Enantiopure 3-methyl-3,4-dihydroisocoumarins and 3-methyl-1,2,3,4-tetrahydroisoquinolines via chemoenzymatic asymmetric transformations†
Juan
Mangas-Sánchez
,
Eduardo
Busto
,
Vicente
Gotor-Fernández
* and
Vicente
Gotor
*
Universidad de Oviedo, Departamento de Química Orgánica e Inorgánica, Instituto Universitario de Biotecnología de Asturias, Avenida Julián de Clavería 8, Oviedo 33006, Spain. E-mail: vicgotfer@uniovi.es; vgs@uniovi.es; Fax: +34 985103451; Tel: +34 985103448
First published on 24th April 2012
Abstract
A new divergent and asymmetric synthetic route has been developed for the production of enantiomerically pure isocoumarin and isoquinoline derivatives. Stereoselective formation of 2-(2-hydroxypropyl)benzonitriles has been identified as the key step. Rhizomucor miehei lipase has displayed from moderate to excellent selectivities in the acetylation of its (R)-enantiomers, while ADH-A from Rhodococcus ruber catalyzed the bioreduction of the corresponding ketones with excellent activities and stereopreferences. Enantiopure alcohols obtained in this manner have been used for the straightforward synthesis of a series of 3-methyl-3,4-dihydroisocoumarins and 3-methyl-1,2,3,4-tetrahydroisoquinolines with good overall yields.
Introduction
The development of new strategies in heterocyclic chemistry continues to be an attractive and challenging task for synthetic chemists. In this context, the preparation of 3,4-dihydroisocoumarins has been gathering increasing attention in recent years because of their structural implications as basic scaffolds in alkaloids and many natural products1 that exhibit a broad range of biological activities. Both non-asymmetric and stereoselective methods for the preparation of this family of oxygenated compounds have been extensively reported in recent years.2 In addition, dihydroisocoumarins are a class of naturally occurring biologically active lactones possessing remarkable antibacterial, anticancer, antifungal or antimalarial properties.3 Asymmetric synthesis and isolation from plant extracts of 3-substituted-3,4-dihydroisocoumarins have particularly attracted attention.4 However, only a few chemoenzymatic routes have been reported until now, among which biocatalytic methods for the preparation of 3-methyl-3,4-dihydroisocoumarins only include chemoenzymatic pathways involving ketone transformations such as Baeyer–Villiger oxidations5 or bioreduction processes.6Here we describe for the first time a divergent synthetic approach for the production of 3-methyl-3,4-dihydroisocoumarins and 3-methyl-1,2,3,4-tetrahydroisoquinolines starting from readily commercially available 2-bromobenzonitriles. The asymmetric actions of several lipases and alcohol dehydrogenases (ADHs) have been considered in the search for the optimal production of enantiopure 2-(2-hydroxypropyl)benzonitriles, which have shown an excellent versatility as key intermediates for the preparation of the target molecules.
Results and discussion
Hydrolase-catalyzed kinetic resolution7 and dynamic kinetic resolution8 of racemic 1-phenylpropan-2-ols have been widely reported through acylation processes using a variety of acyl donors and biocatalysts. In spite of the fact that lipases act with a different degree of selectivity in the resolution of 1-phenylpropan-2-ols,9 their action towards 2-(2-hydroxypropyl)benzonitriles still remains unexplored. The reaction of commercially available 2-bromobenzonitrile (1a) with a solution of butyl lithium in THF as solvent, followed by the addition of propylene oxide in the presence of boron trifluoride etherate, afforded racemic 2-(2-hydroxypropyl)benzonitrile (2a) in 71% yield (Scheme 1).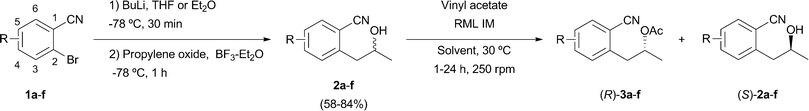 | ||
| Scheme 1 Chemical synthesis and kinetic resolution of racemic alcohols 2a–f using Rhizomucor miehei lipase. | ||
The lipase-catalyzed kinetic resolution of (±)-2a was then analyzed using 3 equiv. of vinyl acetate and variable biocatalyst loadings and solvents at 30 °C. Candida cylindracea lipase (CCL), porcine pancreas lipase (PPL), lipase AK from Pseudomonas fluorescens, Thermomyces lanuginosa lipase (TLL) and alcalase CLEA all displayed very low conversions and poor enantiodiscrimination values, while Candida antarctica lipase type A (CAL-A), Candida antarctica lipase type B (CAL-B) and Burkholderia cepacia lipase (PSL-IM) led to higher reactivities, with close to 50% conversion, but very poor selectivities (E < 20). However, the use of Rhizomucor miehei lipase (RML IM) as a biocatalyst led to excellent enantioselectivities (E > 150) at different temperatures and variable protein loadings (Table 1, entries 1–3). Optimal results were found when using a substrate![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :enzyme ratio of 1:2 at 30 °C, with both substrate and product being recovered in 96% ee after 8 h (entry 3). Similar results in terms of excellent selectivities were achieved with other solvents, such as 1,4-dioxane, THF, toluene or vinyl acetate as both solvent and acyl donor, although with a lower reactivity (entries 4–7).
:enzyme ratio of 1:2 at 30 °C, with both substrate and product being recovered in 96% ee after 8 h (entry 3). Similar results in terms of excellent selectivities were achieved with other solvents, such as 1,4-dioxane, THF, toluene or vinyl acetate as both solvent and acyl donor, although with a lower reactivity (entries 4–7).
| Entry | Substrate | RML IM:2a–fa |
T/°C | Solvent | t/h | eepb (%) | eesb (%) | c (%) | E |
|---|---|---|---|---|---|---|---|---|---|
|
a Ratio enzyme:substrate in weight (w/w).
b Enantiomeric excesses of product (eep) and substrate (ees) determined by HPLC.
c Conversion values calculated as c = ees/(ees + eep).
d Enantiomeric ratio calculated as E = ln[(1 − ees)/1 + (ees/eep)]/ln[(1 + ees)/1 + (ees/eep)].10
|
|||||||||
| 1 | 2a | 1:1 |
30 | TBME | 23 | 99 | 63 | 39 | >200 |
| 2 | 2a | 1:1 |
45 | TBME | 8 | >99 | 35 | 26 | >200 |
| 3 | 2a | 2:1 |
30 | TBME | 8 | 96 | 96 | 50 | 194 |
| 4 | 2a | 2:1 |
30 | 1,4-Dioxane | 23 | >99 | 32 | 24 | >200 |
| 5 | 2a | 2:1 |
30 | THF | 23 | >99 | 14 | 12 | >200 |
| 6 | 2a | 2:1 |
30 | Toluene | 23 | 98 | 85 | 47 | >200 |
| 7 | 2a | 2:1 |
30 | Vinyl acetate | 23 | 98 | 44 | 31 | 153 |
| 8 | 2b (R = 4-Me) | 2:1 |
30 | TBME | 8 | 96 | 99 | 51 | >200 |
| 9 | 2c (R = 5-OMe) | 2:1 |
30 | TBME | 24 | 92 | 80 | 47 | 59 |
| 10 | 2d (R = 5-Me) | 2:1 |
30 | TBME | 9 | 95 | 61 | 39 | 73 |
| 11 | 2e (R = 3-Me) | 2:1 |
30 | TBME | 9 | 94 | 20 | 17 | 39 |
| 12 | 2f (R = 4-F) | 2:1 |
30 | TBME | 24 | 99 | 57 | 37 | >200 |
To examine the scope of RML IM lipase, the kinetic resolution of the substituted benzene derivatives 2b–f was studied. Racemic alcohols 2b–e were prepared in moderate to high yields using an identical synthetic approach. However, for the 4-fluorinated derivative 1f, 2-bromo-4-fluoro-3-(2-hydroxypropyl) benzonitrile was isolated as the unique reaction product due to the fluorine promoted metallation at the C3-position.11 The use of Et2O instead of THF as reaction medium led to the bromine–lithium exchange, the desired alcohol 2f being obtained in 73% isolated yield. Lipase-catalyzed acetylation led to the production of all the acetates (R)-3b–f in a range between 92 and 99% ee using TBME as solvent (entries 8–12). However only non-substituted alcohol 2a (entry 3, E = 194) and 4-substituted 2b,f (E > 200, entries 8 and 12) were acetylated with very good to excellent enantioselectivities. On the other hand, the 5-substituted 2c,d (entries 9 and 10) and the 3-methylated substrate 2e (entry 11) led to moderate selectivities, the reaction rate being much lower for 2e, which posses the substitution closer to the reacting centre.
In order to improve the synthetic access to enantiopure precursors of 3-methyl-3,4-dihydroisocoumarins, and motivated by the high potential shown by redox enzymes in the bioreduction of 1-aryl-2-propanones,12 we decided to explore the potential of alcohol dehydrogenases for the production of enantioenriched 2a–f. To do this, racemic alcohols 2a–f were chemically oxidized, the bioreduction of the ketones 4a–f obtained in this way was then studied. The Dess–Martin reagent was chosen to perform the chemical oxidations due to the mild conditions required, affording 4a–f in good yields (Scheme 2).
 | ||
| Scheme 2 Chemoenzymatic synthesis of 3-methyl-3,4-dihydroisocoumarins through bioreduction processes. | ||
A series of ADHs were tested for the bioreduction of 4a, including alcohol dehydrogenases from Rhodococcus ruber (ADH-A), Candida parapsilosis (ADH-CP) or Lactobacillus kefir (ADH-LK), and also the very well known Baker's yeast.13 Initial experiments were carried out in the mg-scale, obtaining alcohol (S)-2a in high to excellent enantiomeric excess, except for the reaction with ADH-LK which, following the anti-Prelog stereoselectivity rule, led to the opposite enantiomer (Table 2, entries 1–4). On the basis of these results, and bearing in mind the scaling-up of the biocatalytic processes for further synthetic applications, ADH-A overexpressed in E. coli was successfully applied in the bioreduction of 4a, yielding identical results to those of commercially available ADH-A (entry 5).
| Entry | Ketone | ADHa | t/h | eepb (%) | c (%) |
|---|---|---|---|---|---|
| a (S)-Alcohols 2a–f were obtained as final products with the exception of the bioreduction of 4a with ADH-LK. b Determined by HPLC. c Determined by GC. d Reactions in 80 mg scale of substrate. Isolated yields in brackets. | |||||
| 1 | 4a | ADH-A | 24 | >99 | >97 |
| 2 | 4a | Baker's yeast | 24 | 96 | >97 |
| 3 | 4a | ADH-CP | 24 | >99 | >97 |
| 4 | 4a | ADH-LK | 24 | 98 | 38 |
| 5 | 4a | E. coli/ADH-A | 24 | >99 | >97 |
| 6d | 4a | E. coli/ADH-A | 24 | >99 | >97 (>97) |
| 7d | 4b | E. coli/ADH-A | 24 | >99 | >97 (93) |
| 8d | 4c | E. coli/ADH-A | 24 | >99 | >97 (>97) |
| 9d | 4d | E. coli/ADH-A | 24 | >99 | >97 (82) |
| 10d | 4e | E. coli/ADH-A | 72 | >99 | >97 (62) |
| 11d | 4f | E. coli/ADH-A | 24 | >99 | >97 (77) |
Using our knowledge of the best reaction conditions, an extension of this enzymatic study was done for substituted ketones 4b–f. The formation of the enantiopure (S)-alcohols in complete conversion was detected after 24 h for all the substrates, with the exception of the bioreduction of the 3-methyl derivative (4e), which reached 49% conversion (data not shown). This may be explained by the hindering effect of the methyl group which is very close to the carbonyl group. Biocatalytic reactions were subsequently scaled-up to 80 mg of substrate. In all cases complete conversions (entries 6–11) were obtained, longer reaction times (72 h) only being required for ketone 4e (entry 10). Finally, the intramolecular cyclisation of alcohols (S)-2a–f in acidic medium led to the enantiopure 3-methyl-3,4-dihydroisocoumarins (S)-5a–f after purification by extraction with CH2Cl2 (65–90%).14
In order to expand the applicability of these enantiomerically pure alcohol intermediates, we decided to move towards the asymmetric synthesis of structurally similar compounds. The possibility of using the same alcohol precursors highlights the importance of this divergent synthetic approach, especially when the desired targets are isoquinoline derivatives, a class of nitrogenated compounds that are widely distributed in Nature and show a potent physiological action.15 As a result of their importance, a number of research groups have reported the asymmetric synthesis of isoquinoline derivatives.16 Biocatalytic production of these derivatives is nowadays mainly based upon lipase-catalyzed kinetic resolution,17 bioreduction of cyclic imines with metalloenzymes18 or deracemisation experiments using evolved monoamine oxidases combined with non-selective reducing agents.19 However, little attention has been paid to the asymmetric synthesis of 3-substituted tetrahydroisoquinolines,20 a class of compounds that not only shows remarkable biological activity21 but also offers practical applications in asymmetric catalysis.22
Taking advantage of the versatility of (S)-2a–f, our aim was to design a general synthetic pathway for the synthesis of 3-methyl-1,2,3,4-tetrahydroisoquinolines. To do this, alcohols were mesylated in dichloromethane at room temperature, and then different methods to reduce the nitrile group were attempted. The mild reduction procedure with NiCl2–NaBH4,23 followed by an in situ protection of the amine formed, was found to be an ideal strategy for the preparation of the diprotected compounds (S)-7a–f (Scheme 3). The intramolecular cyclisation of (S)-7a–f in the presence of cesium carbonate yielded enantiopure N-Boc amines (R)-8a–f, a minimum presence of the corresponding tert-butyl 2-((1E)-prop-1-en-1-yl)benzyl carbamates (2–10%) being observed in all cases. The chromatographic purification of the reaction crudes afforded the N-protected amines 8a–d in moderate yield.
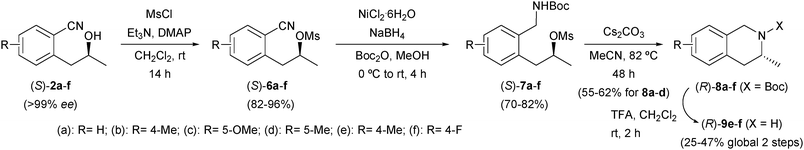 | ||
| Scheme 3 Chemoenzymatic synthesis of 3-methyl-1,2,3,4-tetrahydroisoquinolines. | ||
However, the N-Boc tetrahydroisoquinolines 8e,f decomposed in silica gel, so further transformations were required for the isolation of these amines. Then, (R)-8e,f were chemically deprotected with trifluoroacetic acid in dichloromethane, yielding (R)-9e in 47% overall yield after flash chromatography. Purification of (R)-9f proved unsuccessful, since it shows a high instability in the chromatographic purification step. To avoid this drawback we designed an alternative procedure using p-nitrobenzaldehyde as a scavenger for the remaining amounts of 1-(2-((1E)-prop-1-en-1-yl)-p-fluorophenyl)methanamine, followed by an extraction purification step affording (R)-9f in high chemical purity and low yield. In order to confirm the absolute configuration of the final products, we have synthesized (R)-3-methyl-1,2,3,4-tetrahydroisoquinoline (9a) from 8a. The optical rotation value is in concordance with previously published data for (R)-9a.24
Conclusions
2-(2-Hydroxypropyl)benzonitriles have been found to be adequate building blocks in the divergent asymmetric synthesis of enantiopure substituted 3-methyl-3,4-dihydroisocoumarins and 3-methyl-1,2,3,4-tetrahydroisoquinolines. Rhizomucor miehei lipase has displayed the best results in the lipase-catalyzed acetylation of racemic alcohols. However, enantioselectivity was found to be highly dependent on the ring pattern substitution, with moderate stereoselectivities being found for 3- and 5-monosubstituted alcohols, and very high to excellent values being achieved with none or 4-substituted derivatives. The chemical oxidation of racemic alcohols and subsequent bioreduction of the related ketones provided access to a series of novel enantiopure 2-(2-hydroxypropyl)benzonitriles, ADH-A from Rhodococcus ruber being found as an excellent asymmetric biocatalyst for this reduction step. The enantiopure alcohols so obtained were treated in acidic medium, leading to a family of substituted (S)-3-methyl-3,4-dihydroisocoumarins in moderate to very high yield by intramolecular cyclisation. Taking advantage of their versatility, alcohol precursors were used in the synthesis of enantiopure N-Boc-3-methyl-1,2,3,4-tetrahydroisoquinolines through a three-step synthesis in a very straightforward manner and without loss of the optical purity.Experimental
General procedure for the synthesis of 2-(2-hydroxypropyl)benzonitriles 2a–f
To a solution of the corresponding commercially available 2-bromobenzonitriles 1a–f (1 equiv.) in THF (or Et2O for 1f, 0.2 M), BuLi (1.1 equiv., 1.6 M in hexane) was added dropwise at −78 °C under an inert atmosphere. The solution was stirred for 30 min at −78 °C and then, propylene oxide (1.5 equiv.) and boron trifluoride etherate (1.5 equiv.) were added dropwise. The mixture was stirred at −78 °C for additional 1 h, until the reaction was quenched with water. The mixture was extracted with Et2O (3 × 20 mL), organic layers were collected, dried over Na2SO4, and the solvent evaporated under reduced pressure. The resulting crude was purified by flash chromatography (50% Et2O/hexane), obtaining the corresponding alcohols 2a–f in high yields (69–84%).General procedure for the synthesis of racemic 1-(2-cyanophenyl)propan-2-yl acetates 3a–f
To a solution of the corresponding racemic alcohols 2a–f (1 equiv.) in dry CH2Cl2 (0.1 M), triethylamine (3 equiv.), 4-(N,N-dimethylamino)pyridine (0.3 equiv.) and acetic anhydride (2 equiv.) were successively added at rt under an inert atmosphere. The mixture was stirred and the reaction monitored by TLC analysis (50% Et2O/hexane) until no starting material was detected (30–60 min). Solvent was evaporated afterwards and the crude purified by flash chromatography (50% Et2O/hexane), obtaining the corresponding 1-(2-cyanophenyl)propan-2-yl acetates 3a–f in high to excellent yields (82–98%).General procedure for the synthesis of 2-(2-oxopropyl)benzonitriles 4a–f
To a solution of the corresponding racemic alcohols 2a–f (1 equiv.) in CH2Cl2 (0.1 M), the Dess–Martin reagent (1.5 equiv.) was added at rt under an inert atmosphere. The mixture was stirred for 4 h. Then, the reaction was quenched with an aqueous saturated 1:1 solution of Na2S2O3–NaHCO3. The mixture was extracted afterwards with CH2Cl2, organic layers were collected, dried over Na2SO4, filtered, and the solvent evaporated under reduced pressure. The resulting crude was purified by flash chromatography (50% Et2O/hexane), affording the corresponding ketones 4a–f in high yields (70–81%).
General procedure for the synthesis of 3-methylisochroman-1-ones 5a–f
The corresponding alcohols 2a–f (1 equiv.) were dissolved in a 1:1 solution of MeOH and hydrochloric acid (37%) (0.02 M). The solution was warmed to 66 °C and stirred overnight under reflux. The reaction was quenched with water and extracted with CH2Cl2 (3 × 10 mL). Organic layers were collected, dried over Na2SO4, filtered and the solvent evaporated under reduced pressure, affording the corresponding lactones 5a–f without further purification in high yields (71–90%).
General procedure for the synthesis of 1-(2-cyanophenyl)propan-2-yl methanesulfonates 6a–f
To a solution of the corresponding alcohols 2a–f (1 equiv.) in CH2Cl2 (0.1 M), triethylamine (2 equiv.), 4-(N,N-dimethylamino)pyridine (0.1 equiv.) and methanesulfonyl chloride (2 equiv.) were successively added under an inert atmosphere. The reaction was stirred overnight at rt and then the solvent evaporated under reduced pressure. The resulting crude was purified by flash chromatography (50% Et2O/hexane), to afford the corresponding 1-(2-cyanophenyl)propan-2-yl methanesulfonates 6a–f in good yields (82–96%).General procedure for the synthesis of 1-[2-(((tert-butoxycarbonyl)amino)methyl)phenyl]propan-2-yl methanesulfonates 7a–f
To a solution of the corresponding 1-(2-cyanophenyl)propan-2-yl methanesulfonates 6a–f (1 equiv.) in MeOH (0.13 M), NiCl2·6H2O (0.1 equiv.), di-tert-butyldicarbonate (2 equiv.) were added under an inert atmosphere. Then, the mixture was cooled to 0 °C and NaBH4 (7 equiv.) was added in small portions over 15 min. The reaction was warmed to rt afterwards and stirred for 4 h. Then, the solution was filtered over Celite®, the solvent evaporated under reduced pressure and the resulting crude purified by flash chromatography (50% Et2O/hexane), affording the corresponding protected amines 7a–f with good yields (70–82%).General procedure for the synthesis of tert-butyl 3-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylates 8a–d
To a solution of the corresponding 1-[2-(((tert-butoxycarbonyl)amino)methyl)phenyl]propan-2-yl methanesulfonates 7a–d (1 equiv.) in dry MeCN (0.1 M), cesium carbonate was added (10 equiv.) under an inert atmosphere. The mixture was stirred at 82 °C for 2 days. Then, water was added and the reaction extracted with CH2Cl2 (3 × 10 mL), organic layers were collected, dried over Na2SO4, filtered, and the solvent evaporated under reduced pressure. The resulting crude was purified by flash chromatography (5% EtOAc/hexane), affording the corresponding tert-butyl 3-methyl-3,4-dihydroisoquinoline-2(1H)-carboxylates 8a–d in good yields (55–62%).General procedure for the synthesis of 3-methyltetrahydroisoquinolines 9a,e
To a solution of the corresponding 1-[2-(((tert-butoxycarbonyl)amino)methyl)phenyl]propan-2-yl methanesulfonates 7a,e (1 equiv.) in dry MeCN (0.1 M), cesium carbonate was added (10 equiv.) under an inert atmosphere. The mixture was stirred at 82 °C for 2 days. Then, water was added and the reaction extracted with CH2Cl2 (3 × 10 mL), organic layers were collected, dried over Na2SO4, filtered, and the solvent was evaporated under reduced pressure. The mixture was then dissolved in a 1:1 mixture of CH2Cl2 and TFA (0.025 M), stirred for 2 h, and then basified with NaOH (3 N) and extracted with CH2Cl2. Organic layers were collected, dried over Na2SO4, filtered, the solvent was evaporated under reduced pressure, and the resulting crude purified by flash chromatography (10% MeOH/CH2Cl2), affording the corresponding 3-methyltetrahydroisoquinolines 9a,e in moderate global yields for the two steps (47–54%).
General procedure for the synthesis of (R)-6-fluoro-3-methyl-1,2,3,4-tetrahydroisoquinoline 9f
To a solution of (S)-1-(2-(((tert-butoxycarbonyl)amino)methyl)-5-fluorophenyl)propan-2-yl methanesulfonate 7f (53 mg, 0.15 mmol) in dry MeCN (1.5 mL), cesium carbonate was added (478 mg, 1.46 mmol) under an inert atmosphere. The mixture was stirred at 82 °C for 2 days. Then, water was added and the reaction extracted with CH2Cl2 (3 × 15 mL), organic layers were collected, dried over Na2SO4, filtered, and the solvent evaporated under reduced pressure. The resulting crude was then dissolved in CH2Cl2 (2 mL) and TFA (2 mL) was added. The mixture was stirred for 2 h at rt, and then basified with NaOH (3 N), and extracted with CH2Cl2 (3 × 15 mL). Organic layers were collected, dried over Na2SO4, filtered and the solvent evaporated under reduced pressure. The mixture was then dissolved in CH2Cl2 (10 mL) and 4-nitrobenzaldehyde (15 mg, 0.10 mmol) and MgSO4 (100 mg, 0.83 mmol) were added. The mixture was stirred for 30 min. Then, the mixture was extracted with a pH 4.5 NaOAc/HCl buffer (30 mL). The aqueous phase was afterwards basified with NaOH (3 N) and extracted with CH2Cl2 (3 × 10 mL). Organic layers were collected, dried over Na2SO4, filtered, and the solvent evaporated under reduced pressure, affording pure 9f in low yield (6 mg, 25%).General procedure for the lipase mediated kinetic resolution of alcohols 2a–f
To a suspension of racemic alcohols 2a–f (0.2 mmol, 1 equiv.) and the adequate lipase in dry solvent (2 mL), vinyl acetate (55 μL, 0.6 mmol) was added under an inert atmosphere. The reaction was shaken at 30 °C and 250 rpm, taking regularly aliquots that were analyzed by HPLC until around 50% conversion was reached. Then the reaction was stopped, and the enzyme filtered with CH2Cl2 (3 × 10 mL). The solvent was evaporated and the crude of the reaction purified by flash chromatography on silica gel (50% Et2O/hexane), obtaining the corresponding enantiomerically enriched acetates (R)-3a–f and the alcohols (S)-2a–f (see Table 1 for additional details).General procedure for the bioreduction of ketones 4a–f with ADHs
Different protocols were used depending on the enzyme and have been listed below. See Table 2 for additional details.Acknowledgements
We thank Novozymes for the generous gift of the CAL-B (Novozyme 435), and also Prof. Wolfgang Kroutil for ADH-A overexpressed in E. coli cells. Financial support of this work by the Spanish Ministerio de Ciencia e Innovación (MICINN) through CTQ 2007-61126 and BIO 2008-03683 projects is gratefully acknowledged. V.G.-F. acknowledges MICINN for a postdoctoral grant (Ramón y Cajal Program). J.M.-S. acknowledges MICINN for a predoctoral fellowship (FPU Program).Notes and references
- The Alkaloids Chemistry and Pharmacology, ed. G. A. Cordell, Academic Press, San Diego, 1995 Search PubMed.
- For recent articles about the synthesis of racemic 3,4-dihydroisocoumarins: E. T. da Penha, J. A. Forni, A. F. P. Biajoli and C. R. D. Correia, Tetrahedron Lett., 2011, 52, 6342 CrossRef CAS; M. D. Obushak, V. S. Matiychuk and V. V. Turytsya, Tetrahedron Lett., 2009, 50, 6112 CrossRef; I. Ullah, M. Sher, R. A. Khera, A. Ali, M. F. Ibad, A. Villinger, C. Fischer and P. Langer, Tetrahedron, 2010, 66, 1874 CrossRef; S. K. Mandal and S. C. Roy, Tetrahedron, 2008, 64, 11050 CrossRef; M. Sher, A. Ali, H. Reinke and P. Langer, Tetrahedron Lett., 2008, 49, 5400 CrossRef; S. K. Mandal and S. C. Roy, Tetrahedron Lett., 2007, 48, 4131 CrossRef; T. Suzuki, T. Yamada, K. Watanabe and T. Katoh, Bioorg. Med. Chem. Lett., 2005, 15, 2583 CrossRef.
- C. A. Higgins, Z. Delbederi, K. McGarel, T. Mills, O. McGrath, S. Feutren-Burton, W. Watters, P. Armstrong, P. G. Johnston, D. Waugh and H. van den Berg, Bioconjugate Chem., 2009, 20, 1737 CrossRef CAS and references cited therein.
- For recent literature: C. Zidorn, B. O. Petersen, V. Sareedenchai, E. P. Ellmerer and J. Ø. Duus, Tetrahedron Lett., 2010, 51, 1390 CrossRef CAS; Y. Li, I. Plitzko, J. Zaugg, S. Hering and M. Hamburger, J. Nat. Prod., 2010, 73, 768 CrossRef; R. Haritakun, M. Sappan, R. Suvannakad, K. Tasanathai and M. Isaka, J. Nat. Prod., 2010, 73, 75 CrossRef; S. A. Shahzad, C. Venin and T. Wirth, Eur. J. Org. Chem., 2010, 3465 CrossRef; A. Kumar Sharma, Y. Maheshwary, P. Singh and K. N. Singh, ARKIVOC, 2010,(ix), 54 Search PubMed; B. Cramer, H. Harrer, K. Nakamura, D. Uemura and H.-U. Humpf, Bioorg. Med. Chem., 2010, 18, 343 CrossRef; A. Habel and W. Boland, Org. Biomol. Chem., 2008, 6, 1601 Search PubMed; S. Shinkaruk, B. Bennetau, P. Babin, J.-M. Schmitter, V. Lamothe, C. Bennetau-Pelissero and M. C. Urdaci, Bioorg. Med. Chem., 2008, 16, 9383 CrossRef; K. Uchida, T. Fukuda and M. Iwao, Tetrahedron, 2007, 63, 7178 CrossRef; G. Kerti, T. Kurtán, T.-Z. Illyés, K. E. Kövér, S. Sólyom, G. Pescitelli, N. Fujioka, N. Berova and S. Antus, Eur. J. Org. Chem., 2007, 296 CrossRef; H. B. Mereyala and G. Pathuri, Synthesis, 2006, 2944 CrossRef; Y. Kurosaki, T. Fukuda and M. Iwao, Tetrahedron, 2005, 61, 3289 CrossRef.
- A. Rioz-Martínez, G. de Gonzalo, D. E. Torres Pazmiño, M. W. Fraaije and V. Gotor, J. Org. Chem., 2010, 75, 2073 CrossRef.
- T. Schubert, M.-R. Kula and M. Müller, Synthesis, 1999, 2045 CrossRef CAS.
- L. L. Machado, T. L. G. Lemos, M. C. de Mattos, M. C. F. de Oliveira, G. de Gonzalo, V. Gotor-Fernández and V. Gotor, Tetrahedron: Asymmetry, 2008, 19, 1418 CrossRef CAS; C. Csajági, G. Szatzker, E. R. Tóke, L. Ürge, F. Darvas and L. Poppe, Tetrahedron: Asymmetry, 2008, 19, 237 CrossRef; G. Kerti, T. Kurtán, T.-Z. Illyés, K. E. Kövér, S. Sólyom, G. Pescitelli, N. Fujioka, N. Berova and S. Antus, Eur. J. Org. Chem., 2007, 196 Search PubMed; V. Kiss, G. Egri, J. Bálint, I. Ling, J. Barkóczi and E. Fogassy, Tetrahedron: Asymmetry, 2006, 17, 2220 CrossRef; A. Ghanem and V. Schurig, Tetrahedron: Asymmetry, 2003, 14, 2547 CrossRef; N. S. Hatzakis and I. Smonou, Bioorg. Chem., 2005, 33, 325 CrossRef; A. Ghanem and V. Schurig, Monatsh. Chem., 2003, 134, 1151 CrossRef; D. Bianchi, P. Cesti and E. Battistel, J. Org. Chem., 1988, 53, 5531 CrossRef; K. Laumen, D. Breitgoff and M. P. Schneider, J. Chem. Soc., Chem. Commun., 1998, 1459 Search PubMed; K.-E. Jaeger, B. Schneidinger, F. Rosenau, M. Werner, D. Lang, B. W. Dijkstra, K. Schimossek, A. Zonta and M. T. Reetz, J. Mol. Catal. B: Enzym., 1997, 3, 3 CrossRef; S. Ruppert and H.-J. Gais, Tetrahedron: Asymmetry, 1997, 8, 3657 CrossRef.
- M. Päiviö, D. Mavrynsky, R. Leino and L. T. Kanerva, Eur. J. Org. Chem., 2011, 1452 Search PubMed; S.-B. Ko, B. Baburaj, M.-J. Kim and J. Park, J. Org. Chem., 2007, 72, 6860 CrossRef CAS; B. Martín-Matute, M. Edin, K. Bogár, F. B. Kaynak and J.-E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 8817 CrossRef; M.-J. Kim, H. M. Kim, D. Kim, Y. Ahn and J. Park, Green Chem., 2004, 6, 471 RSC; M.-J. Kim, Y. I. Chung, Y. K. Choi, H. K. Lee, D. Kim and J. Park, J. Am. Chem. Soc., 2003, 125, 11494 CrossRef; J. H. Koh, H. M. Jung, M.-J. Kim and J. Park, Tetrahedron Lett., 1999, 40, 6281 CrossRef.
- J. Mangas-Sánchez, E. Busto, V. Gotor-Fernández and V. Gotor, Org. Lett., 2010, 12, 3498 CrossRef CAS; D. Acetti, E. Brenna, C. Fuganti, F. G. Gatti and S. Serra, Tetrahedron: Asymmetry, 2009, 20, 2413 CrossRef; M. Fujii, S. Yasuhara and H. Akita, Tetrahedron: Asymmetry, 2009, 20, 1286 CrossRef; P. Bachu, J. Sperry and M. A. Brimble, Tetrahedron, 2008, 64, 4827 CrossRef; T. Choshi, Y. Uchida, Y. Kubota, J. Nobuhiro, M. Takeshita, T. Hatano and S. Hibino, Chem. Pharm. Bull., 2007, 55, 1060 CrossRef; T. Ema, M. Yoshii, T. Korenaga and T. Sakai, Tetrahedron: Asymmetry, 2002, 13, 1223 CrossRef.
- C.-S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294 CrossRef CAS; J. L. L. Rakels, A. J. J. Straathof and J. J. Heijnen, Enzyme Microb. Technol., 1993, 15, 1051 CrossRef.
- P. L. Coe, A. J. Waring and T. D. Yarwood, J. Chem. Soc., Perkin Trans. 1, 1995, 2729 RSC.
- G. A. Petkova and V. Král, Bioorg. Med. Chem., 2010, 18, 6651 CrossRef CAS; S. Broussy, R. S. Cheloha and D. B. Berkowitz, Org. Lett., 2009, 11, 305 CrossRef; M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille and R. S. Philips, Org. Biomol. Chem., 2008, 6, 887 Search PubMed; K. I. Ziegelmann-Fjeld, M. M. Musa, R. S. Phillips, J. G. Zeikus and C. Vieille, Protein Eng., Des. Sel., 2007, 20, 47 CrossRef; M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, J. Org. Chem., 2007, 72, 30 CrossRef; M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, Angew. Chem., Int. Ed., 2007, 46, 3091 CrossRef; B. Erdélyi, A. Szabó, G. Seres, L. Birincisk, J. Ivanics, G. Szatzker and L. Poppe, Tetrahedron: Asymmetry, 2006, 17, 268 CrossRef; K. Inoue, Y. Makino and N. Itoh, Tetrahedron: Asymmetry, 2005, 16, 2539 CrossRef; J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef; K. Nakamura and T. Matsuda, J. Org. Chem., 1998, 63, 8957 CrossRef.
- G. Fronza, C. Fuganti, P. Grasselli and A. Mele, J. Org. Chem., 1991, 56, 6019 CrossRef CAS.
- (S)-5a: [α]20D +116.1 (c 1, CHCl3) (>99% ee) in comparison with previously described [α]20D +90.2 (c 0.55, CH2Cl2) in ref. 4 ( K. E. Kövér, S. Sólyom, G. Pescitelli, N. Fujioka, N. Berova and S. Antus, Eur. J. Org. Chem., 2007, 296 Search PubMed ).
- The Chemistry and Biology of Isoquinoline Alkaloids, ed. J. D. Philipson, M. F. Roberts and M. H. Zenk, Springer-Verlag, Berlin, 1985 CrossRef CAS; J. D. Scott and R. M. Williams, Chem. Rev., 2002, 102, 1669 CrossRef CAS.
- P. Siengalewicz, U. Rinner and J. Mulzer, Chem. Soc. Rev., 2008, 37, 2676 RSC; Z. Czarnocki, A. Siwicka and J. Szawkalo, Curr. Org. Synth., 2005, 2, 301 CrossRef CAS; A. B. J. Bracca and T. S. Kaufman, Tetrahedron, 2004, 60, 10575 CrossRef; J. L. Vicario, D. Badía, L. Carrillo and J. Etxeberria, Curr. Org. Chem., 2003, 7, 1775 CrossRef.
- A. J. Blacker, M. J. Stirling and M. I. Page, Org. Process Res. Dev., 2007, 11, 642 CrossRef CAS; M. Stirling, J. Blacker and M. I. Page, Tetrahedron Lett., 2007, 48, 1247 CrossRef; T. A. Paál, E. Forró, F. Fülöp, A. Liljeblad and L. T. Kanerva, Tetrahedron: Asymmetry, 2008, 19, 2784 CrossRef.
- M. Dürrenberger, T. Heinisch, Y. M. Wilson, T. Rossel, E. Nogueira, L. Knörr, A. Mutschler, K. Kersten, M. J. Zimbron, J. Pierron, T. Schirmer and T. R. Ward, Angew. Chem., Int. Ed., 2011, 50, 3026 CrossRef.
- K. R. Bailey, A. J. Ellis, R. Reiss, T. J. Snape and N. J. Turner, Chem. Commun., 2007, 3640 RSC; J. M. Foulkes, K. J. Malone, V. S. Coker, N. J. Turner and J. R. Lloyd, ACS Catal., 2011, 1, 1589 CrossRef CAS.
- D. García, F. Foubelo and M. Yus, Eur. J. Org. Chem., 2010, 2893 CrossRef CAS; D. García, B. Moreno, T. Soler, F. Foubelo and M. Yus, Tetrahedron Lett., 2009, 50, 4710 CrossRef; L. Carrillo, D. Badía, E. Dominguez, E. Anakabe, I. Osante, I. Tellitu and J. L. Vicario, J. Org. Chem., 1999, 64, 1115 CrossRef.
- For biological activity related to 3-substituted-1,2,3,4-tetrahydroisoquinolines: T. Saitoh, A. Yamashita, K. Abe, K. Ogawa, M. Kitabake, K. Taguchi and Y. Horiguchi, Med. Chem., 2008, 4, 531 CrossRef CAS; G. L. Grunewald, M. R. Seim, J. Lu, M. Nakbaud and K. R. Criscione, J. Med. Chem., 2006, 49, 2939 CrossRef; G. L. Grunewald, T. M. Caldwell, Q. Li and K. R. Criscione, J. Med. Chem., 1999, 42, 3315 CrossRef; G. L. Grunewald, V. H. Dahanukar, B. Tok and K. R. Criscione, J. Med. Chem., 1999, 42, 1982 CrossRef; G. L. Grunewald, V. H. Dahanukar, P. Ching and K. R. Criscione, J. Med. Chem., 1996, 39, 3539 CrossRef.
- B. K. Peters, S. K. Chakka, T. Naicker, G. E. M. Maguire, H. G. Kruger, P. G. Andersson and T. Governder, Tetrahedron: Asymmetry, 2010, 21, 679 CrossRef CAS.
- S. Caddick, D. B. Judd, A. K. K. Lewis, M. T. Reich and M. R. V. Williams, Tetrahedron, 2003, 59, 5417 CrossRef CAS.
- (R)-9a: [α]20D −110.0 (c 0.6, CHCl3) (>99% ee) in comparison with previously described [α]20D −113.6 (c 1, CHCl3) in ref. 21 ( T. Saitoh, A. Yamashita, K. Abe, K. Ogawa, M. Kitabake, K. Taguchi and Y. Horiguchi, Med. Chem., 2008, 4, 531 CrossRef CAS ).
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20152f |
| This journal is © The Royal Society of Chemistry 2012 |