Cofactor promiscuity among F420-dependent reductases enables them to catalyse both oxidation and reduction of the same substrate
Gauri V.
Lapalikar
a,
Matthew C.
Taylor
*a,
Andrew C.
Warden
a,
Hideki
Onagi
b,
James E.
Hennessy
b,
Roger J.
Mulder
c,
Colin
Scott
a,
Susan E.
Brown
a,
Robyn J.
Russell
a,
Chris J.
Easton
b and
John G.
Oakeshott
a
aEcosystem Sciences, Commonwealth Scientific and Industrial Research Organisation, PO Box 1700, Canberra, Australian Capital Territory, Australia. E-mail: m.taylor@csiro.au; Fax: +61 2 6246 4094; Tel: +61 2 6246 4404
bARC Centre of Excellence for Free Radical Chemistry and Biotechnology, Research School of Chemistry, Australian National University, Canberra ACT 0200, Australia
cMaterials Sciences and Engineering, Commonwealth Scientific and Industrial Research Organisation, Clayton, Victoria, Australia
First published on 17th May 2012
Abstract
The recently reported F420H2-dependent reductases (FDRs) catalyse the reduction of aflatoxins and coumarin via hydrogenation of the α,β-unsaturated moiety. We report that three FDRs (MSMEG_2027, MSMEG_6848 and MSMEG_3356) from Mycobacterium smegmatis also exhibit a different catalytic function towards some aflatoxins through the use of a different cofactor. When F420 was replaced by FMN in these three enzymes, the aflatoxins AFG1 and AFG2 were oxidised via dehydrogenation, producing the reduced cofactor (FMNH2) and an unstable aflatoxin derivative that hydrolyses to an enol with three distinct structural isomers. Both the oxidation and reduction reactions are discussed in detail. This is the first example of an enzyme showing promiscuity for its cofactor leading to divergence of function against the same substrate.
Introduction
Enzymes that utilise the cofactor F420 are gaining interest because they are able to catalyse otherwise recalcitrant redox reactions. As well as playing a role in methanogenesis, F420 has been found to reduce sulphite,1 activate the antituberculosis drug PA-824,2,3 catalyse reductions on nitrophenols4 and the carcinogenic mycotoxin, aflatoxin,5 and is also involved in the biosynthesis of many antibiotics.6,7 F420 is a deazaflavin and shares the initial stage of its biosynthesis with the riboflavin biosynthetic pathway.8–10 Although structurally similar to riboflavin (Scheme 1), the differences between the two bestow F420 with a lower midpoint redox potential than both the riboflavins and also NAD(P)H (F420, −350 mV;11,12 FMN, −230 mV;13 and NAD(P)H, −320 mV14), but it is only capable of two-electron reductions.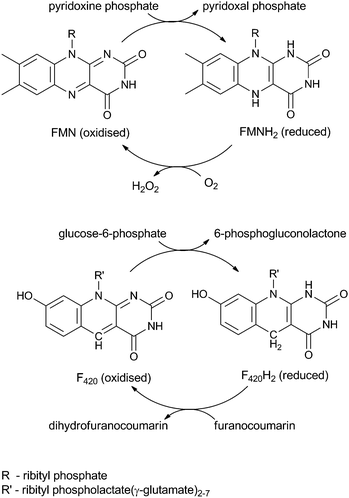 | ||
| Scheme 1 Recycling of FMN (top) in vitamin B6 biosynthesis and F420 (bottom) in aflatoxin degradation. | ||
Two families of F420-dependent reductases (FDRs) have been reported that catalyse the hydrogenation of aflatoxin and other plant derived furanocoumarins, apparently via the reduction of an α,β-unsaturated bond (Scheme 1).5 These FDRs have the same FMN-split barrel fold as the pyridoxine phosphate oxidase (PNPOx) enzymes that utilise FMN to oxidise pyridoxamine or pyridoxine phosphate (PMP/PNP) to pyridoxal phosphate (PLP) (Scheme 1).5,15–18 PLP is essential to all organisms and consequently the FMN-dependent PNPOx enzymes are highly conserved in all domains of life. PNPOx and the two families of FDRs show a low but significant level of sequence identity (<12%) despite their differing cofactor specificities.5 Structural analysis has shown that cofactor binding by both the FDRs and PNPOxs utilise similar, highly conserved amino acid residues.19
The FDRs have been shown to catalyse the reduction of coumarin and other furanocoumarins, acting upon the α,β-unsaturated lactone moieties of these molecules. One enzyme within the FDR-A family, Deazaflavin-dependent nitroreductase (Ddn), has also been shown to catalyse the reduction of nitrosubstrates, including, the anti-tuberculosis drug PA-824. These reactions are similar to the reactions of enoate reductase, old yellow enzyme (OYE)20 and Xenobiotic reductase A (XenA),21 which utilise NADH and FMN to catalyse the asymmetric reduction of alkene bonds in enoates.22 Given the interest in asymmetric synthesis, it is of interest to confirm that the FDR/F420-catalysed reaction on either aflatoxin or coumarin is indeed an alkene reduction.
Previous results show that the FDRs are not capable of utilising oxidised FMN to catalyse the penultimate step in PLP metabolism.5 However, if the FDRs catalyse the stereoselective reduction of coumarin in a similar fashion to XenA and OYE, the question remains as to whether the FDRs are capable of using reduced FMN to catalyse an enoate reduction in the presence of NADH.
We report herein that the FDR enzymes are indeed F420-dependent enoate reductases, albeit mechanistically distinct from the NAD/FMN-dependent OYE/XenA. Moreover, we demonstrate that some FDRs can utilise both F420 and FMN to catalyse different chemistries on the same substrate. To the best of our knowledge, this is the first example of this type of cofactor promiscuity.
Results and discussion
FDRs are enoate reductases
Previous LC-MS ToF analysis has shown that F420H2-bound FDR enzymes reduce all four of the major aflatoxins (AFB1, AFB2, AFG1 and AFG2).5 In that work, mass spectrometry-induced fragmentation of the AFG1 product suggested that the α,β-unsaturated ester bond was reduced by F420H2. Studies with coumarin and other furanocoumarins corroborate this hypothesis,23 although in all cases the products were rapidly further degraded rendering it difficult to confirm the reaction mechanism.To clarify the reaction products of the FDR/F420H2-catalysed reaction, we used coumarin (1, Scheme 2) as a substrate, because an authentic standard of the dihydro- form was commercially available. Analysis by LC-MS confirmed that the first intermediate reaction product of the FDR/F420H2-catalysed reaction was indeed dihydrocoumarin (2, Scheme 2), as indicated by both its retention time and molecular weight (data not shown). To confirm that the dihydrocoumarin product of the enzyme-catalysed reaction was spontaneously hydrolysed, the dihydrocoumarin standard was added to water at 28 °C and the hydrolysis product was confirmed to be 3-(2-hydroxyphenyl) propionic acid (3, Scheme 2) by LC-MS and 1H NMR after 72 hours (data not shown). This supports the proposed mechanism of furanocoumarin hydrolysis.23
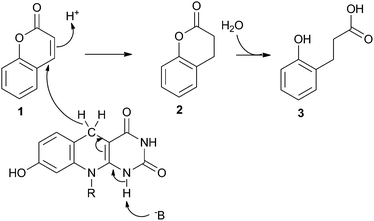
The reduction of coumarin by the OYE, XenA, utilising reduced FMN has previously been reported.21 In this reaction the double bond of the α,β-unsaturated ester is reduced by a hydride from FMNH2 and a solvent derived proton whose transfer is thought to be mediated through a highly conserved tyrosine residue24 which is close to the α-carbon of coumarin,21 although there is evidence that this conserved Tyr residue may not be the proton donor in all cases.25
Structural analysis of the F420-dependent Ddn binding PA-824 shows an analogous tyrosine to the catalytic one in XenA, however this residue is not conserved throughout the FDR-As. There is a second highly conserved tyrosine residue in the binding pocket of all aflatoxin degrading FDR-As. This conserved residue may not directly participate in substrate or cofactor binding in Ddn, but may participate in the formation of the binding pocket.19,23 The role of the conserved Tyr is unknown in the FDR-A family and further structural and mechanistic studies are required to elucidate its role in the reduction of substrates such as coumarin.
The reaction mechanism of OYE/XenA-dependent enoate reduction is responsible for determining the stereochemistry of their reaction products.26 As the FDR-As operate via an alternative mechanism, the stereochemistry of their products may be subject to quite different control, making these enzymes attractive targets for potential biocatalysts in industrial asymmetric syntheses.
FMN dependent oxidation by FDRs
As the FDRs are structurally and phylogentically related to the FMN dependent PNPOx, and they can catalyse the reduction of coumarin in a similar fashion to XenA and OYE, we hypothesised that the FDRs may be capable of using reduced FMN to catalyse an enoate reduction in the presence of NADH. In a preliminary experiment an FDR-B, MSMEG_6848, was found to catalyse the transformation of AFG1 and AFG2 (data not shown) when provided with FMNH2 produced in situ using an NADH-dependent flavin reductase (MsFR from Mycobacterium smegmatis).27 However, LCMS data suggested that the transformation catalysed by MSMEG_6848 increased the mass of AFG1 and AFG2 by 16 Da, which would be more consistent with an oxidation than a reduction.To assess if the transformation was an FMN-mediated oxidation or an FMNH2-dependent reduction, reactions were then carried out under stringent conditions. One reaction (with NADH and MsFR) was sealed with a rubber stopper and kept under nitrogen, in order to minimise spontaneous re-oxidation of FMN. In a second reaction, FMN was not reduced or sealed and was constantly mixed, in order to facilitate incorporation of O2 and oxidation of the FMN. Reaction products were only observed in the reaction containing the oxidised FMN (data not shown), suggesting that MSMEG_6848 was using oxidised FMN. Thus, this FDR behaves as a reductase when provided with F420H2 and as an oxidase when provided with FMN. In contrast, it has no discernable activity if provided with F420 or reduced FMNH2.
The activities of a further five FDR-A and five FDR-B enzymes from M. smegmatis and M. smegmatis PNPOx MSMEG_5675 were then analysed in the presence of oxidised FMN (data not shown). In addition to the FDR-B, MSMEG_6848, two FDR-A enzymes, MSMEG_3356 and MSMEG_2027, were also found to catalyse the oxidative transformation of AFG1 and AFG2 under these conditions. FMN alone, the remaining three FDR-A's (MSMEG_3004, 5998 and 2850), four FDR-B's (MSMEG_0048, 3380, 5717 and 5710) and PNPOx did not show any activity after a three hour incubation. None of the enzymes could utilise oxidised FMN to transform AFB1 or AFB2, nor PNP, as shown previously.5
There are several cases in the literature of individual enzymes using different cofactors for the same reactions. One example of this involves the p-hydroxyphenylacetate hydroxylase–flavin reductase complex isolated from Acinetobacter baumannii, which converts p-hydroxyphenylacetate to 3,4-dihydroxyphenylacetate in the presence of either FMNH2 or FADH2 by addition of a hydroxy group.28,29 A dibenzothiophene monooxygenase–flavin reductase complex has also been described from Mycobacterium goodii X7B which participates in the “4s” pathway,30 converting dibenzothiophene into dibenzothiophene sulphone in the presence of either FMNH2 or FADH2, eventually producing the desulphurisation inhibitor 2-hydroxybiphenyl.31
There are, however, also reports of enzymes using different cofactors for different reactions, albeit these cases are so far limited to metalloenzymes. For example, the dihydroxyacetone kinase from Citrobacter freundii CECT 4626 shows different catalytic activities if its Mg2+ ion is exchanged for a Mn2+ ion.32 In the presence of Mg2+, it catalyses ATP-dependent phosphorylation of dihydroxyacetone to give dihydroxyacetone phosphate and ADP. However, it functions as a cyclase in the presence of Mn2+, catalysing the splitting of an FAD molecule to release riboflavin 4′,5′-cyclic phosphate and an AMP molecule. Conversely, the rat and human FAD-AMP lyases (cyclases), which are distant homologues (40% amino acid identities) of the C. freundii kinases, can also function as dihydroxyacetone kinases depending on the relative concentrations of Mg2+ or Mn2+.33,34
To our knowledge this is the first case of an enzyme catalysing a different reaction (oxidation instead of a reduction) on the same substrate with a different cofactor.
Identification of the oxidation products
The unexpected evidence for two separate mechanisms with F420 and FMN led us to investigate the reactions further. LCMS analysis had already been carried out on the F420-dependent reduction reaction (see above and previously published reports5,23). LCMS of the FMN catalysed oxidation reaction showed that the initial product of AFG1 (Mass + H+ = 329.04 m/z) oxidation with FDR and FMN has a mass (Mass + H+ = 327.10 m/z) that suggests the loss of two hydrogen atoms. This dehydrogenation is rapidly followed by hydrolytic cleavage of the lactone ring giving a second product (Mass + H+ = 345.08 m/z). Unfortunately, the reduction product of the F420 mediated reaction which degraded rapidly and the interpretation of the NMR spectra is further complicated by the requirement for glucose-6-phosphate and F420-glucose-6-phosphate dehydrogenase to regenerate the cofactor. However, the oxidation reaction had no requirement for additional reagents to recycle the cofactor and the final breakdown products appeared to be more stable, enabling a thorough analysis of the mechanism by NMR. 1D and 2D COSY 1H NMR revealed several products from the oxidation of AFG1 by MSMEG_6848. The general reaction of AFG1 is shown in Scheme 3.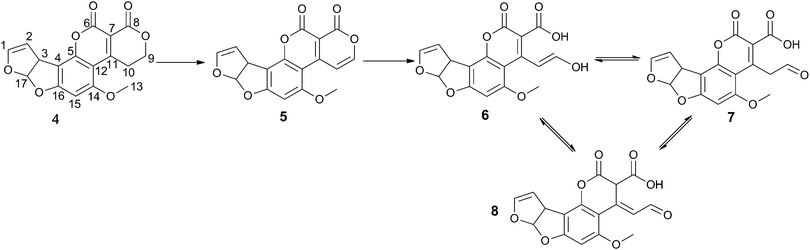 | ||
| Scheme 3 Products of enzyme-catalysed AFG1 breakdown. Atom numbering scheme used throughout is shown on 4. | ||
Upon addition of enzyme to the reaction mixture, 1D 1H NMR spectra were taken approximately every 10 minutes for 21 hours. After the first 10 minutes, two doublets (7.89 and 8.07 ppm) appeared, corresponding to the alkene hydrogens on C9 and C10 in 5 (see Fig. 1). This was accompanied by a concurrent decrease in the triplets associated with the two adjacent CH2 groups (3.62 and 4.51 ppm).
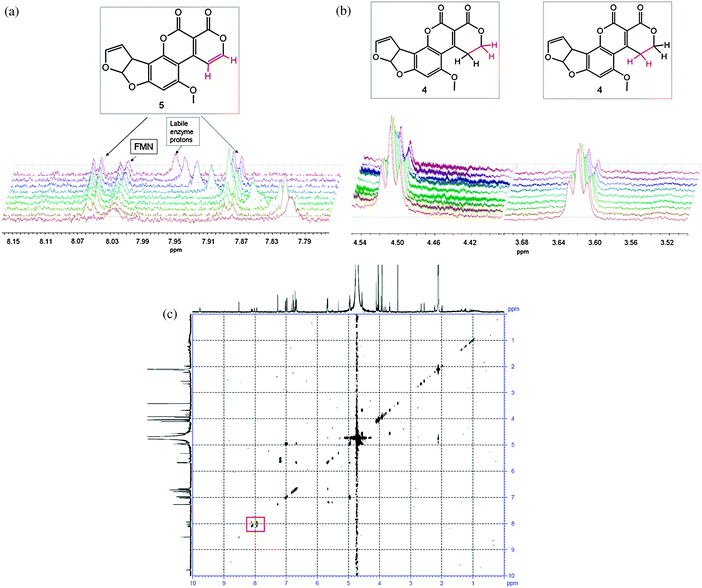 | ||
| Fig. 1 Appearance of doublets from the oxidised AFG1 (top left), the reduction of the triplets on the corresponding carbon atoms (top right) and the COSY spectrum of the final reaction mixture (bottom), with the signals from the alkene hydrogens in 5 indicated by a red square. The broad peak attributable to the labile enzyme protons shifts throughout the course of the reaction due to the change in pH/pD upon hydrolysis of the lactone of 5. The diagnostic groups are shown in red. | ||
The initial extension of the conjugated π-system through the dehydrogenation at C9/C10 caused a downfield shift of the singlets corresponding to hydrogens on C13 and C15 (Fig. 2). Shortly afterwards, appreciable amounts of the ring-opened product appeared, manifesting as three isomers (6, 7 and 8). These were each observed most clearly through two sets of three singlets at around 6.6 and 3.9 ppm attributable to resonances from the aromatic hydrogen on C15 and the methoxy CH3 group (C13), respectively (Fig. 2). There were corresponding shifts in the signals from the hydrogens on the furan moiety however these were less pronounced owing to their distance from the reaction site, and less easily distinguished than those from the hydrogens on C13 and C15.
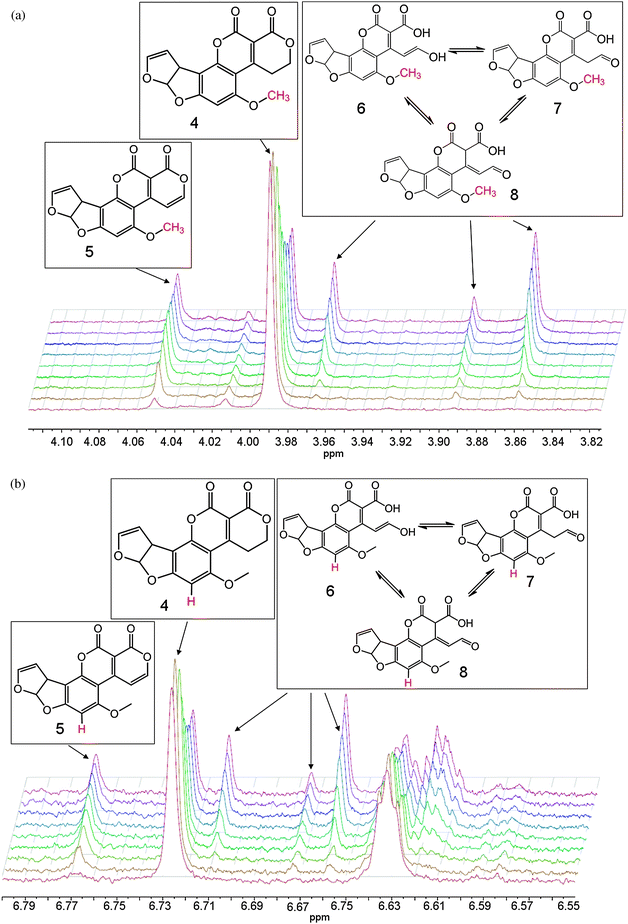 | ||
| Fig. 2 Sets of singlets corresponding to AFG1 (4), the oxidised product (5) and the ring-opened isomers (6, 7 and 8). The diagnostic groups are shown in red. | ||
We propose that two of the three isomers are generated through base-catalysed keto–enol tautomerism with the third being another resonance structure of the aldehyde form which is also produced through a solvent-mediated mechanism (see Scheme 4). This is consistent with the observation of only two products in the LC-MS spectra. Generally, keto–enol tautomerism strongly favours the keto (or aldehyde in this case) form, however it appears that the enol form (6) and the two aldehyde forms (7 and 8) are present at comparable concentrations (i.e. within the same order of magnitude). A plausible explanation for this observation is that the enol form is stabilised through conjugation with the extended π-system. The additional signals that would be expected from the hydrogen atoms on C7 and C10 in the hydrolysis product isomers were not seen in the spectra owing to deuterium exchange with the solvent. A small, slightly broadened singlet appears at 9.72 ppm that corresponds to the aldehyde hydrogen on C7. This signal increases steadily in intensity over the course of the reaction, suggesting a move towards the two aldehyde isomers, most likely driven by the change in pH as hydrolysis of 5 occurs. These two observations provide strong evidence for the proposed mechanism.
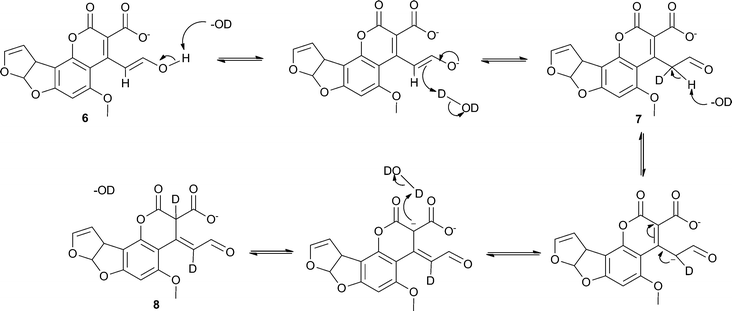 | ||
| Scheme 4 Proposed base-catalysed mechanism for the generation of the three isomers of the hydrolysis product of 5. | ||
Kinetic parameters
The specific activities of the FDR-B (MSMEG_6848) and the two FDR-A enzymes (MSMEG_3356 and MSMEG_2027) utilising FMN against AFG1 were then determined by quantifying substrate loss at defined intervals with saturating concentrations of each of the cofactors using a HPLC (Table 1). Although the differences were not large, the specific activity of the FDR-B MSMEG_6848 against AFG1 was the highest amongst the three FDR enzymes, as well as being higher than that observed with F420H2. The two FDR-A enzymes, however, still had over two-fold higher specific activities with F420H2 than FMN for AFG1 breakdown.| Enzyme | AFG1 | |||
|---|---|---|---|---|
| Rate (μmol/min/μmol enzyme) | K M (μM) | |||
| F420H2 | FMN | F420H2 | FMN | |
| FDR-B | ||||
| MSMEG_6848 | 0.8 ± 0.1 | 1.2 ± 0.2 | 0.76 ± 0.15 | 7.9 ± 1.7 |
| FDR-A | ||||
| MSMEG_3356 | 2.0 ± 0.3 | 0.4 ± 0.04 | 0.41 ± 0.1 | 45.0 ± 13 |
| MSMEG_2027 | 1.3 ± 0.2 | 0.5 ± 0.03 | 0.62 ± 0.28 | 42.8 ± 15 |
The Michaelis constants (KM) of the three enzymes above for F420 and FMN were also estimated (Table 1). The KM of MSMEG_6848 was almost 10-fold lower with F420H2 than with FMN whilst the KM of MSMEG_3356 and MSMEG_2027 were approximately 100-fold lower for F420H2 than with FMN (Table 1). Thus the two FDR-A enzymes, MSMEG_3356 and MSMEG_2027 showed tighter binding and higher AFG1 reduction activity with F420H2 than FMN, while MSMEG_6848 showed tighter binding to F420H2 but faster substrate transformation rate with FMN (Table 1). This indicates an apparent correlation between binding constant and reaction rate, however, the small sample size of enzymes with this promiscuous activity prevents us from drawing any firm conclusions as to whether this is a general characteristic of these enzymes.
The binding constants are also informative in respect of the possible physiological significance of the FMN- and F420-based reactions. F420 is typically present at an intracellular concentration of 0.3 μmol g−1 dry weight,35 (approx. 300 μM) in M. smegmatis. Therefore, the intracellular concentrations of F420 exceed the three enzymes' KM for this cofactor, consistent with their known physiological cofactor requirements. To the best of our knowledge there are no reports on the comparative concentrations of FMN and F420 in M. smegmatis, or reports on the relative concentrations of riboflavin, FMN and FAD concentrations in this organism. However, the concentration of FMN may potentially be 10-fold higher than the F420, based on a total riboflavin content of M. smegmatis of less than 5 μmol g−1 dry weight.36 For most of the F420 dependent enzymes this suggests that FMN is not present at a sufficiently high intracellular concentration to function as a physiologically relevant cofactor. In contrast, both cofactors may compete for the active site of MSMEG_6848, in vitro.
Structural analysis of both the PNPOx and FDR enzymes provides some insight as to why some FDR enzymes have cofactor promiscuity. FMN and F420 share a common synthetic pathway which results in them having a conserved ribityl phosphate chain moiety.10 This common structure of the cofactors has extensive hydrogen bond interactions with a conserved cofactor binding groove formed mainly by the first two β-sheets of the conserved six anti-parallel β-barrel sheets in both the PNPOx and FDR enzymes.17,19 Of the residues that bind the ribityl phosphate chain, there is only one highly conserved residue across all three classes that has extensive hydrogen bonds with both the phosphate and ribityl chains. This is either a lysine in PNPOx/FDR-Bs and tryptophan residue in the FDR-As on the helix between β-sheets 3 and 4.5 No other residues are as highly conserved across all enzyme classes, indicating the importance of this residue in cofactor binding. Additionally, in PNPOx, the active site is partially shielded by the C-terminus of the second chain of the dimer, which is likely to account for the tight binding of FMN (KD 30 nM)37 and the amino acid sequence of the active site is highly conserved between species,17 which is consistent with the conserved function of PNPOx. Indeed, we did not observe any activity of the M. smegmatis PNPOx, MSMEG_5675, with AFG1. Conversely, the FDR's have a truncated C-terminus, leaving the active site open. This open active site would relieve the steric constraints on the cofactor and enable F420 to be diffusible, such that it can be recycled by F420 dependent glucose-6-phosphate dehydrogenase. This more open active site would also enable the enzymes to bind other cofactors with a conserved ribityl chain and account for the cofactor promiscuity shown here. Further investigations into the cofactor range of these promiscuous enzymes are warranted with other riboflavins such as FAD and roseoflavin, deazaflavins, and other cofactors that have a common ribityl phosphate chain moiety.
Materials and methods
Chemicals used
FMN, reduced nicotinamide adenine dinucleotide (NADH), glucose-6-phosphate, coumarin, dihydrocoumarin and aflatoxins B1, G1, B2 and G2 were purchased from Sigma-Aldrich, and used as received. Deuterated solvents were purchased from Cambridge Isotopes Laboratories. F420 was prepared from M. smegmatis mc2155 soluble fractions based on the method of Isabelle et al.35 with modifications.5Cloning, expression and purification
Eleven FDR genes (MSMEG_3380, 6848, 5717, 5170, 5819, 0048, 2850, 2027, 3356, 3004 and 5998) containing 6-his tag sequences cloned into the Gateway™ destination vector pDEST17 were expressed and their protein products purified as described by Taylor et al.5 MsFR was expressed from the pET14b plasmid (a gift from Dr Sutherland) following previously published methods.27Enzyme activity assays
Qualitative experiments utilised a modified version of the LC-MS assay in Taylor et al. (2010)5 to test for the degradation of aflatoxins B1, G1, B2 and G2 by the eleven FDR enzymes with FMNH2 at 37 °C. The reaction components for a 20 μl reaction were: 100 μM AFG1 (1 mg ml−1 stock), 20 μM FMN, 10 mM NADH, 0.5 μl crude extract of M. smegmatis flavin reductase (MsFR),27 10 μM FDR enzyme and 50 mM Tris–HCl, pH 7.5. The reaction was initiated by the addition of the AFG1 and incubated for 180 minutes. Each reaction was stopped by the addition of 10% acetic acid to a final concentration of 5%, and then centrifuged and analysed by LC-MS ToF.The conditions used for quantitation of activities for the eleven FDR enzymes with oxidised FMN in 100 μl reactions were: 100 μM AFG1, 200 μM FMN, 10 μM FDR enzyme and 50 mM Tris–HCl, pH 7.5. The reactions were initiated by addition of AFG1 and were incubated at 22 °C and sampled every 15 minutes for a total of 180 minutes on a HPLC autosampler (Agilent) to monitor AFG1 loss. For the experiments to estimate the Michaelis constants, FMN concentrations ranging from 5 to 200 μM and F420H2 concentrations ranging from 0.1 to 10 μM were used. The KM values were determined by fitting the measured specific activities into the one site saturation module of the Michaelis–Menten equation using Sigma Plot (Systat Software Inc., USA).
LC-MS and HPLC analysis
Products from the reactions using F420H2 or FMN were determined qualitatively on an Agilent 1100 Series Binary LC with diode array detector and in-line Time of Flight (ToF) Mass Spectrometer ESI (+ve), scanning at two fragmentor voltages of 140 V and 225 V. The LC-MS analysis method was the same as described by Lapalikar et al.23 Briefly, mobile phase A was 0.1% formic acid in water (v/v) and mobile phase B was 0.1% formic acid in acetonitrile (v/v). Substrate loss was monitored at 365 nm by gradient elution on a Zorbax XDB C18 column (3.5 μm, 2.1 × 30 mm). The gradient scheme was as follows: 10% solvent B (acetonitrile) for 0.25 minutes, increased to 20% over 1 minute, increased to 40% over 5 minutes, increased to 60% over 5.2 minutes, then decreased to 10% over 5.5 minutes, and finally 10% solvent B for an additional 2 minutes at a flow rate of 0.6 ml/minute. The reaction products were analysed using Analyst QS software (Agilent, Australia).AFG1 loss with the two cofactors was determined quantitatively on an Agilent 1200 series HPLC with an autosampler by injecting the samples at 2 minute intervals on a Zorbax XDB C18 column. The reaction components were separated isocratically with 30% acetonitrile and 0.5% acetic acid for aflatoxin detection at 365 nm and the loss of AFG1 was quantified over 60 and 180 minutes for the F420H2 and FMN reactions respectively, using the Chemstation Software (Agilent).
Nuclear magnetic resonance spectroscopy (1H NMR)
To confirm the spontaneous hydrolysis of dihydrocoumarin, 10 mg dihydrocoumarin was added to 200 ml Milli-Q water and left on a shaker at 37 °C for 72 hours. The reaction mixture was then frozen under liquid nitrogen and freeze dried. The residual powder was dissolved in d3-acetonitrile. The product was analysed at 25 °C on an Inova 300 NMR spectrometer operating at 300 MHz via1H NMR at the Research School of Chemistry, Australian National University, Canberra, Australia.To determine the FDR/FMN-catalysed AFG1 breakdown product, a reaction of 1 ml was conducted consisting of 1 mM AFG1, 20 μM FMN, 2 μM MSMEG_6848 in 50 mM K2HPO4, pH 9.0, and 10% deuterated acetonitrile (to reduce the reaction rate and allow unambiguous identification of reaction products as they appeared). NMR spectroscopy was carried out on a Bruker BioSpin Av600 NMR spectrometer with a 5 mm 1H–13C–15N Cryoprobe operating at 600.27 MHz for 1H. The sample was maintained at 30 °C. 1H NMR spectra of 128 scans (approx. 10 minutes duration) were collected every approx. 10 minutes over a period of 16 hours. After approx. 21 elapsed hours a COSY and an HSQC experiment were collected to aid in determining the structure of the products. Processing of all 1D spectra was carried out using the MestReNova software.
Conclusions
The FDR-A and -B enzymes from the Actinomycetes represent unique families of enzymes, some members of which are able to both oxidise and reduce AFG1 by using different cofactors. 1H NMR and LC-MS analyses have enabled the identification of the products of the oxidation reaction between AFG1 and FMN catalysed by an FDR enzyme, and we have proposed a degradation pathway and mechanism. The reduction of AFG1 via these enzymes utilising F420 was not as easily characterised and was studied vicariously through the analogous reaction using coumarin as a substrate. We confirm that these enzymes catalyse an enoate reduction of coumarin which is spontaneously hydrolysed, and most likely perform a similar reduction on the α,β-unsaturated moiety of AFG1. Further work is underway to determine the mechanism. The precise role of the conserved tyrosine in the active site pocket in both reactions is as yet unknown and future work will focus on clarifying the part it plays in the aflatoxin breakdown pathway through structural analysis. Enzyme kinetic analysis suggests that F420 is the most likely cofactor for these enzymes under physiological conditions. However, the FDR enzymes that display cofactor promiscuity offer a unique opportunity to study the evolution of cofactor usage and also provide an opportunity to help us understand the constraints required for incorporating new cofactors that may enable us to catalyse industrially relevant reactions.Acknowledgements
G.L. was supported by a PhD scholarship through The Commonwealth Scientific and Industrial Research Organisation (CSIRO) Synthetic Enzymes Emerging Science Initiative.Notes and references
- E. F. Johnson and B. Mukhopadhyay, J. Biol. Chem., 2005, 280, 38776–38786 CrossRef CAS.
- K. P. Choi, T. B. Bair, Y. M. Bae and L. Daniels, J. Bacteriol., 2001, 183, 7058–7066 CrossRef CAS.
- U. H. Manjunatha, H. Boshoff, C. S. Dowd, L. Zhang, T. J. Albert, J. E. Norton, L. Daniels, T. Dick, S. S. Pang and C. E. Barry, 3rd, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 431–436 CrossRef CAS.
- S. Ebert, P. Fischer and H. J. Knackmuss, Biodegradation, 2001, 12, 367–376 CrossRef CAS.
- M. C. Taylor, C. J. Jackson, D. B. Tattersall, N. French, T. S. Peat, J. Newman, L. J. Briggs, G. V. Lapalikar, P. M. Campbell, C. Scott, R. J. Russell and J. G. Oakeshott, Mol. Microbiol., 2010, 78, 561–575 CrossRef CAS.
- W. Li, A. Khullar, S. Chou, A. Sacramo and B. Gerratana, Appl. Environ. Microbiol., 2009, 75, 2869–2878 CrossRef CAS.
- T. Nakano, K. Miyake, H. Endo, T. Dairi, T. Mizukami and R. Katsumata, Biosci., Biotechnol., Biochem., 2004, 68, 1345–1352 CrossRef CAS.
- K. P. Choi, N. Kendrick and L. Daniels, J. Bacteriol., 2002, 184, 2420–2428 CrossRef CAS.
- M. Fischer and A. Bacher, Physiol. Plant., 2006, 126, 304–318 CrossRef CAS.
- D. E. Graham, H. Xu and R. H. White, Arch. Microbiol., 2003, 180, 455–464 CrossRef CAS.
- L. M. I. de Poorter, W. J. Geerts and J. T. Keltjens, Microbiology, 2005, 151, 1697–1705 CrossRef CAS.
- F. Jacobson and C. Walsh, Biochemistry (Moscow), 1984, 23, 979–988 CrossRef CAS.
- R. D. Draper and L. L. Ingraham, Arch. Biochem. Biophys., 1968, 125, 802–808 CrossRef CAS.
- K. Kakinuma, M. Kaneda, T. Chiba and T. Ohnishi, J. Biol. Chem., 1986, 261, 9426–9432 CAS.
- B. K. Biswal, K. Au, M. M. Cherney, C. Garen and M. N. James, Acta Crystallogr., Sect. F: Struct. Biol. Cryst. Commun., 2006, 62, 735–742 Search PubMed.
- S. Canaan, G. Sulzenbacher, V. Roig-Zamboni, L. Scappuccini-Calvo, F. Frassinetti, D. Maurin, C. Cambillau and Y. Bourne, FEBS Lett., 2005, 579, 215–221 CrossRef CAS.
- M. K. Safo, I. Mathews, F. N. Musayev, M. L. di Salvo, D. J. Thiel, D. J. Abraham and V. Schirch, Structure, 2000, 8, 751–762 CrossRef CAS.
- M. L. di Salvo, M. K. Safo, F. N. Musayev, F. Bossa and V. Schirch, Biochim. Biophys. Acta, 2003, 1647, 76–82 CAS.
- S. E. Cellitti, J. Shaffer, D. H. Jones, T. Mukherjee, M. Gurumurthy, B. Bursulaya, H. I. Boshoff, I. Choi, A. Nayyar, Y. S. Lee, J. Cherian, P. Niyomrattanakit, T. Dick, U. H. Manjunatha, C. E. Barry, 3rd, G. Spraggon and B. H. Geierstanger, Structure, 2012, 20, 101–112 CrossRef CAS.
- R. E. Williams and N. C. Bruce, Microbiology, 2002, 148, 1607–1614 CAS.
- J. J. Griese, R. P. Jakob, S. Schwarzinger and H. Dobbek, J. Mol. Biol., 2006, 361, 140–152 CrossRef CAS.
- M. Hall, C. Stueckler, W. Kroutil, P. Macheroux and K. Faber, Angew. Chem., Int. Ed., 2007, 46, 3934–3937 CrossRef CAS.
- G. V. Lapalikar, M. C. Taylor, A. C. Warden, C. Scott, R. J. Russell and J. G. Oakeshott, PLoS One, 2012, 7, e30114 CAS.
- R. M. Kohli and V. Massey, J. Biol. Chem., 1998, 273, 32763–32770 CrossRef CAS.
- H. Khan, T. Barna, N. C. Bruce, A. W. Munro, D. Leys and N. S. Scrutton, FEBS J., 2005, 272, 4660–4671 CrossRef CAS.
- K. Durchschein, W. M. Fabian, P. Macheroux, K. Zangger, G. Trimmel and K. Faber, Org. Biomol. Chem., 2011, 9, 3364–3369 CAS.
- T. D. Sutherland, I. Horne, R. J. Russell and J. G. Oakeshott, Appl. Environ. Microbiol., 2002, 68, 6237–6245 CrossRef CAS.
- P. Chaiyen, C. Suadee and P. Wilairat, Eur. J. Biochem., 2001, 268, 5550–5561 CrossRef CAS.
- P. Xu, J. C. Li, J. H. Feng, Q. Li, C. Q. Ma, B. Yu, C. Gao and G. Wu, Bioresour. Technol., 2009, 100, 2594–2599 CrossRef.
- P. Xu, B. Yu, F. L. Li, X. F. Cai and C. Q. Ma, Trends Microbiol., 2006, 14, 398–405 CrossRef CAS.
- H. Chen, W. J. Zhang, Y. B. Cai, Y. Zhang and W. Li, Bioresour. Technol., 2008, 99, 6928–6933 CrossRef CAS.
- I. Sanchez-Moreno, L. Iturrate, R. Martin-Hoyos, M. L. Jimeno, M. Mena, A. Bastida and E. Garcia-Junceda, ChemBioChem, 2009, 10, 225–229 CrossRef CAS.
- F. J. Fraiz, R. M. Pinto, M. J. Costas, M. Aavalos, J. Canales, A. Cabezas and J. C. Cameselle, Biochem. J., 1998, 330(Pt 2), 881–888 CAS.
- A. Cabezas, M. J. Costas, R. M. Pinto, A. Couto and J. C. Cameselle, Biochem. Biophys. Res. Commun., 2005, 338, 1682–1689 CrossRef CAS.
- D. Isabelle, D. R. Simpson and L. Daniels, Appl. Environ. Microbiol., 2002, 68, 5750–5755 CrossRef CAS.
- R. L. Mayer and M. Rodbart, Arch. Biochem., 1946, 11, 49–63 CAS.
- H. Wada and E. E. Snell, J. Biol. Chem., 1961, 236, 2089–2095 CAS.
| This journal is © The Royal Society of Chemistry 2012 |