Synergy between catalysts: enzymes and bases. DKR of non-natural amino acids derivatives
Paola
D'Arrigo
ab,
Lorenzo
Cerioli
ab,
Stefano
Servi
*ab,
Fiorenza
Viani
bc and
Davide
Tessaro
ab
aDipartimento di Chimica, Materiali, Ingegneria Chimica “Giulio Natta”, Politecnico di Milano, Piazza Leonardo da Vinci 32, 20133 Milano, Italy. E-mail: stefano.servi@polimi.it; Fax: +39 0223993081; Tel: +39 0223993047
bThe Protein Factory, Politecnico di Milano, ICRM, CNR and Università degli Studi dell'Insubria, via Mancinelli 7, 20131 Milano, Italy
cICRM, CNR, Via Mario Bianco, Milano, Italy
First published on 23rd April 2012
Abstract
A double catalyst system (protease + base) is applied for the dynamic kinetic resolution (DKR) of amino acid thioesters in hydrolysis, transesterification or transamidation, allowing to obtain L-N-Boc-aminoacids, esters or amides in high yields and ee. This approach not only obviates the tedious recycling steps of the undesired remaining enantiomer, but, more importantly, the enantiomeric excess of the product (ee) is independent of the extent of conversion, and the process becomes more enantiospecific compared to a kinetic resolution. Substrates of different α-C acidity can be transformed by selecting the base of appropriate strength. Conditions have been found to extend the DKR of phenylglycine thioesters to the whole set of aminoacids thioesters fully representative of natural and non-natural amino acid structures.
Dynamic kinetic resolution1
Non-proteinogenic α-amino acids either because of their absolute configuration or for their chemical structure are often defined as non-natural even though some of them occur in Nature.2–4They belong to a group of building blocks required for the preparation of peptide mimetics and new bio active compounds.5–10
They are often chiral and although they can be obtained by an asymmetric synthesis,11–15 the most commonly used method requires in general the preparation of the racemate and a kinetic resolution step. The racemization of the undesired enantiomer can be done in a separate step, increasing however the complexity of the process. The in situ racemization is by far a more efficient procedure. There are several examples in which a first catalyst (often a hydrolytic enzyme) is used for the resolution step, and a second one for the racemization. The second catalyst can be an enzyme (a racemase), a transition metal catalyst (Pt, Pd, Rh, etc.) or a base. In an ideal case two enzymes are used: a hydrolytic enzyme and a racemase. Due to their intrinsic specificity they are completely compatible one with the other. However while hydrolytic enzymes have a wide substrate specificity, racemases (enzymes catalyzing the abstraction-delivery of a proton) are very strict in the choice of their substrates, and thus not of general application.16,17
There is only one widely applicable such enzymatic system for practical applications: the hydantoinase/carbamoylase/racemase system,18 a complex combination of catalytic activities used as a whole cell catalyst for the production of α-AAs of D-configuration.
When the two steps (kinetic resolution + racemization) are performed in a one pot procedure the overall process is referred to as a deracemization.19–26
In particular, dynamic kinetic resolution (DKR) is a deracemization process in which a resolution process is combined with the in situ racemization of the non-transformed enantiomer.
This approach not only obviates the tedious recycling steps of the undesired remaining ester but, more importantly, the enantiomeric excess of the product (ee) is now independent of the extent of conversion, and the process becomes more enantiospecific.27,28
This process is highly desirable and it has been thoroughly investigated.
Recent methods combining enzyme catalyzed resolution and transition metal catalyzed racemization, aimed at the production of chiral secondary alcohols as single enantiomers, have been reported.29–35
Hydrolytic enzymes and bases
A better alternative to Rh or Ru catalyzed reactions, besides an entirely enzyme catalyzed reaction (difficult for the non-availability of racemases of wide substrate specificity), is the combination of the enzymatic resolving step with a base catalyzed racemization. In this case, the reaction conditions must be devised such that the enantioselective catalyst (enzyme) is not affected by the presence of the basic catalyst. There are several cases in which such an approach is possible. The common feature is that the substrate can be racemized by a base, while the product (usually a carboxylate) is not racemized in the same basic conditions.Compounds in Fig. 1 have such properties (although in the non-cyclic thioester a delocalyzed charge stabilization is not possible this is compensated by the higher contribution to acidity due to the softer nature of sulfur compared to oxygen). They are compounds from which α-amino acids can be obtained by enzyme catalyzed hydrolytic steps. In all three compound families, base catalyzed racemization on the substrates and not on the products allows a DKR.36–41
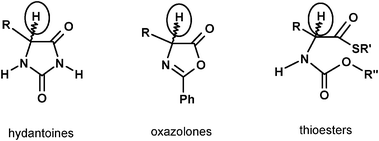 | ||
| Fig. 1 Racemizable substrates for combined enzyme-base DKR. | ||
The similarity in the acidity of the proton in the three compounds in Fig. 1 parallels the structural similarities. As an alternative, it is possible to induce racemization of the AA by reacting it with an aromatic aldehyde and promoting the base catalysed racemization of the formed imine. This procedure mimics the mechanism of PLP dependent racemases in vivo.42,43
α-Aminoacid-thioesters as substrates for DKR
Thiol esters of carboxylic acids are reactive species mimicking the activation occurring in Nature in the transfer of acyl moieties. They have been used as mild activated intermediates in protein synthesis, coupling two parts through a chemical ligation44–47 in the formation of cyclic peptides48,49 or macrolides.50–52However, in some cases they are considered too reactive being prone to react directly with water in an undesired hydrolytic reaction competing with the transfer of acyl group to N or O atom.
They behave as excellent acyl donors in trans-esterification, trans-amidation and hydrolysis reactions, catalyzed by hydrolytic enzymes, lipases and proteases. Many examples of lipase catalyzed kinetic resolution employing thioesters as acyl donors both in hydrolysis and transesterification have appeared.53–58
Thiol esters have another peculiar chemical reactivity: they are considerably more α-C-acidic than the corresponding carboxylates or their oxo-esters. This reactivity towards bases has been exploited in a base–enzyme mixed catalysis leading to a dynamic kinetic resolution of a series of pharmaceutical intermediates, primarily α-methyl propionates of the profen family. In these studies the requirements for a possible and efficient base catalyzed racemization/enzyme catalyzed kinetic resolution have been explored and identified giving access to products in a dynamic kinetic resolution process with high yields and ees.59–61
In order to establish the applicability of thioesters of amino acid as substrates for a protease-base catalyzed DKR, a combination of calculation and experimental study of the acidity of the α-carbon in AA-thioesters was necessary. pKa values of complex molecules are in general not available, although a semi-quantitative evaluation can be obtained by analogy with compounds of similar structure. We applied an energy minimization approach to a series of compounds of interest and we obtained enthalpies for their proton abstraction (Scheme 1).62
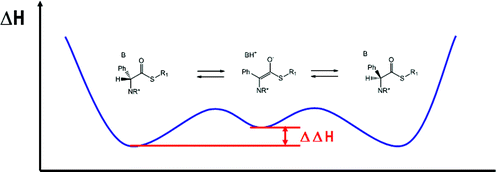 | ||
| Scheme 1 High energy intermediate in the racemization of AAs derivatives. | ||
Calculation of proton abstraction enthalpies from energy minimization
The racemization enthalpies for the reaction in Scheme 1 were calculated for a series of structurally diverse amino acid thioesters. The selected substrates differ from each other for the distance of the aromatic group from the α-carbon, or its absence. Accordingly ΔΔH values for the proton abstraction between compounds of type 1 and model substrates 2–4 were calculated and found to be between 10 and 15 Kcal mol−1. Clearly the racemization of the latter compounds requires non-nucleophilic bases with increasing basic properties.Thus for instance, the enthalpy change of the reaction between trioctylamine (TOA) and
| TOA + DBUH+ ⇔ TOAH+ + DBU |
The thermodynamic data calculated were in good agreement with racemization rates obtained from chiroptical methods. The experimental values obtained showed that, although the nature of the thio-ester strongly influences the carbon acidity which allows racemization to occur at a useful rate (thiobenzyl > thioethyl), a larger effect is associated with the presence of functional groups stabilizing the intermediate anion and present in position α to the stereogenic C-atom.63 The racemization kinetic constants krac for compounds 1–4 were measured. Taking compound 1a as a reference (Fig. 2), the ratio between the kinetic constants tells that compound 1a was racemized 58 times faster than 2a, 580 times faster than 3a, and 950 compared to thioester 4a. Thus, while compounds of type 1 due to the presence of an aromatic ring on the α-C, can be racemized with TOA, stronger bases are required to readily racemize compounds 2–4.62
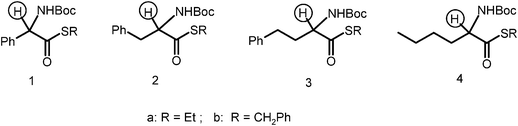 | ||
| Fig. 2 Aminoacid derivatives of decreasing acidity of the α-C. | ||
Among a group of cyclic, non-nucleophilic organic amines of different base strength values in acetonitrile63 (trioctylamine, triethylamine, dimethylamino pyridine, pyridine, 3-quinuclidone, diazobicycloundecene, triazabicyclodecene), TOA and DBU were found to effectively racemize compounds with a wide range of α-C acidity and they were suitable to achieve the projected DKR.
DKR of N-Boc-phenylglycine thioesters
On the basis of the above numbers, phenylglycine derivatives are expected to be excellent substrates for a DKR, also considering the following:• Phenylglycine esters of L-configuration are known to be good substrates for the protease subtilisin.
• Protons in the α-position of thioesters have increased acidity if compared with their oxo-analogues.
• Subtilisin is as an alkaline protease, suitable to withstand alkaline conditions.
A commercial preparation of subtilisin Carlsberg was able to transform N-Boc amino acid thioesters of type 1a (Scheme 2) with reaction rates comparable although inferior to the one observed with the corresponding oxo-esters.64,65 Moreover, compounds 1a could be easily racemized with TOA in methyl-t-butyl ether (MTBE).
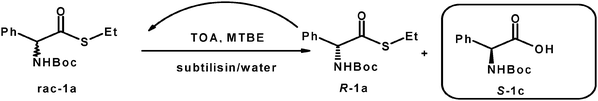 | ||
| Scheme 2 Deracemization of N-Boc-phenylglycine-thioesters with subtilisin. | ||
Racemization conditions were studied by observing the exchange rate of the α-proton of the thioester with deuterium from CD3OD by 1H NMR. In a typical experiment to 20 mg of substrate dissolved in 500 mL of DMSO-d6 and 100 mL of CD3OD, 0.5 equivs of trioctylamine were added at 318 K. The 50% exchange rate varied from a few minutes (for compound 1a) to several hours for the 2-substituted phenylglycines.66
In all cases, no exchange was observed in the absence of trioctylamine. Control experiments with the corresponding oxo-esters and acids showed no measureable proton exchange under similar conditions. This indicated that the thioester, but not the oxo-ester, was suitable for the in situ racemization method and that the product acid was configurationally stable under thioester-racemizing conditions. Further experiments showed that thioesters were not prone to spontaneous hydrolysis under the biotransformation conditions. 1a hydrolysis was initially studied under non-racemizing conditions: 2 g of 1a in 150 mL of a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of H2O:MTBE (approximately 50 mM) at pH 8.0 and 37 °C were treated with 500 U of subtilisin and the pH controlled in a pHstat apparatus. The base addition stopped sharply at 50% conversion (24 h). Addition of TOA (1.5 mL, 3.38 mmol, 0.5 equivs) restored the hydrolysis and stoichiometric base uptake was complete after 48 h. The profile of the reaction as registered on a pHstat before and after TOA addition is reported in Fig. 3.
:1 mixture of H2O:MTBE (approximately 50 mM) at pH 8.0 and 37 °C were treated with 500 U of subtilisin and the pH controlled in a pHstat apparatus. The base addition stopped sharply at 50% conversion (24 h). Addition of TOA (1.5 mL, 3.38 mmol, 0.5 equivs) restored the hydrolysis and stoichiometric base uptake was complete after 48 h. The profile of the reaction as registered on a pHstat before and after TOA addition is reported in Fig. 3.
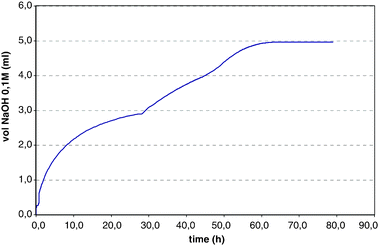 | ||
| Fig. 3 Conversion time course of N-Boc-PheGly-SEt 1a. | ||
Thus compound 1a and analogues66 (Scheme 2) were completely deracemized in quantitative yields giving the S-enantiomers with ee higher than 99%.67
The reaction took place in a biphasic system with up to 1 eq. of base with subtilisin as a catalyst. The efficiency of the deracemization process depends on the rate of enzymatic hydrolysis and the proton abstraction rate. The latter should be of the same order of magnitude of the enzymatic hydrolysis and can be modulated by the trioctylamine concentration which influences the racemization rate with pseudo-first order kinetics.
Attempts to extend the procedure to compounds with non-aromatic R groups failed since they were not racemized in the experimental conditions applied in the DKR of N-Boc-PheGly-SEt and similar compounds. The extension of the method requires designing substrates and conditions suitable for a chemo-enzymatic deracemization.
DKR of less C-acidic N-Boc-aminoacid thioesters: subtilisin and DBU
Fig. 4 shows a set of thioesters of α-amino acid derivatives not bearing an aromatic group in the α-position and not racemizable under the above conditions. On the basis of previous calculations, attempts to racemize them in the presence of DBU were made. However, the experimental conditions applied before (H2O-MTBE) were not suitable when DBU was used as a base since at pH = 8 the base is protonated and no racemization can occur, while at pH 10 at which DBU is catalytically active, rapid substrate hydrolysis occurred.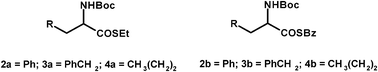 | ||
| Fig. 4 Group of substrates homogeneous for their racemization conditions. | ||
DKR with subtilisin and DBU in 2-propanol
N-Boc-AA-thioesters have good solubility in 2-propanol, a solvent with low nucleophilic power and a relatively low boiling point which allows a simple recovery of the products. Commercial 2-propanol containing 0.5–1.0% water proved to be suitable for the deracemization. In order to control the water amount in the solution, subtilisin immobilized on a solid support was used instead of the water solution used in the previous experiments.Best results were obtained with Subtilisin CLEA®68 for the higher specific activity (allowing faster reaction with possible minimization of chemical hydrolysis in the presence of water) and the mechanical stability in organic solvents.
A new reaction protocol was assembled by employing cross linked subtilisin in 2-PrOH and 1 eq. of DBU as a base and thioester 60 mM. Under these conditions chemical hydrolysis was completely suppressed and transesterification was observed. When the reaction was run in neat isopropanol in the absence of water, the isopropyl ester (SS-3c, Scheme 3) as the main product was observed, in about 93% yield, 99% conversion and 90% ee. The ee, lower than expected, is probably due to some competing chemical transesterification, not negligible under the reaction conditions.
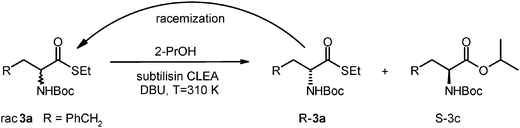 | ||
| Scheme 3 DKR in wet 2-propanol of N-Boc-homoPhe-thioesters 3a with a combination of DBU and cross-linked enzyme aggregates. | ||
DKR with cross-linked enzyme aggregates (CLEA) and DBU in t-butanol
As an alternative, t-BuOH was used as a solvent. In this case no transesterification is expected. Moreover the water content in commercial t-BuOH (from Karl–Fischer analysis the water content in commercial t-BuOH was 0.08% but after the addition of the enzymatic preparate the water content raised to 1.5% corresponding to 20 times the stoichiometric amount required) ensures a good enzymatic activity and remains constant during the reaction course. Compounds 2a–6a were submitted to the dynamic kinetic resolution described in Scheme 4. At the end of the reaction, ees were evaluated by chiral HPLC analysis. Table 1 summarizes the results obtained. The optical purity of N-Boc-L-amino acids obtained was excellent in all cases (99%).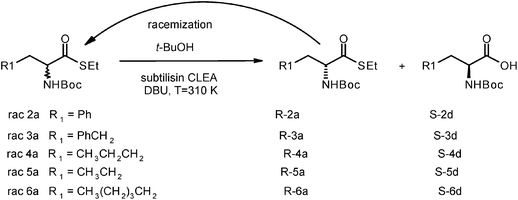 | ||
| Scheme 4 DKR in t-ButOH of aryl and aliphatic thioesters with the double catalysis: protease + base. | ||
| Thioester | N-Boc-AA | R1 | Time/h | Yield (%) | ee (L) % | α D |
|---|---|---|---|---|---|---|
| 2a | 2d | Ph | 12 | 99 | >99 | +34.8 |
| 3a | 3d | PhCH2 | 40 | 94 | 99 | +48.1 |
| 4a | 4d | CH3CH2CH2 | 48 | 91 | 99 | +23.4 |
| 5a | 5d | CH3CH2 | 24 | 98.5 | >99 | +24.8 |
| 6a | 6d | CH3(CH2)3CH2 | 110 | 95.8 | >99 | +25.9 |
Enzymatic amide formation
Esters and thioesters aminolysis is a practical method of amide synthesis avoiding more intensive carboxylate activation.69In recent evaluations of chemical processes trying to comply to the rule of good manufacturing practice (GMP) and green chemistry, amide bond formation was identified as one of the most utilized and problematic synthetic steps in the pharmaceutical industry. From a detailed study it was found that N-acylation reactions for amide formation were used in more than 50% of current syntheses of drug candidates.70
Clearly, the enzymatic procedure even when requiring carboxylate activation (esters as intermediates) avoids the need of condensing agents necessary for chemical amide formation. Lipases are often considered for amoniolysis/aminolysis with the argument that the reverse amide hydrolysis is not efficiently catalyzed by these enzymes. Moreover, the enantioselectivity of lipases can be modulated through substrate engineering and reaction engineering or by selecting the enzyme among the great diversity of these catalysts allowing us to have access to compounds of both stereochemical preference.71,72
Proteases, in contrast, have well defined stereochemical preferences particularly towards amino acid derivatives and the problem of shifting reactions towards reverse-hydrolysis requires a specific solution.73,74
In particular, subtilisin has been applied in ammoniolysis reactions in water75 in genetically modified form76 and in organic solvents77 for the enzyme-catalyzed formation of amides and peptide bonds.78 The stability of this protease in organic solvent and in the presence of bases makes it particularly suitable for the purpose of catalyzing aminolysis reactions with concomitant base-catalyzed racemization of the substrate.79
An effective transamidation process under DKR conditions with highly reactive species like thio-esters must first evaluate the competing enzymatic hydrolysis and chemical ammoniolysis/aminolysis (Scheme 5).
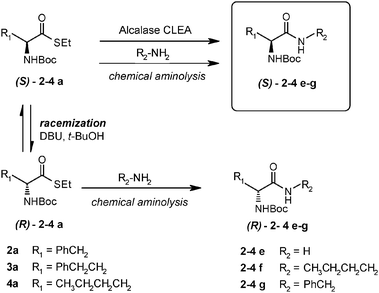 | ||
| Scheme 5 DKR in amide formation and competition with chemical amidation. | ||
Competing chemical/enzymatic hydrolysis
Due to the relatively high reactivity of thioesters, the competing hydrolysis reaction must be suppressed or minimized. While the enzyme-catalyzed hydrolytic step is much slower than the corresponding amoniolysis/aminolysis reaction when water is competing with a stronger nucleophile, the spontaneous chemical hydrolysis is also dependent on water concentration and can significantly compete with the enzyme catalyzed reaction. Therefore t-BuOH containing 0.08% of water was used. After the addition of the enzymatic preparation the water content raised to 1.5%, not always optimal for selectivity.However, it has been observed that the hydrolysis rates of thioester and oxoester in basic solution are very similar while the aminolysis of a thioester with a primary amine to form an amide is more than 100-fold faster than the reaction of the corresponding oxoester.64,80 Therefore, it is expected that also the protease-catalyzed aminolysis reaction should be much faster than the corresponding hydrolysis and that this reaction will not compete with the enzyme catalyzed ammoniolysis.
Competing chemical amoniolysis/aminolysis
Table 2 summarizes the data concerning the transamidation of substrate 2a in the presence of different concentrations of the three nucleophiles object of this study: ammonia, n-propylamine (nPr-NH2), benzylamine (BnNH2).| Nucleophile | Eq. | Unreacted 2a (%) | |||
|---|---|---|---|---|---|
| 1 h | 2 h | 5 h | 25 h | ||
| NH3 | 1.5 | 100 | 100 | 100 | 96 |
| 3 | 100 | 100 | 99 | 95 | |
| 6 | 100 | 100 | 99 | 90 | |
| nPrNH2 | 1.2 | 96 | 91 | 85 | 55 |
| 2.4 | 89 | 86 | 68 | 3 | |
| 4.8 | 74 | 55 | 28 | 0 | |
| BnNH2 | 1.2 | 83 | 74 | 54 | 14 |
| 2.4 | 59 | 46 | 26 | 1 | |
| 4.8 | 64 | 77 | 7 | 0 | |
The spontaneous chemical reaction of the reactive thioesters with these nitrogen nucleophiles is not negligible, and is dependent on their concentration and on the nature of the nucleophile. Thus, while the effect of ammonia is very limited, with the two primary amine the amide formation can go to completion when 5 eq. of amine are used in 25 h. Needless to say, in these conditions no enantiomer discrimination can occur. Therefore, the catalytic efficiency of the biocatalyst is crucial. The effect of the competing non-enzyme catalyzed reaction could be controlled if the lower concentrations of amines were applied (Table 3).
DKR of N-BOC-AA-thioesters in ammoniolysis-aminolysis
In a first experiment, a 40 mM solution of substrate 2a (8 mL) in t-BuOH with 1.2 eq. DBU as the racemization catalyst and 100 mg subtilisin CLEA were treated with 1.5 eq. of propylamine and the reaction mixture was stirred at 30 °C. After 5 h the reaction was complete and the corresponding L-propylamide isolated in 85% yield and 98% ee, proving that under these conditions, base-catalyzed racemization was occurring at the required rate, the chemical amidation was largely suppressed because of the effective catalytic activity of the protease which works with a practically complete enantioselectivity. The reaction was repeated with all the three nucleophiles and the data collected were compared (Table 3) with the one from non-enzymatic reactions (Table 2).The results observed both in terms of isolated yields (∼80%) and ee (>98%) proved that in all the three reactions the base-catalyzed racemization is active, with no or very limited competition from chemical aminolysis which would otherwise affect the ee, and with no or very limited competition from chemical hydrolysis which would otherwise affect the chemical yields. The phenylalanine derivative 2a was efficiently deracemized to give the corresponding amides in high yields and almost absolute stereoselectivity. Under similar experimental conditions compounds 3a and 4a were also transformed although at reduced rates.
In conclusion, this method appears to be of general applicability81 for the preparation of L-amides starting from a racemic N-Boc amino acid-thioester with good yields and excellent optical purity.
Conclusion
Substrate engineering (type of thioester, N-protecting group) and reaction engineering (solvent, nature of the organic base, nature of the biocatalyst) allow us to set up general protocols for the DKR of α-AA-derivatives of different nature: compounds bearing an aromatic group in α-, β-, γ-positions, or purely aliphatic α-AAs. In general a racemic AA-thioester is hydrolyzed with L-selectivity while the D-thioester is continuously racemized by an organic base. In the case of phenylglycine derivatives, the reaction medium is a biphasic (water/MTBE) system where all components are in solution and base (TOA) and protease (soluble subtilisin) coexist. Compounds with a lower acidity of the proton on the α-carbon can still be racemized with a stronger base (DBU) in a t-BuOH solution. In this case the best conditions are obtained with CLEA-subtilisin which is completely stable in the presence of the base. In all cases the product of hydrolysis (L-N-Boc-α-amino acid) is configurationally stable in the presence of the base. Reactions occur in very high isolated yields and ees. Aminolysis and amoniolysis reactions with complete deracemization and amide formation were equally effective. It should be stressed that a DKR process, when compared to the usual kinetic resolution, not only obviates the tedious recycling steps of the undesired remaining ester, but, more importantly, the enantiomeric excess of the product does not depend on the extent of conversion, thus assuring a higher enantiomeric purity of the product,27,28 an argument which is often underevaluated.The cross-linked enzyme can be recovered and recycled. After 15 reaction cycles 80% of activity was recovered by washing the catalyst with buffer and carefully drying it.
Experimental section
General
All chemicals were purchased from Sigma-Aldrich. All solvents were of analytical grade. Silica gel 60 F254 plates (Merck) were used for analytical TLC. Detection was achieved with UV light followed by I2, ninhydrin or potassium permanganate staining. HPLC analysis were performed on a Merck apparatus equipped with an UV detector, fitted with a Chiralcel OD (length 250 mm; diameter 4.6 mm; pore size 5 μm; eluent: hexane/2-propanol), and a Crownpack CR+ (length 250 mm; diameter 4.6 mm; pore size 5 μm; eluent: with H2O/perchloric acid) column.1H-NMR spectra were recorded on a Bruker ARX 400 instrument operating at the 1H resonance frequency of 400 MHz. Chemical shifts (δ, ppm) are reported relative to tetramethylsilane (TMS) as an internal standard. All spectra were recorded in DMSO-d6, or CDCl3 at 305 K. Optical rotations were determined with a Propol Digital Polarimeter Dr Kenchen, and [α]D values are given in units of deg cm2 g−1 at 25 °C.
Enzymes
• Subtilisin Carlsberg from B. subtilis from Fluka and Alcalase 2.5L DX from Novozymes (50 U mL−1) were used in all experiments and gave similar results.• Immobilized subtilisin from B. subtilis Fluka (2.4 U g−1) and Alcalase 3.0 T from Novozymes (3.0 U g−1) were used in all experiments and gave similar results.
• Crosslinked subtilisin from B. subtilis CLEA® was from CLEA technologies (450 U g−1). 1 U is defined as the amount of enzyme which catalyzes the hydrolysis of 1 μmol of N-acetyl-glycine-ethyl ester in 1 min at pH 7.5 and 40 °C.
The L-enantiomers were prepared from optically active material of commercial origin.
D,L-N-Boc-PheGly-SEt (racrac-1a). Starting from 3 g (0.12 mmol) of starting D, L-N-Boc-PheGlycine, 2.9 g (82%) of racrac-1a as a white solid (mp 74 °C) was obtained. Exact mass: 295.12, molecular weight: 295.40, m/z: 295.12 (100.0%), 296.13 (16.6%), 297.12 (4.6%), 297.13 (2.0%), 296.12 (1.2%). For C15H21NO3S: elemental analysis: C, 60.99; H, 7.17; N, 4.74; O, 16.25; S, 10.85%. 1H-NMR (DMSO-d6): δ = 1.13 (t, 3H), 1.41 (s, 9H), 2.80 (q, 2H), 5.27 (br s, 1H), 7.35 (m, 3H), 7.42 (m, 2H), 7.99 (br s, 1H). 13C-NMR (CDCl3): δ = 198.65, 154.65, 136.82, 128.89, 128.50, 127.44, 80.23, 64.50, 28.22, 23.46, 14.25. HPLC analysis (hexane/2-propanol = 99/1, flow = 1.2 mL min−1, T = 308 K): peaks D: 13.5 min; L: 14.5 min.
D,L-N-Boc-PheGly-SBn (racrac-1b). White solid, mp 119–122 °C. Chemical formula: C20H23NO3S, exact mass: 357.14, molecular weight: 357.47. m/z: 357.14 (100.0%), 358.14 (22.9%), 359.14 (5.4%), 359.15 (2.3%), 360.14 (1.0%). Elemental analysis: C, 67.20; H, 6.49; N, 3.92; O, 13.43; S, 8.97%. 1H-NMR δ = 1.41 (s, 9H), 4.068–4.162 (m s, 2H), 5.44 (br s, 2H), 7.24 (m, 5H), 7.33 (m, 5H). 13C-NMR δ = 198.096, 154.534, 136.607, 136.351, 128.757, 128.618, 128.423, 128.348, 127.359, 80.145, 64.392, 33.241, 28.066. HPLC analysis (hexane/2-propanol = 400/1.5, flow = 1.2 mL min−1; T = 308 K): peaks D: 28.8 min; L: 34.9 min.
D,L-N-Boc-Phe-SEt (racrac-2a). White solid, mp 79–81 °C. Chemical formula: C16H23NO3S, exact mass: 309.14, molecular weight: 309.42. m/z: 309.14 (100.0%), 310.14 (18.6%), 311.14 (5.3%), 311.15 (1.5%). Elemental analysis: C, 62.11; H, 7.49; N, 4.53; O, 15.51; S, 10.36%. 1H-NMR, δ = 1.236 (t, J = 7.465 Hz, 3H), 1.4 (s, 9H), 2.874 (q, J = 7.383 Hz, 2H), 3.04 (br s, 1H), 3.06–3.178(m, 1H), 4.62 (br s, 1H), 4.87 (br s, 1H), 7.135–7.19 (m, 2H), 7.205–7.31 (m, 3H). 13C-NMR, δ = 200.799, 154.854, 135.825, 129.243, 128.344, 126.870, 80.010, 60.944, 38.355, 28.163, 23.156, 14.326. HPLC analysis (hexane/2-propanol = 400/1.5, flow = 1.2 mL min−1, T = 308 K): peaks D: 26.4 min; L: 19.5 min.
D,L-N-Boc-Phe-SBn (racrac-2b). White solid, mp 103–104 °C. Chemical formula: C21H25NO3S, exact mass: 371.16, molecular weight: 371.49. m/z: 371.16 (100.0%), 372.16 (23.1%), 373.15 (4.5%), 373.16 (3.4%), 372.15 (1.2%), 374.15 (1.0%). Elemental analysis: C, 67.89; H, 6.78; N, 3.77; O, 12.92; S, 8.63%. 1H-NMR δ = 1.388 (s, 9H), 3.1 (br s, 2H), 4.052–4.158 (m s, 2H), 4.65 (br s, 1H), 4.84 (br s, 1H), 7.02–7.026–7.127 (m, 2H), 7.167–7.330 (m, 8H). 13C-NMR δ = 200.171, 154.822, 137.095, 135.567, 129.211, 128.770, 128.406, 127.120, 126.855, 80.089, 60.828, 38.198, 33.132, 28.128. HPLC analysis (hexane/2-propanol = 99/1, flow = 1.2 mL min−1, T = 308 K): peaks D: 15.4 min; L: 14.6 min.
D,L-N-Boc-homoPhe-SEt (racrac-3a). White solid, mp 85–86 °C. Chemical formula: C17H25NO3S, exact mass: 323.16, molecular weight: 323.45. m/z: 323.16 (100.0%), 324.16 (18.8%), 325.15 (4.5%), 325.16 (2.4%), 324.15 (1.2%). Elemental analysis: C, 63.13; H, 7.79; N, 4.33; O, 14.84; S, 9.91%. 1H-NMR, δ = 1.248 (t, J = 7.415 Hz, 3H), 1.463 (s, 9H), 1.8–1.959 (m, 1H), 2.1–2.26 (m, 1H), 2.6–2.8 (m, 2H), 2.876 (q, J = 7.415 Hz, 2H), 4.382 (br s,1H), 4.960 (br s,1H), 7.179 (m, 2, 5H), 7.276 (m, 2, 5H). 13C-NMR, δ = 201.082, 155.033, 140.572, 128.438, 128.254, 126.024, 79.954, 60.221, 34.365, 31.574, 28.204, 23.033, 14.374. HPLC analysis (hexane/2-propanol = 99/1, flow = 1.2 mL min−1, T = 308 K): peaks D: 13.5 min; L: 32.3 min.
D,L-N-Boc-homoPhe-SBn (racrac-3b). White solid, mp 77–78 °C. Chemical formula: C22H27NO3S, exact mass: 385.17, molecular weight: 385.52. m/z: 385.17 (100.0%), 386.17 (25.0%), 387.17 (4.8%), 387.18 (3.4%), 388.7 (1.1%). Elemental analysis: C, 68.54; H, 7.06; N, 3.63; O, 12.45; S, 8.32%. 1H-NMR δ = 1.460 (s, 9H), 1.907 (br s, 1H), 2.2 (br s, 1H), 2.67 (m, 2H), 4.121 (s, 2H), 4.42 (br s, 1H), 4.947 (br, 1H), 7.120–7.330 (m, 10H). 13C-NMR δ = 200.516, 155.008, 140.446, 137.079, 128.694, 128.437, 128.363, 128.268, 127.133, 126.069, 80.100, 60.158, 34.288, 33.069, 31.529, 28.202. HPLC analysis (hexane/2-propanol = 99/5, flow = 1.2 mL min−1, T = 308 K): peaks D: 10 min; L: 19 min.
D,L-N-Boc-norLeu-SEt (racrac-4a). White solid, mp 37–38 °C. Chemical formula: C13H25NO3S, exact mass: 275.16, molecular weight: 275.41. m/z: 275.16 (100.0%), 276.16 (14.5%), 277.15 (4.5%), 277.16 (1.7%), 276.15 (1.2%). Elemental analysis: C, 56.69; H, 9.15; N, 5.09; O, 17.43; S, 11.64%. 1H-NMR δ = 0.901 (t, J = 7.3 Hz, 3H), 1.250 (t, J = 7.312 Hz, 3H), 1.289–1.416 (br s, 4H), 1.459 (s, 9H), 1.599(br s, 1.5H), 2.2 (br s, 0.5H), 2.88 (q, J = 7.63 Hz, 2H), 4.316 (br s, 1H), 4.902 (br s,1H). 13C-NMR, δ = 201.337, 155.098, 79.694, 60.405, 32.318, 28.140, 27.266, 22.880, 22.065, 14.297, 13.558. HPLC analysis (hexane/2-propanol = 99/1 flow = 1.2 mL min−1, T = 308 K): peaks D: 6 min; L: 7.4 min.
D,L-N-Boc-norLeu-SBn (racrac-4b). White solid, mp 54–55 °C. Chemical formula: C18H27NO3S, exact mass: 337.17, molecular weight: 337.48. m/z: 337.17 (100.0%), 338.17 (20.6%), 339.17 (4.8%), 339.18 (2.5%). Elemental analysis: C, 64.06; H, 8.06; N, 4.15; O, 14.22; S, 9.50%. 1H-NMR δ = 0.863 (t, J = 7.279 Hz, 3H), 1.3 (br s, 4H), 1.423 (s, 9H), 1.562 (br s, 1.5H), 1.825 (br s, 0.5H), 4.088 (s, 2H), 4.332 (br s, 1H), 4.984 (br s, 1H), 7.162–7.293 (m, 5H). 13C-NMR δ = 200.807, 155.109, 137.155, 128.680, 128.409, 128.088, 79.953, 60.383, 33.015, 32.313, 28.191, 27.282, 22.096, 13.616. HPLC analysis (hexane/2-propanol = 99/1, flow = 1.2 mL min−1, T = 308 K): peaks D: 8.4 min; L: 10.3 min.
D,L-N-Boc-norVal-SEt (racrac-5a). White solid, mp 32–34 °C. Chemical formula: C12H23NO3S, exact mass: 261.14, molecular weight: 261.38. m/z: 261.14 (100.0%), 262.14 (14.3%), 263.14 (5.3%). Elemental analysis: C, 55.14; H, 8.87; N, 5.36; O, 18.36; S, 12.27%. 1H-NMR, δ = 0.901 (t, J = 7.3 Hz, 3H), 1.250 (t, J = 7.312 Hz, 3H), 1.289–1.410 (br s, 2H), 1.459 (s, 9H), 1.599 (br s, 1.5H), 2.2 (br s, 0.5H), 2.88 (q, J = 7.63 Hz, 2H), 4.316 (br s, 1H), 4.902 (br s,1H). 13C-NMR, δ = 201.356, 155.070, 79.664, 60.216, 34.651, 28.117, 22.853, 18.470, 14.264, 13.388. HPLC analysis (hexane/2-propanol = 99/5, flow = 1.2 mL min−1, T = 308 K): peaks D: 10 min; L: 19 min.
D,L-N-Boc-amino-octanoic-acid-SEt (racrac-6a). Yellow oil. Chemical formula: C15H29NO3S, exact mass: 303.19, molecular weight: 303.46. m/z: 303.19 (100.0%), 304.19 (17.5%), 305.18 (4.5%), 305.19 (2.1%). Elemental analysis: C, 59.37; H, 9.63; N, 4.62; O, 15.82; S, 10.57%. 1H-NMR δ = 0.901 (t, J = 7.3Hz, 3H), 1.250 (t, J = 7.312 Hz, 3H), 1.2–1,4 (m, 8H), 1.44 (m, 9H), 1.599 (br s, 1.5H), 2.2 (br s, 0.5H), 2.88 (q, J = 7.63 Hz, 2H), 4.316 (br s, 1H), 4.902 (br s,1H). 13C-NMR δ = 201.401, 155.128, 79.816, 60.475, 32.725, 31.392, 28.677, 28.191, 25.128, 22.957, 22.333, 14 61, 13.801. HPLC analysis (hexane/2-propanol = 99/1, flow = 1.2 mL min−1, T = 308 K): peaks D: 9.7 min; L: 11.4 min; naphthalene: 8.1 min.
Racemization procedure
Approximately 20 mg of substrate were dissolved in 2 mL of isopropanol in a polarimetric tube and the rotation value was recorded. 0.5 eq. of diaza-bicyclo-undecene (DBU) were then added at 40 °C and the decrease in optical rotation value was recorded during 30 min.Control experiments proved that no change in the absolute rotation values in time was observed when the substrate was the N-Boc-amino acid or with the thioesters in the absence of bases.
Chemical formula: C13H17NO4, exact mass: 251.12, molecular weight: 251.28. m/z: 251.12 (100.0%), 252.12 (14.4%), 253.12 (1.8%). Elemental analysis: C, 62.14; H, 6.82; N, 5.57; O, 25.47%. 1H-NMR δ = 1.4 (s, 9H), 5.117 (br s, 1H), 5.338 (br s, 0.5H), 5.623 (br s, 0.5H), 7.255–7.453 (m, 5H). 13C-NMR δ = 173.417, 156.860, 138.205, 128.785, 128.387, 127.933, 127.148, 80.104, 58.856, 28.193. [α]D: +149.4 (c = 0.8, 1 N HCl; lit [10] + 156, c = 1, 1 N HCl, 25 °C).
Enantiomeric excess analyzed by chiral HPLC was >99%. SS-1b was deprotected with dioxane saturated with HCl at 60 °C to give 1.106 g of L-PheGly-OH*HCl as a white solid (87%; ee >99%).
HPLC analysis of t0 sample: racrac-3a thioester + naphthalene, peaks: D: 12.5 min, L: 28.5 min, naphthalene: 4.2 min. HPLC analysis of tn sample: SS-3c + naphthalene, peaks: D: 8 min, L: 11 min, naphthalene: 4.2 min. ee (L): 90%. SS-3c: 1H-NMR δ = 1.263 (t, 6H), 1.46 (s, 9H), 1.94–2.30 (m, 2H), 2.73 (m, 2H), 4.3 (br s, 0.5H), 5–5.15 (m, 1.5H), 7.18–7.32 (m, 5H). 13C-NMR δ = 171.883, 155.186, 140.854, 128.282, 128.210, 125.921, 79.495, 68.726, 53.379, 34.345, 31.495, 28.178, 21.576. Chemical formula: C17H25NO4, exact mass: 307.18, molecular weight: 307.38. m/z: 307.18 (100.0%), 308.18 (19.2%), 309.19 (1.7%). Elemental analysis: C, 66.43; H, 8.20; N, 4.56; O, 20.82%. Polarimetric analysis. [α]D = 11.4° (c = 1, CHCl3, 25 °C), ee 90%.
:1.5; flow = 1.2 mL min−1; T = 308 K).
At the end of the reaction (tn), enzyme was filtered and kept for successive use and t-BuOH was evaporated. The residue was dissolved in water (at pH 8), the water phase washed with ethyl ether (3 times). L-N-Boc-AA-OH was recovered by extraction of the water phase with ethyl acetate (3 times) at pH 3. Drying and evaporation of the solvent gave a quantitative amount of L-N-Boc-AA-OH. Enantiomeric excess was analyzed by chiral HPLC, polarimetric analysis or by chiral GC.
Analytical evaluation of enantiomeric excess of the DKR products:
L-N-Boc-AA-OH was reacted with diazomethane and the enantiomeric excess of N-Boc-AA-OMe has been evaluated by chiral GC analysis (ramp 60 °C → (1.5 °C min−1) 180 °C), or by chiral HPLC (Chiralcel OD 5 μm column, λ = 254 nm).
L-N-Boc-AA-OH was deprotected with dioxane saturated with HCl at 60 °C to give L-AA-OH*HCl as a white solid. And the enantiomeric excess of L-AA-OH*HCl has been evaluated by chiral HPLC (Crownpack CR column, eluent H2O/perchloric acid, λ = 210 nm).
By polarimetric analysis on L-AA-OH*HCl.
L-N-Boc-Phe-OH (SS-2d). Yield: 98% (3.165 mmol, 839 mg). 1H-NMR δ = 1.28 (s, 3H), 1.42 (s, 6H), 2.91–3.19 (m, 2H), 4.40 (s, 0.4H), 4.62 (s, 0.6H), 4.99 (s, 0.7H), 6.59 (s, 0.3H), 7.19–7.29 (m, 5H), 10.6 (br s, 1H). 13C-NMR δ = 176.069, 156.294, 135.880, 129.329, 128.421, 126.874, 80.148, 54.201, 37.802, 28.176. Chiral HPLC analysis: peak L: 16.1 min (D: 15.5 min). ee (L-N-Boc-PheAla-OMe): 99%. Polarimetric analysis: [α]D (L-PheAla-OH*HCl) = + 34.8° (HCl 6 N, c = 1, 25 °C).
L-N-Boc-homoPhe-OH (SS-3d). Yield: 94% (2.9 mmol, 811 mg). 1H-NMR δ = 1.396 (s, 9H), 1.951(m, 1H), 2.132 (m, 1H) 2.666 (t, J = 7.8 Hz, 2H), 4.080 (br s, 0.4H), 4.331 (br s, 0.6H), 5.115 (br s, 0.6H), 6.574 (br s, 0.4H), 7.122–7.210 (m, 5H). 13C-NMR δ = 176.936, 155.586, 140.642, 128.404, 128.364, 126.088, 80.197, 53.211, 34.000, 31.358, 28.223. ee (L-N-Boc-homoPhe-OMe): 99%. Polarimetric analysis: [α]D(L-homoPhe-OH*HCl) = + 51.63° (HCl acq, c = 1, 25 °C).
L-N-Boc-norLeu-OH (SS-4d). Yield: 91% (3.31 mmol, 765.56 mg). 1H-NMR δ = 0.90 (t, J = 6.94 Hz, 3H), 1.28–1.41 (m, 4H), 1.45 (s, 9H), 1.60–1.93 (m, 2H), 4.11 (br s, 0.3H), 4.30 (br s, 0.7H), 5.01 (br s, 0.7H), 6.09 (br s, 0.3H). 13C-NMR δ = 177.452, 155.768, 80.088, 53.355, 32.115, 28.206, 27.309, 22.171, 13.678. ee (L-norLeu -OMe): 99%. Polarimetric analysis: [α]D (L-norLeu-OH*HCl) = + 23.4° (HCl 6 N, c = 2, 25 °C).
L-N-Boc-norVal-OH (SS-5d). Yield: 98.5% (3.774 mmol, 820 mg). 1H-NMR δ = 0.95 (t, J = 7.37 Hz, 3H), 1.35–1.45 (m, 2H), 1.44 (s, 9H), 1.59–1.89 (m, 2H), 4.15 (br s, 0.3H), 4.30 (br s, 0.7H), 4.99 (br s, 0.7H), 5.95 (br s, 0.3H). 13C-NMR δ = 177.466, 155.641, 80.53, 34.514, 28.217, 18.530, 13.511. HPLC peaks: D: 9 min; L: 10.6 min; naphthalene: 6.1 min. ee (L-norVal-OMe): 99.9%. Polarimetric analysis: [α]D(L-norVal-OH*HCl) = + 24.8° (HCl 6 N, c = 2, 25 °C).
L-N-Boc-aminooctanoic-OH (SS-6d). Yield: 96% (3.16 mmol, 820 mg). 1H-NMR δ = 0.90 (t, J = 6.94 Hz, 3H), 1.44 (m, 9H), 1.20–1.40 (m, 8H), 1.60–1.70 (m, 1H), 1.83 (br s, 1H), 4.10 (br s, 0.3H), 4.30 (br s, 0.7H), 5.00 (br s, 0.7H), 6.04 (br s, 0.3H), 9.60 (br s, 1H). 13C-NMR δ = 177.529, 155.584, 79.984, 53.349, 32.420, 31.469, 28.732, 28.217, 25.144, 22.413, 13.871. ee (L-aminooctanoic-OMe*HCl): 99.8 %. Polarimetric analysis: [α]D (L-aminooctanoic –OH*HCl) = + 25.87° (HCl 6 N, c = 1, 25 °C).
L-N-Boc-phenylalanine-amide (SS-2e). Compound 2e was obtained as a solid (reaction time 7 h) in 82% yield (70 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 95
:5, flow rate = 1.00 mL min−1, λ = 210 nm) tR = 13.83 min; enantiomeric excess 98.3%; mp 147 °C (AcOEt/Hexane); [α]20D = 10.74 (c 1, MeOH); 1H NMR(400 MHz, DMSO-d6) δ = 1.30 (s, 9H), 2.74 (dd, 1H), 2.97 (dd, 1H), 3.98 (s, 1H), 4.10 (s, 1H), 6.68 (d, 1H), 6.95 (d, 1H), 7.18 (m, 1H), 7.22–7.32 (m, 5H); 13C NMR(400 MHz, DMSO-d6) δ = 33.8, 59.9, 60.4, 83.1, 130.4, 132.3, 133.5, 143.4, 160.3, 178.8; m/z [M + Na]+ calcd for C14H21N3O2 287.1, found 287.0.
L-N-Boc-phenylalanine-propylamide (SS-2f). Compound 2f was obtained as a solid (reaction time 7 h) in 76% yield (75.7 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 98
:2, flow rate = 1.00 mL min−1, λ = 254 nm) tR = 15.51 min; enantiomeric excess >99%; mp 113 °C (CH2Cl2/iPr2O); [α]20D = 2.89 (c 1.5, MeOH); 1H NMR(400 MHz, CDCl3) δ = 0.80 (t, 3H), 1.40 (m, 11H), 3.03 (m, 2H), 3.12 (m, 2H), 4.28 (dd, 1H), 5.12 (s, 1H), 5.80 (s, 1H), 7.18–7.30 (m, 5H); 13C NMR(400 MHz, CDCl3) δ = 11.1, 22.5, 28.2, 38.7, 41.1, 56.1, 80.1, 126.8, 128.6, 129.2, 136.8, 155.4, 171.1; m/z [M + H]+ calcd for C17H26N2O3 307.2, found 307.0.
L-N-Boc-phenylalanine-benzylamide (SS-2g). Compound 2g was obtained as a solid (reaction time 5 h) in 89% yield (102 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 98
:2, flow rate = 1.00 mL min−1, λ = 254 nm) tR = 24.00 min; enantiomeric excess 98.3%; mp 128 °C (AcOEt/iPr2O); [α]20D = −2.7 (c 1, MeOH); 1H NMR(400 MHz, CDCl3) δ = 1.40 (s, 9H), 3.08 (ddd, 2H), 4.34 (m, 3H), 5.02 (s, 1H), 6.06 (s, 1H); 7.08–7.30 (m, 10H); 13C NMR(400 MHz, CDCl3) δ = 28.1, 38.7, 43.3, 50.0, 80.2, 126.7, 127.2, 127.5, 128.4, 129.2, 136.6, 137.6, 155.6, 171.6; m/z [M + H]+ calcd for C12H26N2O3 355.2, found 355.1.
L-N-Boc-homophenylalanine-amide (SS-3e). Compound 3e was obtained as a solid (reaction time 30 h) in 59.9% yield (49.1 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 93
:7, flow rate = 1.00 mL min−1, λ = 210 nm) tR = 19.96 min; enantiomeric excess 98.94%; mp 135–136 °C, (AcOEt/iPr2O); [α]20D = + 4.8 (c 0.5, CHCl3); 1H NMR(400 MHz, CDCl3) δ = 1.44 (s, 9H), 1.94, 2.16 (ddt, 2H), 2.70 (t, 2H), 4.12 (s, 1H), 5.16 (s, 1H), 5.72 (s, 1H), 6.22 (s, 1H), 7.15–7.30 (m, 5H); 13C NMR(400 MHz, CDCl3) δ = 28.3, 31.8, 33.9, 53.8, 80.3, 126.2, 128.3, 128.8, 140.8, 155.7, 174.5; m/z [M + Na]+ calcd for C15H22N2O3 301.2, found 301.2.
L-N-Boc-homophenylalanine-propylamide (SS-3f). Compound 3f was obtained as a solid (reaction time 24 h) in 62.5% yield (61.9 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 98
:2, flow rate = 1.00 mL min−1, λ = 210 nm) tR = 15.13 min; enantiomeric excess 99.04%; mp 97 °C (hexane/iPr2O); [α]20D = −7.7 (c 1, MeOH); 1H NMR(400 MHz, CDCl3) δ = 0.74 (t, 3H), 1.44 (s, 9H), 1.50 (dq, 2H), 1.92–2.12 (ddt, 2H), 2.67 (t, 2H), 3.19 (m, 2H), 4.12 (s, 1H), 5.36 (s, 1H), 6.50 (s, 1H), 7.15–7.28 (m, 5H); 13C NMR(400 MHz, CDCl3) δ = 11.3, 22.7, 28.3, 31.9, 34.2, 41.1, 54.2, 79.9, 126.0, 128.3, 128.4, 141.0, 155.8, 172.0; m/z [M + Na]+ calcd for C22H28N2O3 391.2, found 391.2.
L-N-Boc-homophenylalanine-benzylamide (SS-3g). Compound 3g was obtained as a solid (reaction time 24 h) in 87.1% yield (99.3 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 97
:3, flow rate = 1.00 mL min−1, λ = 210 nm) tR = 20.93 min; enantiomeric excess 99.16%; mp 88 °C (hexane/AcOEt); [α]20D = −14.1(c 1, MeOH); 1H NMR(400 MHz, CDCl3) δ = 1.40 (s, 9H), 1.92–2.17 (ddt, 2H), 2.76 (t, 2H), 4.12 (s, 1H), 4.41 (ddd, 2H), 5.12 (s, 1H), 6.55 (s, 1H), 7.12–7.34 (m, 9H); 13C NMR(400 MHz, CDCl3) δ = 28.3, 31.9, 33.9, 43.4, 54.2, 80.2, 126.1, 127.5, 127.6, 128.4, 128.5, 128.7, 138.0, 140.8, 155.7, 171.8; m/z [M + Na]+ calcd for C22H28N2O3 391.2, found 391.2.
L-N-Boc-norleucine-amide (SS-4e). Compound 4e was obtained as a solid in 65.5% yield (55 mg): HPLC analysis (ChiralCel OD, hexane/2-propanol 95
:5, flow rate = 1.00 mL min−1, λ = 205 nm, 4° C) tR = 11.69 min; enantiomeric excess 99.54%; mp 127 °C (AcOEt/Hexane); [α]20D = + 10.74 (c 1, MeOH); 1H NMR (400 MHz, CDCl3) δ = 0.90 (t, 3H), 1.35 (m, 4H), 1.44 (s, 9H), 1.60 (m, 1H), 1.82 0(m, 1H), 4.12 (s, 1H), 5.16 (s, 1H), 5.87 (s, 1H), 6.36 (s, 1H); 13C NMR(400 MHz, CDCl3) δ = 13.8, 22.3, 27.6, 28.3, 32.2, 54.1, 80.0, 155.7, 175.0; m/z [M + Na]+ calcd for C11H22N2O3 253.2, found 253.1.
L-N-Boc-norleucine-propylamide (SS-4f). Compound 4f was obtained as a solid, mp 74–75 °C (reaction time 30 h), in 65% yield (93 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 99
:1, flow rate = 1.00 mL min−1, λ = 210 nm) tR = 15.71 min; enantiomeric excess 98.36%; [α]20D = −14.5 (c 1, MeOH); 1H NMR(400 MHz, CDCl3) δ = 0.82 (m, 6H), 1.26 (m, 4H), 1.37 (m, 9H), 1.46 (m, 2H), 1.53 (m, 1H), 1.75 (m, 1H), 3.14 (m, 2H), 3.94 (m, 1H), 5.02 (s, 1H), 6.24 (s, 1H); 13C NMR(400 MHz, CDCl3) δ = 11.2, 13.7, 22.3, 22.6, 27.7, 28.2, 32.4, 41.0, 54.6, 79.7, 155.7, 172.3; m/z [M + H]+ calcd for C15H30N2O3 273.2, found 273.0.
L-N-Boc-norleucine-benzylamide (SS-4g). Compound 4g was obtained as a solid (reaction time 20 h) in 69% yield (100 mg): HPLC analysis (Chiralcel OD, hexane/2-propanol 98
:2, flow rate = 1.00 mL min−1, λ = 210 nm) tR = 14.59 min; enantiomeric excess 96.06%; mp 70 °C (AcOEt/Hexane); [α]20D = −19.5 (c 1, MeOH); 1H NMR(400 MHz, CDCl3) δ = 0.88 (t, 3H), 1.32 (m, 4H), 1.41 (s, 9H), 1.86 (m, 2H), 4.06 (d, 1H), 4.43 (ddd, 2H), 4.99 (s, 1H), 6.51 (s, 1H), 7.21–7.34 (m, 5H); 13C NMR (400 MHz, CDCl3) δ = 13.8, 22.3, 27.7, 28.2, 32.3, 43.2, 54.7, 79.8, 127.2, 127.5, 128.5, 138.2, 155.7, 172.3; m/z [M + H]+ calcd for C18H28N2O3 321.2, found 321.1.
Acknowledgements
We are grateful for the financial support COST Action CM0701CASCAT Cascade Chemo-Enzymatic Processes: New Synergies between Chemistry and Biochemistry.Notes and references
- Part of this work has been published as preliminary communication in ref. 67 and 81. Scheme 2 and Table 1 describe entirely unpublished material.
- D. E. Cane, C. T. Walsh and C. Khosla, Science, 1998, 282, 63 CrossRef CAS.
- M. Strieker and M. A. Marahiel, ChemBioChem, 2009, 10, 607 CrossRef CAS.
- S. Caboche, M. Pupin, V. Leclère, P. Jacques and G. Kucherov, BMC Struct. Biol., 2009, 9, 15 CrossRef.
- A. Loffet, J. Pept. Sci., 2002, 8, 1 CrossRef CAS.
- S. Kee and S. D. S. Jois, Curr. Pharm. Des., 2003, 9, 1209 CrossRef.
- S. Hanessian, G. McNaughton Smith and H. G. Lombart, Tetrahedron, 1997, 53, 12789 CrossRef CAS.
- L. Wang and P. G. Schultz, Angew. Chem., Int. Ed., 2005, 44, 34 CrossRef CAS.
- H. Sun, C. J. Zaneta, C. Y. Yang, L. Xu, L. Liu, Y. Tomita, H. Pan, Y. Yoshioka, K. Krajewski, P. P. Roller and S. Wang, J. Am. Chem. Soc., 2004, 126, 16686 CrossRef CAS.
- T. K. Oost, C. Sun, R. C. Armstrong, A. S. Al-Assaad, S. F. Betz, T. L. Deckwerth, H. Ding, S. W. Elmore, R. P. Meadows, E. T. Olejniczak, A. Oleksijew, T. Oltersdorf, S. W. Rosenberg, A. R. Shoemaker, K. J. Tomaselli, H. Zou and S. W. Fesik, J. Med. Chem., 2004, 47, 4417 CrossRef CAS.
- R. M. Williams, Synthesis of optically active α-amino acids, Pergamon, Oxford, 1989 Search PubMed.
- T. Wirth, Angew. Chem., Int. Ed., 1997, 36, 225 CrossRef CAS.
- J. A. Ma, Angew. Chem., Int. Ed., 2003, 42, 4290 CrossRef CAS.
- M. J. Burk, Acc. Chem. Res., 2000, 33, 363 CrossRef CAS.
- R. Noyori and T. Ohkuma, Angew. Chem., Int. Ed., 2001, 40, 40 CrossRef CAS.
- S. Yamaguchi, H. Komeda and Y. Asano, Appl. Environ. Microbiol., 2007, 73, 5370 CrossRef CAS.
- D. Tessaro, G. Molla, L. Pollegioni and S. Servi, Chemo-enzymatic deracemization methods, in Modern Biocatalysis, ed. W.-D. Fessner and T. Anthonsen, 2009, Wiley-VCH, p. 195 Search PubMed.
- A. S. Bommarius, M. Kottenhahn, H. Klenk and K. Drauz, in Microbial Reagents in Organic Synthesis, ed. S. Servi, NATO-ASI Series, Kluwer Academic Publisher, 1992, p. 161 Search PubMed.
- K. Faber, Chem.–Eur. J., 2001, 7, 5004 CrossRef CAS.
- S. Servi, D. Tessaro and G. Pedrocchi-Fantoni, Coord. Chem. Rev., 2008, 252, 715 CrossRef CAS.
- M. T. El Gihani and J. M. J. Williams, Curr Opin. Chem. Biol., 1999, 3, 11 CrossRef CAS.
- R. Azerad and D. Buisson, Curr. Opin. Biotechnol., 2000, 11, 565 CrossRef CAS.
- R. Noyori, M. Tokunaga and M. Kitamura, Bull. Chem. Soc. Jpn, 1995, 68, 3623 CrossRef.
- C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789 CrossRef CAS.
- P. M. Dinh, J. A. Howarth, A. R. Hudnott, J. M. J. Williams and W. Harris, Tetrahedron Lett., 1996, 37, 7623 CrossRef CAS.
- R. M. Williams and J. A. Hendrix, Chem. Rev., 1992, 92, 889 CrossRef CAS.
- G. Fülling and C. J. Sih, J. Am. Chem. Soc., 1987, 109, 2845 CrossRef.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294 CrossRef CAS.
- A. L. E. Larsson, B. A. Persson and J. E. Backvall, Angew. Chem., Int. Ed. Engl., 1997, 36, 1211 CrossRef CAS.
- F. F. Huerta, A. B. E. Minidis and J. E. Backvall, Chem. Soc. Rev., 2001, 30, 321 RSC.
- J. Petzold and J. E. Bäckvall, J. Am. Chem. Soc., 2005, 127, 17620 CrossRef.
- B. Martin-Matute, M. Edin, K. Bogàr and J.E. Bäckvall, Angew. Chem., Int. Ed., 2004, 116, 6697 CrossRef.
- O. Pamies and J. E. Backvall, Trends. Biotechnol., 2004, 22, 130 CrossRef CAS.
- O. Pamies and J. E. Backvall, Chem. Rev., 2003, 103, 3247 CrossRef CAS.
- M.-J. Kim, Y. Ahn and J. Park, Curr. Opin. Chem. Biotechnol., 2002, 13, 578 CrossRef CAS; J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999 CrossRef; Y. K. Choi, Y. Kim, K. Han, J. Park and M.-J. Kim, J. Org. Chem., 2009, 74, 9543 CrossRef.
- R. L. Gu, I. S. Lee and I. C. J. Sih, Tetrahedron Lett., 1992, 33, 1953 CrossRef CAS.
- J. Z. Crich, R. Brieva, P. Marquart, R. L. Gu, S. Flemming and C. J. Sih, J. Org. Chem., 1993, 58, 3252 CrossRef CAS.
- H. S. Bevinakatti, R. V. Newadkar and A. A. Banerji, J. Chem. Soc., Chem. Commun., 1990, 1091 RSC.
- H. S. Bevinakatti, A. A. Banerji, R. V. Newadkar and A. A. Mokashi, Tetrahedron: Asymmetry, 1992, 3, 1505 CrossRef CAS.
- N. J. Turner, J. R. Winterman, R. McCague, J. S. Parrat and S. J. C. Taylor, Tetrahedron Lett., 1995, 1113 CrossRef CAS.
- S. A. Brown, M. C. Parker and N. J. Turner, Tetrahedron: Asymmetry, 2000, 11, 1687 CrossRef CAS.
- K. Toth and J. P. Richard, J. Am. Chem. Soc., 2007, 129, 3013 CrossRef CAS.
- V. S. Parmar, A. Singh, K. S. Bisht, N. Kumar, Y. N. Belokon, K. A. Kochetkov, N. S. Ikonnikov, S. A. Orlova, V. I. Tararov and T. F. Saveleva, J. Org. Chem., 1996, 61, 1223 CrossRef CAS.
- M. Schnoelzer and S. B. H. Kent, Science, 1992, 256, 221 CAS.
- P. E. Dawson, T. W. Muir, I. Clark-Lewis and S. B. H. Kent, Science, 1994, 266, 776 CAS.
- J. B. Blanco-Canosa and P. E. Dawson, Angew. Chem., Int. Ed., 2008, 47, 6851 CrossRef CAS.
- R. Gill, E. Lopez-Fandiiio and E. Vulfson, J. Am. Chem. Soc., 1995, 117, 6175 CrossRef.
- S. Santra, M. E. Bucks, S. N. Savinov, Abstracts of Papers, 240th ACS National Meeting, Boston, MA, United States, August 22–26 2010, 2010.
- J. P. Tam and Y. A. Lu, Tetrahedron Lett., 1997, 38, 5599 CrossRef CAS.
- S. Masamune, S. Kamata and W. Schilling, J. Am. Chem. Soc., 1975, 97, 3515 CrossRef CAS.
- E. J. Corey and K. C. Nicolaou, J. Am. Chem. Soc., 1974, 96, 5614 CrossRef CAS.
- A. Thalmann, K. Oertle and H. Gerlach, Org. Synth., 1985, 63, 192 CAS; A. Thalmann, K. Oertle and H. Gerlach, Org. Synth., 1990, Coll. 7, 470 Search PubMed.
- A. Zaks and A. M. Klibanov, Proc. Natl. Acad. Sci. U. S. A., 1985, 82, 3192 CrossRef CAS.
- K. C. Sproull, G. T. Bowman, G. Carta and J. L. Gainer, Biotechnol. Prog., 1997, 13, 71 CrossRef CAS.
- M. Caussette, A. Marty and D. Combes, J. Chem. Technol. Biotechnol., 1997, 68, 257 CrossRef CAS.
- D. Bianchi and P. Cesti, J. Org. Chem., 1990, 55, 5657 CrossRef CAS.
- H. Frykman, N. Ohrner, T. Norin and K. Hult, Tetrahedron Lett., 1993, 8, 1367 CrossRef.
- S. Iriuchijima and N. Kojima, J. Chem. Soc., Chem. Commun., 1981, 185 RSC.
- P. J. Um and D. G. Drueckhammer, J. Am. Chem. Soc., 1998, 120, 5605 CrossRef CAS.
- S. D. Tan, M. M. Giinter and D. G. Drueckhammer, J. Am. Chem. Soc., 1995, 117, 9093 CrossRef.
- C. N. Lin and S. W. Tsai, Biotechnol. Bioeng., 2000, 69, 31 CrossRef CAS.
- P. D'Arrigo, D. Arosio, L. Cerioli, D. Moscatelli, S. Servi, F. Viani and D. Tessaro, Tetrahedron: Asymmetry, 2011, 22, 851 CrossRef CAS.
- T. L. Amyes and J. P. Richard, J. Am. Chem. Soc., 1992, 114, 10197 CrossRef.
- K. A. Connors and M. L. Bender, J. Org. Chem., 1961, 26, 2498 CrossRef CAS.
- In experiments with model substrates in ref. 61 it was found that the rate of hydrolysis of the oxoester was about 20% faster than that of the corresponding thioester. In the enzyme catalyzed hydrolysis we found that the ratio of the specific activities of the protease catalyzed hydrolysis L-N-Boc-Phg-OEt/ L-N-Boc-Phg-SEt was 2.52 (ref. 61).
- A number of analogues of compound 1a were also successfully deracemized (see ref. 66).
- D. Arosio, A. Caligiuri, P. D'Arrigo, G. Pedrocchi-Fantoni, C. Rossi, C. Saraceno, S. Servi and D. Tessaro, Adv. Synth. Catal., 2007, 349, 1345 CrossRef CAS.
- Subtilisin Carlsberg from B. subtilis from Fluka and Alcalase 2.5L DX from Novozymes (50 U mL−1) were used in all experiments and gave similar results. Immobilized subtilisin from B. subtilis Fluka (2.4 U g−1) and Alcalase 3.0 T from Novozymes (3.0 U g−1) were found equivalent. Crosslinked subtilisin from B. subtilis CLEA® was from CLEA technologies (450 U g−1).
- J. J. Godfrey, U.S. Patent 3086049, 1963 CrossRef CAS; S. Horvat, S. Tomic and Z. Jericevic, Tetrahedron, 1984, 40, 1047 CrossRef CAS; S. Horvat and D. J. Keglevic, Carbohydr. Res., 1982, 1, 71 Search PubMed; M. Yoshimatsu, K. Machida, T. Fuseya, H. Shimizu and T. Kataoka, J. Chem. Soc., Perkin Trans 1, 1996, 15, 1839 RSC.
- S. van Pelt, R. L. M. Teeuwen, M. H. A. Janssen, R. A. Sheldon, P. J. Dunn, R. M. Howard, R. Kumar, I. Martínez and J. W. Wong, Green Chem., 2011, 13, 1791 RSC; D. J. C. Constable, P. J. Dunn, J. D. Hayler, G. R. Humphrey, J. L. Leazer, R. J. Linderman, K. Lorenz, J. Manley, B. A. Pearlman, A. Wells, A. Zaks and T. Y. Zhang, Green Chem., 2007, 9, 411 RSC; J. S. Carey, D. Laffan, C. Thomson and M. T. Williams, Org. Biomol. Chem., 2006, 4, 2337 Search PubMed.
- U. T. Bornscheuer and R. J. Kazlauskas, Hydrolases in Organic Synthesis Regio- and Stereoselective Biotransformations, Wiley-VCH, Weinheim, 2nd edn, 2006 CrossRef CAS; V. Gotor, in Microbial Reagents in Organic Synthesis, ed. S. Servi, Kluwer Academic Publisher, NATO-ASI Series, 1992, p. 199 CrossRef CAS; A. Torres-Gavilán, E. Castillo and A. López-Munguía, J. Mol. Catal. B: Enzym., 2006, 41, 136 CrossRef CAS; K. Ditrich, Synthesis, 2008, 2283 CrossRef; M. A. P. J. Hacking, M. A. Wegman, J. Rops, F. van Rantwijk and R. A. Sheldon, J. Mol. Catal. B: Enzym., 1998, 5, 155 CrossRef; F. van Rantwijk, M. A. P. J. Hacking and R. A. Sheldon, Monatsh. Chem., 2000, 131, 549 CrossRef; V. Gotor-Fernández, E. Busto and V. Gotor, Adv. Synth. Catal., 2006, 348, 797 CrossRef; E. Gotor-Fernández and V. Gotor, Curr. Org. Chem., 2006, 10, 1125 CrossRef; P. Tufvesson, A. Annerling, R. Hatti-Kaul and D. Adlercreutz, Biotechnol. Bioeng., 2007, 97, 447 CrossRef; M. Fernandez-Perez and C. Otero, Enzyme Microb. Technol., 2003, 33, 650 CrossRef; A. Baldessari and C. P. Mangone, J. Mol. Catal. B: Enzym., 2001, 11, 335 CrossRef.
- A DKR process based on a lipase resolution/Pd-catalyzed racemization leading to L-amino acid amides and peptides has been reported. The process is limited to phenylglycines derivatives: Y.-K. Choi, Y. Kim, K. Han, J. Park and M.-J. Kim, J. Org. Chem., 2009, 74, 9543 CrossRef CAS.
- K. Kitaguchi, P. Fitzpatric, J. E. Huber and A. M. Klibanov, J. Am. Chem. Soc., 1989, 111, 3094 CrossRef CAS; H. D. Jakubke, in Microbial Reagents in Organic Synthesis, ed. S. Servi, Kluwer Academic Publisher, NATO-ASI Series, 1992, 189 Search PubMed; S. Groothuys, B. H. M. Kuijpers, P. J. L. M. Quadflieg, H. C. P. F. Roelen, R. W. Wiertz, R. H. Blauw, F. L. van Delft and F. P. J. T. Ruttjes, Synthesis, 2006, 3146 Search PubMed.
- For the production of D-amino acid derivatives from D,L-amino acid amide, the use of a hydrolytic enzyme with D-specificity is required. When coupled with a racemase the result is a DKR: Y. Asano and S. Yamaguchi, J. Am. Chem. Soc., 2005, 127, 7696 CrossRef CAS.
- A. Schwarz, D. Steinke, M. R. Kula and C. Wandrey, Biotechnol. Appl. Biochem., 1990, 12, 188 Search PubMed; M. Y. Gololobov, T. L. Voyushina, V. M. Stepanov and P. Adlercreutz, Biochim. Biophys. Acta, 1992, 1160, 188 CrossRef CAS; S. T. Chen, C. L. Kao and K. T. Wang, Int. J. Pept. Protein Res., 1995, 46, 314 CrossRef; C. G. Boeriu, A. E. Frissen, E. Boer, K. van Kekem, D. J. van Zoelen and I. F. Eggen, J. Mol. Catal. B: Enzym., 2010, 66, 33 CrossRef.
- Z. P. Wu and D. Hilvert, J. Am. Chem. Soc., 1989, 111, 4513 CrossRef CAS; K. Khumtaveeporn, A. Ullmann, K. Matsumoto, B. G. Davis and J. B. Jones, Tetrahedron: Asymmetry, 2001, 12, 249 CrossRef.
- A. Joensson, E. Wehtje, P. Adlercreutz and B. Mattiasson, J. Mol. Catal. B: Enzym., 1996, 2, 43 CrossRef CAS; A. L. Margolin, D. F. Tai and A. M. Klibanov, J. Am. Chem. Soc., 1987, 109, 7885 CrossRef; A. Ferjancic, A. Puigserver and H. Gaertner, Appl. Microbiol. Biotechnol., 1990, 32, 651 CrossRef; S. T. Chen, S. Y. Chen and K. T. Wang, J. Org. Chem., 1992, 57, 6960 CrossRef; S. T. Chen, S. Y. Chen, H. J. Chen, H. C. Huang and K. T. Wang, Bioorg. Med. Chem. Lett., 1993, 3, 727 CrossRef; V. Cerovsky and H. D. Jakubke, Biotechnol. Bioeng., 1996, 49, 553 CrossRef; M. V. Sergeeva, V. M. Paradkar and J. S. Dordick, Enzyme Microb. Technol., 1997, 20, 623 CrossRef.
- T. Nuijens, E. Piva, J. A. W. Kruijtzer, D. T. S. Rijkers, R. M. J. Liskamp and P. J. L. M. Quaedflieg, Adv. Synth. Catal., 2011, 353, 1039 CrossRef CAS.
- In a recent paper the DKR of phenylglycine amide was reported: Y. Kim, J. Park and M.-J. Kim, ChemCatChem, 2011, 3, 271 CrossRef CAS.
- W. Yang and D. G. Drueckhammer, J. Am. Chem. Soc., 2001, 123, 11004 CrossRef CAS.
- D. Tessaro, L. Cerioli, S. Servi, F. Viani and P. D'Arrigo, Adv. Synth. Catal., 2011, 353, 2333 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |