4) |
| Ti(OPri)4 + H3CCOCH2COCH3 → Ti(OPri)3H3CCOCHCOCH3 + PriOH | (5) |
| Ti(OPri)4 + xAmiOH → Ti(OPri)4−x(OAmi)x + xPriOH | (6) |
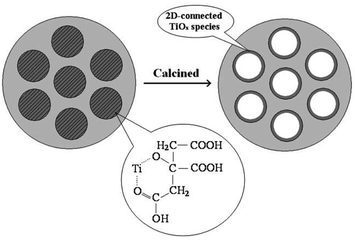 | ||
| Fig. 1 Proposed model for the formation process of mesostructured TiO2–SiO2 materials through titanium–citric acid complexes. Reprinted with permission from ref. 56. Copyright 2009, American Chemical Society. | ||
The silica phase may also play a role in stabilizing dispersed TiOx species in the TiO2–SiO2 materials and minimize agglomeration as a result. A pH below 2 was critical to obviate the phase separation, and promote the formation of relatively stable and uniformly dispersed titanium–CA complexes.
Other complexing and pore generating agents with abundant OH groups have been investigated to enhance the surface and pore geometries such as hexylene glycol, dimethyl sulphate,57 and β-cyclodextrin (β-CD).58 Other sugar derivatives that have found application as pore forming agents in the sol–gel processing of advanced materials are glucose, tartaric acid, and chitosan.59 The presence of the NH2 group offers chitosan unique properties that enhance its solubility and reactivity through hydrogen bonding between the amino groups and inorganic species.
With a similar amine mechanism approach, Oki et al. demonstrated the ability to vary the porosities and surface areas, and disperse titania effectively in the silica matrix by the use of various n-alkyl amines as structure and pore directing templates such as hexylamine (HXA), dodecylamine (DDA), octadecylamine (ODA),60 triethanol amine (TEA), cyclohexyl amine,61 octylamine,62 and polyethylenimine (PEI).63 The numerous –NH groups in PEI attach to inorganic networks by hydrogen bonds and form molecular hybrids as depicted in Fig. 2.
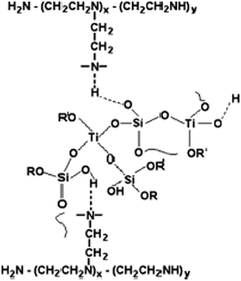 | ||
| Fig. 2 Compatibilization scheme of the PEI (polyethylenimine-modifying agent) and inorganic oligomer phases. Reprinted with permission from ref. 63. Copyright 2007, Springer. | ||
Narrow pore size distributions are achieved and use of constant PEI concentration ensures that the pore diameter of the mixed oxide remains relatively constant with an increase in the Si/Ti ratio in the mixed oxides. However, the surface area and pore volume increase with polymer concentration in the precursor hybrid. This allows a considerable degree of control of porosity in the synthesized titania–silica materials.
Pore characteristics have been manipulated by using more intricate approaches involving micellar processes through which pre-mixed TEOS and a separately prepared colloidal suspension of titania nanoparticles were added to a basic NH4OH solution of surfactants such as cetyltrimethylammonium bromide (CTABr).64 The use of surfactants as templates has facilitated the formation of materials with ordered pore structures.
Recent research has reported more advanced surfactant based materials prepared using block copolymers as templates for the formation of titania containing mesoporous oxides via a co-assembly approach as shown in Fig. 3.
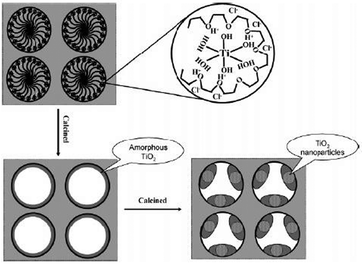 | ||
| Fig. 3 Model of the synthesis of mesoporous TiO2–SiO2 materials. The triblock copolymer acts as a structure-directing agent and complex ligand. Reprinted with permission from ref. 65. Copyright 2004, American Chemical Society. | ||
The block copolymer serves as a structure directing agent and a moderator of the hydrolysis rate of titanium alkoxide in acidic media. The alkylene oxide moieties form crown-ether-type complexes with inorganic ions through weak coordination bonds and hydrogen bonds that ensure the dispersion of metal atoms throughout the nascent mesophase. Materials of large pore geometries with titania well dispersed inside the silicate framework are feasible by this approach. The monoliths were prepared by dissolving the metal precursors in alcohol solutions of Pluronic F108 (EO130PO60EO130) (where EO = ethylene oxide, PO = propylene oxide)65 or Pluronic F127 (EO106PO70EO106; Mav = 12600)66 under vigorous stirring.
A key property is the ability of these copolymers to bind strongly with the metal; however, the interactions are highly dependent on the solvent used. The acid and water content ratios are also crucial in the optimization of interactions between the metal ions and the polymers at the interface to prevent their segregation. The alcohol may act as a co-surfactant and result in small units of titania formed in hydrophilic domains of the folded block copolymers, and silica species grow surrounding the preformed titania particles. The removal of the template by calcination resulted in a mixed oxide with the titania particles located mainly at the surface of the pore walls.
To utilize existing efficient and benchmark photocatalysts, some researchers have incorporated commercially available Degussa P25 into an acid catalysed and pre-hydrolysed alcohol solution of TEOS.67,68 The use of HF as a catalyst for hydrolysis and condensation in a similar reaction resulted in the formation of mixed oxide pellets.69 Conversely, dispersion of titania was achieved by incipient wetness impregnation of the commercially available Aerosil 200,49 Cab-O-Si1 EH-5 and Degussa Alon C support with a solution of Ti(OCH3)4, and conc. HNO370,71 in an acidic alcohol solution of titania precursor.
The effect of precursor on the dispersion of titania supported on silica was later investigated by Tanaka et al. The catalysts were prepared by a simple impregnation method of double distilled TEOS with three kinds of titanium complexes comprising different ligands namely bis(isopropylato)-bis(pivaroylmethanato) titanium(IV) (Ti(DPM2(OPri)2), bis(acetylacetonato)oxo-titanium(IV) (TiO(acac)2) and tetrakis(isopropylato) titanium(IV) (Ti(OPri)4) in toluene under reflux conditions.72
Another simple approach to regulate particle size distribution was adopted in which an adsorption layer on a support surface was utilized to prepare titania–silica nanocomposites as illustrated in Scheme 1.
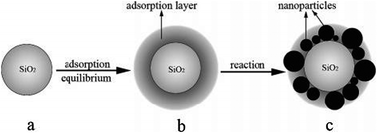 | ||
| Scheme 1 Scheme of preparation of TiO2 in the adsorption layer at low water concentration: (a) dispersion of support in the binary liquids system; (b) adsorption equilibrium; and (c) particle distribution after reaction. Reprinted with permission from ref. 74. Copyright 2007, American Chemical Society. | ||
Dehydrated hydrophilic SiO2 was dissolved in absolute alcohol under magnetic stirring, and the effects of different amounts of water were investigated. The selective adsorption capacity of SiO2 gradually created a water-rich layer on the surface. TBOT dissolved in ethanol was added after the adsorption equilibrium was reached. TBOT undergoes hydrolysis and condensation reactions with water in the adsorption layer, forming TiO2 nanoparticles that may link with silanol groups on the silica surface.73–76
Owing to their unique structural properties, hybrid organic–inorganic sol–gel derived materials have attracted attention in various applications such as molecular imprinting, microfilters, absorbents, and catalysis. It has been determined that hydrophobicity plays a crucial role in organic contaminant adsorption, and hydroperoxide mediated catalytic epoxidation of olefins by titania–silica xerogels. Larsen and co-workers demonstrated an enhancement in the hydrophobicity of titania–silica materials using a non-cyclic silicon precursor as the cyclic compounds (permethylated cyclo-oligosiloxane rings) are prone to low hydrolysis reactivity.77 The materials were prepared in ethanol by acid pre-hydrolysis of TEOS and dimethyl-dimethoxysilane SiMe2(OMe)2, followed by reaction with TiCl4 in isopropanol that results in an in situ formation of a dichloro-diisopropyl titanium complex TiCl2(OPri)2.78,79 In a similar approach, Mariscal et al. improved the hydrophobic character of the xerogel by mixing the Ti and Si alkoxide precursor with a hexane solution of drying chemical compound additive (DCCA) such as trimethylchlorosilane (TMCS) in a N2 atmosphere. The reaction of the hydroxyl groups of the xerogel with the TMCS protects the texture from collapse during drying and preserves hydrophobicity.80 A less elaborate approach was used by Dagan et al. Silica was modified with functional groups such as methyl and phenyl moieties in order to manipulate the polarity, hydrophobicity, ion-exchange capacity, and concentration of silanol groups of the xerogel surface. The resultant organically modified silica sols i.e. R–Si–(OCH3)3 (R = methyl or phenyl) were dissolved in ethanol followed by a slow addition of Ti(OPri)4 to obtain mixed oxide gels.81 The selection of modifiers was such that they facilitated strong adsorption and the diffusion of the organic contaminants from the adsorption sites to the photocatalytic centres was not hindered.
The crystallization enrichment process during HTS of mixed oxide materials is indeterminate. Bein and co-workers91 utilized electron microscopy, dynamic light scattering (DLS) and X-ray diffraction (XRD) to understand the crystallization of amorphous gel during the hydrothermal synthesis. This study illustrated the significance of crystallinity in harnessing the catalytically active features of porous gel materials. Zeolite Y nucleation on colloidal gel particles was monitored at 100 °C under high resolution transmission electron microscopy (HRTEM). Crystals were observed to nucleate in amorphous aluminosilicate gel particles containing islands of nuclei randomly dispersed in the semi-ordered network. HTS facilitated reversible condensation reactions that break M–O–M bonds and harness the attraction of energetically favourable coordination of oxy-species. This results in the morphological transformation of random gel structures to islands of regular crystal lattice from several weeks to as little as half a day. Evidently, HTS provides a quick mechanism through which TiO2 and SiO2 species interact to improve homogeneity, and form more catalytically active Ti–O–Si hetero-linkages in the resultant mixed oxide materials.
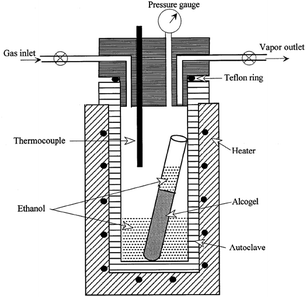 | ||
| Scheme 2 Schematic illustration of an autoclave for supercritical drying with ethanol as the solvent. Reprinted with permission from ref. 92. Copyright 2002, American Chemical Society. | ||
In this process, the wet gel is heated in the autoclave reactor, so that the pressure and temperature exceed the critical temperature, Tc, and critical pressure, Pc, of the solvent, such as alcohol or CO2, occupying the gel pores.92 A plot describing the phase diagram of CO2 is shown in Fig. 4.
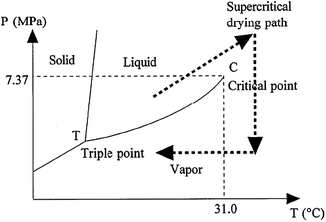 | ||
| Fig. 4 Supercritical drying path in the pressure (P)–temperature (T) phase diagram of CO2. Reprinted with permission from ref. 92. Copyright 2002, American Chemical Society. | ||
The enhanced textural properties of titania–silica aerogels promise higher adsorption capacities, and the availability of active titania species dispersed in the interstitial cavities of the thermally stable silica or on the surface of the matrix. The textural properties of aerogels are dependent on the method of drying. Since conventional ambient drying conditions result in shrinkage and cracking of the gel structure, supercritical drying has been utilized to minimize capillary stresses, and preserve the large pore geometries that exist in the wet gel. The pores might collapse due to the formation of a liquid–vapour interface that is formed during the course of emptying of the wet gels.92–94
Wet gels have been prepared using various methods with precursors such as tetraethylorthosilicate (TEOS),95 tetramethoxysilane (TMOS),96 and methyltrimethoxysilane (MTMS)97 as the silica sources, and tetrabutylorthotitanate (TBOT),97,98 titanbisacetyl-acetonatdiisopropoxide (TIBADIP)96,99 as the titania precursors of choice. The pre-hydrolysis of silica before addition of the titania precursor is simple and has been employed in various aerogel preparation studies.50,90,100 Even though the reactivity of Si(OEt)4 is low and can be promoted by an acidic catalyst i.e. CH3COOH, HNO3, or HCl, moderation of the rapid reactivity of Ti(OPri)4 with water is necessary to ensure that polymeric gels are obtained instead of dense precipitates of unreacted alkoxides in the sol–gel process.90
Alternatively, the hydrolysis of the titania precursor may be delayed by chelating Ti atoms with slow hydrolysing multidentate ligands such as acetylacetone (acac) that allows for partial substitution of the Ti alkoxy groups.18,97,101 Although the application of these modifying co-solvents resulted in lower surface area aerogels, these materials were found to be more active than those prepared using a non-modified titania precursor.101,102
One of the most critical components for the enhancement of aerogel function is to tailor the interconnectivity of the pore network. Sun et al. described a procedure for the preparation of TiO2–SiO2 mixed oxides via the use of semi-crystalline polypropylene (PP) polymer in an in situ sol–gel process with the aid of SC CO2 as a swelling agent. It has been documented that SC CO2 can effectively swell the amorphous region of polyolefin without altering the crystalline structure. The inorganic species dissolved in the SC CO2 were infused and dispersed in the polymer matrix, thus forming inorganic oxides upon release of CO2 and pyrolysis of the polymer.103
Equally intricate approaches consisted of the introduction of well-defined hierarchal mesoporous structures via an appropriate template of triblock polymer (Pluronic P123) in the presence of acidified solutions of the alkoxide precursors,104 or Pluronic 17R4 surfactant105 to allow the preparation of crack-free titania–silica aerogels of high titanium loading and large surface areas with more ordered mesophase structures. A schematic representation of the interaction between the semiconductors phases and the polymer is shown in Scheme 3.
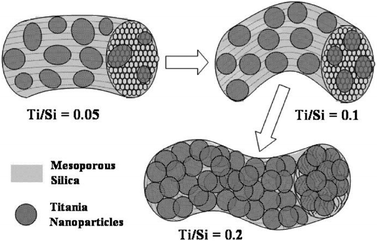 | ||
| Scheme 3 Schematic of the described particle growth and morphology evolution of the TiO2–SiO2 composites with different Ti/Si ratios (the gray parts: mesoporous SiO2, probably Ti-doped; the dark particles: TiO2 nanoparticles, probably Si-doped). Reprinted with permission from ref. 132. Copyright, 2009 American Chemical Society. | ||
Various polar organic compounds have been incorporated into the aerogel matrix with the aim of tuning the acid–base and redox surface properties and consequently improving the selectivity and activity in liquid phase oxidation reactions. Samantaray and Parida suggested that the surface acidity and the hydrophilic character of the surface silanol groups can be tailored and side reactions can be suppressed in this manner.106 The control of the surface properties was investigated by Kochkar and Figueras, who determined that the partial substitution of tetraethoxysilane by phenyl or methyltriethoxysilane permitted the preparation of titania–silica aerogels of hydrophobic character. The attachment of large phenyl moieties improved the hydrophobicity evident in the higher hexane adsorption percentage, which resulted in enhanced epoxidation activities.95
In complementary approaches, amines are often utilized as functional groups because of their ability to coordinate to transition metals such as Ti. Amine (basic) additives such as urea, tetra-butylammonium hydroxide (TBAOH), isoquinoline, s-triazine, dimethyl butyl amine, and 1,4-diazine107 have been utilized to suppress non-oxidative secondary reactions and improve the selectivity towards epoxidation. As a result, epoxidation activity of titania–silica that is attributed to the Lewis acidity of the (Si–O)4Ti sites of the mixed oxide materials is enhanced.
Structural and catalytic properties may also be tailored by functionalizing pre-hydrolyzed silicon alkoxides precursors with trialkyl precursors RSi(OMe)3 in the presence of amine modifiers such as ethylenediaminopropyl (EDAP) and propylenediaminopropyl (PDAP). An alcohol is first protected by acylation to prevent the formation of a Si–O bond followed by hydrosilylation to yield alcohol trialkyl precursors like acetoxybutyl (AcOB) or diacetoxybutyl (DAcOB). Subsequently the amine or alcohol modifier in iPrOH was added, followed by trihexylamine (THA) to induce gelation.99 Modified aerogels demonstrated considerably more improved olefin selectivity than that reported by Maier32 and Figueras.108
The wet gels prepared are then subjected to the drying process aforementioned. The two methods in practice are the high temperature supercritical drying (HT-SCD) and the low temperature supercritical drying (LT-SCD) that eliminate the liquid–vapour interface inside the gel network. The capillary stress is evaded by transferring the solvent into the supercritical state in HT-SCD and replacing the alcohol solvent with CO2 in LT-SCD. Temperature programmed reduction (TPR) and X-ray photoelectron spectroscopy (XPS) results revealed that the Ti-concentration on the surface increased, and LT-SCD yielded high titania surface concentration (lower Si enrichment), suggesting a relative contribution to surface Ti–O–Si linkages.47 Brodsky demonstrated that increasing the drying temperature from 70 °C to 200 °C did not change the surface area but increased the pore volume, suggesting an increase in average pore diameter.109 High temperature drying mixtures are undesirable for maintaining homogeneity in mixed oxide aerogels. A lower drying temperature at 70 °C resulted in materials that were more susceptible to phase transformation due to smaller crystallites that have more surface defects than the materials dried at 200 °C. Although pre-hydrolysis of the silica precursor was used in an effort to promote better mixing and generate more acid sites, a high drying temperature retarded homogeneous mixing because crystallization of titania segregates it from silica leading to a phase separated material of fewer Brønsted sites and consequently lesser activity towards the isomerization of 1-butene.109 Dutoit et al.18 subjected alkoxide based titania–silica sol gels to a high temperature of 260 °C using HT-SCD and realized anatase segregation that marked growth of mesopores to macropores. The structural changes were attributed to the enhanced solvent reactivity and accelerated aging of the gel. For this reason, drying a mixed oxide with supercritical alcohols may not be desirable in preserving homogeneity. Drying at high temperatures (T > 200 °C) is viable using certain alcohols as solvents due to their high critical temperatures that enable dissolution, re-precipitation of particles, de-polymerization, alkoxylation, and enhanced syneresis (network densification), leading to structural network rearrangement in these materials. These processes may result in coarsening, sintering of particles, syneresis or effectively preserve large pore geometries, thus rendering the choice of solvent crucial for attaining desired properties.
2.2. Non-hydrolytic method
The use of inorganic derivatives of titanium and silicon precursors in a non-hydrolytic sol–gel process has attracted attention as it offers a simple, efficient one-pot procedure for the preparation of highly active mixed oxides. Contrary to the conventional hydrolytic process, it epitomizes the possibility to simultaneously enhance the Ti dispersion, maintain the mesostructure, and impart homogeneity in the resultant material under solvent free conditions without the use of chemical additives.54,110,111 Hybrid titania–silica gels were prepared by reaction of chloride precursors; TiCl4 and SiCl4,112 MeSiCl3 or Me3SiCl,113 with diisopropyl ether (iPr2O) as an oxygen donor under Schlenck line conditions. This synthetic procedure is based on the intermediate formation of isopropoxide groups by reaction of iPr2O with chloride groups, followed by non-hydrolytic condensation between chloride and alkoxide groups, leading to the formation of M–O–M bridges, and the elimination of isopropyl chloride. The materials generated are of high surface areas and pore volumes, thus discarding the need for a sophisticated drying procedure such as supercritical conditions.It seems that the non-hydrolytic procedure imparts a high degree of condensation, resulting in mesopores instead of micropores observed typically in materials prepared using hydrolytic methods. The silica support provides a versatile surface that allows for modification with chloride and alkoxide groups instead of hydroxyl groups, thus minimizing and weakening the interactions between the pore surface and the liquid phase. The robust nature of silica is epitomized in ensuring a rigid gel oxide network with minimal collapse under capillary stresses during solvent evaporation.112 Although the resultant materials have enhanced physical properties, their applicability to catalysis is limited.
Titania supported on silica materials were prepared by the impregnation of commercial silica powder (CARiACT),114 homogeneous precipitation of Aerosil 200,115 and methanol solutions of sodium silicate precursor (Na2SiO3).116 The various titania precursors used were Ti(OPri)4 and titanium sulphate (Ti(SO4)2), titanium trichloride (TiCl3) and TBOT in acidic media respectively. The surface properties of the silica microspheres are influenced by the titania loading and were characterized by XPS studies. If the active sites are predominantly on the surface of these coated microspheres, the photocatalytic yields may be superior at specified loadings. The loading ratios are critical as low or too high inhomogeneous deposition may result in agglomeration of titania species on the surface of the silica microspheres.
Non-balanced oxygen atoms emerge at the binary oxide interface, resulting in a charge imbalance that requires acidic bridging hydroxyl groups that are referred to as Brønsted acid sites and characterized by Ti–O–Si bonds. It has been demonstrated that grafting titanium species onto Aerosil 200 through the –OSi(OtBu)3 (trialkoxy–siloxy) ligand introduced isolated catalytically active –O–Ti(–O–Si)3 sites on the oxide surface that are deemed responsible for the epoxidation of cyclohexene in the presence of a peroxide oxidant.49
With the continued postulation that Ti–O–Si are the active sites in these mixed oxide materials, researchers have endeavoured to utilize precursors in which these heterolinkages pre-exist. The use of single source precursors (non-hydrolytically) is a promising method to curb the inherent phase separation prone to hydrolysis sol–gel processing of alkoxide mixtures in the preparation of mixed oxide materials.19,117 Torma et al. describe an approach in which 3-oxoethyl-6-trimethoxysilyl-hexan-2-one (OTH-H) is reacted with Ti(OPri)4 in a dry argon atmosphere. The β-diketonate moiety in OTH-H reacts by alkoxo ligand substitution resulting in a precursor, (iPrO)3Ti(OTH), with an organic spacer between Si and Ti alkoxide moieties. In a complementary experiment, similar conditions were maintained followed by the addition of appropriate amounts of TEOS diluted in methanol in an effort to circumvent the limitation of fixed stoichiometry apparent in the use of single source precursors. It was noted that tethering between these moieties was destroyed by calcination at ca. 800 °C, thus producing titania–silica gels of improved homogeneity. A different set of materials was subjected to drying by SC CO2 to produce aerogels.
The investigators utilized a comparative experiment, and small angle X-ray scattering (SAXS) studies to postulate the interactions that facilitated the gelation behaviour of (iPrO)3Ti(OTH). An equimolar mixture of (acac)Ti(OPri)3, prepared in situ from Ti(OPri)4 and acetylacetone (acac-H), was reacted with (propyl)Si(OMe)3 in the presence of methanol and water under the same conditions, resulting in untethered alkoxide moieties. The absence of gelation even after a 6 week period suggested that the formation of (iPrO)3Ti(OTH) was attributed to the tethering of the alkoxide moieties. This notion is derived from the percolation theory that proposes that the gelation phenomenon is more eminent in (iPrO)3Ti(OTH) than in the mixture of (acac)Ti(OPri)3 and RSi(OMe)3, owing to the preformed linkages in the reacting species.19
Coles et al. investigated the pyrolytic conversion and solution phase thermolysis of a tris(tert-butoxy)siloxide precursor such as Ti[OSi(OtBu)3]4 to stoichiometrically form TiO2·4SiO2 materials.49 They demonstrated that variations in the drying methods had an effect on the physical and catalytic properties. The extraction of toluene from the “wet gel” with SC CO2 at low temperatures afforded an aerogel of high surface area (677 m2 g−1), and enhanced catalytic activity (49%) for the selective oxidation of cyclohexene to cyclohexene oxide using cumene hydroperoxide as the oxidant. The improved activity of the aerogel compared to the xerogels prepared in this study is attributed to the silica that provides a support onto which the titania species are highly dispersed.
Rice husk ash and TiCl4 were used as silica and titania precursors, respectively, in the presence of NaOH and H2O. The surfaces of the titania–silica aerogels produced were modified via acid treatment using H3PO4, HCl, and H2O4. Although the role of silica was not elucidated, the ability to manipulate the acidity and enhance the photocatalytic activity while preserving the mesostructure was noted. It is notable that destruction of the structure was observed in the H3PO4 treated materials. Brønsted sites were detected in the H3PO4 and H2SO4 treated samples as the phosphate and sulphate groups made contact with the Si–OH group, thus providing bifunctional oxidative and acidic catalysts in the latter materials. An increment of Lewis acidity was observed in the HCl treated material without formation of any Brønsted acid sites.118
Ionic liquids (ILs) have been of great interest as templates for the fabrication of nanostructured materials. This is owed to tunable solvent properties that can readily interact with various surfaces to facilitate the distribution of particles by in situ formation of metal oxide nanoparticles on the mesoporous supports. Gopala et al.119 demonstrated a facile method of introducing Ti centres into the mesoporous silica support by impregnation of an acidic solution of TiCl4 mixed in N,N-dialkylimidazolium chloride type ILs such as 1-hexadecyl-3-methylimidazolium chloride. It forms a mesophase in aqueous solution with the TEOS precursor as well as interacts with the TiCl4 to form an imidazole based chlorometalate complex, Ti–I16 IL. The choice of tail length and head group size influences the volume of the micelle structures and mesoporosity can be tuned. Table 1 is a summary of the synthetic methodologies describing the tangible and intangible components expected in the preparation of titania based silica materials.
| Characteristics | Hydrolytic | Non-hydrolytic | ||
|---|---|---|---|---|
| Xerogel | Aerogel | Hydrothermal | ||
| Surface area | Low | Very high | High | High |
| Crystallinity | Low | High | High | Moderate |
| Porosity | Low | Very high | High | High |
| Advantages | Simple synthesis process; no need for sophisticated drying procedure or expensive equipment | Drying procedure minimizes capillary stress and preserves the large pore geometries present in the wet gel | Mild temperature and pressure, minimized phase separation; facile and quick method for increasing rate of sol–gel reactions and improving homogeneity | Simple efficient one pot procedure; allows solvent free conditions without use of chemical additives; no need for sophisticated drying procedure |
| Disadvantages | Dense low porous materials with low crystallinity of the titania phase | Requires expensive equipment operating at high pressures | Requires additional steps of washing and drying | Requires Schlenck line equipment and controlled inert environments |
3. Structural and textural properties
3.1. Surface characterization
However, correlation studies to unequivocally prove this phenomenon are still inconclusive and require extensive investigation. Researchers exploring the efficacies of titanium based silicates acknowledge the complexity in determining the origin of the acidic properties, however, they reach broad agreement that the Brønsted acidity is associated with Ti–O–Si hetero-linkage and the surface hydroxyl groups play a significant role in the development of these sites. Fig. 5 illustrates the two possible doping configurations and the calculations required to predict the acid site type.
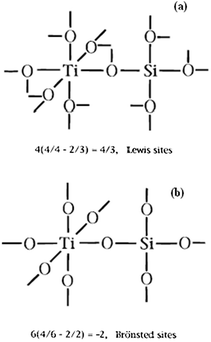 | ||
| Fig. 5 Illustration of the Ti–O–Si linkages proposed in Tanabe's model to describe the increased surface acidity of binary metal oxides. (a) Silicon in titania, and (b) titanium in silica. Reprinted with permission from ref. 164. Copyright 1996, American Chemical Society. | ||
Detailed FT-IR spectroscopy studies have been carried out to elucidate the acidic properties of mesoporous titanium containing silicate materials using adsorbed probe molecules. Weakly interacting probe molecules such as CO and N2 are preferred because they are much more specific than intermediate (CD3CN) or the classical strongly interacting probes (NH3 and pyridine) and provide more information about the nature of the active sites. The acidic catalyst surface of titania–silica materials may have protic (Brønsted) sites of typically hydroxyl groups, and aprotic (Lewis) sites. The latter are due to framework Ti at the surface and/or any metal present in the catalyst.
These physico-chemical properties are influenced globally by the preparation method, synthesis parameters, and chemical composition which are in turn related to the Ti–O–Si connectivity, the degree of surface hydroxylation, and the titanium distribution in the silicate matrix.52 Avendaño et al. evidenced the presence of only Lewis sites in hydrothermally synthesized materials compared to both Lewis and Brønsted sites in the xerogels probably due to the consumption of surface hydroxyl groups by the high temperature autoclave synthesis of the former materials.85
Trukhan et al. revealed that TiO2–SiO2 xerogels exhibited the highest Brønsted acidity among the different titanium–silicates studied.52 Adsorption of CO occurred at two additional sites on the surface in comparison to pure silicate, characterized by change in ν(CO) stretching from 2185 (high pressure) to 2178 (low pressure, 0.1 mbar) cm−1 and from 2174 (high pressure) to 2170 cm−1 (low pressure) as shown in Fig. 6.
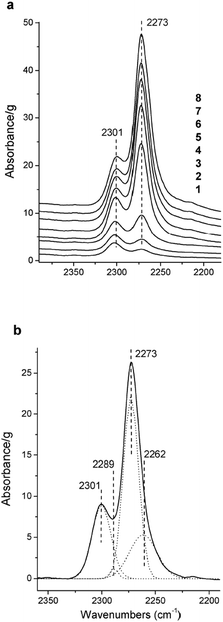 | ||
Fig. 6 FT-IR difference spectra of adsorbed CD3CN at 293 K on TiO2–SiO2 xerogel (7.29% Ti) (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N stretching region): (1) outgassing for 60 min, (2) outgassing for 20 min, (3) outgassing for 5 min, (4) 0.1 mbar, (5) 0.5 mbar CD3CN, (6) 1 mbar, (7) 2 mbar, and (8) 5 mbar. (b) Spectrum at 0.5 mbar CD3CN with deconvolution. The spectrum of TiO2–SiO2 xerogel (7.29% Ti) before interaction with adsorbate molecules. Reprinted with permission from ref. 52. Copyright 2005, American Chemical Society. N stretching region): (1) outgassing for 60 min, (2) outgassing for 20 min, (3) outgassing for 5 min, (4) 0.1 mbar, (5) 0.5 mbar CD3CN, (6) 1 mbar, (7) 2 mbar, and (8) 5 mbar. (b) Spectrum at 0.5 mbar CD3CN with deconvolution. The spectrum of TiO2–SiO2 xerogel (7.29% Ti) before interaction with adsorbate molecules. Reprinted with permission from ref. 52. Copyright 2005, American Chemical Society. | ||
These bands are due to CO adsorbed on isolated titanium cations in the surrounding silica or having one Ti4+ cation in their second coordination sphere, and CO interactions with Ti–OH groups, respectively. CD3CN adsorption similarly revealed the existence of two additional sites, which were not detected for pure silicate. The band at 2289 cm−1 is due to CD3CN interaction with weaker acid titanol groups and a second band is observed at either 2306 (low pressure) or 2300 cm−1 (high pressure) and distinguished after spectral deconvolution. These absorption bands are due to deuterated acetonitrile interaction with isolated framework titanium cations with probably one Ti4+ cation in their second coordination shell.52
With a strongly basic probe molecule, Ren et al. utilized pyridine-FT-IR studies in a high-temperature flow infrared cell-reactor to relate the intensity of the IR band and the amount of pyridine adsorbed on the materials to the nature and degree of acidity.42 The absorption band around 1540 cm−1 is attributed to the vibrational modes of Brønsted coordinated pyridine and that at 1480 cm−1 is associated to both Brønsted and Lewis acid sites. The band at 1445 cm−1 corresponds to vibration of pyridine chemisorbed on specifically Lewis acid sites. The material with equimolar compositions elucidated that there are theoretically no Si or Ti rich regions in the bulk structure and it is free from charge imbalance in the Ti–O–Si bonds and hence no acidic sites are generated. It was also determined that the population of acidic sites of the material with highest band intensity was dependent on the calcination temperature. The mixed oxides calcined at 450–550 °C show four bands at 1610, 1540, 1480, and 1445 cm−1, and a sharp decrease in intensity of these bands was observed after calcination at 600 °C. The disappearance of apparent absorbance in the FT-IR spectra of the mixed oxide when calcined at 650 °C is noteworthy. This result suggests the removal of surface hydroxyl groups and the dissociation of existing Ti–O–Si linkages.
NH3 Temperature Programmed Desorption (TPD) provides a measure of the acid strength at the sites in heterogeneous catalysts derived from the amount of NH3 desorbed from the catalyst. The non-existence of an NH3 peak in the TPD profile suggests the absence of acidic sites in pure silica as shown in Fig. 7. This implies that the presence of weak acidic sites in the TiO2–SiO2 materials should be mainly ascribed to the presence of titanium oxides species.56
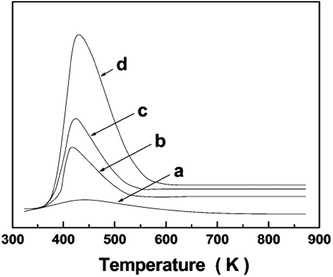 | ||
| Fig. 7 NH3-TPD patterns of: (a) SiO2-ref and (b) TiO2–SiO2 materials calcined at 873 K: 6% TiO2–SiO2, (c) 12% TiO2–SiO2, and (d) 25% TiO2–SiO2. Reprinted with permission from ref. 56. Copyright 2009, American Chemical Society. | ||
The desorption peak due to NH3 shifts slightly toward relatively high temperature with an increase in the Ti content, indicating an increment in the acidic strength. The peak area also increases, suggesting an increase in the amount of acid sites as well. In a different study, Ren et al. correlated TPD profiles of NH3 and related peaks to that of pyridine FT-IR and it was noted that the strength of the acid sites is fairly constant and the surface acidity originates from the presence of weak acid sites. Contrary to the absence of acidic sites in the material of equimolar composition inferred by pyridine-FTIR, the NH3 desorption peak in the TPD profile of the same material clearly indicated the significance of weak acid sites. Such observations may be a source of confusion and caution must be observed since the probe molecules and experimental conditions for pyridine-FT-IR and ammonia TPD are generally different. However both methods are in agreement when only 0.01 mmol g−1 of NH3 is desorbed, indicative of none to very low acid density observed in the materials calcined at 650 °C. Overall, the authors concluded that the independent techniques used are in good agreement and are reliable for estimating the changes in surface acidic sites with increasing calcination temperature. A continuous decrease in the amount of NH3 desorbed, indicative of the relative abundance of Ti–O–Si connectivity, when calcination temperature is increased from 500 to 650 °C is evident. TGA studies also credit this decline to the disintegration of Ti–O–Si linkages and the removal of surface hydroxyls.42
The source of acidity and its impact on catalytic activity is still the subject of stiff debate. Tanabe et al. hypothesized with high validity that Lewis acidity is derived from the presence of an excess of positive charge on the impurity cation while an excess of negative charge is responsible for Brønsted acidity in chemically mixed binary oxides. These notions were proposed under the assumptions that the coordination number of each metal cation is maintained in its pure oxide structure, and the coordination of oxygen is that of the pure oxide that is in abundance. This suggests that Lewis and Brønsted acidity will be most prominent in titania-rich and silica-rich mixed oxides respectively.120 In spite of the distinction of these sites in such materials, this hypothesis was challenged when applied to microporous silica supported titania materials. Liu et al. revealed the presence of unanticipated Brønsted acid sites and concurrent suppression of Lewis acidity on Ti-rich mixed oxdies by using FT-IR spectroscopy of adsorbed NH3. They proposed the probability for electroneutrality whereby hydroxyl groups balance the postive charge difference, which is apparent when tetrahedral Si mixes chemically with octrahedral Ti, thus producing Brønsted acidity. Another concern was that the charge difference calculated by Tanabe et al. for Si-rich mixed oxides was based on the assumption that Ti maintains its octahedral coordination environment, a depiction unlikely under experimental conditions since such materials exhibit high acidity.120 On the contrary, microstructural investigations by spectroscopic studies such as Extended X-ray Absorption Fine Structure (EXAFS), X-ray Absorption Near-Edge Structure (XANES), Diffuse Reflectance UV-Vis spectra, FT-IR and Raman spectroscopic techniques indicate that at least 70% of the Ti atoms reside in sites of tetrahedral coordination whereas the remaining atoms reside in non-tetrahedral sites, and the apparent absence of Ti atoms in the octahedral sites in the Si-rich material (Ti:Si = 1:8) was verified.121 A significant decrease in surface reactivity was observed as a result of minimal acidity, consistent with the charge balance after isomorphic substitution. This low activity is due to weak bonding interaction of Si-rich materials evidenced by low intensity of the NH3 TPD peaks. Overall, the profiles indicated that Brønted acid sites adsorbed the greatest amount of NH3 (ca. 80%) on the Ti–Si mixed oxides at 100 °C and the adsorption capacity decreased with increasing silica content as the silica suppressed a significant fraction of the Lewis acid sites normally associated with titania.122
In a separate study, the data collected by Odenbrand et al. suggested that acidity on titania–silica materials was significantly weaker than titania doped by sulfate, phosphate, tungstate, and molybdate ions as well as sulfated and phosphated silica. Titania–silica materials are found to possess substantially weak Brønsted acid solids with the inability to protonate a relatively weak base such as pyridine, and strong bases such as ammonia and n-butylamine.123 Pyridine-FT-IR spectra of silica supported titania showed the presence of only Lewis acid sites on the surface. Exposure of the oxidized material to water vapour resulted in the generation of some weak Brønsted acid sites, shown by a decrease in intensity of the band at 1545 cm−1 upon evacuation near 150 °C.
Kataoka and Dumesic124 proposed a model using Pauling's electrostatic bond strength based on the notion that the formal charge of an ion should be equivalent to the sum of the electrostatic bond strengths around it. This model serves as a means to predict the existence of Brønsted acid sites on the surface of metal oxides by evaluating the degree of under-saturation for surface oxygen atoms. An under-saturation of 0.1–0.4 valence units (v.u.) has been deemed ideal for creating Brønsted acidity. The possibilities that span this model are with respect to the coordination of Ti4+. No Brønsted acidity should be expected if tetrahedral coordination is assumed. The bridging oxygen atoms between Ti and Si are under-saturated by 0.33 v.u. and create Brønsted acidity if Ti4+ is in octahedral coordination. This scenario is postulated for the oxidized materials exposed to water vapour. No evidence for Brønsted acid sites was observed for the reduced (Ti3+ and Ti2+) form of the supported materials irrespective of the coordination environment of the Ti species.124 Overall, the silica phase was shown to stabilize the supported Ti4+ in sites of low coordination and this leads to acid sites.
A consensus to this debate was reached by Galan-Fereres et al. providing explanation to the co-existence of Brønsted acidity and Lewis acidity in silica supported titania materials. The larger titania phases assume octahedral coordination at higher titania loadings and bond with Si (Ti–O–Si) at the interface to induce Brønsted acidity. The strong Lewis acidity detected is attributed to the existing bulk titania. Much higher titania loading results in segregation of both oxide phases thus reducing interface hetero-linkages of TiO2–SiO2 and this subsequently diminishes Brønsted acidity.115 More clarity may be achieved by investigation of in situ experiments with a constant and continuous flow of reactants over catalysts of precisely known geometries.
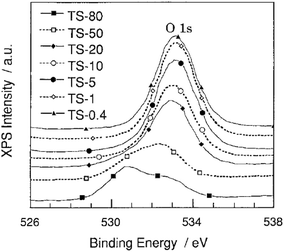 | ||
| Fig. 8 X-ray photoelectron spectra of the O 1s level for Ti/Si binary oxides. TS-0.4–TS-80 samples are Ti/Si binary oxides of 0.4–80 wt% TiO2, respectively. Reprinted with permission from ref. 38. Copyright 1998, American Chemical Society. | ||
The band due to the O ion in the TiO2 matrixes (Ti–O–Ti) and SiO2 matrixes (Si–O–Si) was observed at around 530.5 eV and 533.5 eV respectively.38 An increase in the Ti content results in a shift of the O 1s peak from 533.5 eV to a lower energy of around 532.5 eV, reflecting a substitution of the Si atoms by less electronegative and more polarizable Ti atoms in the SiO2 matrixes.125 At higher Ti contents (50–80 wt%), the two O 1s bands can be observed at around 530.5 and 532.5 eV, thus indicating phase separation with titania-rich regions and regions with titanium oxide species embedded into the SiO2 matrices.
Similarly, Xie et al. showed detailed curve fitting results that allude to four different oxygen coordinations in the O 1s spectra for TiO2–50% SiO2 calcined at 600 °C. The peaks at ca. 529.6, 531.0, and 533.3 eV may be attributed to the oxygen in Ti–O–Ti, Ti–O–Si and Si–O–Si linkages, respectively, while the peak at ca. 532.0 eV can be assigned to surface hydroxyl groups. The results also indicate high surface hydroxyl content for the material of higher SiO2 content. However for materials of lower SiO2 content, the surface hydroxyl content decreased as the calcination temperature was increased from 400 to 800 °C.51 A subsequent study by Yang et al.53 demonstrated the effect of calcination temperature on the interaction of metal species in a high Ti content TiO2–9.1% SiO2 material. The peak of Si 2p for the material calcined at 400 °C appeared around 102.3 eV with silica primarily existing in the form of Si–O–Si linkages. An increase in the calcination temperature to 600 °C resulted in the emergence of the peak at 103.6 eV suggesting the restructuring of the matrix. It was concluded from the XPS spectra and curve fitting results of the O 1s region that calcination at 400 °C was insufficient to facilitate strong interactions between titania and silica and leads to phase separation depicted as Ti–O–Ti and Si–O–Si linkages while calcination at 600 °C promotes the formation of Ti–O–Si hetero linkages.
Fig. 9 shows the XPS spectra of the Ti 2p band for titania–silica mixed oxides of varying Ti content. The binding energy of the Ti(2p3/2) and Ti(2p1/2) bands shifts to higher values when the Ti content decreases, especially with materials of lower TiO2 content.
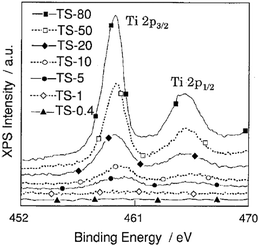 | ||
| Fig. 9 X-ray photoelectron spectra of the Ti 2p levels for Ti/Si binary oxides. TS-0.4–TS-80 samples are Ti/Si binary oxides of 0.4–80 wt% TiO2, respectively. Reprinted with permission from ref. 38. Copyright 1998, American Chemical Society. | ||
These shifts can be attributed to the smaller relaxation energy for the highly dispersed titanium oxide species as compared to the powdered bulk TiO2 catalysts.38,126 Yang et al. noted an increase in the intensity of the Ti 2p3/2 peak at 458.5 eV resulting from the Ti–O–Si hetero linkages formed from the interaction between silica and titania.53 This increase in intensity is attributed to an increase in coordination number of Ti and the shortening of the Ti–O bond, thus implying incorporation of Ti4+ into the tetrahedral sites of the silica network. This shift can also be accounted by the change in the electronic structure of Ti species in titania–silica mixed oxide. An increase in the effective positive charge on the Ti species due to a decrease in the electron density around Ti species developed from the greater electronegativity of Si via O–Ti.53,83
3.2. Bulk characterization
X-ray Absorption Near-Edge Structure (XANES). The height and position of the pre-edge peak in X-ray Absorption Near-Edge Structure (XANES) is a valuable indicator of the coordination of an absorbing metal atom. It provides information about the geometry of the metal site and the data can be interpreted qualitatively hitherto by comparison with published near edge structures of reference compounds. XANES analysis of titania–silica mixed oxides by Liu et al. exhibits pre-edge peaks that are more distinct than the peaks for TiO2, indicating that a portion of Ti resides in tetrahedral locations when mixed with Si as shown in Fig. 10.121 However, the pre-edge peaks for the mixed oxides are consistent with the presence of mixtures of 4-, 5-, and 6-coordinated Ti denoted [4]Ti, [5]Ti, and [6]Ti, respectively. Since these materials have large specific surface areas, a significant fraction of Ti atoms will be exposed to the surface and interact with water molecules. Initially, these materials contain isolated and distorted [6]Ti sites. Dehydration at temperatures between 300 and 750 °C of materials of low Ti content rapidly converts [6]Ti to [4]Ti (TiO6 to TiO4) as depicted by an increase in the height of the pre-edge peak. Heating for longer periods or at higher temperatures such as 750 °C created stable [4]Ti that is incorporated into the bulk of the SiO2 network.
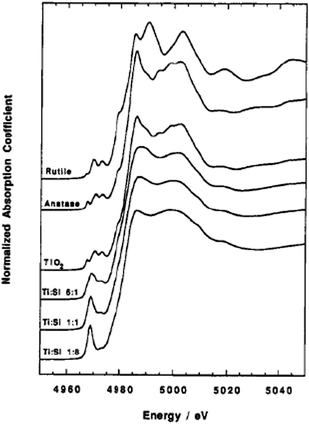 | ||
| Fig. 10 Ti K edge XANES for Ti oxides and Ti–Si mixed oxides. Reprinted with permission from ref. 122. Copyright 1994, American Chemical Society. | ||
The [4]Ti reverted to [6]Ti upon cooling and this phenomenon is attributed to the interactions with ambient moisture.44,45,122,129 Xerogels with low TiO2 content may have Ti dispersed in porous regions of the SiO2 network and hence accessible to water groups forming isolated, distorted [6]Ti. In situ experiments suggest the presence of [5]Ti coordination as well.130 The large pre-edge feature in the dehydrated material indicates that the majority of Ti occupies sites of tetrahedral symmetry, and suggests that adsorbed water disrupts the tetrahedral geometry associated with the dehydrated sample.
The xerogels with higher TiO2 content exhibited Ti initially present as phase separated [6]Ti i.e. TiO2, and heat treatment at temperatures as high as 500 °C resulted in the formation of anatase. In situ treated materials demonstrate similar patterns, however, it is suggested from Ti K-edge XANES that only stable [4]Ti was detected under ambient moisture (ex situ) whereas both stable and unstable [4]Ti was detected under in situ conditions.44 Farges et al.129 predicted that the Ti atoms in the dehydrated higher titania content material (12% TiO2/SiO2) are predominantly [5]Ti coordinated, and hydration increases their coordination to [6]Ti.130 These differences provide information to understand the type of [4]Ti or any other coordination that emerges as a major contributor to catalytic activity of mixed oxide materials.
Such structural information is relevant and promising in relating Ti coordination to the catalytic properties of these materials. For example, Imamura et al.36 utilized XANES analysis to propose that epoxidation of alkenes occurs due to the presence of [4]Ti which act as Lewis acid sites. In this study, the pre-edge feature of titania–silica with Ti content below 40 mol% corresponds to Ti species with tetrahedral configuration, whereas octahedral Ti species predominate above 50 mo1% of Ti. The main assumption was that the area of the pre-edge peak is a measure of tetrahedral content and as such an estimate of the amount of Lewis acid sites. The authors illustrate a correlation between the estimated number of Lewis acid sites and the XANES pre-edge peak height (obtained without peak fitting). They postulate that the amount of tetrahedrally coordinated titanium and as such the amount of Lewis acid sites increased with a decrease in the titanium content.
Extended X-ray Absorption Fine Structure (EXAFS). EXAFS analysis is vital for probing the nearest environment of the titanium atoms present in the sol–gel matrix. The plots in Fig. 11 are results obtained from EXAFS experiments by Liu et al.121 using radial structure function (RSF) that shows a single prominent peak associated with the first coordination shell of Ti bonded to O.
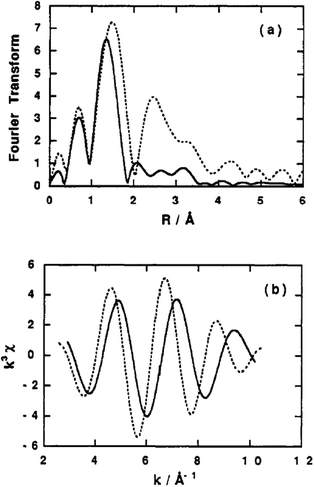 | ||
| Fig. 11 Radial structure functions: (a) phase shifts and (b) Fourier filtered EXAFS functions for Ti:Si 1:8 (solid lines) and synthesized TiO2 (dotted lines). Reprinted with permission from ref. 121. Copyright 1994, American Chemical Society. | ||
A comparison between the RSF of TiO2 and a Si-rich material (Ti:Si = 1:8) reveals shortening of the first shell of Ti–O bond distance in the mixed oxide from 1.94 to 1.82 Å. The absence of higher shell peaks between 2 and 4 Å in the mixed oxide is attributed to the distortion of Ti structure that occurs with the substitution of Ti for Si atoms in the second coordination shell. Si is also known to have lesser effective backscatter of the photoelectron wave than more electron rich transition elements.
It is also likely that a larger fraction of the Ti is exposed to the surface which diminishes the higher shell contributions as confirmed by XANES data in the same study. It is notable that the coordination numbers are less than 6, suggesting that Ti loses coordination from octahedral to possibly tetrahedral.
Anderson et al.131 studied the effect of three calcination temperatures (250, 500, and 750 °C) on the local environment of different loadings of 8, 18 and 41 mol% TiO2. At these temperatures, the materials of low Ti content appear to have [4]Ti and the phase separated materials demonstrate a large portion of [6]Ti with detectable 4-fold coordinated Ti, and probable traces of 5-fold coordination at the interfacial regions as well. Calcination at the two lower temperatures, 250 and 500 °C, indicates that the materials of high Ti content have lower ratios of [6]Ti to [4]Ti. The latter coordination appears to predominate when the calcination temperature is increased to 750 °C and this was attributed to the relaxation of the Ti sites and confirmed by a decrease in Debye–Waller factor on heating. This relaxation suggests the vulnerability of the 6-fold coordination and its pseudo existence with other coordinations.
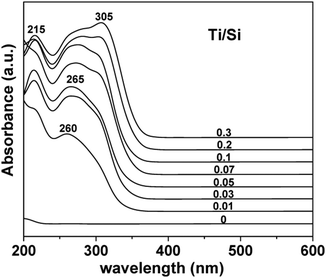 | ||
| Fig. 12 UV-Vis DR spectra of calcined TiO2–SiO2 composites synthesized with different Ti/Si ratios. Reprinted with permission from ref. 132. Copyright 2009, American Chemical Society. | ||
The band at ca. 210–230 nm, which is assigned to a ligand-to-metal charge transfer (LMCT) transition from O to Ti in isolated TiO4 as shown in eqn (7) or HOTiO3 units is associated with the tetrahedral (or non-octahedral84) incorporation of titanium in the SiO2 framework i.e. [Ti–(O–Si)4] as titanium aggregates interact strongly with the silica matrix.
| [Ti4+ − OL2−] ⇄ [Ti3+ − OL−]* | (7) |
Murata et al. and Aguado et al. showed that the absorption edge was shifted to longer wavelength (red shift) with increasing Ti content and appeared close to coincide with the bare TiO2 due to an increase in size of the aggregated species and an increase in the coordination number of Ti.22,84 Using various in situ spectroscopic techniques, Gao et al. proposed that isolated TiO4 units exist at low Ti content, which, when increased, leads to the formation of 1-D polymerized TiO4 units and 2-D polymerized TiO5 units, followed by the development of small particles comprised of TiO6 units, and finally followed by an increment in the particle size of TiO2.130 It was demonstrated that the UV absorption edge shifted to higher energies (blue shift) as the content of Si was increased in the mixed oxides.121,133 Lafond et al. attributes this blue shift in titania silica materials to the presence of TiO2 nano-domains (quantum size effect) or the presence of Ti species with different first and/or second coordination spheres (matrix or support effect).112 In combination with XANES experiments, it was also evident that the dehydration of a low Ti content material resulted in a further blue shift in the position of the absorption edge compared to the hydrated material.134 On the contrary, Chen et al.68 incorporated P25 into TEOS and showed that the blue-shift in sol–gel derived materials is not achievable with P25 based systems.
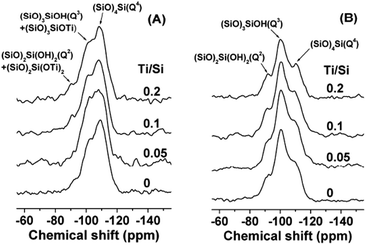 | ||
| Fig. 13 Solid-state 29Si MAS and 1H–29Si CP/MAS NMR spectra of the mesoporous TiO2–SiO2 composites synthesized with different Ti/Si molar ratio. Reprinted with permission from ref. 132. Copyright 2009, American Chemical Society. | ||
The chemical shift of the 29Si signal is a function of the local environment and can be used to distinguish neighbouring –OSi groups from neighbouring –OTi groups. The enhancement of the signals in the Q2 and Q3 regions of the mixed oxides in comparison with pure SiO2 was suggestive of the number of –OTi groups around a Si atom, thus supporting the concept of mixed phases in such materials. For example, since enhanced adsorption of R-6G on titania–silica is attributed to the presence of Ti–O–Si phase, Anderson et al. determined TiO2/SiO2 = 30/70 as the optimum ratio for achieving the largest Ti–O–Si interface area. The authors proposed that as the amount of TiO2 is decreased, more framework Ti is formed in the SiO2 matrix while bulk TiO2 in minimized. An increase in TiO2 led to the segregation of the TiO2 and SiO2 phases resulting in a diminished interface region.57,136
Similarly, it was determined that the Ti–O–Si interface is critical in the stabilization of the anatase phase by the surrounding amorphous SiO2 phase. Hirano et al. explained that the Ti atoms are substituted into the tetrahedral positions of the SiO2 lattice that lock the Ti–O species at the interface to form tetrahedral Ti sites.135 The interaction was believed to prevent the diffusion of anatase crystallites and retard the nucleation and growth that is necessary for the phase transformation to rutile.
Dirken et al. used NMR spectroscopy to compare the degree of polymerization in two synthesis routes.137 The one-stage (addition of the Ti(OPri)4 to the TEOS) and two-stage (partial hydrolysis of the TEOS before addition of the Ti(OPri)4) procedures were followed. 29Si MAS NMR spectra of the resultant amorphous gels indicate that the more polymerized Qn species are formed as the temperature increases irrespective of the synthesis route. The materials consist of Q2, Q3, and Q4 SiO4 species that eventually become Q4 after heat treatment at 600 °C due to loss of organic groups, thus indicating that Ti replaces Si in the network at this temperature.
17O MAS NMR is a valuable tool in the characterization of amorphous oxides and it is more informative than 29Si MAS NMR.17O NMR directly reveals the phase separated nature in the gels through OTi3 and OTi4 sites, indicative of Ti–O–Ti bonds. Similarly, homogeneity between titania and silica is established by signals depicting the presence of Ti–O–Si hetero-linkages and the absence of Ti–O–Ti bonds. The studies were confirmed by Ti–O–Si resonances from fresnoite (Ba2TiSi2O8). Ti–O–Si bonds were still observed even after heating the materials to 600 °C, suggesting that homogeneity of the titania and silica was achieved and sustained.137 Pickup et al. utilized spectroscopic studies to determine the atomic structure of (TiO2)x(SiO2)1−x (x = 0.18, 0.30, and 0.41) xerogels prepared using an acetylacetone stabilized Ti(OPri)4 precursor. 17O MAS NMR analysis of (TiO2)0.41(SiO2)0.59 materials indicated a significant amount of Ti–O–Si bridges, suggesting that there is a high level of atomic mixing in materials prepared with acac.45 The use of acac significantly increases the amount of Ti–O–Si bonding at this composition due to improved solubility of precursors in that media.
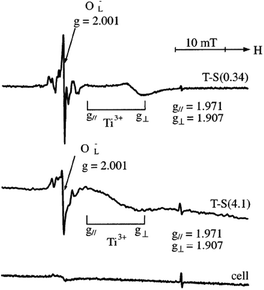 | ||
| Fig. 14 ESR spectra at 77 K of the T-S(0.34) and T-S(4.1) samples and cell after the photo-irradiation in vacuo at 77 K. Reprinted with permission from ref. 22. Copyright 2003, American Chemical Society. | ||
However under UV irradiation, materials in vacuo at such cryogenic temperatures gave rise to signals with an axial symmetric g tensor assigned to Ti3+ (g∥ = 1.971 and g⊥ = 1.907), and a signal due to OL− (g = 2.001), a hole center on the lattice oxygen due to LMCT from O2− to Ti4+. These signals were stable at 77 K even after photo-irradiation was ceased but disappeared upon warming to room temperature. ESR spectra with signals at g∥ = 2.002 and g⊥ = 2.008 showed that O3− and O2− were generated over the TiO2–SiO2 samples irradiated at 77 K in the presence of oxygen, while only O2− was observed at room temperature.
Similarly, Yamashita et al.38 utilized an ESR measurement to investigate the local structural environment of the titanium oxide species in mixed oxides. The Ti3+ ions formed by the reduction of the oxide with H2 were monitored by ESR studies as shown in eqn (8).
| (Ti4+ − O2−) + H2 → (Ti3+ − OH−) + H+ | (8) |
4. Photocatalytic reactions
Photocatalysis has emerged as a promising technology for the degradation of organic pollutants and photosplitting of water. In this process, organic pollutants are degraded in the presence of semiconductor photocatalysts such as TiO2, ZnO, CdS, Fe2O3, etc.,136,140 UV light, and an oxidizing agent such as hydrogen peroxide (H2O2), oxygen, ozone or air. Irradiation of semiconductors with photons of energies greater than their band gap results in the excitation of valence band (VB) electrons to the conduction band (CB), leaving behind positive holes.141 In aqueous media, the positive holes may oxidize pollutants directly or oxidize water to produce ˙OH radicals. The electrons in the conduction band reduce the oxygen adsorbed on the photocatalyst surface to produce superoxide radical anion species, O2˙−, and other highly reactive oxidative species (ROS) such as hydroxyl and peroxyl radicals, and hydroxide ions as shown in Scheme 4.The photocatalytic activity of the semiconductors depends on the structural properties such as crystal composition, surface area, particle size distribution, porosity, band gap, and surface hydroxyl density, to name a few parameters.142,143 Although, the formation of ROS on the catalyst surface remains constant for a given light intensity, catalyst amount, and duration of irradiation, it was determined that the degradation decreases when the concentration of the pollutant is increased.144
In addition, the pH of the solution plays a vital role on the photocatalytic degradation of organics since it determines the surface charge of the photocatalyst, the electrostatic interaction between the semiconductor surface and the substrate, and the amount of charged radicals formed during the photocatalytic oxidation process.
In this section, we will first discuss the applications of titania–silica oxide materials for photocatalytic degradation in aqueous and gas phase. In Section 4.2, gas phase photocatalytic reactions using TiO2–SiO2 photocatalysts will be extensively reviewed. Finally, water splitting reactions will be discussed.
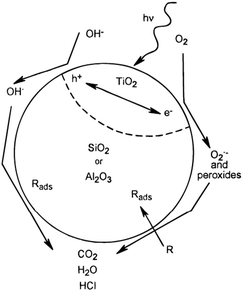 | ||
| Scheme 4 Schematic representation of the TiO2–SiO2 or TiO2–Al2O3 photocatalyst with no interaction between the TiO2 and SiO2 or Al2O3 phases. Reprinted with permission from ref. 136. Copyright 1997, American Chemical Society. | ||
TiO2 has been studied extensively for photocatalytic applications due to its high activity, low cost, and stability under irradiation conditions. Among the three common TiO2 polymorphs, anatase and rutile have been vastly explored as photocatalysts than brookite owing to their ease of preparation. In general, anatase is proven to be more photoactive than rutile. The semiconductor is activated by UV irradiation through the Advanced Oxidation Process (AOP) and electrons are excited to higher energy states resulting in the formation of ROS that are fundamental for heterogeneous photocatalysis as described by the equations summarized below.
| TiO2 → h+ + e− | (9) |
| H2O + h+ → HO˙ + H+ | (10) |
| O2 + e− → O2−˙ | (11) |
| O2−˙ + H+ → ˙O2H | (12) |
| 2˙O2H → H2O2 + O2 | (13) |
| H2O2 + e− → HO˙ + −OH | (14) |
4.1. Aqueous phase reactions
Various types of phenolic compounds, pesticides, and cationic and anionic dyes were studied using AOP.144,145 A recent publication reviews the photocatalytic degradation reactions in aqueous media using periodic mesoporous materials.146 In this section, the various photocatalytic reactions mainly conducted using aperiodic titania–silica are discussed.Benzyl compounds. The photocatalytic activity of TiO2/SiO2 nanoparticles were studied by monitoring the decomposition of 1,4-dichlorobenzene.24,40 The rate constant of the samples prepared with an ethanol-rich solvent was higher than that of Degussa P25. This was attributed to the abundant anatase phase and inhibition of the formation of the rutile phase due to the presence of SiO2. The authors determined that 20 mol% silica was the optimum loading for the most efficient photocatalytic activity i.e. about 1.2 times higher than that of Degussa P25.147
SiO2 loaded TiO2 has been utilized effectively for the degradation of benzyl trimethyl ammonium chloride (BTMA) and propionic acid under UV irradiation.148 It was notable that the TiO2–SiO2 surface is more negatively charged than bare TiO2 at pH 6. Among the compounds studied, propionic acid showed the least degradation rates because of the repulsion between the negatively charged propionic acid and the negatively charged catalyst surface. The enhanced activity was due to the electrostatic interaction between the anionic TiO2 surface and the cationic pollutant. Vohra and Tanaka also determined that the surface charge of TiO2 may be influenced by adjusting the pH, and as a result observed a difference in the photocatalytic activity.149 Enhanced photocatalytic activity was attained for cationic pollutants owing to the presence of silica that resulted in increased specific surface area and introduction of Si–O− groups, which in turn facilitated substrate adsorption onto TiO2. However, the efficiency of the supported catalysts decreased with an increase in substrate concentration, suggesting an occlusion of active sites. It was interesting to note that that degradation of anionic and non-ionic compounds such as acetate and phenol remained unaffected.
A key parameter in sol–gel chemistry is the nature of solvent utilized in the synthesis procedure. Xu et al. propose that during the hydrolysis of titanium n-butoxide, the alcohol co-solvent participates in the ligand exchange and influences the diffusion of the positively-charged Ti(OH)x(OR)yn+ species. Easier hydrolysis and diffusion is attained when a highly polar solvent of lower viscosity such as methanol is used, resulting in TiO2 of larger particle sizes compared to those that are obtained from the use of 2-pentanol. The investigators observed a steady increase in the activity of the resultant photocatalysts for the degradation of acetophenone. The solvent used in preparation was changed from methanol to ethanol, 2-propanol, n-butanol, and 2-pentanol. This was accompanied by a higher anatase content and smaller crystallite size of TiO2.150 On the contrary, an opposite effect was observed when SiO2 was loaded onto TiO2 due to the suppression of crystallinity of the highly dispersed anatase phase. Interestingly, supported TiO2 prepared using higher amounts of alcohol showed increased photocatalytic activity. The effect of different silica particle size on the activity was also examined. It was concluded that the materials of smaller silica particle sizes resulted in a higher activity within one hour compared to the larger particles. It is predicted that other than providing improved adsorptivity and a charged framework for the attachment of organics, the silica support serves as a dispersing agent for preventing agglomeration of the active TiO2 crystallites.
Phenolic compounds are being released into the aquatic environment from industrial wastes and they continue to contribute to the pollution of water sources. In a study comparing three different photocatalysts, Alemany and co-workers probed the role of the support and the support–substrate interactions. They determined that TiO2–SiO2 activated the oxidation of phenol due to the Ti–O–Si phase via strong Brønsted acid sites whereas TiO2–Al2O3 was determined to be inactive. However enhanced photocatalytic activity was observed in the decomposition of salicylic acid with TiO2–Al2O3 clearly suggesting that the activity of the photocatalyst is unique to the nature of substrate. Hydroquinone, catechol, and maleic acid were identified as the main intermediates formed initially, and further oxidation produced acetic acid and formic acid as ring opening products.140 Jammaer et al. prepared high surface area TiO2–SiO2 composites using a non-conventional procedure involving a non-aqueous reaction of Ti and Si alkoxides in formic acid and SC CO2. Phenol solutions were subjected to degradation using the resultant materials under UV irradiation.105 Degussa P25 showed higher photoactivity (>95%) than the composites due to the highly mixed structure of anatase and rutile phases that minimized charge-carrier recombination resulting from effective band bending between the two phases. A correlation between the titania loading, the support, and the photocatalytic activity of these composites was not established. However, it is noteworthy that composite materials prepared using SC CO2 showed higher activity than those prepared using Pluronic 17R4 because of the occlusion of active sites by the surfactant.
Photocatalytic degradation of phenolic compounds under UV irradiation has been performed by numerous investigators using synthesized and commercial TiO2. There are only a handful of studies addressing the mechanistic pathways for the degradation of phenol. However, the literature is still inconclusive of the reactions between phenol and ROS to form the intermediates. An understanding of the photoactivity of TiO2 towards degradation of phenol is necessary to provide a platform over which intermediates due to TiO2–SiO2 can be identified. Such studies have been carried out by Bahnemann et al., who investigated the influence of pH and the initial concentration of 4-chlorophenol (4-CP) in the photocatalytic degradation using two commercial catalysts, Sachtleben Hombikat UV 100 and Degussa P25.151 The degradation of 4-CP in the presence of Degussa P25 is slightly faster than that with Hombikat UV 100, but the main intermediates observed using the two catalysts are identical. In acidic solution, hydroquinone and benzoquinone are the main intermediates identified when Hombikat UV 100 catalyst was employed. Seven additional intermediates, hydroxyhydroquinone, hydroxybenzoquinone, phenol, 4-chlorocatechol, 4-hydroxyphenylbenzoquinone, 2,5,4′-trihydroxybiphenyl, and 5-chloro-2,4′-dihydroxybiphenyl were identified with the use of Degussa P25.
Another mechanistic investigation on the photodegradation of phenolic compounds was carried out by Madras and Priya. Combustion synthesized TiO2 was reported to be a more effective catalyst compared to Degussa P25 for the degradation of a mixture of 4-chlorophenol and 4-nitrophenol. The improved activity was attributed to the inhibition of phase transformation from anatase to rutile and brookite by this unique synthetic approach.152 In addition, the TiO2 particles prepared had small size, large surface area and more surface hydroxyl content compared to Degussa P25. A faster degradation was observed with 4-chlorophenol than 4-nitrophenol due to their higher susceptibility to hydroxyl radical attack. Degradation of a binary mixture of 4-chlorophenol and 4-nitrophenol was slightly faster than 4-nitrophenol alone due to competition for the catalyst active site and the interaction between these compounds. Hydroquinone and hydroxyhydroquinone were identified as by-products during 4-chlorophenol degradation, whereas nitrocatechol, hydroquinone, and hydroxyhydroquinone were formed as a result of the breakdown of 4-nitrophenol. Succinic acid was also observed as an intermediate when the mixture of chloro and nitrophenol was degraded.147 In this study, Lee et al.147 investigated the role of silica and determined that the mixed oxides showed better activity than pure TiO2 for the degradation of p-nitrophenol due to the improved adsorptivity of substrate provided by the presence of SiO2.
We have reported the photocatalytic activity of titania–silica xerogels34 and hydrothermally prepared materials.89 Mechanistic studies have also been attempted recently by our research group using the hydrothermally synthesized materials. The photocatalytic degradation of phenol for a series of titania–silica mixed oxides was evaluated using a Shimadzu TOC-V CSH total organic analyser and High Performance liquid Chromatography (HPLC). All the catalysts show more than 90% degradation after 6 h of solar simulated irradiation even though the specific surface area varied from 65 to 1033 m2 g−1 as more SiO2 was added. The pore volume also increases as the silica amount increases. This result proposes that the overriding function of silica is to stabilize the anatase phase and prevent the phase transformation to the fairly inactive rutile phase as well as facilitate the effective dispersion of the highly active TiO2 species. Adsorption results of phenol on the titania–silica catalysts showed that the amount of phenol adsorbed is similar for all catalysts and reaches a maximum of ∼9 × 10−5 mol g−1 within one hour.
It opens insightful debate to the misconception that only high surface areas and improved adsorption due to SiO2 are the key factors in the degradation process. We acknowledge that the phenomenon may be different for other substrates and this observation provides opportunity for probing into the adsorption–activity relationships between various types of organic substrates and these semiconductors. Table 2 illustrates the textural properties and the total organic carbon (TOC) concentration with respect to time. We achieved complete degradation of organics (∼0 ppm) after 6 h of solar simulated irradiation as depicted by the TOC results. No discernible trends in the TOC values with titania content were evident from our studies. A detailed analysis of the data obtained is under progress to derive a structure–activity relationship. Concentrations of phenol remaining in solution from HPLC studies are shown in Fig. 15.
| Sample | Surface area (m2 g−1) | Pore volume (cm3 g−1) | TOC (ppm) | |||||
|---|---|---|---|---|---|---|---|---|
| 30 min | 60 min | 90 min | 120 min | 180 min | 360 min | |||
| T1-E-T | 65 | 0.23 | 11.1 | 6.4 | 2.9 | 1.2 | 0.92 | < 0.1 |
| T1S1-E-T | 423 | 0.73 | 16.3 | 12.9 | 10.4 | 8.1 | 3.3 | 0.8 |
| T1S1-E-H | 426 | 0.58 | 17.9 | 13.9 | 10.8 | 7.8 | 3.1 | 0.4 |
| T1S1-E-M | 431 | 0.61 | 17.0 | 13.9 | 11.6 | 8.2 | 3.8 | 0.3 |
| T1S3-E-T | 671 | 1.61 | 13.6 | 11.6 | 11.4 | 5.9 | 1.9 | 0.6 |
| T1S4-E-T | 1033 | 1.77 | 12.9 | 10.4 | 9.7 | 6.2 | 3.4 | 0.2 |
 | ||
| Fig. 15 Photocatalytic efficiencies of the TiO2–SiO2 mixed oxide materials showing the concentration of phenol remaining at different time intervals. | ||
TiO2 prepared in ethanol only showed good degradation efficiency due to the active anatase phase. After 180 min, no phenol is detected from HPLC in all these six catalysts. The change in the co-solvent composition from aliphatic to aromatic (hexane to mesitylene) has no significant impact on the surface area or on the photocatalytic activity. HPLC studies indicate that pyrogallol (PG), hydroquinone (HQ), catechol (CC), hydroxybenzoquinone (HBQ), and benzoquinone (BQ) are the major intermediates under our experimental conditions. The catalysts seem to favour the formation of catechol over hydroquinone which is indicative of the preferred mechanistic pathway. This may be due to the availability of two ortho positions for attack by hydroxyl radicals or the possibility that intramolecular H-bonding favours the stability of CC rather than HQ. The attack of further hydroxyl radicals results in the formation of ring opening products. Maleic acid (MA) and fumaric acid (FA) have been identified as minor intermediates in our studies as demonstrated in Scheme 5. Further cleavage of these aliphatic acids leads to the formation of acetic acid in a trace amount after 6 hours.
 | ||
| Scheme 5 Photocatalytic degradation of phenol over titania–silica mixed oxides. | ||
Apart from the degradation of the organic pollutants, titania–silica mixed oxides have also been employed in environmentally benign methods for selective oxidation of organic compounds. Kholdeeva et al. reported an effective and novel method for the conversion of trimethylphenol (TMP) to trimethylbenzoquinone (TMBQ) using aqueous H2O2 as an oxygen donor.50 The accessibility of highly dispersed TiO2 plays a crucial role in the conversion. In fact TiO2–SiO2 aerogels showed more improved catalytic activity than the xerogels of similar titania content. Due to the low porosities and the occlusion of the active centers embedded in the SiO2 phase, xerogels showed less activity.
Dyes. Large volumes of coloured dye effluents are produced by textile industries and are toxic and non-biodegradable. Some of these dyes prevent the penetration of light into water, resulting in biological development disorders in aquatic life. The World Bank estimates that 17 to 20% of industrial water pollution comes from textile dyeing and treatment. They have also identified 72 toxic chemicals in water solely from textile dyeing, 30 of which are persistent and require intricate methods for removal.
Various ingenious approaches are used for the removal of dyes such as electro-oxidation, advanced oxidation processes (AOPs), adsorption, biological degradation, and membrane based processes.144,153 Chemical techniques are often expensive and may cause secondary pollution problems due to excessive usage of chemicals. However, adsorption and photocatalytic degradation are promising methods for industrial applications.
Methyl Orange (MO), a member of the azo dye family, is a source of pollution, which releases toxic and potential carcinogenic substances into the aqueous phase. Photocatalytic activity for MO degradation in solution using TiO2–SiO2 composites under light irradiation has been studied by several groups recently.153,154 These composite materials at low loadings of TiO2 show higher photocatalytic activity than bare TiO2 as a result of effective dispersion facilitated by the silica phase.
Excess silica content was shown to hinder crystallinity and retard the catalytic activity. Further increase in the amount of TiO2 led to their aggregation, phase separation, and decreased the photocatalytic activity. Addition of silica to TiO2 also enhanced the surface area, which provided more adsorption sites for MO. Furthermore, at the appropriate calcination temperature, materials containing highly crystalline anatase showed high activity due to the reduced number of defect sites which minimizes the electron–hole recombination. Chen et al. found that 450 °C was the optimum calcination temperature for TiO2–SiO2 to achieve high photocatalytic activity and calcination above 800 °C decreased the activity due to the structural change from anatase to rutile.68 But Cheng and co-workers claimed that the silica doped titania showed the best activity when the catalyst was calcined at 800 °C probably due to the proportion of both phases that promote effective band bending.155 Other factors that played a role in activity were the addition of H2SO4 that increases the acidity on the surface, and the oxidants such as KIO4, (NH4)2S2O8 and H2O2 that assisted in the inhibition of electron–hole recombination. Similar results were observed in the photodegradation of methylene blue (MB) by nanoporous TiO2–SiO2 particles.156
Titania was deposited inside the pores of mesoporous supports such as MCF (Meso Cellular Foams) and SBA-15.157 Due to the higher pore volume and pore diameter, more anatase and rutile nanoparticles diffused inside the pores of MCF than in SBA-15. The close contact between these two phases of TiO2 consequently resulted in an improved photodegradation activity of rhodamine 6G (R-6G). Rutile particles were mainly formed when the nanoparticles were synthesized at 70 °C using SBA-15.
Due to their large size, the rutile nanoparticles were located outside the pores, thus minimizing the chance for close contact with anatase particles formed inside, and resulting in a decrease in activity of TiO2-SBA-15.
The photocatalytic degradation of rhodamine B (RhB) was performed by Kang et al. using mesoporous SiO2 modified nanocrystalline TiO2 under solar simulated illumination.87 A great enhancement in thermal stability of the nanocrystalline anatase TiO2 was achieved, which enabled effective inhibition of the growth of anatase nanocrystals to form less active rutile phase. Moreover, an appropriate amount of mesoporous SiO2-modified nanocrystalline TiO2 samples exhibited much higher photocatalytic activity than the commercially available Degussa P25 TiO2 owing to the large surface area and the high charge separation rate. In addition, high adsorption of rhodamine molecules on sol–gel synthesized TiO2–SiO2 has been shown to facilitate good photocatalytic activity than bare TiO2.136,158 The high adsorption increases the concentration of substrate molecules nearer to the active TiO2 sites relative to the solution concentration.
Pesticides and herbicides. Floating TiO2–SiO2 photocatalyst beads prepared by a dip coating method were used for the photocatalytic degradation of organophosphorus pesticides such as dichlorvos, monocrotophos, parathion, and phorate.145 Complete degradation was achieved after 420 min of sunlight irradiation. It was determined that an optimal silica amount (Si/Ti = 0.20) and calcination temperature of 650 °C produced the most active catalysts. Addition of a small amount of Cu2+ (electron scavenger) increases the photodegradation efficiency of organophosphorus pesticides rapidly via reduced electron–hole recombination and increased opportunity for the formation of hydroxyl radicals. The formation of these reactive species on the TiO2 surface is favoured in acidic and alkaline solutions. Acids and bases facilitate the photocatalytic degradation of phosphate esters, converting them into trimethyl phosphate ester, formic acid, and acetic acid. The trimethyl phosphate ester is photocatalytically degraded directly into PO43− and formic acid. However, the use of higher concentrations of Cu2+ leads to the formation of [Cu(H2O)4]2+ complex ions that absorb UV light and results in reduced photoefficiencies. The role of silica is not elucidated; however, we believe that it may provide buoyancy to the photocatalyst.
Brigante and Schulz studied the absorption of herbicide paraquat, PQ2+ (1,1′-dimethyl-4,4′-dipyridinium chloride), on the TiO2–SiO2 binary system.159 The results of this study are interesting and exemplify the role played by silica different from other studies that we have encountered in this review. The adsorption of PQ2+ on the bare silica surface is low, only detectable at pH 9.5, and negligible in all experimental conditions on non-supported titania. The neutral framework of SiO2 (lack of acid sites) and the low surface area of titania agglomerates seem to be the main factors that prevent the attachment of this herbicide. However, the presence of TiO2 on the silica surface strongly enhances the adsorption capacity to PQ2+. The homogeneous dispersion of TiO2 nanoparticles on the SiO2 support may offer more acid sites (heterolinkages) for adsorption to occur. In addition, the increase in surface acidity (negative sites) of titania due to SiO2 may enhance its interaction with several cationic species that include PQ2+ ions. The investigators propose that the direct binding between PQ2+ and supported TiO2 generates ternary surface species SiO2–TiO2–PQ2+, whose formation is mainly driven by PQ2+–TiO2 electrostatic interactions, where negatively charged groups of titania could bind the dication by forming ionic pairs or outer-sphere complexes as deduced from adsorption experiments performed at different ionic strengths and temperatures. The increase in adsorption by increasing pH increased the degree of dissociation of TiO2 functional groups, leading to an increased electrostatic attraction between PQ2+ and TiO2–SiO2 and a higher, fast, endothermic, and spontaneous adsorption.
Sharma and co-workers utilized a styrene-acrylic acid emulsion as a latex polymer template for the synthesis of titania–silica composite materials. The resultant material was used for the degradation of isoproturon (N,N-dimethyl-N-[4-(1-methylethyl)phenyl. This model pollutant is used for the control of annual grasses and broad leaved weeds in cereal crops and is identified as a hazardous class III herbicide by the World Health Organization (WHO). Effective degradation was achieved at an optimal 5 wt% TiO2 under solar light, and the approach was extended successfully for the degradation of commercially available pesticides i.e. imidacloprid and phosphadium.160,161 Solar degradation of imidacloprid with TiO2–SiO2 was also carried out at a pilot-plant scale by Malato and co-workers.162 They studied the effect of pH and the influence of metals such as Fe3+ on the photodegradation of the pesticide. The results showed that the pH is the most influential variable, followed by the initially added dose of iron, and finally the TiO2 concentration. Initial reaction rate decreases with an increase in the titania content, due to the detrimental effect produced by competition for photons. However, more increments in titania loading, above a critical point, increases the overall reaction rate due to direct photocatalytic degradation of the pesticide, or due to beneficial titania–iron interactions, improving the iron redox cycle of the photo-Fenton catalytic mechanism.
The photodegradation of cyanide using TiO2–SiO2 xerogels and aerogels has been carried out by several authors.84,100 Higher degradation efficiency of CN− was observed for aerogels than the xerogels. Marked increases in activity were also noted for catalysts treated at higher calcination temperatures and these effects were observed by Ismail et al. with a threshold beyond which the activity tapers off.165 In addition, the authors studied the effects of varying the reaction conditions such as the ratios of Ti/Si, ethanol/TEOS, H2O/TEOS, amount of acid catalyst and the reaction temperature on the degradation of CN−. It was noted that the CN− removal was independent of the surface area but more responsive to the titania content. The amount of titania loading was also deemed crucial in the oxidation of 1-octanol in liquid phase. A notable increase in the activity was observed in the catalysts of low Ti content due to the charge-transfer excited state of the isolated titanium oxide species in tetrahedral coordination. From these results Yamashita et al. concluded that the Ti/Si binary oxides with low Ti content prepared by the sol–gel method are valuable and promising photocatalysts in the liquid phase.38 It is evident from literature searches that degradation of toxic non-aromatic compounds is an area that requires more exploration.
4.2. Gas phase reactions
Volatile organics such as benzene, toluene, formaldehyde etc. are toxic to human and animal health. Thus, degradation of these organics from the air stream is crucial to minimize respiratory diseases. Wang et al. studied the photodegradation of gaseous toluene with nano TiO2 prepared by the Adsorption Phase Synthesis (APS) and monitored the degradation by gas chromatography (GC) at different reaction intervals.75 Another study on the removal of volatile organic with toluene was conducted by Zou and co-workers with the TiO2–SiO2 catalyst prepared by the sol–gel method. In the presence of UV irradiation, 25% destruction efficiency was achieved in 20 h, but 55% removal efficiency was observed when the system operated under adsorption/regeneration mode.166 Liu et al. suggested that the quantum confined TiO2 nanoparticle within the silica film matrix is responsible for the high photocatalytic reduction of carbon dioxide.30 In the presence of acetonitrile and 2-propanol, stoichiometric ratios of reduction and oxidation products can be achieved in the form of carbon monoxide, formate, and acetone respectively.A control experiment to elucidate the role of silica was conducted by using pure bulk TiO2 in identical experimental conditions. The results indicated that bulk TiO2 exhibits sluggish photocatalytic activity in the absence of the SiO2 support. The extremely high photoactivity of the quantum sized TiO2 embedded SiO2 can be rationalized by the shift of the conduction band edge of TiO2 towards greater reductive potentials, which was proposed using the quantum size effect theory. The presence of 2-proponal as the hole scavenger helps in suppressing the charge recombination.
Moreover, the effect of solvents on the photocatalytic reaction was also discussed in this work. It was suggested that the polarity of the solvents used in the photocatalytic reduction of carbon dioxide influenced the nature of the product formation since the photocatalytic reduction reaction was initiated through the formation of CO2 anion radicals. Solvents with low polarity have little solvation effect on the radical species and are conducive for the diffusion of the CO2 anion radicals onto the photocatalytic surface which are further reduced to carbon monoxide. In contrast, in strongly polar solvents, CO2 radicals can hardly access the Ti surface active sites for further reduction to CO.
The photocatalytic epoxidation of propene was studied by Murata et al.22 from which a degradation mechanism was proposed (Scheme 6) with reference to ESR studies and stoichiometric reaction tests. Over the isolated tetrahedral Ti species, a [Ti3+–OL−]* radical pair is formed by UV irradiation while maintaining the geometry of the original Ti4+ species.
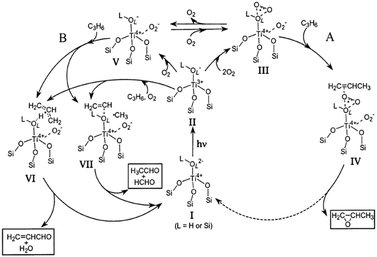 | ||
| Scheme 6 Proposed mechanism for the formation of propylene oxide (PO), ethanal, and acrolein over isolated tetrahedral Ti species. Route A is the cycle for PO production through species I–II–III–IV–I. Route B is the cycle for acrolein or ethanal production through species I–II–(V)–VI or VII–I. L denotes either H or Si. Reprinted with permission from ref. 22. Copyright 2003, American Chemical Society. | ||
The Ti3+ moiety reacts with O2 to form O2−, which could not activate propene by itself. The OL− moiety, a hole centre on lattice oxygen, reacts with O2 to form O3−, and the O3− reacts with propene to yield propylene oxide (PO). It is first suggested that the O3− is the electrophilic oxygen species effective for the epoxidation of propene. However, the OL− moiety reacts with propene to form allyl, methyl, and alkyl radical species that produce acrolein or ethanal through H abstraction or C–C bond fission by photo-irradiation at 77 K. The lower selectivity of the aggregated titanium oxide species was attributed to the lower stability of the O3−. The overall study reveals that the isolated tetrahedral Ti species on the TiO2–SiO2 samples are active for the photo-epoxidation of propene, while the aggregated titanium oxide species are active mainly for the side reactions.
Obuchi et al. investigated the photocatalytic decomposition of acetaldehyde over TiO2 loaded porous silica under UV irradiation.71 The specific surface area increased significantly with incorporation of TiO2 into the SiO2 matrix, thus enabling the accommodation of more substrate molecules onto surface active sites on the TiO2.
The utilization of Pt as co-catalyst created a Schottky barrier at the Pt–TiO2 interface and enabled the metal to function as an electron-sink. Therefore, an enhanced photocatalytic activity was observed in the Pt/TiO2–SiO2 material. Other metals such as Cu were incorporated into the mixed oxide matrix in optimum amounts, and high activities were realized for the photoreduction of carbon dioxide under UV light irradiation.167 Meanwhile, recycling studies were paramount for evaluating the applicability of the Pt–TiO2–SiO2 prepared by Obuchi et al. Since harmful intermediates were generated and firmly captured by TiO2–SiO2 during the photocatalytic decomposition of acetaldehyde, the activity of the photocatalyst is gradually lost with more coverage of the intermediates on the surface. However, after the heat treatment at an appropriate temperature, the photocatalytic activity of the spent catalyst was recovered.
Tanaka et al. reported the photocatalytic oxidation of propane using TiO2–SiO2 prepared from different titanium precursors.72 TiO2 formed from tetrakis(isopropylato) titanium(IV) that was chemically mixed with the SiO2 support displayed higher photocatalytic activity than that of bulk TiO2. In addition, the supported material showed a significant degree of reactive selectivity during photo-oxidation, producing acetone as the predominant product. Nevertheless, the other two catalysts prepared using bis(isopropylato)-bis(pivaroylmethanato) titanium(IV) and bis(acetylacetonato)oxo-titanium(IV) as Ti sources indicated that an indispensable mixture of acetaldehyde and acetone was present as the product.
Xie et al. assessed the photocatalytic activity of heptane and toluene oxidation over TiO2–SiO2.51,163 The material possesses higher photooxidation efficiencies than bulk TiO2. The enhancement was strongly linked to the chemical properties of the TiO2 introduced into the SiO2 host. Firstly, the TiO2 species in the SiO2 matrix were of anatase phase, which has been well established as a better photoactive phase than rutile. Also, the high crystallinity lessens surface defects sites which normally act as trapping sites, aiding the photogenerated electron–hole pair recombination on the TiO2 surface. Moreover, the advent of the quantum size effect with the entrapment of TiO2 into the silica matrix as well as the surface acidity contributes to an overall increase of the photocatalytic efficiency as discussed earlier in this review.
Cao et al. harnessed TiO2 modified SiO2 aerogels for the photocatalytic oxidation of trichloroethylene (TCE).168 Various preparation methods such as TiO2 entrapment in silica gel, post-impregnation of TiO2 onto silica gel, two-step co-condensation synthesis, and evaporation induced self-assembly (EISA) method have been adopted. The results in this study indicated that different preparation methods lead to diverse degradation efficiencies. The materials prepared by the EISA approach exhibited the highest activity for TCE photo-oxidation under UV light irradiation. The physicochemical properties of TiO2 are crucial since the photoactive sites in the mixed oxide reside on the semiconductor. The TiO2 species in the most active material possess high crystallinity, resulting in the retardation of electron–hole pair recombination. Also, the high surface area is conducive for the accommodation of more active sites and this feature was optimized at a Ti/Si ratio of 0.75. It is noteworthy that the highest photooxidation activity can partly be attributed to the periodic arrangement of the mesoporous silica matrix.
The effect of the photocatalytic removal of nitrogen oxide pollutants from the air over TiO2–SiO2 binary metal oxides was evaluated by Kominami et al.88 The results suggested that the crystallite size of TiO2 and surface area of the photocatalyst were impetus for improved photoactivity. A control experiment was carried out to illustrate the significance of surface area on the photoactive TiO2. Initially, the authors determined that TiO2 prepared by the thermal decomposition method with larger surface area of 149 m2 g−1 presented considerable enhancement in the photocatalytic removal of nitrogen oxides than the TiO2 P25 with relatively small surface area (50 m2 g−1). This observation revealed that the amount of TiO2 is critical since the TiO2 is the main photoactive portion of the mixed oxides. The composite with 74% weight content of TiO2 was the most efficient for this purpose. A decrease in TiO2 content resulted in the sharp reduction in the efficiency probably due to considerable depreciation of active sites. However, further increase of TiO2 caused a loss in activity, clearly indicating the indispensable role of the SiO2 support. The SiO2 host encapsulated the TiO2 within its matrix and confined the crystallite size of TiO2 to a few nanometres ideal for photocatalytic enhancement. In addition, the surface area of the composite photocatalyst enlarges dramatically with the presence of SiO2, which is also conducive for strong adsorption of the nitrogen oxide gas molecules. The reactants can be captured and concentrated around the photoactive sites on the TiO2 surface, and subsequently improvements in photocatalytic activity can be obtained.
TiO2–SiO2 mixed oxides containing different sizes of TiO2 nanoparticles were compared for the photocatalytic efficiency over the decomposition of toluene.75 The results distinctly illustrated that the mixed metal oxides with amorphous TiO2 in the silica support exhibited poor activity under UV light irradiation. This again highlights the influence of crystallinity on resultant activity. The authors established a TiO2 particle size of 6.2 nm as optimum for achieving the highest activity for the decomposition of toluene.
In summary, it is widely acknowledged that the photocatalytic degradation or reduction activity of the gaseous organics over TiO2–SiO2 mixed oxides closely relates to their physico-chemical properties. First, the supporting SiO2 offers mechanical stability and this increases the robustness of the TiO2 incorporated binary metal oxides. With the shielding by the silica matrix, relatively high temperatures can be applied to calcine the TiO2–SiO2 composite in order to achieve highly crystalline anatase phase of TiO2 within the silica matrix.
Moreover, due to the rise of quantum size effect, the silica support confines the TiO2 particles in the nanosize range in its matrix. Relatively small sized crystalline TiO2 nanoparticles exhibit better performance in photocatalytic reactions. Shrinkage of the particle size shortens the pathway through which the photo-excited electrons and holes need to migrate to the TiO2 surface active sites. This enhances the photocatalytic efficiency by suppressing the photogenerated charge recombination (volume charge carrier recombination). As the particle size is decreased further, the rate of surface charge carrier recombination increases and thus there is a critical size at which recombination in the volume and surface is minimized and the resulting photocatalytic activity is the highest. In this study, the optimum crystallite size of TiO2 was determined to be in the range of 6 to 9 nm.
Besides, an increase in the silica content in the mixed oxide may convert the geometry of Ti ions from octahedral to tetrahedral as confirmed by UV-vis spectroscopy. Ti ions with the latter chemical environment have been shown to exhibit higher activity in several photocatalytic reactions. In addition, by embedding the TiO2 into SiO2, the specific surface area of the photoactive species can be significantly improved. The extended surface area subsequently harnesses increased contact between the TiO2 and reactant gas molecules. Also, SiO2 acts as a good adsorbent conducive for the capture of more gaseous reactants around the TiO2 active sites. It is worth mentioning that, in addition to the markedly improved physical properties of the TiO2–SiO2 binary metal oxides, the chemical properties also play a profound role in the catalytic performance. The robustly resident TiO2 existing in the SiO2 lattice structure can be confirmed by the emergence of the “Ti–O–Si” hetero-linkage species conformed by spectroscopic studies. The stable Ti–O–Si linkage can further cause charge imbalance around the Ti ions due to the different coordination numbers of the original Ti and Si metal centre. Consequently, the negatively imbalanced charges over the Ti–O species are compensated by the protons and the surface OH− groups are generated. Hence, the rise of the surface acidity on the Ti terminal atoms dramatically enhances the photocatalytic efficiency of TiO2–SiO2.
4.3. Photosplitting of water
Energy is the mainstay and basic guarantee for sustainable development of modern civilization. Over the past few centuries, power supplied by the sun served as the primary source of energy. The rise of the industrial revolution marked a transition towards the dependence on crude oil as the main power source. Meanwhile, the expending of these fossil fuels cannot be excluded from the global environmental concerns, and the debate about its exhaustibility is on-going. Indeed, combustion of fossil fuels is one of the main contributors of greenhouse gas emissions. Recent research has targeted these activities to the accumulation of the carbon dioxide in the atmosphere and the exacerbation of the greenhouse effect and global warming. Therefore, it is indispensable to establish green systems, in which the energy source is recyclable and ultimately clean.Hydrogen, with its non-polluting nature, has been pointed out as a suitable candidate for the blue-print of the proposed ideal energy system.169 However, current industrial production of hydrogen by steam reforming and thermal cracking of natural gas cannot suffice the growing energy demand and alleviate the adverse environmental impact.170
Since the ground breaking work published by Fujishima and Honda in 1972,171 a revolutionary idea of generating hydrogen by splitting water via the unlimited solar energy source has attracted massive research interest.120,172–176 Photocatalytic cleavage of water to produce hydrogen and oxygen is also acknowledged as artificial photosynthesis, which occurs in the green plants, converting the solar energy into sugar and oxygen. For the semiconductor photocatalysts that harness “solar to hydrogen” energy conversion, their photocatalytic activity and the quantum yield relies mostly on their intrinsic band gap structure. The bottom of the conduction band within the photocatalyst should be located at more negative values than the hydrogen reduction potential and the top of the valence band of the semiconductor needs to be at more positive values than the water oxidation potentials (Fig. 16).
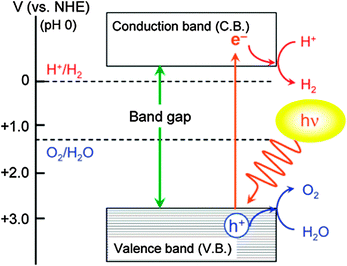 | ||
| Fig. 16 Basic principle of overall water splitting on a heterogeneous photocatalyst. Reprinted with permission from ref. 177. Copyright 2007, American Chemical Society. | ||
Nguyen and Yang determined that the solar hydrogen evolution rate from water decomposition can be boosted from trace amounts with P25 to 25.5 μmol after 24 h UV light irradiation using TiO2 mediated SiO2 prepared by sol–gel processes.35 The results obtained from employing the mixed oxides are attributed to the textural and electronic properties of the materials. Confinement of TiO2 particles to the SiO2 matrix prevents their growth into bulk species. These effects allow the shift in band structure and the flat band potential can reach as high as −1.045 eV, and as such the titania–silica possesses greater reduction potential than bare TiO2. Furthermore, the dispersion of the TiO2 components into the SiO2 host increases the surface area of the mixed oxides, which is also conducive for the photocatalytic processes.
The effect of chelating agents on the textural property of TiO2–SiO2 prepared by a sol–gel process was investigated.178 The material prepared using acetic acid exhibited the highest photocatalytic activity with 45.36 μmol of hydrogen evolved after 24 h of UV light irradiation. TiO2–SiO2 prepared without introducing any chelating agents displays much less photocatalytic hydrogen production in the same time interval (25.5 μmol).35 The enhancement is attributed to the participation of acetic acid in the sol–gel process in which the condensation proceeds via the pathway of forming an edge-shared octahedral polymer that results in a larger surface area mixed oxide material. The higher specific surface area is beneficial to the dispersion of TiO2 nanoparticles within the SiO2 host. This accommodates a larger number of surface active sites that are responsible for the superior photocatalytic activity.
Nguyen and Yang have demonstrated the concurrent evolution of hydrogen and oxygen by using TiO2–SiO2 impregnated with RuS2 semiconductors via a sol–gel approach. The newly formed composite photocatalyst showed appreciable hydrogen evolution rate (25.9 μmol h−1), while binary metal oxides produced a mere 1.2 μmol h−1. The impressive increase of the solar hydrogen production may be credited to the effective charge transfer mechanism facilitated by a narrower band gap RuS2. The photogenerated electrons can transfer from the conduction band of TiO2–SiO2 to that of RuS2. Since the reduction potential of the conduction band in RuS2 is more negative than the proton reduction potential, electrons can further reduce the protons to evolve H2 gas. Oxygen generation has also been observed in this work, however, due to the existence of the reductive RuS2, the stoichiometric ratio of hydrogen and oxygen was not obtained.179
Owing to the limited research attention, reaction mechanisms over TiO2 mediated SiO2 photocatalysts still remain unclear and hence worth receiving more research interest. For instance, the local environment of the Ti species within a SiO2 matrix, the optimum dopant ratio between TiO2 and SiO2, the utilization of the co-catalyst and sacrificial reagent are all areas that can be probed for renewable energy practitioners. Titania–silica photocatalyst undeniably presents fascinating physico-chemical properties as well as their great potential for the photocatalytic hydrogen evolution from the decomposition of water. Indeed, titania incorporated periodic silica host materials have exhibited impressive performance on the photocatalytic water splitting.92,129,180–183 It is also notable that the strong surface acidity generated by the formation of hydroxyl groups anchored on the TiO2–SiO2 surface are accessible to photogenerated holes. Consequently, the effective separation of electron–hole pairs immensely improves the photocatalytic activity.
5. Emerging opportunities
5.1. Advanced material synthesis
In recent years, more intricate synthetic approaches have been adopted to improve the development of core–shell microspheres consisting of titania and silica for catalysis applications. Established methods such as layer by layer self-assembly are tedious and time consuming as they require repeated adsorption of polyelectrolyte layers. Batch processes require mechanical stirring that causes fluctuations in concentration and temperature, and results in uneven coating and irreproducibility. The great need for a single step technique for the synthesis of such hybrid materials has resulted in the development of a single emulsion technique formed by liquid–liquid two-phase flow, through which solidification of the outermost surface to form core–shell microparticles is preceded by the generation of uniform droplets. This method presents simplicity in use compared to the double-emulsion method. Lan et al. utilized a novel and simple microfluidic method for fabricating monodisperse titania–silica core–shell microspheres in one step. The microfluidic approach provides versatility and simplicity to attain good reproducibility, uniformity of coating, and better control of sphere size and surface composition. Uniform droplets of silica sol were dispersed into an oleic acid (chemical modifier) phase of tetrabutyl titanate dissolved in liquid paraffin, via a coaxial microfluidic device. The presence of water droplets facilitated hydrolysis of the Ti alkoxide at the water–oil interface on the surface of the silica sol droplets moving in the channel. Titanium hydroxide gel formed around the droplets and resulted in titania–silica core–shell microspheres after calcination. The investigators determined that the dispersity and size of the microspheres could be manipulated by changing the microfluidic flow parameters. The uniform loading of titania was adjusted from almost 1.0 to 98.0 mol% by changing the oleic acid concentration in the continuous phase and the residence time. In addition, the microsphere average diameter was improved from 100 to 300 μm by changing the continuous phase flow rate. Monodisperse microspheres of uniform crystalline titania coatings were confirmed by X-ray diffraction, scanning electron microscopy (SEM), and energy dispersive spectrometry characterization.185
Demirörs et al.186 reported a fast and novel method of fabricating eccentric core–shell particles by taking advantage of porous titania particles that, upon sintering, shrink much more than the silica shell enclosing them as depicted in Scheme 7.
 | ||
| Scheme 7 Schematic representation of the procedure for fabrication of comp-TiO2@SiO2, core–shell TiO2@SiO2, and eccentric-TiO2@SiO2 particles. Reprinted with permission from ref. 186. Copyright 2009, American Chemical Society. | ||
This approach produces eccentric particles directly from titania microspheres coated with silica, eliminating the need for costly and time-consuming sacrificial shell-removing steps associated with other procedures. The access of silica to the pores in titania may be controlled, resulting in various types of particles through which particles properties like density and refractive index are tuned for applications such as photonic crystals and photocatalysis. The TEM images in Fig. 17 illustrate the morphology of the particles obtained from such a delicate synthetic process.
 | ||
| Fig. 17 TEM images of the titania particles: (a) titania particles as synthesized and (b) eccentric titania–silica core–shell particles after calcination. Reprinted with permission from ref. 186. Copyright 2009, American Chemical Society. | ||
Polycarbonate (PC) has been the substrate of choice owing to its higher transparency to visible light, thus exhibiting better light transmission characteristics than many types of silica glass. Fateh et al.188 have recently pre-coated PC substrates with SiO2 followed by the TiO2 film formation to enhance the acidity of the films as depicted in Fig. 18.
 | ||
| Fig. 18 Illustration of photocatalytic self-cleaning using TiO2–SiO2 nanoparticles on polycarbonate substrate. Reprinted with permission from ref. 188. Copyright 2011, American Chemical Society. | ||
Subsequently, water molecules that lead to the formation of hydroxyl groups are adsorbed preferentially to the pollutant species. It is postulated that the increase in acidity as well as hydrophilicity of the coatings (vide infra) are critical components for improved photocatalytic performance and in self-cleaning technologies. UV(A) exposure resulted in a decrease of the water-contact angle of the coated slides to zero within a few hours after which the surfaces were found to be superhydrophilic. This is proposed to lead to a considerable increase in the photonic efficiency due to the high amount of hydroxyl groups on the surface.
Permpoon et al. postulated that homogeneous mixing results in interfacial Ti–O–Si hetero-linkages that promote charge imbalances due to the formation of TiO62− or SiO44/3+ units. These deprotonated units can favour adsorption of H3O+ and/or −OH groups that induce enhanced molecular or dissociative water adsorption. Super-hydrophilicity arises from surface oxygen vacancies (O2*) that are a result of redox reactions of TiO2 induced by photogenerated charge carriers. The −OH groups then saturate the O2* vacancies, resulting in a super-hydrophilic surface, showing a water contact angle of zero. Several authors have reported the preparation of TiO2–SiO2 thin films with sustainable photo-induced super-hydrophilicity even in the absence of UV irradiation.189–191
In a different method, Takeuchi et al. described the preparation of Ti–Si binary oxide thin films using TEOS and Ti(OPri)4 alkoxide precursors that were determined to achieve highly dispersed 4-fold coordinated Ti-oxide species within the SiO2 matrices.192 An ionized cluster beam (ICB) was applied using multi-ion sources to deposit the TiO2 species. In respect to the known fast hydrolysis, the reactive Ti-alkoxide precursor was coordinated by the hydrophilic head group of a surfactant molecule. This ligand served as a structure directing agent and lowered the reactivity of the alkoxide.193
Surface modification has paved the way for inducing functionality on these binary thin film materials. More recently, mesoporous materials with anisotropic refractive index and birefringence have been applied in optical devices such as liquid crystal displays, light modulators, tuneable lasers, and phase plates. The synthesis of such materials has been of major success using organic uniaxially stretched polycarbonate; however, such substrates are unstable and limited to mild conditions. A simple procedure to prepare stable inorganic based transparent binary thin films of high refractive index, Δn, is in demand. Miyata et al. reported the preparation of a mesoporous titania–silica composite film with a uniaxially oriented (highly cylindrical) porous structure by the sol–gel method using a substrate with structural anisotropy. The resultant film shows a remarkable birefringence with a Δn value over 0.06 at a wavelength of 550 nm, which is due to the large refractive index of titania that spans the pore walls and forms the highly anisotropic mesoporous structure. This value was unachievable with the conventional mesoporous silica films with uniaxially aligned mesoporous structure even though the alignment of the pores in the films was perfected. The in-plane orientation of the meso-channels was controlled by using a rubbing-treated polyimide as an alignment-controlling layer that provides a narrow alignment distribution in the plane of the film regardless of the fast condensation rate of titania precursors. The structure was retained after template removal by exposing the as-deposited film to a vapour treatment of tetramethoxysilane (TMOS) prior to the calcination process. The obtained film exhibited good optical quality with high transparency. This method utilized by Miyata et al. could be applied for the preparation of aligned mesoporous films with various compositions, which promises a myriad of practical applications of these anisotropic mesoporous films in the field of optics.194
5.2. Other applications
Nanomaterials are of great interest for application in sensors and other electronic devices because of their size-dependent optical, electrical, and magnetic properties. The non-toxicity and susceptibility to surface modification allows for their application in self-cleaning, biomaterials and bio-related therapies. The inertness and relatively low toxicity of titania–silica based mixed oxide materials can contribute to meeting these opportunities. | ||
| Fig. 19 Reprinted with permission from ref. 200. Copyright 2011, American Chemical Society. | ||
The resultant titania–silica mesoporous coating ensures enhanced and sustainable surface cleaning properties.200 Most recently, Jafry and co-workers developed a procedure though which physical mixing of fumed silica and P25 resulted in a material that inactivated bacteriophage MS2 virus under UV irradiation.201 The highlight of this procedure is that in situ preparation is not necessary to achieve degradation efficiencies three times higher than P25 using an optimum silica loading of 2.5 wt%. The enhanced photocatalysis was attributed to greater adsorptive capacity of MS2 to the catalyst and/or band-bending at the mixed oxide interface that may have minimized the charge-carrier recombination. These results demonstrate viability of titania–silica mixed oxides for use as disinfectants to curb pathogen transmission.
Recurrent seizures and crippling side effects in epilepsy patients present challenges in the administration of conventional pharmacological therapies using anti-convulsant drugs. The inefficacy of the current treatment may be attributed to failure to target the delivery of these anticonvulsive agents towards the impaired site in the brain and release the drug effectively for action. Lopez et al. developed an implantable titania–silica xerogel functionalized with sodium phenytoine (DPH) or valproic acid (VPA) as the anticonvulsive drug. These polymers of high reproducible release kinetics are biocompatible with brain tissue and show potential in the provision of sustained drug release that inhibits the spread of seizure activity in the brain cortex.202 The novel sol–gel functionalized TiO2–SiO2 were surgically implanted into the basolateral amygdale of a rat and the biocompatibility studies revealed phenytoine was slowly released in vitro from the binary TiO2–SiO2 xerogel over a period of 500 h. Most importantly, the structure of the brain parenchyma and reservoir interface was preserved, and no evidence of inflammation or gross tissue reaction was detected. These studies provide promise for sol–gel derived titania–silica systems to play a significant role in the development of new generation biomaterials for controlled drug release.
Most recently, in-depth studies on the surface interactions on nanoparticles have been carried out to provide knowledge of the factors affecting the efficacy of detecting and responding to low concentrations of biomarkers. Biomolecules have found application as recognition elements owing to their ability to differentiate subtleties in chemical structure. Peptides that bind inorganic surfaces and template the formation of nanometre-sized inorganic particles are of great interest for the self- or directed assembly in sensors and diagnostic applications. Mirau et al. studied the relationship between the primary sequence of peptides and their ability to recognize titania and silica surfaces. High resolution Nuclear Magnetic Resonance (NMR) refinement and saturation transfer difference (STD) studies confirmed that the conformation and orientation of related peptides are similar between the silica and titania surfaces, suggesting a common binding motif of a compact “C”-shaped structure. Such information determined by peptide exchange kinetics and magnetization transfer from the nanoparticle surface to the nearby peptide protons is crucial for research and design of effective biomaterial interactions for drug delivery using titania–silica based systems.203 The surface properties of silica will certainly play a role in the types of biomolecules that bind or interact with the nanoparticles.
6. Structure–activity relationships
Titania–silica aerogels demonstrated high activity and selectivity in the epoxidation of a variety of bulky cyclic alkenes, alkenones, and alkenols48,107,204 requiring the use of alkyl hydroperoxides as oxidant such as cumene hydroperoxide because the employment of H2O2 deactivates the hydrophilicity of the materials due to either inhibition of the catalyst by water or to leaching of Ti from the solids.18 Kochkar et al. also revealed that the hydrophobic gel is far more selective to the epoxidation reactions when organic hydroperoxide (tert-butyl hydroperoxide TBHP in tert-butanol solvent) is used as the oxidizing agent attaining yields of cyclohexene oxide >95%.Optimizing the solvent, a selectivity of about 80% for the attack of the double bond on cyclohexene was achieved in the presence of H2O2. The hydration of the resulting hydroxide is fast and can be slightly decreased but not suppressed, even on hydrophobic surfaces. This is consistent with the idea that although the site active for epoxidation is a strong Lewis site, the selective inhibition of the secondary reaction is then impractical.95
The enhanced performance of organically modified titania–silica aerogels has been attributed to the interaction of the amino and acetoxy alkyl groups with the Lewis acidic Ti active sites and the Brønsted acidic Si–OH functions. In the former case, the amino and acetoxy moieties act as electron donor ligand of Ti, which can modify the acidity and thus the activity of the Ti–peroxo complex. On the other hand, the H-bonding interaction with the surface silanol groups can block these acidic sites and prevent undesired acid catalyzed side reactions of the reactant and/or product epoxide.95,99 Dusi et al. proposed that these basic amines interact mainly with surface silanol groups at low concentrations, and deactivate these Brønsted sites, thus limiting the secondary reaction of hydration of the epoxide. However at higher concentrations, the interaction between amines and Ti atoms is prominent, blocking the Lewis sites, and resulting in lower epoxidation activity.107
Kim and co-workers94 demonstrated that the improved textural properties of titania–silica did not always translate to improved photocatalytic activity of materials prepared using chloride precursors. The adsorption capacity of titania–silica aerogels was higher than that of pure titania aerogel. However, the methanol conversion for titania aerogels was about 98%, approximately equal to that of titania–silica aerogels (TiO2:SiO2 = 1:1) at 96%. In this scenario, the silica phase in the mixed oxide seemed to play a significant role in improving adsorption of the reactant but not necessarily having a similar impact on the degradation as often professed.
Amongst the family of transition metal oxide semiconductors examined, TiO2 has been widely studied due to its suitable band gap position and attractive chemical and physical properties such as relatively high activity and stability under UV light irradiation. Nevertheless, the main drawback that constrains its activity in photocatalytic processes is the fast recombination of photogenerated electrons and holes. This significantly decreases the quantum efficiency of the photocatalytic reaction and the photocatalytic hydrogen generation rate over the pure TiO2 is fairly low at around 0.013 μmol h−1 gTiO2−1.180
Considerable efforts have been devoted to overcome this drawback and increase the quantum efficiency of TiO2 photocatalyst.205–210 Doping TiO2 into another metal oxide as supporting material to form binary metal oxides photocatalyst has been extensively studied to meet this purpose.36,211 The major contributor to photocatalytic efficiency in titania–silica composite systems is indeterminate owing to the inert and amorphous silica phase. This section addresses some of the factors that need to be considered when evaluating the performance of these catalysts.
It is proposed that the photocatalytic activity of the binary metal oxides photocatalyst (TiO2–SiO2) will enhance the efficiencies owing to the increased surface acidity in these materials.212 The surface of TiO2–SiO2 is prone to attract and anchor more hydroxyl groups that eventually act as hole-scavengers and readily oxidize the adsorbed water molecules. The formation of the surface hole-scavenger active sites can effectively enhance the charge separation and minimize the recombination of photogenerated electrons and holes.
Generally, titania–silica composites possess high surface acidity due to the charge imbalance created by the homogeneous chemical mixing of the two components. Octahedrally coordinated TiO2 dopant species have been incorporated into the host tetrahedral SiO2 matrix. The different coordination number of these two metal oxides gives rise to the negative charge imbalance in the metal oxide mixtures.94 These negatively uneven charges on the surface oxygen terminals can further be compensated by surface hydroxyl groups. Therefore, these hydroxyl groups play the role of a “hole-sink”.
It has been documented extensively that the confinement of nano-sized TiO2 particles in other metal oxide matrices will cause a shift of the band edges, and result in a change in the band gap. The conduction band of the quantum confined TiO2 particles will shift to more negative values of the hydrogen reduction potential while the valence band moves to more positive values relative to the water oxidation potential. It is well known that the band gap energy of bulk TiO2 (anatase) is ∼3.3 eV, however, the band gap of TiO2–SiO2 could be as high as 4.1 eV.124 This enlargement in the band gap of the nano-sized TiO2 particles enables the electrons and holes to possess stronger reduction and oxidation abilities. With the enhancement of the redox capabilities of the electrons and holes, photocatalytic activity and the quantum efficiency of the TiO2–SiO2 photocatalysts was found to increase dramatically. In addition, smaller particle size provides a shorter pathway for the photogenerated electrons and holes to migrate to the surface active sites.172 In other words, the electrons and holes can be more accessible for the participation in photocatalytic reactions. Alemany et al.140 demonstrated clearly that the small particle size of titania provided a large surface-to-volume ratio which increases the chance of electrons and holes to reach the surface and interact with substrate molecules such as phenol and this leads to improved photocatalytic activity. Conversely, although it appeared as though greater crystallite size resulted in higher photoactivity for the decomposition of TCE, Jung et al. attributed the result to the production of anatase phase of high crystallinity that effectively reduced the bulk defects which serve as the recombination centers.29
It has been determined that embedding TiO2 species into the SiO2 host may result in an increase in the specific surface area of the photocatalyst.22,113,213,214 The surface area of pure bulk TiO2 can reach up to 100 m2 g−1, nonetheless, the TiO2–SiO2 binary metal oxides possess surface areas above 300 m2 g−1 due to the presence of the SiO2 matrix which results in increased porosities and good dispersion of the titania species. Some studies have acknowledged surface area to be an important factor in the photocatalytic reaction. It is believed that more surface acidified active sites are accommodated if more surface area is accessible to reactant molecules. The dispersion of the photoactive species is also critical to the overall photocatalytic quantum efficiency as well and has been proven,215 however, quantitative methods for elucidating and evaluating the amount of acid sites and extent of dispersion of active sites are still lacking in the literature. This is an area that can be explored to better the understanding of structural features of such composite materials.
The advantages of TiO2–SiO2 mixed oxides as photocatalysts for the photocatalytic splitting of water, epoxidation and organic substrate degradation include:
1. the strong surface acidity that is responsible for the redox active sites on the surface and is conducive for suppressing the charge carrier recombination,
2. the band structure shift arising from the quantum size effect to harvest the photogenerated electrons and holes with higher redox potentials,
3. the enlarged specific surface area and high dispersion of the titania that enable the catalyst to accommodate a greater amount of active sites on the surface that are more accessible to the reactant molecules, and
4. enhanced crystallinity that lessens surface defects sites which normally act as trapping sites and aid the photogenerated electron–hole pair recombination on the TiO2 surface.
Noble metals (Pt, Au, Ag) have been frequently adopted as co-catalysts in the photocatalytic water splitting and organic substrate degradation. The addition of the precious metal is designed to limit the recombination of the electrons and holes (Fig. 20). Platinum possesses higher work function and different Fermi levels from the n-type TiO2 semiconductor. Hence, electrons have the tendency to migrate from TiO2 towards Pt until the Fermi levels in both species reach equilibrium. Eventually, a Schottky barrier is formed as a result of abundant electrons gathering on the surface of Pt and the positive charges reside on TiO2. The Schottky barrier acts as electron-sink, which can trap the photogenerated electrons and as such minimize the electron–hole recombination. Domen et al. examined the photocatalytic activity over Pt loaded TiO2–SiO2 binary metal oxides and a hydrogen evolution rate of 132 μmol h−1 was achieved with apparent quantum yield of 35% at an optimal loading of Pt.216
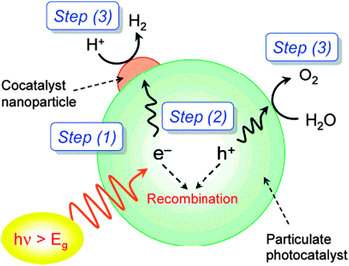 | ||
| Fig. 20 Processes involved in photocatalytic overall water splitting on a heterogeneous photocatalyst. Reprinted with permission from ref. 177. Copyright 2007, American Chemical Society. | ||
We have recently demonstrated that the dispersion of Pt of small crystallite sizes is beneficial for achieving enhanced photocatalytic efficiencies for the degradation of phenol in solar simulated conditions.217 The presence of an inert UV transparent silica phase allows for higher catalyst concentration without loss of effective irradiation. Silica also provides economic benefits as well as improved mechanical properties that harness application of such materials in fluidized-bed reactors and other advanced technologies.
7. Conclusions
Titania–silica mixed oxides have been used extensively in catalytic oxidation reactions, however, they are increasingly used in photocatalytic reactions for environmental remediation. Surprisingly, only limited studies have been reported for photocatalytic splitting of water. In general the activity of TiO2–SiO2 photocatalysts is higher in comparison with bare TiO2 and this has been attributed to the high dispersion of titania crystallites that can harness the incident radiation effectively, the enhanced surface acidity, and quantum size effects that confine the crystallite (particle) size of titania and shift the conduction and valence band edges to more negative and positive values respectively. A big drawback of the TiO2–SiO2 photocatalysts is that the absorption of titania is shifted further into the UV region, necessitating the use of higher energy photons. One method to alleviate this problem is to incorporate noble metals such as Pt, as demonstrated by us recently.217Acknowledgements
This work was supported by NSF-CHE-0722632, NSF-EPS-0903804, DOE-DE-EE0000270 and State of SD.Notes and references
- P. V. Kamat, J. Phys. Chem. C, 2007, 111, 2834–2860 CAS.
- F. Zaera, J. Phys. Chem. Lett., 2010, 1, 621–627 CrossRef CAS.
- A. H. K. Fujishima, Nature, 1972, 238, 37–38 CrossRef CAS.
- C. McCullagh, N. Skillen, M. Adams and P. K. J. Robertson, J. Chem. Technol. Biotechnol., 2011, 86, 1002–1017 CrossRef CAS.
- J. A. Byrne, P. A. Fernandez-Ibanez, P. S. M. Dunlop, D. M. A. Alrousan and J. W. J. Hamilton, Int. J. Photoenergy, 2011 Search PubMed , Article ID 798051.
- A. E. Mohamed and S. Rohani, Energy Environ. Sci., 2011, 4, 1065–1086 CAS.
- H. Tada, M. Fujishima and H. Kobayashi, Chem. Soc. Rev., 2011, 40, 4232–4243 RSC.
- K. T. Ranjit, H. Cohen, I. Willner, S. Bossmann and A. M. Braun, J. Mater. Sci., 1999, 34, 5273–5280 CrossRef CAS.
- K. T. Ranjit, E. Joselevich and I. Willner, J. Photochem. Photobiol., A, 1996, 99, 185–189 CrossRef CAS.
- K. T. Ranjit, T. K. Varadarajan and B. Viswanathan, J. Photochem. Photobiol., A, 1996, 96, 181–185 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 73–78 CrossRef CAS.
- K. T. Ranjit and B. Viswanathan, J. Photochem. Photobiol., A, 1997, 108, 79–84 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. Bossmann and A. Braun, J. Phys. Chem. B, 1998, 102, 9397–9403 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. Bossmann and A. Braun, Res. Chem. Intermed., 1999, 25, 733–756 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. H. Bossmann and A. M. Braun, J. Catal., 2001, 204, 305–313 CrossRef CAS.
- K. T. Ranjit, I. Willner, S. H. Bossmann and A. M. Braun, Environ. Sci. Technol., 2001, 35, 1544–1549 CrossRef CAS.
- S. G. Kumar and L. G. Devi, J. Phys. Chem. A, 2011, 115, 13211–13241 CrossRef CAS.
- D. C. M. Dutoit, M. Schneider and A. Baiker, J. Catal., 1995, 153, 165–176 CrossRef CAS.
- V. Torma, H. Peterlik, U. Bauer, W. Rupp, N. Hüsing, S. Bernstorff, M. Steinhart, G. Goerigk and U. Schubert, Chem. Mater., 2005, 17, 3146–3153 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- B. E. Yoldas, J. Non-Cryst. Solids, 1980, 38–39, 81–86 CrossRef CAS.
- C. Murata, H. Yoshida, J. Kumagai and T. Hattori, J. Phys. Chem. B, 2003, 107, 4364–4373 CrossRef CAS.
- A. S. Soult, D. F. Carter, H. D. Schreiber, L. J. van de Burgt and A. E. Stiegman, J. Phys. Chem. B, 2002, 106, 9266–9273 CrossRef CAS.
- Y. K. Kim, E. Y. Kim, C. M. Whang, Y. H. Kim and W. I. Lee, J. Sol-Gel Sci. Technol., 2005, 33, 87–91 CrossRef CAS.
- S. Lisinski, J. Krause, D. Schaniel, L. Ratke and T. Woike, Scr. Mater., 2008, 58, 553–555 CrossRef CAS.
- G. Mountjoy, J. S. Rigden, R. Anderson, G. W. Wallidge, R. J. Newport and M. E. Smith, J. Mater. Res., 2000, 15, 1998–2005 CrossRef CAS.
- M. A. Holland, D. M. Pickup, G. Mountjoy, E. S. C. Tsang, G. W. Wallidge, R. J. Newport and M. E. Smith, J. Mater. Chem., 2000, 10, 2495–2501 RSC.
- S. Rajesh Kumar, C. Suresh, A. K. Vasudevan, N. R. Suja, P. Mukundan and K. G. K. Warrier, Mater. Lett., 1999, 38, 161–166 CrossRef CAS.
- K. Y. Jung and S. B. Park, J. Photochem. Photobiol., A, 1999, 127, 117–122 CrossRef CAS.
- B.-J. Liu, T. Torimoto, H. Matsumoto and H. Yoneyama, J. Photochem. Photobiol., A, 1997, 108, 187–192 CrossRef CAS.
- R. J. Davis and Z. Liu, Chem. Mater., 1997, 9, 2311–2324 CrossRef CAS.
- S. Klein and W. F. Maier, Angew. Chem., Int. Ed. Engl., 1996, 35, 2230–2233 CrossRef CAS.
- B. E. Handy, M. Maciejewski, A. Balker and A. Wokaun, J. Mater. Chem., 1992, 2, 833–840 RSC.
- S. Budhi, H. S. Kibombo, D. Zhao, A. Gonshorowski and R. T. Koodali, Mater. Lett., 2011, 65, 2136–2138 CrossRef CAS.
- T.-V. Nguyen and O. B. Yang, Catal. Today, 2003, 87, 69–75 CrossRef CAS.
- S. Imamura, T. Shiomi, S. Ishida, K. Utani and H. Jindai, Ind. Eng. Chem. Res., 1990, 29, 1758–1761 CrossRef CAS.
- T. Fernandez, G. Jose, S. Mathew, P. Rejikumar and N. Unnikrishnan, J. Sol-Gel Sci. Technol., 2007, 41, 163–168 CrossRef.
- H. Yamashita, S. Kawasaki, Y. Ichihashi, M. Harada, M. Takeuchi, M. Anpo, G. Stewart, M. A. Fox, C. Louis and M. Che, J. Phys. Chem. B, 1998, 102, 5870–5875 CrossRef CAS.
- D. A. Ward and E. I. Ko, Ind. Eng. Chem. Res., 1995, 34, 421–433 CrossRef CAS.
- E. Kim, C. Whang, W. Lee and Y. Kim, J. Electroceram., 2006, 17, 899–902 CrossRef CAS.
- S. Vives and C. Meunier, Ceram. Int., 2008, 34, 37–44 CrossRef CAS.
- J. Ren, Z. Li, S. Liu, Y. Xing and K. Xie, Catal. Lett., 2008, 124, 185–194 CrossRef CAS.
- A. Pirson, A. Mohsine, P. Marchot, P. Machaux, O. Cantfort and J. P. Pirard, J. Sol-Gel Sci. Technol., 1995, 4, 179–185 CrossRef CAS.
- G. Mountjoy, D. M. Pickup, G. W. Wallidge, J. M. Cole, R. J. Newport and M. E. Smith, Chem. Phys. Lett., 1999, 304, 150–154 CrossRef CAS.
- D. M. Pickup, G. Mountjoy, G. W. Wallidge, R. Anderson, J. M. Cole, R. J. Newport and M. E. Smith, J. Mater. Chem., 1999, 9, 1299–1305 RSC.
- J. M. Miller and L. J. Lakshmi, J. Phys. Chem. B, 1998, 102, 6465–6470 CrossRef CAS.
- D. C. M. Dutoit, U. Göbel, M. Schneider and A. Baiker, J. Catal., 1996, 164, 433–439 CrossRef CAS.
- R. Hutter, T. Mallat, A. Peterhans and A. Baiker, J. Mol. Catal. A: Chem., 1999, 138, 241–247 CrossRef CAS.
- M. P. Coles, C. G. Lugmair, K. W. Terry and T. D. Tilley, Chem. Mater., 1999, 12, 122–131 CrossRef.
- O. A. Kholdeeva, N. N. Trukhan, M. P. Vanina, V. N. Romannikov, V. N. Parmon, J. Mrowiec-Białoń and A. B. Jarzębski, Catal. Today, 2002, 75, 203–209 CrossRef CAS.
- C. Xie, Z. Xu, Q. Yang, B. Xue, Y. Du and J. Zhang, Mater. Sci. Eng., B, 2004, 112, 34–41 CrossRef.
- N. N. Trukhan, A. A. Panchenko, E. Roduner, M. S. Mel'guno, O. A. Kholdeeva, J. Mrowiec-Białoń and A. B. Jarzebski, Langmuir, 2005, 21, 10545–10554 CrossRef CAS.
- Q. Yang, C. Xie, Z. Xu, Z. Gao, Z. Li, D. Wang and Y. Du, J. Mol. Catal. A: Chem., 2005, 239, 144–150 CrossRef CAS.
- A. N. Murashkevich, A. S. Lavitskaya, O. A. Alisienok and I. M. Zharskii, Inorg. Mater., 2009, 45, 1146–1152 CrossRef CAS.
- J. M. Stokke and D. W. Mazyck, Environ. Sci. Technol., 2008, 42, 3808–3813 CrossRef CAS.
- G. Liu, Y. Liu, G. Yang, S. Li, Y. Zu, W. Zhang and M. Jia, J. Phys. Chem. C, 2009, 113, 9345–9351 CAS.
- M. Toba, F. Mizukami, S.-i. Niwa, T. Sanoa, K. Maedaa, A. Annila and V. Komppa, J. Mol. Catal., 1994, 91, 277–289 CrossRef CAS.
- W. Xu, L. Wei and M. Luo, Key Eng. Mater., 2008, 368–372, 1497–1499 CrossRef CAS.
- E. Pabón, J. Retuert, R. Quijada and A. Zarate, Microporous Mesoporous Mater., 2004, 67, 195–203 CrossRef.
- A. R. Oki, Q. Xu, B. Shpeizer, A. Clearfield, X. Qiu, S. Kirumakki and S. Tichy, Catal. Commun., 2007, 8, 950–956 CrossRef CAS.
- F. Garbassi and L. Balducci, Microporous Mesoporous Mater., 2001, 47, 51–59 CrossRef CAS.
- K. Kosuge and P. S. Singh, J. Phys. Chem. B, 1999, 103, 3563–3569 CrossRef CAS.
- E. Pabón, J. Retuert and R. Quijada, J. Porous Mater., 2007, 14, 151–158 CrossRef.
- B. Cojocaru, V. I. Parvulescu, E. Preda, G. Iepure, V. Somoghi, E. Carbonell, M. Alvaro and H. García, Environ. Sci. Technol., 2008, 42, 4908–4913 CrossRef CAS.
- H. Zhu, Z. Pan, B. Chen, B. Lee, S. M. Mahurin, S. H. Overbury and S. Dai, J. Phys. Chem. B, 2004, 108, 20038–20044 CrossRef CAS.
- V. Zeleňák, V. Hornebecq, S. Mornet, O. Schäf and P. Llewellyn, Chem. Mater., 2006, 18, 3184–3191 CrossRef.
- A. Jaroenworaluck, W. Sunsaneeyametha, N. Kosachan and R. Stevens, Surf. Interface Anal., 2006, 38, 473–477 CrossRef CAS.
- L.-C. Chen and C.-M. Huang, Ind. Eng. Chem. Res., 2004, 43, 6446–6452 CrossRef CAS.
- E. Pitoniak, C.-Y. Wu, D. W. Mazyck, K. W. Powers and W. Sigmund, Environ. Sci. Technol., 2005, 39, 1269–1274 CrossRef CAS.
- M. G. Reichmann and A. T. Bell, Langmuir, 1987, 3, 563–567 CrossRef CAS.
- E. Obuchi, T. Sakamoto, K. Nakano and F. Shiraishi, Chem. Eng. Sci., 1999, 54, 1525–1530 CrossRef CAS.
- T. Tanaka, K. Teramura, T. Yamamoto, S. Takenaka, S. Yoshida and T. Funabiki, J. Photochem. Photobiol., A, 2002, 148, 277–281 CrossRef CAS.
- X. Jiang, T. Wang and Y.-w. Wang, Colloids Surf., A, 2004, 234, 9–15 CrossRef CAS.
- X. Jiang and T. Wang, Environ. Sci. Technol., 2007, 41, 4441–4446 CrossRef CAS.
- T. Wang, X. Jiang and Y. Wu, Ind. Eng. Chem. Res., 2009, 48, 6224–6228 CrossRef CAS.
- X. Jiang and T. Wang, J. Am. Ceram. Soc., 2008, 91, 46–50 CrossRef CAS.
- E. Lotero, D. Vu, C. Nguyen, J. Wagner and G. Larsen, Chem. Mater., 1998, 10, 3756–3764 CrossRef CAS.
- G. Larsen, M. Buechler-Skoda, C. Nguyen, D. Vu and E. Lotero, J. Non-Cryst. Solids, 2001, 279, 161–168 CrossRef CAS.
- G. Larsen, H. S. Silva and R. Venturini de Silva, J. Non-Cryst. Solids, 2000, 271, 1–11 CrossRef CAS.
- R. Mariscal, M. López-Granados, J. L. G. Fierro, J. L. Sotelo, C. Martos and R. Van Grieken, Langmuir, 2000, 16, 9460–9467 CrossRef CAS.
- G. Dagan, S. Sampath and O. Lev, Chem. Mater., 1995, 7, 446–453 CrossRef CAS.
- M. Hirano, K. Ota, M. Inagaki and H. Iwata, J. Ceram. Soc. Jpn., 2004, 112, 143–148 CrossRef CAS.
- Z. Li, B. Hou, Y. Xu, D. Wu and Y. Sun, J. Colloid Interface Sci., 2005, 288, 149–154 CrossRef CAS.
- J. Aguado, R. van Grieken, M.-J. López-Muñoz and J. Marugán, Appl. Catal., A, 2006, 312, 202–212 CrossRef CAS.
- R. G. Avendaño, J. A. d. l. Reyes, J. A. Montoya and T. Viveros, Superficies Vacio, 2006, 19, 1–6 Search PubMed.
- F. Sayilkan, M. Asilturk, S. Sener, S. Erdemoglu, M. Erdemoglu and H. Sayilkan, Turk. J. Chem., 2007, 31, 211–221 CAS.
- C. Kang, L. Jing, T. Guo, H. Cui, J. Zhou and H. Fu, J. Phys. Chem. C, 2008, 113, 1006–1013 Search PubMed.
- H. Kominami, K. Yukishita, T. Kimura, M. Matsubara, K. Hashimoto, Y. Kera and B. Ohtani, Top. Catal., 2008, 47, 155–161 CrossRef CAS.
- H. S. Kibombo, D. Zhao, A. Gonshorowski, S. Budhi, M. D. Koppang and R. T. Koodali, J. Phys. Chem. C, 2011, 115, 6126–6135 CAS.
- Z. Deng, J. Wang, Y. Zhang, Z. Weng, Z. Zhang, B. Zhou, J. Shen and L. Cheng, Nanostruct. Mater., 1999, 11, 1313–1318 CrossRef CAS.
- S. Mintova, N. H. Olson and T. Bein, Angew. Chem., Int. Ed., 1999, 38, 3201–3204 CrossRef CAS.
- A. C. Pierre and G. M. Pajonk, Chem. Rev., 2002, 102, 4243–4266 CrossRef CAS.
- M. Schneider and A. Baiker, Catal. Today, 1997, 35, 339–365 CrossRef CAS.
- W.-I. Kim, D. Suh, T.-J. Park and I.-K. Hong, Top. Catal., 2007, 44, 499–505 CrossRef CAS.
- H. Kochkar and F. Figueras, J. Catal., 1997, 171, 420–430 CrossRef CAS.
- C. A. Müller, M. Maciejewski, T. Mallat and A. Baiker, J. Catal., 1999, 184, 280–293 CrossRef.
- S. Yoda, D. J. Suh and T. Sato, J. Sol-Gel Sci. Technol., 2001, 22, 75–81 CrossRef CAS.
- S. Kim and S. H. Ehrman, Langmuir, 2007, 23, 2497–2504 CrossRef CAS.
- A. Gisler, C. A. Müller, M. Schneider, T. Mallat and A. Baiker, in Studies in Surface Science and Catalysis, ed. A. Corma, F. V. Melo, S. Mendioroz and J. L. G. Fierro, Elsevier, 2000, vol. 130, pp. 1637–1642 Search PubMed.
- I. A. Ibrahim, A. A. Ismail, M. S. Ahmed and A. K. Ismail, Egypt. J. Chem., 2002, 45, 193–203 CAS.
- K. Brodzik, J. Walendziewski, M. Stolarski, L. Van Ginneken, K. Elst and V. Meynen, J. Porous Mater., 2007, 14, 219–226 CrossRef CAS.
- K. Brodzik, J. Walendziewski, M. Stolarski, L. Van Ginneken, K. Elst and V. Meynen, J. Porous Mater., 2008, 15, 541–549 CrossRef CAS.
- D. Sun, Y. Huang, B. Han and G. Yang, Langmuir, 2006, 22, 4793–4798 CrossRef CAS.
- N. Yao, S. Cao and K. L. Yeung, Microporous Mesoporous Mater., 2009, 117, 570–579 CrossRef CAS.
- J. Jammaer, C. Aprile, S. W. Verbruggen, S. Lenaerts, P. P. Pescarmona and J. A. Martens, ChemSusChem, 2011, 4, 1457–1463 CrossRef CAS.
- S. K. Samantaray and K. Parida, Catal. Commun., 2005, 6, 578–581 CrossRef CAS.
- M. Dusi, C. A. Muller, T. Mallat and A. Baiker, Chem. Commun., 1999, 197–198 RSC.
- F. Figueras, H. Kochkar and S. Caldarelli, Microporous Mesoporous Mater., 2000, 39, 249–256 CrossRef CAS.
- C. J. Brodsky and E. I. Ko, J. Non-Cryst. Solids, 1995, 186, 88–95 CrossRef CAS.
- J. N. Hay and H. M. Raval, Chem. Mater., 2001, 13, 3396–3403 CrossRef CAS.
- V. Lafond, P. H. Mutin and A. Vioux, J. Mol. Catal. A: Chem., 2002, 182–183, 81–88 CrossRef CAS.
- V. Lafond, P. H. Mutin and A. Vioux, Chem. Mater., 2004, 16, 5380–5386 CrossRef CAS.
- O. Lorret, V. Lafond, P. H. Mutin and A. Vioux, Chem. Mater., 2006, 18, 4707–4709 CrossRef CAS.
- Y. Shul, H. Kim, S. Haam and H. Han, Res. Chem. Intermed., 2003, 29, 849–859 CrossRef CAS.
- M. Galan-Fereres, L. J. Alemany, R. Mariscal, M. A. Banares, J. A. Anderson and J. L. G. Fierro, Chem. Mater., 1995, 7, 1342–1348 CrossRef CAS.
- Y. Mikushina, A. Shishmakov, V. Matskevich, N. Zhuravlev, O. Koryakova, V. Kharchuk and L. Petrov, Russ. J. Inorg. Chem., 2008, 53, 1557–1560 CrossRef.
- W. Rupp, N. Husing and U. Schubert, J. Mater. Chem., 2002, 12, 2594–2596 RSC.
- S. L. Lee, H. Nur, P. W. Koh, J. M. Ekhsan and S. C. Wei, Int. J. Appl. Phys. Math., 2011, 1, 43–47 Search PubMed.
- D. S. Gopala, R. R. Bhattacharjee, R. Haerr, B. Yeginoglu, O. D. Pavel, B. Cojocaru, V. I. Parvulescu and R. M. Richards, ChemCatChem, 2011, 3, 408–416 CrossRef CAS.
- K. Tanabe, T. Sumiyoshi, K. Shibata, T. Kiyoura and J. Kitagawa, Bull. Chem. Soc. Jpn., 1974, 47, 1064–1066 CrossRef CAS.
- Z. Liu and R. J. Davis, J. Phys. Chem., 1994, 98, 1253–1261 CrossRef CAS.
- Z. F. Liu, J. Tabora and R. J. Davis, J. Catal., 1994, 149, 117–126 CrossRef CAS.
- C. U. Ingemar Odenbrand, J. G. M. Brandin and G. Busca, J. Catal., 1992, 135, 505–517 CrossRef.
- T. Kataoka and J. A. Dumesic, J. Catal., 1988, 112, 66–79 CrossRef CAS.
- A. Y. Stakheev, E. S. Shpiro and J. Apijok, J. Phys. Chem., 1993, 97, 5668–5672 CrossRef CAS.
- M. Anpo, H. Nakaya, S. Kodama, Y. Kubokawa, K. Domen and T. Onishi, J. Phys. Chem., 1986, 90, 1633–1636 CrossRef CAS.
- J. P. Espinós, G. Lassaletta, A. Caballero, A. Fernández, A. R. González-Elipe, A. Stampfl, C. Morant and J. M. Sanz, Langmuir, 1998, 14, 4908–4914 CrossRef.
- L. Soriano, G. G. Fuentes, C. Quirós, J. F. Trigo, J. M. Sanz, P. R. Bressler and A. R. González-Elipe, Langmuir, 2000, 16, 7066–7069 CrossRef CAS.
- F. Farges, G. E. Brown, Jr and J. J. Rehr, Phys. Rev. B: Condens. Matter, 1997, 56, 1809–1819 CrossRef CAS.
- X. Gao, S. R. Bare, J. L. G. Fierro, M. A. Banares and I. E. Wachs, J. Phys. Chem. B, 1998, 102, 5653–5666 CrossRef CAS.
- R. Anderson, G. Mountjoy, M. E. Smith and R. J. Newport, J. Non-Cryst. Solids, 1998, 232–234, 72–79 CrossRef CAS.
- Z. Y. Wu, Y. F. Tao, Z. Lin, L. Liu, X. X. Fan and Y. Wang, J. Phys. Chem. C, 2009, 113, 20335–20348 CAS.
- S. Imamura, S. Ishida, H. Tarumoto, Y. Saito and T. Ito, J. Chem. Soc., Faraday Trans., 1993, 89, 757–762 RSC.
- P. H. Mutin and A. Vioux, Chem. Mater., 2009, 21, 582–596 CrossRef CAS.
- M. Hirano, K. Ota and H. Iwata, Chem. Mater., 2004, 16, 3725–3732 CrossRef CAS.
- C. Anderson and A. J. Bard, J. Phys. Chem. B, 1997, 101, 2611–2616 CrossRef CAS.
- P. J. Dirken, M. E. Smith and H. J. Whitfield, J. Phys. Chem., 1995, 99, 395–401 CrossRef CAS.
- F. Amano, T. Yamaguchi and T. Tanaka, J. Phys. Chem. B, 2005, 110, 281–288 CrossRef.
- H. Kanai, M. Shono, K. Hamada and S. Imamura, J. Mol. Catal. A: Chem., 2001, 172, 25–31 CrossRef CAS.
- L. J. Alemany, M. A. Banares, E. Pardo, F. Martin, M. Galán-Fereres and J. Blasco, Appl. Catal., B, 1997, 13, 289–297 CrossRef CAS.
- D. Chen, M. Sivakumar and A. K. Ray, Dev. Chem. Eng. Miner. Process., 2000, 8, 505–550 Search PubMed.
- S. Ahmed, M. Rasul, W. Martens, R. Brown and M. Hashib, Water, Air, Soil Pollut., 2011, 215, 3–29 CrossRef CAS.
- S. Sakthivel, M. Janczarek and H. Kisch, J. Phys. Chem. B, 2004, 108, 19384–19387 CrossRef CAS.
- D. W. Bahnemann, M. Muneer and M. M. Haque, Catal. Today, 2007, 124, 133–148 CrossRef.
- C. Shifu and C. Gengyu, Sol. Energy, 2005, 79, 1–9 CrossRef.
- R. T. Koodali and D. Zhao, Energy Environ. Sci., 2010, 3, 608–614 CAS.
- M. S. Lee, G.-D. Lee, S. S. Park, C.-S. Ju, K. T. Lim and S.-S. Hong, Res. Chem. Intermed., 2005, 31, 379–389 CrossRef CAS.
- Y. Arai, K. Tanaka and A. L. Khlaifat, J. Mol. Catal. A: Chem., 2006, 243, 85–88 CrossRef CAS.
- M. S. Vohra and K. Tanaka, Water Res., 2003, 37, 3992–3996 CrossRef CAS.
- Y. Xu, W. Zheng and W. Liu, J. Photochem. Photobiol., A, 1999, 122, 57–60 CrossRef CAS.
- J. Theurich, M. Lindner and D. W. Bahnemann, Langmuir, 1996, 12, 6368–6376 CrossRef CAS.
- G. Madras and M. H. Priya, Ind. Eng. Chem. Res., 2005, 45, 482–486 Search PubMed.
- Y. Guo, S. Yang, X. Zhou, C. Lin, Y. Wang and W. Zhang, J. Nanomater., 2011, 2011 Search PubMed Article ID 296953.
- J. Zou and J. Gao, J. Hazard. Mater., 2011, 185, 710–716 CrossRef CAS.
- P. Cheng, M. Zheng, Y. Jin, Q. Huang and M. Gu, Mater. Lett., 2003, 57, 2989–2994 CrossRef CAS.
- S. K. Kim, H. Chang, K. Cho, D. S. Kil, S. W. Cho, H. D. Jang, J.-W. Choi and J. Choi, Mater. Lett., 2011, 65, 3330–3332 CrossRef CAS.
- E. Beyers, E. Biermans, S. Ribbens, K. De Witte, M. Mertens, V. Meynen, S. Bals, G. Van Tendeloo, E. F. Vansant and P. Cool, Appl. Catal., B, 2009, 88, 515–524 CrossRef CAS.
- C. Anderson and A. J. Bard, J. Phys. Chem., 1995, 99, 9882–9885 CrossRef CAS.
- M. Brigante and P. C. Schulz, J. Colloid Interface Sci., 2011, 363, 355–361 CrossRef CAS.
- M. V. Phanikrishna Sharma, G. Sadanandam, A. Ratnamala, V. Durga Kumari and M. Subrahmanyam, J. Hazard. Mater., 2009, 171, 626–633 CrossRef.
- M. V. Phanikrishna Sharma, V. Durga Kumari and M. Subrahmanyam, Chemosphere, 2008, 73, 1562–1569 CrossRef CAS.
- J. Marugán, M.-J. López-Muñoz, W. Gernjak and S. Malato, Ind. Eng. Chem. Res., 2006, 45, 8900–8908 CrossRef.
- C. Xie, Z. Xu, Q. Yang, N. Li, D. Zhao, D. Wang and Y. Du, J. Mol. Catal. A: Chem., 2004, 217, 193–201 CrossRef CAS.
- X. Fu, L. A. Clark, Q. Yang and M. A. Anderson, Environ. Sci. Technol., 1996, 30, 647–653 CrossRef CAS.
- A. A. Ismail, I. A. Ibrahim, M. S. Ahmed, R. M. Mohamed and H. El-Shall, J. Photochem. Photobiol., A, 2004, 163, 445–451 CrossRef CAS.
- L. Zou, Y. Luo, M. Hooper and E. Hu, Chem. Eng. Process., 2006, 45, 959–964 CrossRef CAS.
- Y. Li, W.-N. Wang, Z. Zhan, M.-H. Woo, C.-Y. Wu and P. Biswas, Appl. Catal., B, 2010, 100, 386–392 CrossRef CAS.
- S. Cao, N. Yao and K. Yeung, J. Sol-Gel Sci. Technol., 2008, 46, 323–333 CrossRef CAS.
- B. E. Logan, Environ. Sci. Technol., 2004, 38, 160A–167A CrossRef CAS.
- D. Das and T. N. Veziroglu, Int. J. Hydrogen Energy, 2001, 26, 13–28 CrossRef CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS.
- A. Kudo and Y. Miseki, Chem. Soc. Rev., 2009, 38, 253–278 RSC.
- X. Chen, S. Shen, L. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS.
- M. Ashokkumar, Int. J. Hydrogen Energy, 1998, 23, 427–438 CrossRef CAS.
- T. Takata, A. Tanaka, M. Hara, J. N. Kondo and K. Domen, Catal. Today, 1998, 44, 17–26 CrossRef CAS.
- A. Kudo, Int. J. Hydrogen Energy, 2006, 31, 197–202 CrossRef CAS.
- K. Maeda and K. Domen, J. Phys. Chem. C, 2007, 111, 7851–7861 CAS.
- T.-V. Nguyen, D.-J. Choi and O. B. Yang, Res. Chem. Intermed., 2005, 31, 483–491 CrossRef CAS.
- T.-V. Nguyen, S. Kim and O. B. Yang, Catal. Commun., 2004, 5, 59–62 CrossRef CAS.
- D. Zhao, S. Budhi, A. Rodriguez and R. T. Koodali, Int. J. Hydrogen Energy, 2010, 35, 5276–5283 CrossRef CAS.
- S. Shen and L. Guo, Catal. Today, 2007, 129, 414–420 CrossRef CAS.
- Q. Li, Z. Jin, Z. Peng, Y. Li, S. Li and G. Lu, J. Phys. Chem. C, 2007, 111, 8237–8241 CAS.
- S. S. Rayalu, N. Dubey, N. K. Labhsetwar, S. Kagne and S. Devotta, Int. J. Hydrogen Energy, 2007, 32, 2776–2783 CrossRef CAS.
- B.-S. Lee, D.-J. Kang and S.-G. Kim, J. Mater. Sci., 2003, 38, 3545–3552 CrossRef CAS.
- W. Lan, S. Li, J. Xu and G. Luo, Langmuir, 2011, 27, 13242–13247 CrossRef CAS.
- A. F. Demirörs, A. van Blaaderen and A. Imhof, Chem. Mater., 2009, 21, 979–984 CrossRef.
- T. L. Hanley, V. Luca, I. Pickering and R. F. Howe, J. Phys. Chem. B, 2002, 106, 1153–1160 CrossRef CAS.
- R. Fateh, A. A. Ismail, R. Dillert and D. W. Bahnemann, J. Phys. Chem. C, 2011, 115, 10405–10411 CAS.
- S. Permpoon, G. Berthomé, B. Baroux, J. Joud and M. Langlet, J. Mater. Sci., 2006, 41, 7650–7662 CrossRef CAS.
- S. Permpoon, M. Houmard, D. Riassetto, L. Rapenne, G. Berthomé, B. Baroux, J. C. Joud and M. Langlet, Thin Solid Films, 2008, 516, 957–966 CrossRef CAS.
- M. Houmard, D. Riassetto, F. Roussel, A. Bourgeois, G. Berthomé, J. C. Joud and M. Langlet, Appl. Surf. Sci., 2007, 254, 1405–1414 CrossRef CAS.
- M. Takeuchi, S. Dohshi, T. Eura and M. Anpo, J. Phys. Chem. B, 2003, 107, 14278–14282 CrossRef CAS.
- N. Hüsing, B. Launay, D. Doshi and G. Kickelbick, Chem. Mater., 2002, 14, 2429–2432 CrossRef.
- H. Miyata, Y. Fukushima, K. Okamoto, M. Takahashi, M. Watanabe, W. Kubo, A. Komoto, S. Kitamura, Y. Kanno and K. Kuroda, J. Am. Chem. Soc., 2011, 133, 13539–13544 CrossRef CAS.
- N. S. Allen, M. Edge, G. Sandoval, J. Verran, J. Stratton and J. Maltby, Photochem. Photobiol., 2005, 81, 279–290 CrossRef CAS.
- T. Ishikawa, Physicochem. Probl. Miner. Process., 2010, 57–71 CAS.
- J. M. Watson, A. T. Cooper and J. R. V. Flora, Environ. Eng. Sci., 2005, 22, 666–675 CrossRef CAS.
- J. M. Coronado, J. Soria, J. C. Conesa, R. Bellod, C. Adan, H. Yamaoka, V. Loddo and V. Augugliaro, Top. Catal., 2005, 35, 279–286 CrossRef CAS.
- V. Smitha, K. Manjumol, K. Baiju, S. Ghosh, P. Perumal and K. Warrier, J. Sol-Gel Sci. Technol., 2010, 54, 203–211 CrossRef CAS.
- L. Pinho and M. J. Mosquera, J. Phys. Chem. C, 2011, 115, 22851–22862 CAS.
- H. R. Jafry, M. V. Liga, Q. Li and A. R. Barron, Environ. Sci. Technol., 2010, 45, 1563–1568 CrossRef.
- T. Lopez, J. Manjarrez, D. Rembao, E. Vinogradova, A. Moreno and R. D. Gonzalez, Mater. Lett., 2006, 60, 2903–2908 CrossRef CAS.
- P. A. Mirau, R. R. Naik and P. Gehring, J. Am. Chem. Soc., 2011, 133, 18243–18248 CrossRef CAS.
- R. Hutter, T. Mallat and A. Baiker, J. Catal., 1995, 153, 177–189 CrossRef CAS.
- J.-H. Chae, J.-H. Lee, J.-H. Jeong and M.-S. Kang, Bull. Korean Chem. Soc., 2009, 30, 302–308 CrossRef CAS.
- M. Zalas and M. Laniecki, Sol. Energy Mater. Sol. Cells, 2005, 89, 287–296 CrossRef CAS.
- R. Sasikala, V. Sudarsan, C. Sudakar, R. Naik, T. Sakuntala and S. R. Bharadwaj, Int. J. Hydrogen Energy, 2008, 33, 4966–4973 CrossRef CAS.
- S. Xu, J. Ng, X. Zhang, H. Bai and D. D. Sun, Int. J. Hydrogen Energy, 2010, 35, 5254–5261 CrossRef CAS.
- Q. Yuan, Y. Liu, L.-L. Li, Z.-X. Li, C.-J. Fang, W.-T. Duan, X.-G. Li and C.-H. Yan, Microporous Mesoporous Mater., 2009, 124, 169–178 CrossRef CAS.
- K. Lalitha, J. K. Reddy, M. V. Phanikrishna Sharma, V. D. Kumari and M. Subrahmanyam, Int. J. Hydrogen Energy, 2010, 35, 3991–4001 CrossRef CAS.
- J. Papp, S. Soled, K. Dwight and A. Wold, Chem. Mater., 1994, 6, 496–500 CrossRef CAS.
- H. Tada, M. Akazawa, Y. Kubo and S. Ito, J. Phys. Chem. B, 1998, 102, 6360–6366 CrossRef CAS.
- S. Klein, J. A. Martens, R. Parton, K. Vercruysse, P. A. Jacobs and W. F. Maier, Catal. Lett., 1996, 38, 209–214 CrossRef CAS.
- B. Notari, Adv. Catal., 1996, 41, 253–334 CrossRef CAS.
- H. Nur, Mater. Sci. Eng., B, 2006, 133, 49–54 CrossRef CAS.
- K. Domen, Y. Sakata, A. Kudo, K. Maruya and T. Onishi, Bull. Chem. Soc. Jpn., 1988, 61, 359–362 CrossRef CAS.
- H. S. Kibombo and R. T. Koodali, J. Phys. Chem. C, 2011, 115, 25568–25579 CAS.
| This journal is © The Royal Society of Chemistry 2012 |