Multi-enzymatic cascade synthesis of D-fructose 6-phosphate and deoxy analogs as substrates for high-throughput aldolase screening†
Wolf-Dieter
Fessner
*,
Dirk
Heyl
and
Madhura
Rale
Technische Universität Darmstadt, Institut für Organische Chemie und Biochemie, Petersenstr. 22, 64287 Darmstadt, Germany. E-mail: fessner@tu-darmstadt.de; Fax: +49 6151 166636; Tel: +49 6151 166666
First published on 11th June 2012
Abstract
We have evaluated different strategies for the one-pot synthesis of D-fructose 6-phosphate and its 1-deoxy and 3-deoxy analogs from readily available starting materials, by building up “artificial metabolisms” in vitro. The first consisted of an aldol cleavage–aldol formation cascade, in which glyceraldehyde 3-phosphate as the central intermediate is generated from fructose 1,6-bisphosphate and consumed in situ for a consecutive carboligation step catalyzed by fructose 6-phosphate aldolase (FSA). The second approach consisted of an aldolase–kinase coupling, in which the unphosphorylated ketose was produced in situ by FSA-catalyzed carboligation, followed by a hexokinase-catalyzed phosphorylation step. While both approaches profited from the high stereoselectivity and stability of the aldolases used, the first approach proved to be the most practical, effective and economical, whereas the second strongly depends on the substrate specificity of hexokinase, which shows inferior catalytic efficiency with the 1-deoxy substrate. Phosphorylation of 3-deoxyfructose failed because, contradictory to a literature report, this compound was found not to be acceptable as a substrate of yeast hexokinase.
Introduction
The development of novel aldolases remains a current focus of biocatalysis research because such enzymes have been proven to be formidable tools for asymmetric synthesis due to their high stereoselectivity for up to two chiral centers and their broad substrate tolerance for the aldol acceptor component.1 Recently, we have discovered that the fructose 6-phosphate aldolase (FSA; EC 4.1.2.–) and an engineered variant of transaldolase B (TalBF178Y; EC 2.2.1.2), both from E. coli, show a complementary substrate tolerance for the aldol donor component.2 Together, these catalysts are valuable new entries to an expanding toolbox of biocatalytic carboligation, which complement each other well in their addressable constitutional space for the stereospecific preparation of deoxysugars. While TalBF178Y has a natural preference for dihydroxypropanone as the nucleophile, FSA prefers hydroxypropanone. Further rational protein engineering of the double-site variant TalBF178Y,S176A was able to improve the acceptance for hydroxypropanone but precluded a structural enlargement to 1-hydroxybutanone,2 which in contrast is acceptable for FSA.2,3Yet, the carboligation toolbox currently lacks some important complementary catalysts. Firstly, it contains no biocatalyst having a donor specificity for propanone as a generic nucleophile; the known side reactivity of deoxyribose 5-phosphate aldolase with propanone suffers from extremely low reaction rates.4 Secondly, there is no natural aldolase known that offers selectivity for different stereoisomer configurations; an exception is the catalytic aldolase antibody 38C2 that suffers from low specific activity, however.5 It seems plausible to develop aldolase variants with such novel catalytic abilities from the TalB or FSA scaffold by directed evolution.6 However, adaption of an enzyme's active site to major structural changes of the substrate, such as the transition from dihydroxypropanone to propanone or inversion of configuration at the nucleophilic center, will plausibly require severe structural changes in the immediate vicinity of the catalytic machinery, with potentially dramatic consequences for substrate binding and protein stability. Consequently, we have proposed that biocatalyst evolution towards such demanding changes of substrate binding motifs should best be performed by stepwise adaptation along a gradual “substrate morphing” (SUMO) approach,6 also known as “substrate walking”.7 Plausible SUMO routes towards “acetone aldolases” or aldolases displaying inverted stereoselectivity may be envisioned along the paths illustrated in Scheme 1.
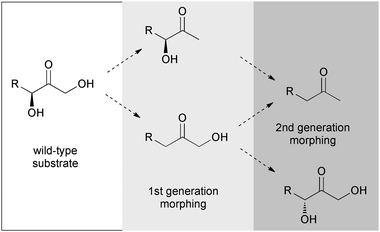 | ||
| Scheme 1 Substrate morphing pathways for conversion of dihydroxypropanone specificity towards propanone (double deletion) or inversion of stereoconfiguration (deletion/addition). | ||
Developing TalB or FSA variants with novel catalytic abilities by directed evolution crucially depends on reliable screening methods to discover mutated protein variants that have acquired the desired capacity. An established screening method for TalB (Scheme 2) is based on the formation of glyceraldehyde 3-phosphate (Ga3P) upon aldol cleavage of D-fructose 6-phosphate (Fru6P).8 This technique should be directly adaptable to the screening for different donor selectivities and other stereoconfigurations because of the uniform release of Ga3P for detection.6 The appropriate SUMO substrates thus transliterate to a requirement for the 6-phosphates of the 1-deoxy, 3-deoxy, and 1,3-dideoxy ketosugar derivatives. Herein we report our efforts to prepare the first generation screening compounds in an economical fashion.
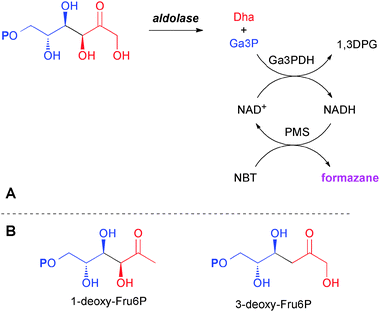 | ||
| Scheme 2 (A) Screening principle for retro-aldolase activity using D-fructose 6-phosphate; (B) target 1-deoxy and 3-deoxy analogs as first generation SUMO substrates. P denotes phosphate ester. | ||
Results and discussion
The first generation substrates for SUMO screening are the regioisomeric 1-deoxy-D-fructose 6-phosphate (1dFru6P) and 3-deoxy-D-fructose (3-deoxy-D-erythro-hexulose) 6-phosphate (3dFru6P), respectively. Synthesis of sugar phosphates is usually performed best by biocatalytic phosphoryl transfer because the high selectivity of enzymes and the very mild reaction conditions render tedious and costly protecting group manipulations unnecessary.9–13Attempts to prepare 1dFru6P on analytical scale have been reported before starting from D-erythrose 4-phosphate (Ery4P) and pyruvate using 1-deoxy-D-xylulose-5-phosphate synthase (DXS) activity,14 or from DL-Ga3P and hydroxypropanone using FSA catalysis.15 However, product formation was deduced only from appearance of a new peak upon HPLC analysis while the corresponding material was never isolated, and only in the former case its constitution was validated after dephosphorylation.14 Anyway, both published approaches were inappropriate to fulfill our demand for sufficient quantity of screening material due to the high costs of phosphorylated precursors (Ery4P, Ga3P). Interestingly, both 1-deoxyfructose (1dFru)16 and 3-deoxyfructose (3dFru)17 were shown to be substrates for yeast hexokinase by enzymatic assay but again, the corresponding phosphorylated products were never isolated. However, these deoxysugars are not available from commercial sources and would need to be synthesized. The only method published so far for chemical synthesis of 3dFru6P requires delicate starting materials and suffers from a lack of diastereoselectivity and regioisomeric specificity.18
Synthesis of 1-deoxy-D-fructose 6-phosphate
With respect to this background we envisioned two different coupled enzymatic approaches for the synthesis of 1dFru6P. The first would be based on the FSA catalyzed equilibrium formation of the target 1dFru6P from the D-enantiomer of Ga3P (Scheme 3A), which could be generated in situ by the cleavage of fructose 1,6-bisphosphate (Fru1,6P2) using fructose 1,6-bisphosphate aldolase (FruA; EC 4.1.2.13) in concert with triose phosphate isomerase (TPI; EC 5.3.1.1); due to the all-equilibrium nature of this procedure product formation might need to be propelled by using an excess amount of inexpensive hydroxypropanone. The second would be based on the FSA catalyzed equilibrium formation of 1dFru2,19 by hydroxypropanone addition to glyceraldehyde that could be coupled to a hexokinase (EC 2.7.1.1) catalyzed phosphorylation (Scheme 3B); the formation of a phosphate ester as the last step with consumption of ATP would provide sufficient driving force to render the total process practically irreversible. In both approaches, it was envisioned that the combined enzymatic steps could occur simultaneously as a one-pot cascade reaction as the individual components were not expected to mutually interfere with either of the enzymatic conversions. However, to exclude any ambiguity arising from unexpected product properties or equilibrium effects, reaction conditions were initially developed for the generic model synthesis of natural Fru6P from Fru1,6P2 in the presence of dihydroxypropanone along the aldol–aldol coupling route, and from dihydroxypropanone addition to glyceraldehyde along the aldol–kinase route (Scheme 3A and B).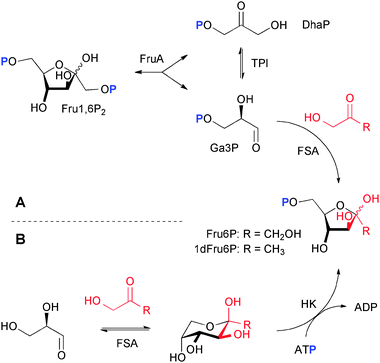 | ||
| Scheme 3 Enzymatic cascade conversions for the synthesis of screening compounds along alternative routes. (A) Sequence consisting of retroaldolization followed by aldol synthesis, and (B) sequence of aldol synthesis followed by phosphorylation; respective yields for Fru6P 94%/88%, and for 1dFru6P 96%/73%. P denotes phosphate ester. | ||
Route A: aldol cleavage and carboligation
For the first strategy (Scheme 3A), Ga3P was needed as the enantiomerically pure D-isomer. For preparative scale applications Ga3P may be produced by chemical synthesis.20 Problematic, however, is the fact that Ga3P is rather unstable and readily decomposes in solution to give methylglyoxal and inorganic phosphate.21 Hence, Ga3P is preferably generated and consumed in situ to avoid the build-up of high stationary concentrations. This can be achieved either by using TPI catalyzed isomerization of dihydroxyacetone phosphate (DhaP)22 or, more conveniently, by retro-aldol cleavage of Fru1,6P2 in the presence of FruA.23 The cleavage yields a mixture of Ga3P and DhaP, from which equilibration by TPI allows utilizing two equivalents of either component to be chosen for subsequent conversions. Indeed, we have previously shown for numerous multi-enzymatic examples that this general synthetic module can be readily exploited in combination with consecutive conversions and nonphysiological components (“artificial in vitro metabolisms”24) for either utilization of DhaP as a nucleophile for synthetic aldolase reactions,9,10,24,25 or of Ga3P as an electrophile for the preparation of D-xylulose 5-phosphate by transketolase catalysis.26Utilization of Ga3P produced in situ for FSA catalysis is facilitated by the fact that FSA prefers phosphorylated substrates, which are bound with high affinity.27 Also, both aldolases share a similar activity optimum around pH 7.5, but otherwise show a distinct and mutually exclusive, high substrate specificity (DhaP and 1-phosphorylated ketoses in the case of FruA, 1-unphosphorylated ketoses in the case of FSA),1 which ought to exclude any interference from substrates, intermediates, or products.
In practice, model reactions were conducted first for Fru6P synthesis to guide in the development of suitable reaction conditions. Thus, Fru1,6P2 was incubated with FruA, TPI and FSA in the presence of dihydroxypropanone with conversion monitoring by tlc. It appeared that the final FSA-catalyzed step was rate limiting, which could be compensated by higher enzyme dosage, but also might be improved otherwise by substituting wild-type FSA for the FSAA126S or TalBF178Y variants that show an improved affinity to the dihydroxylated nucleophile. Although the overall equilibrium should be shifted towards a singly phosphorylated product by the increase in charge entropy alone, this tendency was further promoted by using a threefold excess of dihydroxypropanone, which effected complete consumption of Fru1,6P2 and formation of a practically quantitative yield of the Fru6P aldol product as judged by tlc monitoring.
For the synthesis of 1dFru6P, Fru1,6P2 was incubated with FruA, TPI and FSA in the presence of hydroxypropanone under otherwise unchanged conditions. Initially, smooth conversion took place with formation of a single new sugar phosphate but conversion was less rapid and the reaction did not reach complete conversion. The latter could be improved by using up to a fivefold excess of the nucleophile and extending the reaction time. As a key to practical reactions at a preparative scale, however, it proved to be most economical to replace the commercial FruA from rabbit muscle, which is known to have rather limited stability upon extended reaction times and against organic cosolvents,23 by the corresponding recombinant FruA enzyme from Staphylococcus carnosus,28 which is known to be highly stable in solution and in the present context was found also to tolerate high fractions of hydroxypropanone.
The product from these reactions was isolated in pure form by ion exchange techniques,10 but on larger scale can also be effectively obtained by simple barium salt precipitation. According to its NMR spectra, 1dFru6P exists in aqueous solution as three equilibrating isomeric forms, among which the cyclic β-anomer (1H/13C δ = 1.49/23.8 ppm; 42%) is the most abundant, followed by the open-chain form (δ = 2.32/25.7 ppm; 37%) and the α-anomer (δ = 1.44/21.7 ppm; 21%), respectively.
Route B: carboligation and phosphorylation
Although 1dFru had been prepared by chemical synthesis before,29 none of the reported multi-step preparations is appropriate for direct coupling to an enzymatic phosphorylation. Thus, the second strategy (Scheme 3B) started out from the known stereospecific formation of 1dFru from addition of hydroxypropanone to D-glyceraldehyde (Ga) upon FSA catalysis.19 Again, the enantiomerically pure D-isomer was preferred to using the less expensive racemic aldehyde in order to avoid complications from (reversible) formation of the undesired L-sorbose analog.Reaction conditions for coupling aldol formation to kinase catalyzed phosphorylation again were first evaluated on an analytical scale for the fully hydroxylated substrate case using a mixture of D-Ga, Dha and ATP which was incubated in the presence of FSA and hexokinase. Under standard conditions the desired direct formation of Fru6P could be monitored, while transient formation of Fru was hardly detectable. Apparently, the rate of the kinase conversion of Fru as its natural substrate was considerably higher, so that the initial aldol formation became the rate-limiting step. Indeed, higher overall rates were observed with higher relative dosage of FSA.
For 1dFru6P synthesis, the same reaction system was employed but using hydroxypropanone as a replacement nucleophile instead. Indeed, reaction monitoring of the coupled one-pot process revealed smooth build-up of 1dFru, which was slowly consumed to yield a product with identical Rf to that from the aldol–aldol coupling approach. In contrast to the test system, aldol synthesis now obviously became the faster step, whereas in the coupled step the accumulating 1dFru was consumed only very slowly by the hexokinase catalyzed phosphorylation. We also tested the fructokinase (EC 2.7.1.4) from Zymomonas mobilis as an alternative catalyst,30 but 1dFru was not acceptable as a substrate. A certain difficulty in this approach is that commercial yeast hexokinase shows a less than 2% relative rate with 1dFru as compared to fructose,16 which required much higher dosage of enzyme to compensate for the loss in activity.
Formally, no aldol substrate excess was needed to drive the equilibrium formation of 1dFru because of the energetic advantage of the subsequent ATP-powered phosphorylation. In practice it proved to be beneficial, however, to apply two equivalents of hydroxypropanone for two reasons: (i) the KM of hexokinase for 1dFru16 at >40 mM makes higher concentrations of this intermediate desirable to increase overall reaction rates, and (ii) concentration of the highly electrophilic aldehyde in the equilibrium was kept at a minimum to reduce nonspecific protein inactivation.
Enzymatic phosphorylations are usually performed using an in situ ATP regeneration scheme for economic reasons, for which the pyruvate kinase/phosphoenolpyruvate (PEP) system is considered state-of-the-art because of the reagent's superior stability and phosphoryl donor capacity.10,11,13 An inherent disadvantage of this procedure is the necessity for separation of additional charged compounds, remaining PEP and pyruvate arising as a byproduct, from the phosphorylated target compound. Indeed, purification of 1dFru6P by ion exchange chromatography proved much less cumbersome when ATP was used directly as a stoichiometric reagent.10 From our experience, the somewhat higher costs of equimolar ATP easily outweigh the extra efforts required for more laborious separation of additional components having similar ionic nature, especially for syntheses performed on smaller scale.
Synthesis of 3-deoxy-D-fructose 6-phosphate
The regioisomeric 3dFru is not available from natural sources, although an enzyme catalyzing the reduction of 3-deoxyglucosone to 3dFru had been isolated from porcine livers.31 For our purpose, a coupled enzymatic synthesis was clearly unattractive as neither this NADPH-dependent reductase nor its substrate was readily available. From the several reported chemical methods for 3dFru synthesis,17,18,32,33 the Barton–McCombie deoxygenation protocol34 starting from 1,2:4,5-di-O-isopropylidene-β-D-fructopyranose35 followed by acid catalyzed acetal deprotection is the most straightforward.17,32In practice, attempted hexokinase catalyzed phosphorylation of synthetic 3dFru (Scheme 4) proceeded extremely sluggishly.17 Although reactions were run at high (>1 M) substrate concentration to compensate for the reported KM value of 750 mM for 3dFru with yeast hexokinase, and for its alleged reduced 24% relative rate with respect to natural fructose,17 faint formation of a phosphorylated product required several days even with very high enzyme loadings. However, no product of bona fide 3dFru6P nature could be isolated for characterization, and hexokinase clearly is inappropriate for preparation of this screening compound. More useful kinetic data had been reported for 3dFru conversion by the fructokinase from bovine livers,17 but in our hands using the related fructokinase from Z. mobilis no conversion was detectable. Currently, we are unable to present a conclusive explanation as to the discrepancy between the reported positive assay results for yeast hexokinase and our own negative observations from synthetic studies using the same enzyme. In general, enzymatic assays are unreliable for poor substrates because such assays may not function properly in the presence of high substrate concentrations required to give measurable rates, but false positive results are more likely due to contamination by reactive impurities in a non-substrate. In this case, traces of contaminating natural fructose might have been present in the sample of 3dFru used for the original substrate assessment. Such contamination could have stemmed from incomplete chemical deoxygenation, and might plausibly explain the erroneously positive assay data.
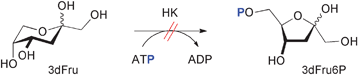 | ||
| Scheme 4 Attempted phosphorylation of 3-deoxyfructose. | ||
These observations are quite unlike our earlier findings for L-rhamnulose kinase, an enzyme which was found to offer a relatively broad tolerance for structural modifications of its natural deoxysugar substrate L-rhamnulose, requiring only absolute (3R)-stereochemistry for activity but allowing for inversion, (de)oxygenation or substitution at the remaining positions.9,10 Although hexokinase accepts various modifications around the anomeric center distant to the O6 reaction site, such as those present in aldose, ketose, anhydrocyclitol, and 2-deoxy structural variations that have tested positive as substrates,13,36 deoxygenation of a ketose substrate seems to pose particular difficulties.
Conclusions
In summary, we have demonstrated that multi-enzyme systems constructed around FSA catalyzed aldol additions are practical and efficient techniques to achieve complex transformations with high stereocontrol for the synthesis of carbohydrate derivatives by simple one-pot operations. In particular, we have shown that Fru6P and 1dFru6P can be prepared from readily available starting materials using two alternative approaches based on FSA-catalyzed carboligation as the crucial stereogenic step. The aldol cleavage–aldol formation cascade proved to be the most efficient and most economical, in which Ga3P as the central intermediate is generated and consumed in situ from stable Fru1,6P2 in a manner similar to our earlier strategy of building “artificial metabolisms” in vitro.9,10,24–26,28,37 The process profits from overall high conversion rates and from the high stability of both the aldolases, FruA from S. carnosus and FSA from E. coli, against electrophilic organic substrates at relatively high concentrations. The latter is important to drive the overall equilibrium towards product formation, which considerably facilitates the product isolation to yield high purity material required for screening purposes. We may note that this strategy should be readily amenable to further nucleophiles that are acceptable to FSA, such as 1-hydroxybutanone or similar structural variations, in the pursuit of more demanding evolutive SUMO targets.In contrast, the alternative approach of aldolase–kinase coupling was found to strongly depend on the substrate specificity of hexokinase, with excellent results for the preparation of Fru6P but much lower efficacy for 1dFru6P due to the inferior catalytic efficiency of hexokinase with the deoxygenated 1dFru substrate. Preparation of 1dFru6P by this route may be reconsidered if new kinases might become available showing improved reactivity with 1dFru.
Contradictory to the reported substrate quality of 3dFru for yeast hexokinase, this route completely failed to yield the anticipated 3dFru6P product. The unexpected absence of reactivity of 3dFru might be due to a misinterpretation in the earlier assay studies caused by impurities in the synthetic material that had been used for the determinations. This failure leaves the reported chemical preparation of 3dFru6P as the only currently viable procedure.18
Experimental
General
Aldolases (FSA from E. coli, FruA from S. carnosus) were produced from recombinant expression strains and purified as described earlier.2,28 Fructokinase from Z. mobilis was prepared following a literature protocol.30 Assay enzymes, hexokinase (yeast, type F-300), FruA (rabbit muscle), TPI (rabbit muscle, type X), and sugar phosphates were obtained from Sigma. Enzyme activity was determined by standard photometric assays. ATP disodium salt and Tris buffer were purchased from Gerbu. Other commercial reagents and solvents were used as received without further purification. D-Glyceraldehyde was prepared according to a literature procedure.38 Ion exchange resins (100–200 mesh) were from Bio-Rad. Column chromatography was performed on Merck 60 silica gel (0.063–0.200 mesh); analytical thin layer chromatography (tlc) was performed on Merck silica gel plates 60 GF254 using a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of satd. ammonia–ethanol for development, and anisaldehyde stain for detection. NMR spectra were recorded on a Bruker DRX500 spectrometer; chemical shifts are referenced to HOD, δ = 4.8 ppm. Mass spectra were recorded on a Bruker-Franzen Esquire LC mass spectrometer. Assays were measured using a Shimadzu UV-1800 spectrophotometer.
:1 mixture of satd. ammonia–ethanol for development, and anisaldehyde stain for detection. NMR spectra were recorded on a Bruker DRX500 spectrometer; chemical shifts are referenced to HOD, δ = 4.8 ppm. Mass spectra were recorded on a Bruker-Franzen Esquire LC mass spectrometer. Assays were measured using a Shimadzu UV-1800 spectrophotometer.
Route A: aldol–aldol cascade
:1 EtOH/aq. NH3 conc.); 1H NMR (500 MHz, D2O) δ 1.44 (s, 3H, 1-Hα), 1.49 (s, 3H, 1-Hβ), 2.32 (s, 3H, 1-Ho), 4.20–3.86 (complex m, ca. 14H, 3-Hα, 3-Hβ, 4-H, 5-H, 6-H), 4.64 (d, 1H, J3,4 = 1.0 Hz, 3-Ho); 13C NMR (125 MHz, D2O) δ = 21.70 (C1α), 23.84 (C1β), 25.65 (C1o), 63.39 (d, J = 4.5 Hz, C6α), 64.84 (d, J = 5.0 Hz, C6β), 66.08 (d, J = 5.0 Hz, C6o), 69.62 (d, J = 7.5 Hz, C5o), 70.02 (C4o), 74.54 (C4β), 76.24 (C4α), 76.78 (C3o), 79.02 (d, J = 8.4 Hz, C5β), 79.85 (C3β), 80.02 (d, J = 8.2 Hz, C5α), 82.17 (C3α), 101.66 (C2β), 105.25 (C2α), 213.49 (C2o); 31P NMR (121 MHz, D2O) δ 4.73 (o), 3.78 (β), 2.44 (α); signals are denoted α/β for the corresponding anomer, and o for the open-chain isomer, respectively; ESI-MS: m/z calcd. for [C6H12O8P] = 243.0, found; 243.3.
Route B: aldol–kinase cascade
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the framework of SPP1170 (grant Fe244/7-2) and by ESF project COST CM0701. The authors thank M. Lemaire (UBP Clermont-Ferrand) for exchange of information prior to publication and G.A. Sprenger (Univ. Stuttgart) for an expression plasmid of Z. mobilis fructokinase.Notes and references
- (a) W.-D. Fessner, in Modern Aldol Reactions, ed. R. Mahrwald, Wiley-VCH, Weinheim, 2004, vol. 1, pp. 201–272 Search PubMed; (b) W.-D. Fessner, in Asymmetric Organic Synthesis with Enzymes, ed. V. Gotor, I. Alfonso and E. Garcia-Urdiales, Wiley-VCH, Weinheim, 2008, pp. 275–318 Search PubMed; (c) W.-D. Fessner, in Enzyme Catalysis in Organic Synthesis, ed. K. Drauz, H. Gröger and O. May, Wiley-VCH, Weinheim, 2012, 3rd edn, vol. 2, pp. 857–917 Search PubMed; (d) P. Clapes, W.-D. Fessner, G. A. Sprenger and A. K. Samland, Curr. Opin. Chem. Biol., 2010, 14, 154–167 CrossRef CAS.
- M. Rale, S. Schneider, G. A. Sprenger, A. K. Samland and W.-D. Fessner, Chem.–Eur. J., 2011, 17, 2623–2632 CrossRef CAS.
- M. Sugiyama, Z. Hong, P.-H. Liang, S. M. Dean, L. J. Whalen, W. A. Greenberg and C.-H. Wong, J. Am. Chem. Soc., 2007, 129, 14811–14817 CrossRef CAS.
- L. Chen, D. P. Dumas and C. H. Wong, J. Am. Chem. Soc., 1992, 114, 741–748 CrossRef CAS.
- T. Hoffmann, G. Zhong, B. List, D. Shabat, J. Anderson, S. Gramatikova, R. A. Lerner and C. F. Barbas III, J. Am. Chem. Soc., 1998, 120, 2768–2779 CrossRef CAS.
- A. K. Samland, M. Rale, G. A. Sprenger and W.-D. Fessner, ChemBioChem, 2011, 12, 1454–1474 CrossRef CAS.
- C. K. Savile, J. M. Janey, E. C. Mundorff, J. C. Moore, S. Tam, W. R. Jarvis, J. C. Colbeck, A. Krebber, F. J. Fleitz, J. Brands, P. N. Devine, G. W. Huisman and G. J. Hughes, Science, 2010, 329, 305–309 CrossRef CAS.
- S. Schneider, T. Sandalova, G. Schneider, G. A. Sprenger and A. K. Samland, J. Biol. Chem., 2008, 283, 30064–30072 CrossRef CAS.
- (a) W.-D. Fessner, J. Badia, O. Eyrisch, A. Schneider and G. Sinerius, Tetrahedron Lett., 1992, 33, 5231–5234 CrossRef CAS; (b) W.-D. Fessner, A. Schneider, O. Eyrisch, G. Sinerius and J. Badia, Tetrahedron: Asymmetry, 1993, 4, 1183–1192 CrossRef CAS.
- W.-D. Fessner and G. Sinerius, Bioorg. Med. Chem., 1994, 2, 639–645 CrossRef CAS.
- H. K. Chenault, E. S. Simon and G. M. Whitesides, Biotechnol. Genet. Eng. Rev., 1988, 6, 221–270 CAS.
- H. Zhao and W. A. van der Donk, Curr. Opin. Biotechnol., 2003, 14, 583–589 CrossRef CAS.
- H. K. Chenault, R. F. Mandes and K. R. Hornberger, J. Org. Chem., 1997, 62, 331–336 CrossRef CAS.
- J. Querol, C. Grosdemange-Billiard, M. Rohmer, A. Boronat and S. Imperial, Tetrahedron Lett., 2002, 43, 8265–8268 CrossRef CAS.
- M. Schürmann, M. Schürmann and G. A. Sprenger, J. Mol. Catal. B: Enzym., 2002, 19–20, 247–252 CrossRef.
- W. L. Dills, Jr., T. R. Covey, P. Singer, S. Neal and M. S. Rappaport, Carbohydr. Res., 1982, 99, 23–31 CrossRef.
- W. L. Dills, Jr., Carbohydr. Res., 1990, 208, 276–279 CrossRef.
- T. Gefflaut, C. Martin, S. Delor, P. Besse, H. Veschambre and J. Bolte, J. Org. Chem., 2001, 66, 2296–2301 CrossRef CAS.
- J. A. Castillo, C. Guerard-Helaine, M. Gutierrez, X. Garrabou, M. Sancelme, M. Schuermann, T. Inoue, V. Helaine, F. Charmantray, T. Gefflaut, L. Hecquet, J. Joglar, P. Clapes, G. A. Sprenger and M. Lemaire, Adv. Synth. Catal., 2010, 352, 1039–1046 CrossRef CAS.
- (a) C. E. Ballou and H. O. L. Fischer, J. Am. Chem. Soc., 1955, 77, 3329–3331 CrossRef CAS; (b) H. Li, J. Tian, H. Wang, S.-Q. Yang and W.-Y. Gao, Helv. Chim. Acta, 2010, 93, 1745–1750 CrossRef CAS.
- J. P. Richard, J. Am. Chem. Soc., 1984, 106, 4926–4936 CrossRef CAS.
- W.-D. Fessner and G. Sinerius, Angew. Chem., Int. Ed. Engl., 1994, 33, 209–212 CrossRef.
- M. D. Bednarski, E. S. Simon, N. Bischofberger, W. D. Fessner, M. J. Kim, W. Lees, T. Saito, H. Waldmann and G. M. Whitesides, J. Am. Chem. Soc., 1989, 111, 627–635 CrossRef CAS.
- W.-D. Fessner and C. Walter, Angew. Chem., Int. Ed. Engl., 1992, 31, 614–616 CrossRef.
- (a) O. Eyrisch and W.-D. Fessner, Angew. Chem., Int. Ed. Engl., 1995, 34, 1639–1641 CrossRef CAS; (b) M. Petersen, M. T. Zannetti and W.-D. Fessner, Top. Curr. Chem., 1997, 186, 87–117 CrossRef CAS.
- F. T. Zimmermann, A. Schneider, U. Schorken, G. A. Sprenger and W.-D. Fessner, Tetrahedron: Asymmetry, 1999, 10, 1643–1646 CrossRef CAS.
- M. Schürmann and G. A. Sprenger, J. Biol. Chem., 2001, 276, 11055–11061 CrossRef.
- M. T. Zannetti, C. Walter, M. Knorst and W.-D. Fessner, Chem.–Eur. J., 1999, 5, 1882–1890 CrossRef CAS.
- (a) K. James and S. J. Angyal, Aust. J. Chem., 1972, 25, 1967–1977 CrossRef CAS; (b) N. A. Jones, S. F. Jenkinson, R. Soengas, M. Fanefjord, M. R. Wormald, R. A. Dwek, G. P. Kiran, R. Devendar, G. Takata, K. Morimoto, K. Izumori and G. W. J. Fleet, Tetrahedron: Asymmetry, 2007, 18, 774–786 CrossRef CAS; (c) F. J. Lopez Aparicio, M. Gomez Guillen and I. Izquierdo Cubero, An. Quim., 1976, 72, 938–945 CAS; (d) J. Sanchez Ballesteros, Rev. Soc. Quim. Mex., 1982, 26, 86–91 CAS.
- (a) P. Weisser, R. Kraemer and G. A. Sprenger, Appl. Environ. Microbiol., 1996, 62, 4155–4161 CAS; (b) B. Zembrzuski, P. Chilco, X. L. Liu, J. Liu, T. Conway and R. Scopes, J. Bacteriol., 1992, 174, 3455–3460 CAS.
- Z. Q. Liang, F. Hayase and H. Kato, Eur. J. Biochem., 1991, 197, 373–379 CrossRef CAS.
- W. A. Szarek, R. J. Rafka, T.-F. Yang and O. R. Martin, Can. J. Chem., 1995, 73, 1639–1644 CrossRef CAS.
- (a) J. Thiem, D. Rasch and H. Paulsen, Chem. Ber., 1976, 109, 3588–3597 CrossRef CAS; (b) R. Kuhn, H. J. Haas and A. Seeliger, Chem. Ber., 1961, 94, 2534–2535 CrossRef CAS.
- J. R. Rasmussen, C. J. Slinger, R. J. Kordish and D. D. Newman-Evans, J. Org. Chem., 1981, 46, 4843–4846 CrossRef CAS.
- R. F. Brady Jr., Carbohydr. Res., 1970, 15, 35–40 CrossRef.
- (a) E. E. Machado de Domenech and A. Sols, FEBS Lett., 1980, 119, 174–176 CrossRef CAS; (b) A. Sols, G. de la Fuente Sanchez, C. Villar-Palasi and C. Asensio, Biochim. Biophys. Acta, 1958, 30, 92–101 CrossRef CAS.
- (a) W.-D. Fessner and O. Eyrisch, Angew. Chem., Int. Ed. Engl., 1992, 31, 56–58 CrossRef; (b) O. Eyrisch, G. Sinerius and W. D. Fessner, Carbohydr. Res., 1993, 238, 287–306 CrossRef CAS.
- C. R. Schmid, J. D. Bryant, M. Dowlatzedah, J. L. Philips, D. E. Prather, R. D. Schantz, N. L. Sear and C. S. Vianco, J. Org. Chem., 1991, 56, 4056–4058 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR spectra of 1dFru6P. See DOI: 10.1039/c2cy20092a |
| This journal is © The Royal Society of Chemistry 2012 |