Effect of zirconium on the structure and activity of Cu/Ti1−xZrxO2 catalysts for selective catalytic reduction of NO with C3H6
Jie
Liu
a,
Xinyong
Li
*ab,
Qidong
Zhao
a and
Dongke
Zhang
*b
aKey Laboratory of Industrial Ecology and Environmental Engineering and Key Laboratory of Fine Chemical, School of Environmental Sciences and Technology, Dalian University of Technology, Dalian, China. E-mail: xinyongli@hotmail.com; Fax: +86-411-8470-8084; Tel: +86-411-8470-7733
bCentre for Energy (M473), The University of Western Australia, 35 Stirling Highway, Crawley, WA 6009, Australia. E-mail: dongke.zhang@uwa.edu.au; Fax: +61-8-6488-7235; Tel: +61-8-6488-7600
First published on 19th April 2012
Abstract
Mixed oxides of 5 wt% Cu/Ti1−xZrxO2 (x = 0, 0.3, 0.5, 0.7, 1.0) were synthesized using the impregnation–urea coprecipitation method and the catalysts prepared were assessed for the selective catalyst reduction (SCR) of NO with C3H6. A significant improvement of the catalytic activity was found over the Zr-doped catalysts with the highest N2 yield of 67.2% achieved over Cu/Ti0.7Zr0.3O2 at 275 °C. Based on the characterizations using BET, ICP, XRD, H2-TPR, FTIR of adsorbed pyridine, XPS, and TEM, the enhanced SCR activity of Cu/Ti0.7Zr0.3O2 was attributed to the large surface area, appropriate structural disorder, abundant Lewis acid sites, increase in the amount of active sites, and greater lattice oxygen mobility. The specific physico-chemical properties were caused by the distortion of the crystal structure and the change of the electron charge density (ECD) due to the introduction of Zr species. The generation of mixed Ti–O–Zr bond was beneficial for the adsorption of NO and the oxidation of NO to NO2, which favored the SCR reaction.
1. Introduction
Due to its contributions to the formation of acid rain and photochemical smog, NOx is regarded as one of the most dangerous atmospheric pollutants during the combustion of fossil fuels in the sectors of industry and transport.1,2 The increasingly stringent environmental regulations limiting atmospheric NOx emissions encourage research into the abatement of NOx in more efficient ways. The selective catalytic reduction of NOx by hydrocarbons (HC-SCR) under lean burn conditions, which can eliminate NOx and unburnt hydrocarbons simultaneously, has proven to be an effective and economical technique for the removal of NOx, especially NOx originating from the mobile sources.3As a support or a component, TiO2 has been employed in a number of commercially important heterogeneous catalyst systems. It has been widely utilized as a support in the SCR of NO. Due to the unique physico-chemical properties and the possible synergistic effects of multicomponent systems, mixed oxides have been researched as novel catalysts with higher specific surface area, mechanical strength and thermal stability, which influence the efficiency and the durability of catalysts.4 The mixed oxide catalysts have exhibited potential in the field of catalysis, energy storage, and electronic applications.5,6 In view of its good redox and acid–base amphoteric properties, ZrO2 has been extensively used as co-catalyst or additive to inhibit the crystal transformation.7 Owing to the different ionic radii of Zr4+ (0.87 Å) and Ti4+ (0.64 Å), the incorporation of Zr4+ into TiO2 lattice leads to the distortion of lattice and the formation of defects, which strongly impact the physical and redox properties of materials, and therefore benefits the process of NO reduction.8,9 In addition, the ZrO2 based catalysts have been shown to exhibit excellent sulfur tolerance in C3H6-SCR of NO.10
Among the options available, transition metal oxides are probably the best candidates for SCR of NOx,11,12 and the copper-based catalysts have become very attractive from the viewpoint of cost as well as efficacy.13 Catalysts that contain highly dispersed CuO on a variety of supports have demonstrated competitive levels of activity, stability, and durability under simulated operating conditions.14 There is much evidence that isolated copper ions are the active sites for the HC-SCR, and small CuO crystallites can accelerate the direct oxidation of hydrocarbons.13,15 Moreover, due to the interaction between the supports and the active species, the properties of the supported active components are often different from those of the corresponding bulk metal oxides. Therefore, the catalytic properties of the active copper phase can be greatly influenced by the dispersion of copper species and the nature of the supports.16,17 Owing to the advantages of mixed oxides mentioned above, many studies have been carried out to develop novel supports by adding additives to promote the dispersion of copper species.18
Based on the idea of combining the advantages of Cu-based catalysts and TiO2–ZrO2 mixed oxides support, we fabricated the Cu/Ti1−xZrxO2 catalysts for C3H6-SCR, and the performance of the Cu/Ti1−xZrxO2 catalysts with different Zr/(Ti+Zr) atomic ratios was investigated systematically. The present study focused on the influence of Zr introduction on the structure of the catalysts and the chemical state of copper species. Moreover, important information about the changes caused by the Zr-doping in the crystal structure and the electron charge density were investigated using the density functional theory (DFT) calculations, which offered a better understanding of the structure–activity relationship.
2. Experimental
2.1. Catalysts preparation
Ti1−xZrxO2 with different amounts of Zr substitution (x = 0, 0.3, 0.5, 0.7, 1) were synthesized using a facile urea precipitation method with TiOSO4·2H2O and ZrOCl·8H2O as the precursors and CO(NH2)2 as the precipitator. The mixed solution was continuously stirred at 90 °C for 8 h. After filtration and washing with deionized water and ethanol, the obtained precipitant was dried at 100 °C for 12 h and calcined at 500 °C for 4 h under static air conditions.The Cu/Ti1−xZrxO2 catalysts were prepared using the wet impregnation method with Cu(NO3)2 solution. The catalyst was dried at 100 °C for 12 h and calcined at 400 °C for 4 h in air condition. The Cu loading was 5 wt%.
2.2. Catalyst characterization
The Brunauer–Emmett–Teller (BET) surface areas of the catalysts were measured by physical adsorption of N2 at −196 °C using NOVA 1200 (Quanta Chrome). The sample was degassed at 300 °C for 4 h under vacuum prior to the BET determination.Inductively coupled plasma-optical emission spectrometry (ICP-OES) was performed to determine the actual copper loading of various catalysts on a Perkin-Elmer Optima 3300 DV apparatus.
The powder X-ray diffraction (XRD) was performed on a Rigaku D/Max 2550VB/PC X-ray powder diffractometer using a Cu-Kα radiation (λ = 0.154056 nm). The diffraction patterns of the samples were recorded at room temperature in the range of 2θ between 10 and 80°.
The temperature-programmed reduction of H2 (H2-TPR) was carried out on a Chembet PULSAR TPR/TPD (p/n 02139-1). Prior to the measurement, the sample (50 mg) was pretreated at 250 °C for 1 h in an Ar flow followed by cooling to room temperature. The TPR measurement was carried out from 50 °C to 700 °C at a heating rate of 10 °C min−1 in a stream of 10 vol% H2/Ar at 50 mL min−1. The H2 consumption was monitored by a TCD detector.
FTIR of adsorbed pyridine was performed to study the nature of surface acid sites using a VERTEX 70-FTIR spectrometer with a smart collector. Prior to each experiment, the sample (20 mg) was degassed at 250 °C for 1 h under vacuum. Pyridine molecules were adsorbed at room temperature, and the spectra were recorded at different temperatures desired.
X-ray photoelectron spectra (XPS) of the catalysts were recorded on a using Al-Kα radiation (1486.7 eV). Binding energies of Cu 2p, O 1s and Ti 2p were calibrated using C 1s (BE = 285.0 eV) as a standard.
TEM and high-resolution transmission electron microscopy (HR-TEM) measurements were carried out using a JEOL-2100 microscope.
The in situ FTIR experiments of NO adsorption over Cu/Ti1−xZrxO2 catalysts were performed on a VERTEX 70-FTIR. Prior to NO adsorption, the sample (approximately 20 mg) was pretreated at 250 °C in a flow of He for 1 h. The spectra of NO adsorption at different temperatures were selected over various catalysts. The reaction condition was controlled as follows: 1000 ppm NO and He as balance, and the flow rate was 30 mL min−1.
2.3. Catalytic performance test
The C3H6-SCR catalytic tests were performed in a fixed-bed quartz tube reactor. Prior to a experiment, a catalyst (200 mg) was pretreated at 250 °C for 1 h in an Ar stream, and the activity measurement was carried out from 150 °C to 350 °C at a heating rate of 10 °C min−1. The reaction conditions were as follows: 1000 ppm NO, 1000 ppm C3H6, 2.5 vol% O2 and He as balance, the total gas flow rate was 100 mL min−1 and the GHSV was 30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1. The concentrations of N2 and C3H6 in the effluent gas were analyzed on-line using a gas chromatograph. The conversion of NO to N2 and the C3H6 conversion were calculated as follows:
000 h−1. The concentrations of N2 and C3H6 in the effluent gas were analyzed on-line using a gas chromatograph. The conversion of NO to N2 and the C3H6 conversion were calculated as follows:| NO conversion to N2 = 2[N2]out/[NO]in × 100% | (1) |
| C3H6 conversion = (1 − [C3H6]out/[C3H6]in) × 100% | (2) |
2.4. Computational details
The atomic positions of anatase TiO2 and Ti0.7Zr0.3O2 and the corresponding electronic structure were obtained by DFT calculation from the CASTEP code.19 The Perdew–Burke–Ernzerh of gradient corrected functional (PBE-GGA) was used.20 The lattice parameters are a = 3.784 Å, c = 9.514 Å for anatase TiO2, and a = 4.711 Å, b = 5.494 Å, c = 4.996 Å for Ti0.7Zr0.3O2. Two different supercells of TiO2 (1 × 1 × 1) and Ti0.7Zr0.3O2 (2 × 1 × 1) were considered to test the possible interactions between the Ti and Zr atoms.3. Results and discussion
3.1. Catalytic test
The N2 yield and C3H6 conversion as a function of temperature over the Cu/Ti1−xZrxO2 catalysts are shown in Fig. 1. The Cu/TiO2 achieved the maximum N2 yield of 36.7% at 250 °C, while the activity of ZrO2 doped catalysts was enhanced significantly, though the temperature corresponding to the maximum N2 yield shifted to a higher temperature of 275 °C. Among the tested catalysts, Cu/Ti0.7Zr0.3O2 exhibited satisfactory catalytic performance, with a maximum N2 yield of 67.2%. In addition, the Cu/ZrO2 catalyst had poor activity based on both N2 yield and C3H6 conversion. It has been suggested that oxygenated hydrocarbon formation is crucial to the production of NCO species, which have been considered as important intermediates for HC-SCR.21 The unsatisfactory performance of Cu/ZrO2 may be attributed to the poor activity of activating C3H6. From Fig. 1(b), it can be observed that the Cu/TiO2 catalyst showed the highest C3H6 conversion. Combining the result of N2 yield, it can be assumed that the Zr-doping inhibited the side reaction of C3H6 oxidation, which occurred more readily over Cu/TiO2, and facilitated the activation of C3H6 to react with NO, and therefore promoted the conversion of NO to N2.![N2 yield (a) and C3H6 conversion (b) as a function of temperature over different Cu/Ti1−xZrxO2 catalysts. Reaction conditions: [NO] = [C3H6] = 1000 ppm, [O2] = 2.5 vol%, He as balance gas, total gas flow rate 100 mL min−1, GHSV = 30 000 h−1.](/image/article/2012/CY/c2cy20121f/c2cy20121f-f1.gif) | ||
| Fig. 1 N2 yield (a) and C3H6 conversion (b) as a function of temperature over different Cu/Ti1−xZrxO2 catalysts. Reaction conditions: [NO] = [C3H6] = 1000 ppm, [O2] = 2.5 vol%, He as balance gas, total gas flow rate 100 mL min−1, GHSV = 30000 h−1. | ||
3.2. BET measurement and elemental composition analysis
The physical properties of various catalysts including specific surface areas, pore volumes, average pore diameters and the actual copper loading measured by ICP-OES are summarized in Table 1. The physical properties of the suite of catalysts demonstrate that the addition of Zr helped to increase the active surface area of catalyst significantly, which favored the heterogeneous reaction. Cu/Ti0.7Zr0.3O2 showed the largest surface area of 327.2 m2 g−1, nearly twice that of Cu/TiO2. The most dramatic losses of specific surface area and pore volume were observed on the Cu/ZrO2 sample, and Cu/TiO2 showed the largest average pore diameter. It has been reported that the large pore volume might be beneficial for the catalytic activity, while a large average pore diameter might be one of the reasons for the declining catalytic activity.22 Thus, the excellent performance of Cu/Ti0.7Zr0.3O2 is considered to be related to the larger surface area and pore volume, and the moderate pore diameter.| Catalyst | Surface area (m2 g−1) | Pore volume (m3 g−1) | Average pore diameter (nm) | Cu concentration (wt%) |
|---|---|---|---|---|
| Cu/TiO2 | 179.2 | 0.3022 | 6.746 | 4.873 |
| Cu/Ti0.7Zr0.3O2 | 327.2 | 0.3668 | 4.484 | 4.786 |
| Cu/Ti0.5Zr0.5O2 | 316.9 | 0.3156 | 3.984 | 4.772 |
| Cu/Ti0.3Zr0.7O2 | 234.6 | 0.2662 | 4.540 | 4.441 |
| Cu/ZrO2 | 17.8 | 0.0298 | 3.788 | 4.168 |
The concentrations of Cu in the different catalysts are also displayed in Table 1. The Cu contents were similar and almost identical to the theoretical content in Cu/TiO2, Cu/Ti0.7Zr0.3O2 and Cu/Ti0.5Zr0.5O2, while a small amount of Cu was lost in the impregnation process for Cu/Ti0.3Zr0.7O2 and Cu/ZrO2. Since a larger surface area promotes the dispersion of copper species, it is rational to conclude that the high activity is related to the concentration of highly dispersed copper species, but not to the total content of copper.
3.3. XRD
The XRD patterns of the Cu/Ti1−xZrxO2 catalysts are shown in Fig. 2. Cu/TiO2 and Cu/ZrO2 featured the typical anatase phase of titania (ICSD 009852) and tetragonal phase of zirconia (ICSD 070014). For the Zr-doping samples, a broad diffraction peak with weak intensity centering at 31.7° was evident, indicating that the mixed oxides were in highly amorphous or poorly crystalline states. Thus, it may be concluded that the Zr-doping inhibited the crystal nucleus growing of TiO2, and consequently helped to increase the surface areas and the pore volumes, beneficial to the enhancement of the SCR activity.23,24 Moreover, ZrO2 can react with TiO2 to form a discontinuous solid solution, which is demonstrated by the diffraction peaks pertaining to ZrTiO4.25 For the Cu/ZrO2 catalyst, the diffraction peak attributed to CuO at 38.7° was observed, suggesting the formation of small CuO particles. However, no characteristic peaks of crystalline CuO were observed on the other catalysts, suggesting that the copper species were highly dispersed on the surface of the supports. According to the suggestion that more defect sites can exist when the crystallization degree is poor,26 it is assumed that copper species have strong interactions with the Zr-doping supports which have a poor degree of crystallization, and the content of CuO in free form increased on the ZrO2 support. | ||
| Fig. 2 X-ray diffraction patterns of Cu/Ti1−xZrxO2 catalysts (▽ ZrTiO4; ◇ CuO). | ||
3.4. H2-TPR
H2-TPR is performed to investigate the reducibility of copper species supported on the different supports, and the results are summarized in Fig. 3. For the samples of x = 0.3, 0.5, and 0.7, the presence of two partially overlapped reduction peaks was observed in the range of 50–300 °C as denoted as α and β, respectively, indicating the existence of different copper species. The α peak split into two peaks on the Cu/TiO2 catalyst, and only one peak noted as γ appears at 310 °C on Cu/ZrO2. The α peak can be attributed to the reduction of highly dispersed copper oxide species strongly interacting with the support, and the β peak can be assigned to small two- and three-dimensional clusters of copper oxide weakly associated with the support.27 In addition, the γ peak can be ascribed to the reduction of Cu2+ ions in the bulk-type CuO particles,28 and the formation of bulk-type CuO particles can be attributed to the lower surface dispersion and the agglomeration of copper oxide caused by the limited surface area. Moreover, the δ reduction peak appearing at 450 °C on the Cu/Ti0.7Zr0.3O2 sample was assigned to the reduction of the support. As the reduction of TiO2 and ZrO2 alone was difficult at temperatures below 600 °C,13,29 the easy reduction of the support may be caused by the proper substitution of Ti by Zr, which increased the oxygen mobility, as proved by XPS in Section 3.6. The improvement in the reduction of copper species and the support was considered as one reason for the excellent activity of Cu/Ti0.7Zr0.3O2 sample.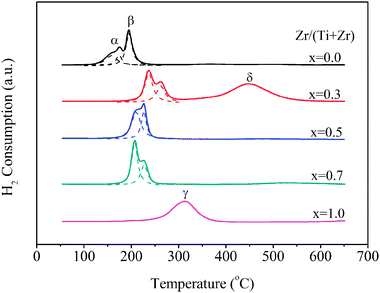 | ||
| Fig. 3 H2-TPR profiles of Cu/Ti1−xZrxO2 catalysts. | ||
The peak areas of H2 consumption over the various catalysts calculated using Gaussian are listed in Table 2. Compared with Cu/TiO2, the H2 consumption according to the α peak increased and that of the β peak decreased significantly over the Zr-doping samples. This suggests that the addition of ZrO2 increased the amount of highly dispersed copper oxide species acting as active sites, which can be attributed to the formation of defects and the promotion of oxygen mobility by Zr doping.13
| Catalyst | T/°C | H2 consumption (a.u.) | ||||
|---|---|---|---|---|---|---|
| α | β | γ | α | β | γ | |
| Cu/TiO2 | 174 | 195 | — | 7812 | 9556 | — |
| Cu/Ti0.7Zr0.3O2 | 237 | 263 | — | 10825 |
6478 | — |
| Cu/Ti0.5Zr0.5O2 | 210 | 226 | — | 11718 |
6371 | — |
| Cu/Ti0.3Zr0.7O2 | 207 | 227 | — | 12105 |
6459 | — |
| Cu/ZrO2 | — | — | 310 | — | — | 23486 |
3.5. Py-IR analysis
FTIR spectra of pyridine chemisorbed on the samples evacuated at different temperatures are presented in Fig. 4. The bands around 1444, 1579 and 1603 cm−1 are attributed to the pyridine coordinated on Lewis acid sites, and the band around 1490 cm−1 corresponds to the Lewis and Brønsted acid complex.30,31 Moreover, no Brønsted acid site was observed emerging at 1545 cm−1.32 It appears that the intensities of the above bands decreased with an increase in temperature, indicating the partial desorption of pyridine. The acidity of Cu/Ti0.7Zr0.3O2 and Cu/Ti0.5Zr0.5O2 was enhanced significantly compared with that of Cu/TiO2, and Cu/Ti0.7Zr0.3O2 appeared to possess the strongest concentration of Lewis acid sites. However, the acidity of catalysts decreased obviously with further doping of ZrO2, in agreement with the result that the ZrO2 possessed predominantly weak Lewis acid sites.33 According to the previous reports,34,35 the enhancement of surface acidity may be due to the increase in oxygen vacancy defect sites accompanying with the Zr doping and the growing charge imbalance based on the generation of Ti–O–Zr bonding. Moreover, considering the interaction between the active component and the acid sites,36 it may be considered that the higher acidity would prevent the copper aggregation, improve the dispersion of copper species, and thus favor the SCR reaction. Several studies have concluded that the acid sites promote the oxidation of NO during HC-SCR, which is the first and rate-determining reaction step on a Ti–Zr mixed oxides support.37,38 Therefore, the high catalytic performance of Cu/Ti0.7Zr0.3O2 was believed to be related to the high surface acidity, which played a key role in the promotion of copper species dispersion and the oxidation of NO. | ||
| Fig. 4 FTIR spectra of pyridine adsorption at different temperature over (a) Cu/TiO2, (b) Cu/Ti0.7Zr0.3O2, (c) Cu/Ti0.5Zr0.5O2, (d) Cu/Ti0.3Zr0.7O2, (e) Cu/ZrO2. | ||
3.6. XPS
The representative peakfits of the Cu 2p core level and their corresponding shake-up satellites are shown in Fig. 5(a). The Cu2+ species can be characterized by a Cu 2p3/2 binding energy of 934.6 eV with a shake-up or satellite peak located at 938.5–946.5 eV, and the binding energy at 933.0 eV is assigned to Cu+.13,39 The ratio of the sum of the areas of peaks 2–4 to that of peak 1 was used as a measure of the relative amount of Cu2+ to Cu+ on the surface. As shown in Fig. 5(a), the ratio of Cu2+/(Cu2++Cu+) was higher over Cu/Ti0.7Zr0.3O2 with approximately 83.9%, while only 57.0% Cu2+ was found on the Cu/TiO2 surface. It has been confirmed by in situ FTIR that the chemisorbed NO occurred on the Cu2+ species.40 Iwamoto41 has proved rigorously that the Cu2+–O− or Cu2+–O2− species act as the active sites on the Cu-MFI zeolite catalyst for C2H4-SCR. Furthermore, a widely accepted interpretation in the literature is that an active center is a [Cu–O–Cu]2+ dimer species and a [Cu–O–Cu]2+/Cu+ redox cycle existed in the NO adsorption process at a high temperature (∼550 K),42 at which the maximum N2 yield was obtained in the present study. Hence a reasonable hypothesis is that the Cu2+ species are able to adsorb and activate NO molecules on the catalyst surface, and the higher Cu2+ ratio favors the C3H6-SCR reaction. | ||
| Fig. 5 XPS spectra of (a) Cu 2p, (b) O 1s and (c) Ti 2p of Cu/TiO2 and Cu/Ti0.7Zr0.3O2 catalysts. | ||
The XPS results of O 1s are shown in Fig. 5(b). The peak at 530.3 eV corresponds to the lattice oxygen O2− (denoted as OL), and the peak at 531.4 eV is attributed to the surface adsorbed oxygen (denoted as OA), such as O22− or O− belonging to a defect oxide or hydroxyl-like group.43–45 Compared to Cu/TiO2, the relative concentration ratio of OA/(OA+OL) on the Cu/Ti0.7Zr0.3O2 catalyst increased from 18.7% to 33.8%. According to the previous study,46 it can be assumed that the increase of surface adsorbed oxygen was attributed to the formation of defects, including the charge imbalance, vacancies, and unsaturated chemical bonds, caused by the corporation of Zr4+ cation into the TiO2 lattice. In addition, the surface adsorbed oxygen was more highly active in the oxidation reaction due to its higher mobility compared with the lattice oxygen, and was beneficial to the NO oxidation to NO2 in the SCR reaction.47,48
Fig. 5(c) shows the XPS results of Ti 2p in the samples. The binding energy of Ti 2p3/2 at 458.7 eV and Ti 2p1/2 at 464.6 eV were observed, indicating that Ti exists in the Ti4+ oxidation state.22,48,49 Moreover, the doping of Zr did not affect the redox behavior of TiO2, as no shift in binding energies was apparent.
3.7. TEM studies of the catalysts
Fig. 6 shows the low and high resolution TEM images of Cu/TiO2 and Cu/Ti0.7Zr0.3O2. From the low magnification image, it can be seen that the Cu species was better dispersed on Cu/Ti0.7Zr0.3O2 than Cu/TiO2, which confirmed that the Zr-doping inhibited the agglomeration of catalyst efficiently. Further investigation of the crystal structure and exposed plane was performed using HR-TEM, and the results are present in Fig. 6(c) and (d). No lattice fringe of CuO was observed on either catalyst, suggesting the copper species were finely dispersing on the surface of the supports, in agreement with the XRD results. For Cu/TiO2, lattice fringes with a spacing of 0.35 nm and 0.24 nm were observed, corresponding to the (101) and (001) planes of TiO2, respectively.40 The attached selective area electron diffraction (SAED) also confirms that the Cu/TiO2 sample was in the highly crystallized anatase. For Cu/Ti0.7Zr0.3O2, neither TiO2 nor ZrO2 related crystallite plane was observed, and the shape of the electron diffraction rings demonstrates the sample was in a noncrystalline structure, indicating that Zr doping destroyed the structure of anatase TiO2 and led to the amorphous phase of the mixed oxides. | ||
| Fig. 6 TEM images of (a) Cu/TiO2 and (b) Cu/Ti0.7Zr0.3O2, HR-TEM images and the corresponding SAED patterns of (c) Cu/TiO2 and (d) Cu/Ti0.7Zr0.3O2 catalysts. | ||
3.8. In situ FTIR of NO adsorption
Fig. 7 shows the results of in situ FTIR of NO adsorption on Cu/TiO2 and Cu/Ti0.7Zr0.3O2 catalysts at different temperatures. According to the literature, the bands of a bridging nitrate (1616 cm−1), bidentate nitrate (1597, 1569, 1543 and 1284 cm−1) and NO2 stretch of a distorted [NO3−]2 adsorbed on Cu2+ (1385 and 1350 cm−1) were observed over Cu/TiO2 catalyst.21,33,50 The bands of a bridging nitrate (1607 cm−1), bidentate nitrate (1578 and 1547 cm−1) and NO2 stretch of a distorted [NO3−]2 (1390 and 1359 cm−1) over Cu/Ti0.7Zr0.3O2 were slightly shifted, and the bands assigned to the bidentate nitrate at 1597 and 1284 cm−1 were not observed, suggesting the changes in the nitrate distribution were caused by the introduction of Zr. The intensity of nitrate adsorption decreased gradually with increasing temperature, indicating the desorption of nitrate species at high temperature. Compared with Cu/TiO2 catalyst, the Cu/Ti0.7Zr0.3O2 sample exhibited much stronger adsorption towards nitrate species. Besides, the negative peaks of nitrate species appeared at 300 °C over Cu/TiO2, suggesting the instability of nitrate species at high temperature, while there were still obvious nitrate species on the surface of Cu/Ti0.7Zr0.3O2. The in situ FTIR results indicated that the Zr addition accelerated the formation and accumulation of nitrate species on the surface of catalyst, meanwhile, the stability of nitrates was also improved. The enhancement in the NO adsorption may be attributed to the increase in the surface area, the Lewis acidity and the oxygen mobility caused by the Zr incorporation.![FTIR spectra of nitrate adsorption species at different temperatures over (a) Cu/TiO2 and (b) Cu/Ti0.7Zr0.3O2 catalysts. Conditions: [NO] = 1000 ppm, He as balance.](/image/article/2012/CY/c2cy20121f/c2cy20121f-f7.gif) | ||
| Fig. 7 FTIR spectra of nitrate adsorption species at different temperatures over (a) Cu/TiO2 and (b) Cu/Ti0.7Zr0.3O2 catalysts. Conditions: [NO] = 1000 ppm, He as balance. | ||
3.9. Analysis of the Zr–O–Ti bond by quantum mechanics
Calculations based on density functional theory (DFT) were performed to analyze the changes in the crystal and electronic structure of TiO2 due to the introduction of ZrO2, which may give some insights into the higher dispersion of copper species and the improvement of SCR activity over Zr-doped TiO2. Fig. 8 shows the optimized configuration of the TiO2 and Ti0.7Zr0.3O2 supports and the electron charge density (ECD) of pattern slice from the model. From Fig. 8, it is observed that the lattice type of anatase TiO2 was tetragonal, while the lattice type changes to an orthorhombic one due to the Zr-doping. In addition, for the anatase TiO2, each Ti atom was coordinated to six O atoms, two of which were along the c-axis and were distant from Ti by 1.99 Å. The other four Ti–O distances were all 1.93 Å. In the Zr-doping sample, each Ti or Zr atom was still coordinated to six O atoms, while the Ti–O distances changed to 2.00, 2.09, 1.88, 1.91, 1.88 and 2.11 Å, resulting in strong distortions in the nuclear structure caused by the substitution of smaller Ti4+ (0.64 Å) with larger Zr4+ (0.87 Å). | ||
| Fig. 8 Representation of the structure and electron density difference of (a) TiO2 and (b) Ti0.7Zr0.3O2 models. | ||
From the map of ECD, a delocalization of ECD around the Zr4+ cation and an enrichment of the ECD around the Ti4+ cation were observed, resulted from the generation of a mixed Ti–O–Zr bond. The charge density concentration on the Ti–O band led to the enhancement of the interaction between the Cu2+ and the Ti–O bond on Ti0.7Zr0.3O2. Consequently, the Cu–O bond of CuO in Ti0.7Zr0.3O2 became weaker and the CuO species acting as active sites were easier to reduce, in agreement with the results of H2-TPR. Moreover, the charge imbalance was confirmed according to the ECD map, providing one reason for the increase in the Lewis acidity. According to the modification in the ECD, the Ti–O–Zr link might have acted as an adsorption center for the catalytic reaction, and the NOx− bond would form on the Zr4+ ion, in agreement with the previous report.38 And this inference was confirmed by the in situ FTIR of NO adsorption results. The improvement of NO adsorption would promote the SCR reaction of NO over the Cu/Ti0.7Zr0.3O2 catalyst.
4. Conclusions
Serial Cu/Ti1−xZrxO2 (x = 0, 0.3, 0.5, 0.7, 1.0) catalysts were facilely synthesized using the impregnation–coprecipitation method and tested for C3H6-SCR of NO. The coexistence of titanium and zirconium led to an increase in N2 yield, and the highest catalytic activity was obtained over Cu/Ti0.7Zr0.3O2. The introduction of ZrO2 significantly enhanced the Lewis acidity and greatly increased the concentration of Cu2+ and surface adsorbed oxygen, which accelerated the oxidation of NO to NO2 and/or NO3−, and thus further benefited the SCR reaction.On the other hand, the specific Ti–O–Zr bond led to the generation of defects, including charge imbalance, vacancies and unsaturated chemical bonds. The structural distortion of the support played a significant role in controlling the dispersion of the active copper species as well as the metal–support interaction, which was proposed as one possible reason for the improved catalytic activity of the Cu/Ti0.7Zr0.3O2 catalyst for C3H6-SCR of NO.
Acknowledgements
This work was supported financially by the National Nature Science Foundation of China (NSFC-RGC 21061160495), the National High Technology Research and Development Program of China (863 Program) (2010AA064902), the Excellent Talents Program of Liaoning Provincial University (LR2010090), China's One Thousand Chinese Talents Award and the Australia Research Council under the Linkage Project Scheme (Project Number: LP0989368).Notes and references
- X. D. Hu, J. J. Zhang, S. Mukhnahallipatna, J. Hamann, M. J. Biggs and P. Agarwal, Fuel, 2003, 82, 1675–1684 CrossRef CAS.
- D. López, R. Buitrago, A. Sepúlveda-Escribano, F. Rodríguez-Reinoso and F. Mondragón, Langmuir, 2007, 23, 12131–12137 CrossRef.
- M. Shelef, Chem. Rev., 1995, 95, 209–225 CrossRef CAS.
- B. M. Reddy, B. Chowdhury, I. Ganesh, E. P. Reddy, T. C. Rojas and A. Fernández, J. Phys. Chem. B, 1998, 102, 10176–10182 CrossRef CAS.
- Q. Yuan, L. L. Li, S. L. Lu, H. H. Duan, Z. X. Li, Y. X. Zhu and C. H. Yan, J. Phys. Chem. C, 2009, 113, 4117–4124 CAS.
- S. Polisetti, P. A. Deshpande and G. Madras, Ind. Eng. Chem. Res., 2011, 50, 12915–12924 CrossRef CAS.
- B. Neppolian, Q. Wang, H. Yamashita and H. Choi, Appl. Catal., A, 2007, 333, 264–271 CrossRef CAS.
- P. E. Lippens, A. V. Chadwick, A. Weibel, R. Bouchet and P. Knauth, J. Phys. Chem. C, 2008, 112, 43–47 CAS.
- N. D. Petkovich, S. G. Rudisill, L. J. Venstrom, D. B. Boman, J. H. Davidson and A. Stein, J. Phys. Chem. C, 2011, 115, 21022–21033 CAS.
- N. Hickey, P. Fornasiero, J. Kašpar, M. Graziani, G. Martra, S. Coluccia, S. Biella, L. Prati and M. Rossi, J. Catal., 2002, 209, 271–274 CrossRef CAS.
- S. Bennici, A. Gervasini, N. Ravasio and F. Zaccheria, J. Phys. Chem. B, 2003, 107, 5168–5176 CrossRef CAS.
- H. Hong, C. B. Zhang and Y. B. Yu, Catal. Today, 2004, 90, 191–197 CrossRef.
- R. Zhang, W. Y. Teoh, R. Amal, B. H. Chen and S. Kaliaguine, J. Catal., 2010, 272, 210–219 CrossRef CAS.
- R. Kydd, W. Y. Teoh, K. Wong, Y. Wang, J. Scott, Q. H. Zeng, A. B. Yu, J. Zou and R. Amal, Adv. Funct. Mater., 2009, 19, 369–377 CrossRef CAS.
- P. Carniti, A. Gervasini, V. H. Modica and N. Ravasio, Appl. Catal., B, 2000, 28, 175–185 CrossRef CAS.
- Z. Liu, M. D. Amiridis and Y. Chen, J. Phys. Chem. B, 2005, 109, 1251–1255 CrossRef CAS.
- K. V. R. Chary, G. V. Sagar, D. Naresh, K. K. Seela and B. Sridhar, J. Phys. Chem. B, 2005, 109, 9437–9444 CrossRef CAS.
- D. Yang, J. H. Li, M. F. Wen and C. L. Song, Catal. Today, 2008, 139, 2–7 CrossRef CAS.
- M. D. Seagall, P. J. D. Lindan, M. J. Probert, C. J. Pickard, P. J. Hasnip, S. J. Clark and M. C. Payne, J. Phys.: Condens. Matter, 2002, 14, 2717 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- L. Q. Nguyen, C. Salim and H. Hinode, Appl. Catal., B, 2010, 96, 299–306 CrossRef CAS.
- B. Guan, H. Lin, L. Zhu and Z. Huang, J. Phys. Chem. C, 2011, 115, 12850–12863 CAS.
- Z. B. Wu, B. Q. Jiang and Y. Liu, Appl. Catal., B, 2008, 79, 347–355 CrossRef CAS.
- F. D. Liu, H. Hong, Y. Ding and C. B. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- Y. S. Shen, Y. F. Ma and S. M. Zhu, Catal. Sci. Technol., 2012, 2, 589–599 CAS.
- Y. Q. Song, H. M. Liu and D. H. He, Energ. Fuel, 2010, 24, 2817–2824 CrossRef CAS.
- E. Moretti, L. Storaro, A. Talon, M. Lenarda, P. Riello and R. Frattini, Appl. Catal., B, 2011, 102, 627–637 CrossRef CAS.
- S. Watanabe, X. L. Ma and C. S. Song, J. Phys. Chem. C, 2009, 113, 14249–14257 CAS.
- G. Avgouropoulos and T. Ioannides, Appl. Catal., A, 2003, 244, 155–167 CrossRef CAS.
- L. Zhao, X. Y. Li, X. Quan and G. H. Chen, Environ. Sci. Technol., 2011, 45, 5375–5379 Search PubMed.
- N. Jagtap, S. B. Umbarkar, P. Miquel, P. Granger and M. K. Dongare, Appl. Catal., B, 2009, 90, 416–425 CrossRef CAS.
- J. H. Li, Y. Q. Zhu, R. Ke and J. M. Hao, Appl. Catal., B, 2008, 80, 202–213 CrossRef CAS.
- G. H. Jing, J. H. Li, D. Yang and J. M. Hao, Appl. Catal., B, 2009, 91, 123–134 CrossRef CAS.
- J. C. Wu, C. S. Chung, C. L. Ay and I. Wang, J. Catal., 1984, 87, 98–107 CrossRef CAS.
- I. X. Green, C. Buda, Z. Zhang, M. Neurock and J. T. Yates, J. Phys. Chem. C, 2010, 114, 16649–16659 CAS.
- A. Auroux, D. Sprinceana and A. Gervasini, J. Catal., 2000, 195, 140–150 CrossRef CAS.
- M. Haneda, Y. Kintaichi, M. Inaba and H. Hanada, Catal. Today, 1998, 42, 127–135 CrossRef CAS.
- R. P. Hernández, D. M. Anaya, M. E. Fernández and A. G. Cortés, J. Mol. Catal. A: Chem., 2008, 281, 200–206 CrossRef.
- C. C. Chusuei, M. A. Brookshier and D. W. Goodman, Langmuir, 1999, 15, 2806–2808 CrossRef CAS.
- C. Z. Sun, J. Zhu, Y. Y. Lv, L. Qi, B. Liu, F. Gao, K. Q. Sun, L. Dong and Y. Chen, Appl. Catal., B, 2011, 103, 206–220 CrossRef CAS.
- H. Yahiro and M. Iwamoto, Appl. Catal., A, 2001, 222, 163–181 CrossRef CAS.
- W. Gruenert, N. W. Hayes, R. W. Joyner, E. S. Shpiro, M. R. H. Siddiqui and G. N. Baeva, J. Phys. Chem., 1994, 98, 10832–10846 CrossRef.
- Z. Wu, R. Jin, Y. Liu and H. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS.
- T. F. Garetto, E. Rincón and C. R. Apesteguía, Appl. Catal., B, 2007, 73, 60–72 CrossRef.
- F. D. Liu, K. Asakura, H. He, W. P. Shan, X. Y. Shi and C. B. Zhang, Appl. Catal., B, 2011, 103, 369–377 CrossRef CAS.
- X. Gao, Y. Jiang, Y. Zhong, Z. Y. Luo and K. F. Cen, J. Hazard. Mater., 2010, 174, 734–739 CrossRef CAS.
- A. Q. Wang, D. B. Liang, C. H. Xu, X. Y. Sun and T. Zhang, Appl. Catal., B, 2001, 32, 205–212 CrossRef CAS.
- F. D. Liu and H. He, J. Phys. Chem. C, 2010, 114, 16929–16936 CAS.
- Z. Q. Zou, M. Meng, L. H. Guo and Y. Q. Zha, J. Hazard. Mater., 2009, 163, 835–842 CrossRef CAS.
- Y. W. Chi and S. S. C. Chuang, J. Catal., 2000, 190, 75–91 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |