Rational design of heterogeneous catalysts for biodiesel synthesis
Karen
Wilson
* and
Adam F.
Lee
Cardiff Catalysis Institute, School of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, UK CF10 3AT. E-mail: wilsonk5@cardiff.ac.uk; Fax: +44 (0) 29 208 74030; Tel: +44 (0)29 208 70827
First published on 16th February 2012
Abstract
Dwindling oil reserves and growing concerns over carbon dioxide emissions and associated climate change are driving the utilisation of renewable feedstocks as alternative, sustainable fuel sources. Catalysis has a rich history of facilitating energy efficient, selective molecular transformations, and contributes to 90% of current chemical manufacturing processes. In a post-petroleum era, catalysis will be pivotal in overcoming the scientific and engineering barriers to economically feasible bio-fuels. This perspective highlights some recent developments in heterogeneous catalysts for the synthesis of biodiesel from renewable resources, derived from plant and aquatic oil sources. Particular attention will be paid to the importance of catalyst pore architecture, surface polarity and acid and base properties, in meeting the challenge of transforming highly polar and viscous bio-based reactants.
1. Introduction
The application of heterogeneous catalysis and use of renewable feedstocks in chemical synthesis are core themes behind the principles of Green Chemistry.1 Worldwide concern over dwindling fossil fuel reserves and impact of CO2 emissions on climate change means there is an urgent need to reduce our dependence on oil based sources of fuels and chemicals. Oil is the most important source of energy worldwide, accounting for some 35% of primary energy consumption and the majority of the chemical feedstocks; the quest for sustainable resources to meet demands of a constantly rising global population is one of the main challenges for mankind this century.2 Global energy demand for transportation is expected to grow by two percent per year, with energy use and greenhouse gas emissions (GHG) for the year 2030 predicted to be 80% higher than 2002 levels.3 The Copenhagen Accord4 states that greenhouse gas concentrations in the atmosphere should be stabilised at a level that would prevent dangerous anthropogenic interference with the climate system, and target the scientific view that the increase in global temperature should be <2 °C against pre-industrial temperature levels. To meet these ambitions there is a technology gap that needs to be addressed to facilitate emission reductions by 2020, and avoid dangerous climate change by mid-21st century.5 It is estimated that addressing climate change and energy development needs at the global level may require an investment on the order of US$45 trillion between now and 2025.To be truly viable, any new alternative energy sources must be sustainable, that is “have the ability to meet 21st century energy needs without compromising those of future generations”, and it is predicted that by 2030 renewable energy usage will increase from 5% to 18%.6 While wind, solar, hydroelectric and nuclear power currently receive heavy investment for stationary energy sources,7 the most easily implemented and low cost solutions for transportation needs, particularly that of aviation, are those arising from liquid biomass-derived fuels. Predictions suggest that ∼9% of transportation energy needs will be met from liquid biofuels by 2030.6
Judicious biomass selection is crucial for ensuring the sustainability of a renewable resources based economy. Despite initial promise, first-generation bio-based fuels derived from edible plant materials provoked much criticism over competition between land usage for fuel crops versus traditional agricultural cultivation.8 Of equal concern are associated deforestation practices, notably in Indonesia, wherein vast tracts of rainforest and peat land are being cleared to support palm oil plantations.9 To be sustainable, so called 'second generation' bio-based fuels and chemicals should use biomass sourced from non-edible components of crops, such as stems, leaves and husks or cellulose from agricultural or forestry waste. Alternative non-food crops such as switchgrass or Jatropha Curcas, which require minimal cultivation, can also be used. There is also growing interest in using oil from aquatic biomass, which can yield 80–180 times the annual volume of oil per hectare than that obtained from plants.10
Biodiesel, comprising fatty acid methyl esters (FAMEs), derived from microalgae is another promising avenue,11 with land requirements for oil production from microalgae only a small fraction of that required for oil seed crops. However, current microalgae production is significantly more expensive than for arable crops. Cost estimates for microalgal oil from photobioreactors or open pond reactors are ∼US$9.74 or $12.54 per kilo respectively,12 considerably higher than crude palm oil, which in 2006 was only ∼US$0.52 per litre.13 The cost of raw feedstocks and their conversion to biodiesel through existing homogeneously catalysed routes14 has restricted biodiesel uptake, and has proved unviable in certain countries without subsidies. It is interesting to note that rising oil prices are driving a shift in investment in the US, where once uneconomic biodiesel plants are now viewed as viable, with tax incentive reinstatement expected to increase biodiesel production and displace foreign petroleum with domestic Advanced Biofuels.15
Despite this encouragement it is important that the overall net energy balance of biodiesel production is considered. Of all the potential transportation biofuels, biodiesel provides one of the highest energy quotients,16 however, when considering life cycle analyses of biodiesel production, fertiliser and pesticide usage, and energy required for cultivation, are key parameters to consider. Non-food crops such from the Euphorbiaceous or Spurge family such as Jatropha have the major advantage of growing on arid waste land and requiring little cultivation. However it is important to consider that a change in land use (e.g. via deforestation) can introduce a carbon debt17 and thus net CO2 emissions for different oil sources will depend on the production methods employed Fig. 1. There is however a clear advantage of using e.g. Jatropha as an oil seed source, meaning biofuels can contribute to GHG mitigation in the transport sector.
 | ||
| Fig. 1 Impact of land usage and plant oil source on CO2 emissions during biodiesel production in China.17 | ||
Second generation biofuels are currently at a pre-commercial phase, and if they are to meet targets for implementation by 2015–2020 significant technical hurdles to the chemical transformation of biomass need to be overcome. Catalysis has a rich history of facilitating energy efficient selective molecular transformations and contributes to 90% of chemical manufacturing processes and to more than 20% of all industrial products. In a post-petroleum era, catalysis will be central to overcoming the engineering and scientific barriers to economically feasible routes to bio-fuels. There are numerous reviews on the application of solid base and acid catalysts for biodiesel production.18–25 Rather than attempt to review the myriad of publications across the field, this perspective focuses on the challenges in rationally designing new high activity catalysts required to deliver biodiesel as a major player in the 21st century sustainable energy portfolio.
2. Current biodiesel production
Oleochemical feedstocks are obtained from triglycerides (TAG) and free fatty acids (FFA) found within plant oil seeds, animal fats and algae. Plant oils are conventionally obtained from seeds by pressing, or solvent extraction. Algal oil harvesting and extraction requires a combination of sedimentation, centrifugation, filtration, ultra-filtration, sometimes with an additional flocculation step to separate algae from water, followed by a drying and then solvent extraction step to remove the oil.10 Cultivation of algal sources using wastewater and CO2 from flue gas could are proposed as a means to improve the energy efficiency of algal oil production.26Table 1 summarises the oil content of various plant and algal oil sources, highlighting the potential enhanced oil yields from algae. However, there are a wide range of algal sources, and the optimum productivity will depend on both oil content and growth rate. While Botryococcus braunii has one of the highest oil contents, it is quite slow growing, and the Chlorella family is proposed as better suited as a biodiesel source.10 The percentage TAG or FFA, length of alkyl chain and degree of unsaturation all depend on the oil feedstock.27 The appropriate oil selection for fuels is critical, as alkyl chain length and unsaturation affect pour and cloud points, as well as stability of the final fuel.28 Incomplete combustion of trace longer chain FAMEs can also result in their long-term accumulation within engines.| Oil source | Seed oil content (% in dry biomass) | Oil yield (L oil/ha year) | FFA content/wt (%) |
|---|---|---|---|
| Soybean29 | 18 | 636 | 2 |
| Jatropha30,31 | 28 | 741 | 14–14.9 |
| Rapeseed32 | 41 | 974 | 2 |
| Sunflower33 | 40 | 1070 | 0.3 |
| Palm31,34 | 36 | 5366 | 2.3–6.6 |
| Microalgae (Low oil content e.g. Pavlova salina)35 | 30 | 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 700 700 |
2 |
| Microalgae (Medium oil content e.g. Chlorella sp.) | 50 | 97800 |
2 |
| Microalgae (High oil content e.g. Botryococcus braunii) | 70 | 136900 |
2 |
TAGs and FFAs from oils can both be converted to biodiesel by respective transesterification with a light alcohol (typically methanol) or esterification, using simple acid or base catalysis.36 FFAs in plant oils and animal fats are problematic for conventional biodiesel manufacturing routes.37,38 The negative influence of high FFA feedstocks on base-catalysed triglyceride transesterification is well known,28,39,40 with saponification resulting in viscous gels which hamper esterification and increase product separation costs. Consequently there are tight specifications on feedstock compositions for transesterification, with FFA contents <0.5% required.29 Moreover, anhydrous alcohols (and catalysts) are required in order to avoid hydrolysis of the desired alkyl esters into FFAs and associated soap formation. Commercial biodiesel manufacture utilises soluble bases such as KOMe, NaOMe for the transesterification step, sometimes employing an acid catalysed pre-esterification41 to remove any FFAs (Scheme 1), before the oil is contacted with the soluble base catalyst. While acid catalysed transesterification is slower, requiring higher reaction temperatures than those typical for base catalysts, a single step acid catalysed route may be more economical for oils high in FFAs, such as those obtained from animal fats or waste oils.
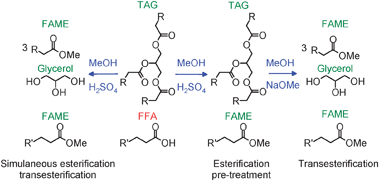 | ||
| Scheme 1 Synthesis of biodiesel via acid or base catalysed routes. (Typically R = C14–C16; R′ = C13–C15). | ||
Unfortunately, homogeneous acid and base catalysts can corrode reactors (and engine manifolds if carried through to the fuel), and their removal from the resulting biofuel is particularly problematic and energy intensive, requiring aqueous quench and neutralisation steps, which themselves result in stable emulsions and soap formation.42–45 Furthermore, the glycerine by-product, of significant potential value to the pharmaceutical and cosmetic industries, obtains in a dilute aqueous phase heavily contaminated by inorganic salts. Glycerol derivatives are widely used in the fine chemicals sector, and its selective conversion to mono- or diglycerides is desirable for use in the cosmetics industry; its upgrading from the contaminated form arising from homogeneous biodiesel processes is not economically viable.
Development of a heterogeneous biodiesel process will dramatically improve the efficiency of fuel production by eliminating the need for quenching steps and allowing continuous operation. In addition the glycerol by-product purity will be improved, adding value to the overall process. Technical advances in catalyst and reactor design and introduction of non-food based feedstocks are thus required to ensure that biodiesel remains a key player in the renewable energy sector for the 21st century. Here we discuss the process consideration for the design of solid acid and base catalysts for biodiesel synthesis. A number of factors need to be considered when developing solid catalysts for esterification/transesterification reactions including water tolerance,46 pore size and dimensionality of the channel system.47,48 A particular focus of this perspective will be the impact of acid/base strength, support hydrophobicity and the benefits of using materials with interconnected hierarchical macro-mesoporous networks to enhance mass-transport of viscous plant oils during reaction.
3. Catalyst design parameters
3.1 Effect of solid acid strength
The use of solid acid catalysts for oil pre-treatment via FFA esterification would be desirable to avoid costly neutralisation and separation steps, and reduce the overall number of processing steps in biodiesel synthesis.48 Despite the wide range of solid acids commercially available, the corresponding direct transesterification of oils into biodiesel by solid acid catalysts has not been extensively explored. This in part reflects the lower activities for acid catalysed transesterification compared with base-catalysed routes45 which in turn necessitates higher reaction temperatures to achieve acceptable reaction rates. While their activities are generally low, solid acids have the advantage that they are less sensitive to contaminants then their basic analogues, and are able to function well with unrefined feedstocks containing 3–6 wt% FFAs.45 In contrast to solid bases which require pre-treatment's to remove free fatty acids, solid acids are able esterify FFAs impurities through to FAME while simultaneously transesterifying the major triglyceride oil components, all without risk of soap formation.A comparison of different commercial acid catalysts, including Amberlyst A26, A27, revealed these to be virtually inactive for FAME synthesis from sunflower oil49 at 60 °C when compared with NaOH. However, Lopez et al.50 subsequently showed that solid acid catalysts (inc. Amberlyst-15, sulfated zirconia, Nafion NR 50 and tungstated zirconia) perform favourably relative to H2SO4 in TAG transesterification at 60 °C, indicating that they could act as alternatives to homogeneous acid catalysts without corrosion or saponification problems. The authors also highlighted the importance of internal mass transfer limitations, which impaired the performance of microporous heterogeneous catalysts such as ETS-10 (H) and zeolite H-Beta.
Several studies have focused on SO4/ZrO2 in biodiesel synthesis, due to its strong (super) acidity and regeneration capacity.51–53 While commercial SO4/ZrO2 is very active towards tricaprylin transesterification at high temperature and pressure (120 °C, 6.8 atm), with 84% conversion observed in only two hours, subsequent re-cycle tests suggest catalyst deactivation occurs due to sulphate loss into solution. In contrast, other groups find SO4/ZrO2 to be stable and an effective catalyst for FFA esterification,53,54 with negligible leaching of sulphate groups when only a small amount of water is present in the organic phase. While clearly a promising solid acid, further understanding regarding the nature of catalyst deactivation and factors affecting stability (e.g. catalyst preparation conditions) and optimisation of the porosity are required to promote the wider use of SO4/ZrO2 catalysts in biodiesel synthesis.
Low temperature transesterification requires a robust solid acid catalyst with strong acidity. Heteropolyacids are an interesting class of well-defined superacids (materials with pKH+ > 12),55 possessing flexible structures and tuneable (super) acidity. However, in their native form heteropolyacids are unsuitable for biodiesel applications due to their high solubility in polar media,56 and although heteropolyacid dispersion onto high area supports can enhance the number of available acid sites,55,57–59 this cannot overcome the solubility issue. To address these issues a number of groups have explored using amine functionalised supports to tether the Keggin units,60,61 or constructing the tungstate clusters around surface PO42− groups on zirconium phosphate.62 Ion-exchanged Keggin type phospho- and silicotungstic acids63 are another means to circumvent this problem, as the Cs+ and NH4+ doped variants are found to be insoluble in water.64 Cs salts of the general formulae CsxH(3−x)PW12O40 and CsyH(4−y)SiW12O40 also have the added benefit of dramatically boosted surface areas relative to their fully protonated forms (Scheme 2).65,66 CsxH(3−x)PW12O4065 and CsyH(4−y)SiW12O4066 catalysts show a high activity for methylpalmitate formation via palmitic acid esterification. Fig. 2 shows the variation in activity with Cs content for CsxH(3−x)PW12O40, revealing optimum performance for both esterification and transesterification occurs for x = 2.1–2.4. Similar trends are observed for CsyH(4−y)SiW12O40 catalysts, with the maximum in activity occurring over the range y = 2.8–3.4.
 | ||
| Scheme 2 Cs-exchanged H3PW12O40 generates CsxH(3−x)PW12O40 which is insoluble in polar media. | ||
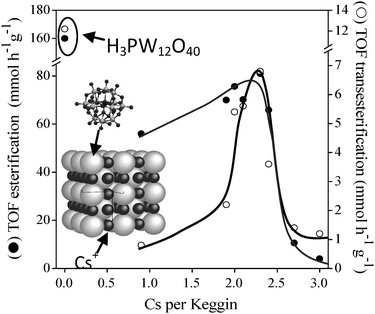 | ||
| Fig. 2 Performance of Cs doped H3PW12O40 in C4 TAG transesterification and C16 FFA esterification. (Reprinted from ref. 64 with permission from Elsevier). | ||
This optimal composition was rationalised in terms of a corresponding maximum in the density of accessible surface acid sites. In the case of CsyH(4−y)SiW12O40, wherein C4 and C8 TAG transesterification were compared, the absolute reaction rates were faster for the shorter chain triglyceride, which may evidence either worse miscibility, or slow in-pore diffusion of the longer chain TAG in MeOH. Furthermore, the TOF for the optimum Cs-doped catalyst was greater than that of highly soluble H4SiW12O40, which operates homogeneously. This latter observation may reflect reports that CsxSiW12O40 salts are more hydrophobic than the parent H4SiW12O40. Ester activation of the more lipophilic C8 TAG (the first step in acid catalysed transesterification) will thus be favoured over the more hydrophobic CsxSiW12O40 catalyst. Reactant/product polarity and associated mass transport to and from the active acid centres thus play an important role in controlling reactivity, even under homogeneous conditions. Indeed a decrease in esterification rate has been reported with acid chain length from (C2–C8) using both H2SO4 and Nafion based SAC-13 which is attributed to the polar and steric effects of the hydrocarbon chain.67 The impact of polarity on catalyst activity will be discussed in more detail in Section 3.3.
Mesoporous silicas from the SBA family have also been examined for biodiesel synthesis and include materials grafted with sulfonic acid groups68–70 or SO4/ZrO2 surface coatings.71 Phenyl and propyl sulfonic acid SBA-15 catalysts, synthesised as shown in Scheme 3, are particularly attractive catalysts with comparable activities to Nafion and Amberlyst acidic resins in palmitic acid esterification.69 Phenylsulfonic acid functionalised catalysts were found to be more active than the corresponding propyl systems, in line with their respective acid strengths.
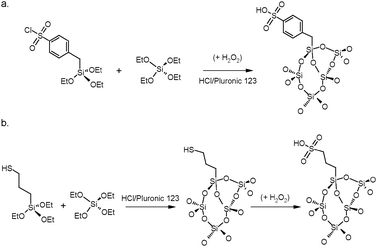 | ||
| Scheme 3 Synthesis of sulfonic acid SBA-15 solid acids from (a) phenyl or (b) propyl thiol precursors. Aromatic sulfonic acid centres provide stronger acid sites. | ||
The impact of tailoring acid site distributions has also been investigated using designer sulfonic acid precursors which offer control over the spatial location of acid centres, and by incorporation of inert organic groups to alter the hydrophilicity of the surface. Grafting of disulfide or sulfonate ester functionalities72 on silica surfaces can be used as a means to generate spatially located alkylsulfonic acid groups. While the use of a bis(trimethoxysilyl)propyl disulfide precursor to functionalise SBA-15 reveals an increase in acid strength for the resulting spatially located sulfonic acid groups, unfortunately when evaluated in palmitic acid esterification,73 acid strength could not be correlated with catalytic activity. Phenylsulfonic acid catalysts have proven particularly promising for biodiesel production from low-grade oils and fats containing a number of impurities including unsaponifiable matter, water, phosphorous and group I metals.74 However, these impurities are found to have a negative effect on biodiesel yield, which was attributed to the strong interaction of unsaponifiable matter with the acid sites. Feedstock pretreatment with a cation exchange resin can improve catalyst longevity; however more work is required to improve the robustness of solid acid catalysts toward these cheaper but low grade oil sources.
The synthesis of Zr-SBA-15 materials from zirconocene dichloride has also been investigated to yield a strong Lewis acid. These confer high catalytic activity in the transesterification of crude palm oil with methanol for biodiesel synthesis, achieving FAME yields > 70% at 200 °C in 3 h. Zr-SBA-15 materials also display excellent stability in palm oil transesterification, and can be regenerated via 550 °C calcination.75
While the solid acid catalysts described in this section are promising for reactions of high FFA-containing oils in a single step high temperature process, they are also applicable for oil pre-treatment via FFA esterification.76 This would be desirable to avoid costly neutralisation/separation steps, and reduce the overall number of processing steps in biodiesel synthesis.48
3.2 Effect of solid base strength
Base catalysts generally exhibit higher activity in transesterification, so would be particularly suitable for high purity oils with low FFA content. Biodiesel synthesis using a solid base catalyst would facilitate separation of both catalyst (via a continuous flow, packed bed arrangement) and glycerol by-product from the final reaction mixture, thereby reducing production costs and enabling catalyst re-use. A variety of solid base catalysts are known, including alkali or alkaline earth oxides, supported alkali metals, basic zeolites and clay minerals (such as hydrotalcites) and supported organic bases.47 The origin of basicity in alkaline earth oxides has been previously reviewed, and is generally believed to arise from the presence of M2+–O2− ion pairs in different coordination environments,77 with the base strength of Group 2 oxides and hydroxides increasing in the order Mg < Ca < Sr < Ba.78 The strongest base site occur at low coordination sites at defects, corners, edges or on high Miller index surfaces as illustrated in Scheme 4. | ||
| Scheme 4 Origin of basic sites in alkaline earth oxides: (a) generation of cationic defect sites; (b) surface Mg2+–O2− ratio varies with crystal facet termination leading to increasing base strength from (100) < (110) < (111). | ||
Such classic heterogeneous base catalysts have been extensively tested for triglyceride transesterification,79 and there are a number of reports of the application of commercial and microcrystalline CaO in rapeseed, sunflower or vegetable oil transesterification with methanol.48,80–83 While promising results are obtained, with 97% oil conversion82 reported at low temperatures ∼75 °C, there is concern in the literature81,84 over partial leaching of Ca2+ from CaO under reaction conditions, and associated homogeneous catalytic contributions, which could limit its utility in biodiesel production. Alkali-doped CaO and MgO have also been investigated for transesterification.85–87 These exhibit enhanced basicity which is generally associated with the formation of O− centres when M2+ is replaced by M+, resulting in charge imbalance and associated defect generation. In the case of Li doped CaO, the optimum activity occurs for dopant levels sufficient to create a saturated Li+ monolayer, however care must be taken during catalyst synthesis to ensure that leaching of promoters does not occur.88
It it widely accepted that the catalytic activity of alkaline earth oxide catalysts is very sensitive to their preparation,89 which can influence surface morphology or defect density. The impact of the exposed MgO crystal facet on base site density and catalyst activity in sunflower oil transesterification is nicely demonstrated via a comparison of nanoparticulate MgO and MgO nanosheets,90 the latter of which exposes mainly (111) surfaces91 as shown in Fig. 3. The results of base site titrations of (111) oriented MgO (I) nanosheets, hydrothermally treated MgO (II) and nano-particulate MgO (III) are shown in Fig. 4. All samples possess a bimodal distribution of base strengths, however the nano-sheets are observed to exhibit a very well defined distribution of medium-strong base sites which would be consistent with uniformity of exposed facets. Higher yields of FAME are obtained during sunflower oil transesterification over the nano-sheets compared to the higher surface area nanoparticulate MgO (III).
 | ||
| Fig. 3 HRTEM and fourier transforms of an isolated (111) oriented MgO nano-sheet. (Reproduced from ref. 91 with permission from Wiley). | ||
 | ||
| Fig. 4 CO2TPD for MgO nano-sheets—MgO(I); hydrothermally treated MgO—MgO(II) and MgO nanoparticles—MgO(III). (Reproduced from ref. 90 with permission of the Royal Society of Chemistry). | ||
Amongst solid base catalysts, hydrotalcites have received considerable attention in recent years because of their high activity and robustness in the presence of water and free fatty acids.92–94 Hydrotalcites have a general formulae [M2+(1−x)M3+x(OH)2]x+(Ax/n)n−·yH2O which adopts a layered double hydroxides structure with brucite like (Mg(OH)2) hydroxide layers containing octahedrally coordinated M2+ and M3+ cations and An− counter anions which reside in the interlayer space to balance the residual positive charge of the hydroxide layers (Scheme 5). Isomorphous substitution of M3+ by M2+ enables the acid/basic properties of hydrotalcites to be readily tuned.95,96
![Layered structure of [Mg(1−x)Alx(OH)2]x+(Ax/n)n−·yH2O hydrotalcite.](/image/article/2012/CY/c2cy20038d/c2cy20038d-s5.gif) | ||
| Scheme 5 Layered structure of [Mg(1−x)Alx(OH)2]x+(Ax/n)n−·yH2O hydrotalcite. | ||
TAG transesterification employing Mg–Al hydrotalcites has been reported using both poor and high quality oil feeds;93 including refined and acidic cottonseed oil, and high water content animal fat feed. For acidified cottonseed oil (9.5 wt% FFA) and animal fat oil (45 wt% water), 99% triglyceride conversion was achieved within three hours at 200 °C. However the reliability of much of the catalytic data for hydrotalcites is of concern due to the ill-advised use of Na or K hydroxide/carbonate solutions during precipitation of the hydrotalcite phase. Complete removal of trace alkali residue from the hydrotalcite surface is inherently difficult, resulting in uncontrolled and unquantifiable homogeneous catalytic activity from leached Na or K.97,98 To circumvent this problem, use of an alkali-free precipitation route using NH3OH/NH3CO3 is advised, and provides more reliable materials for structure-reactivity correlations within thermally activated and rehydrated Mg–Al hydrotalcites.94 [Mg(1−x)Alx(OH)2]x+ (CO3)2−x/n materials with compositions spanning x = 0.25–0.55 reveal that base strength could be controlled by increasing the Mg:Al ratio, and that materials with increased base strength exhibit higher activity in TAG transesterification (Fig. 5).
 | ||
| Fig. 5 Performance of Mg:Al hydrotalcites in transesterification as a function of Mg content. | ||
Xi et al.99 investigated the influence of water on the activity and stability of activated Mg–Al hydrotalcites (Mg/Al molar ratio of 4). Brønsted base sites were active in the presence of water, but significant hydration was found to result in rapid catalyst deactivation, presumably through ester hydrolysis to butyric acid and subsequent irreversible neutralisation of surface base sites. As previously outlined, while solid base catalysts are very effective for transesterification, unfortunately they cannot esterify any free fatty acids in the oil to FAME. Such feedstocks therefore require pre-treatment by a solid acid to ensure that fuel compositions meet legislated standards.
Other mixed oxides have been investigated in biodiesel including zinc100 and lanthanum101 doped hydrotalcites, La2O3/ZrO2,102 CaO–La2O3103 and zinc aluminate (ZnAl2O4), which is the most notable of these and was developed by IFP in the heterogeneously catalysed continuous Ester-fip process.104–106 Calcined Li/Al hydrotalcites107 reportedly possess higher Lewis basicity than Mg/Al hydrotalcites or MgO, and as a consequence, higher activity in the transesterification of soybean oil to fatty acid methyl esters.108,109 While these catalysts facilitate high FAME yields and potential for recycling, catalyst stability requires further verification as small levels of Li leaching are reported.110
Dolomitic rock comprising alternating layers of Mg(CO3)–Ca(CO3) is structurally very similar to calcite (CaCO3), and also a promising solid base precursor. Calcination at 900 °C transforms the mineral into a material comprising MgO nanocrystallites dispersed over larger (>60 nm) CaO particles.111 This calcined material exhibits promising activity and stability in the transesterification of a range of C4–C18 triglycerides under mild conditions (Fig. 6). Recent studies have also revealed calcined dolomite to be highly efficient in canola oil transesterification under mild conditions.112
 | ||
| Fig. 6 Comparative transesterification rates of TAGs to FAMEs for dolomite versus representative solid acids and bases. | ||
Literature suggests that such mixed MgO:CaO systems also show improved resistance to deactivation by CO2 when compared to CaO alone.113 While the origin of this synergy requires further investigation, it may go some way to explaining why calcined dolomite exhibits a higher activity in plant oil transesterification than the separate CaO or MgO components. The genesis of highly dispersed MgO nanocrystallites on the surface of CaO may also contribute to the performance of calcined dolomite, since recent reports reveal that the activity of MgO in TAG transesterification also shows a strong size-dependency; with nanocrystalline MgO significantly outperforming commercial microcrystalline forms.90,114
Despite these findings, the effects of solid base strength on catalytic activity in biodiesel synthesis remains poorly understood, and there is much variability in the literature concerning the activity of different classes of solid base in transesterification. The morphologies of alkaline earth oxide catalysts and their corresponding basicities are very sensitive to their preparative route.89 Since the active surface sites of fresh CaO catalysts are unavoidably poisoned by exposure to atmospheric H2O and CO2, a thermal activation treatment is often required to remove surface Ca(OH)2 and CaCO3. Consequently, reported activities for CaO catalysed transesterification are highly variable, with turnover frequencies (TOFs) for sunflower oil transesterification reported to range from 2.5 to 45 g(oil) h−1 g(cat)−1, depending on sample pre-treatment and microcrystallinity.115 CaO is generally found to be more active than MgO for rapeseed oil transesterification, with respective TOFs of 1.66 g(oil) h−1 g(cat)−1versus 0.14 g(oil) h−1 g(cat)−1 at 65 °C.116
To improve our understanding of factors influencing solid base activity in biodiesel synthesis, a simple surface spectroscopic method employing Auger parameter measurements provides a powerful quantitative determination of surface basicity, independent of adsorption probes.114,117 Oxygen 1s XP and Auger measurements offer a direct measure surface polarisability,118 in which the O KL23L23 and O KL1L23 Auger separation (ΔEk), is found to be a quantitative measure of Lewis basicity.119,120 The validity of this approach was tested with a series of size-controlled MgO nanocrystals prepared via a supercritical drying method.121,122 HRTEM reveals that as-prepared nanocrystalline MgO comprises 3 nm, (100)-terminated crystallites, which sinter on calcination to larger crystallites (≥7 nm) favouring (111) and (110) facets. Such restructuring exposes higher surface densities of polarisable, electron-donating O2− centres, and creates bulk and surface defects, postulated as superbasic sites in solid base catalysts. In contrast, conventional commercial MgO samples are comprised of larger ∼50 nm particles dominated by less basic (100)-oriented facets. A decrease in ΔEk, was observed over this series which was attributed to an increase in MgO surface base strength with calcination temperature (Fig. 7).
 | ||
| Fig. 7 Correlation between surface polarisabilty of nanoparticulate MgO catalysts with their activity in triglyceride transesterification. | ||
It is likely that this dependence reflects the correspondence between particle size and preferential surface termination. The size dependent evolution of surface electronic structure correlates directly with the associated catalytic activity of these MgO nanocrystals in transesterification. This study thus highlights the importance of correlating the surface structure and basicity of solid base materials with their catalytic activity; the absence of systematic materials characterisation and detailed kinetics has to date hampered catalyst optimisation and thus the implementation of new heterogeneous processes in biodiesel synthesis.
3.3 Effect of support hydrophobicity
The design of heterogeneous catalysts with tuneable surface polarity is another key goal in the development of solid acid catalysts. Such properties are the key for controlling adsorption properties, reactant activation and product selectivity in both vapour and liquid phase catalysis. Particular concerns for biodiesel synthesis centre on the hydrophobic nature of long chain esters relative to methanol and the appreciable water contents encountered in oils, particularly waste oil, which can be problematic by promoting undesired ester hydrolysis reactions. The impact of reactant polarity on catalyst performance has been demonstrated in H2SO4 and Nafion-SAC-13 catalysed esterification of C2–C8 organic acids67 and transesterification of C2–C16 ethyl alkanoate esters.123 These studies revealed that in addition to steric hindrance, changes in the polarity of the ester chain with size had a significant effect on rate of both reactions. For esterification, a similar decrease in rate for C2–C8 acids is observed using both liquid and solid acids, attributed to the effect of increased reactant hydrophobicity (Fig. 8). | ||
| Fig. 8 Reactivity of different C2–C8 organic acids in H2SO4 catalysed esterification with methanol at 60 °C. (Reproduced from ref. 67 with permission of Elsevier). | ||
Likewise, transesterification of C2–C16 linear alkyl esters123 shown in Fig. 9 reveals a decrease in activity with chain length, however in this instance a plateau in TOF is observed for ethyl hexanoate when using H2SO4, whereas SAC-13 exhibits a continuous, albeit non-linear, in activity with increasing chain length. Given the differences in chain length used in these two studies it would be interesting to see how esterification rates evolve for > C8 organic acids. In the case of transesterification, it is suggested that steric effects from increased carbon chain length are modulated by changes in the configuration of the hydrocarbon chain via e.g. folding to minimize these constraints during catalysis by H2SO4.
 | ||
| Fig. 9 Reactivity of different C2–C16 ethyl esters in H2SO4 catalysed transesterification with methanol at 60 °C. (Reproduced from ref. 123 with permission of Elsevier). | ||
Considering the mechanism of solid acid catalysed transesterification, such conformational changes are however unlikely in the vicinity of a surface (Scheme 6). Although both the ester and methanol can competitively chemisorb at the acid centre, it is widely believed the surface reaction proceeds via an Eley-Rideal mechanism involving nucelophilic attack of methanol on the activated acid/ester.124 However, we should note that esterification have also been successfully modelled by a Langmuir Hinshelwood mechanism,125,126 highlighting the difficulty in discriminating these bimolecular reaction mechanisms. Similar hypotheses are advanced in mechanistic studies of base catalysed transesterification over MgO127,128 and hydrotalcites,129 wherein Eley-Rideal or Langmuir Hinshelwood models have respectively been used to simulate kinetic data. Regardless of the mechanism, the observed trends in activity can be attributed to repulsive interactions between the alkyl chains and methanol/polar groups of the acid catalyst. Any change in surface polarity that will aid adsorption of the ester at the active site should thus favour reaction by increasing its surface coverage. However, since SAC-13 has a bimodal pore size distribution with average diameters of 8 and 19 nm,130 the impact of hindered accessibility of some acid sites on reaction rate can also not be ruled out.
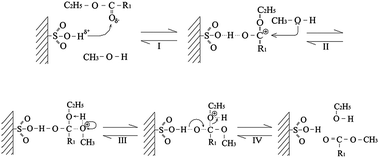 | ||
| Scheme 6 Proposed mechanism for Brønsted acid catalysed transesterification (Reproduced from ref. 123 with permission of Elsevier). | ||
The hydrophilic nature of polar silica surfaces means it is not ideal for reactions of apolar organic molecules. This can be problematic due to differential diffusion or preferential adsorption of more polar components from mixtures of hydrocarbons. Furthermore, surface hydroxyl groups favour H2O adsorption, which if formed during esterification can thereby inhibit adsorption of organic substrates at active sites. Surface modification via the incorporation of spectator organic moieties into the oxide surface or dehydroxylation can overcome this polarity issue to increase initial reaction rates in acid catalysed transformations of liquid phase organic molecules.131 Surface polarity can also be tuned by incorporating alkyl groups directly into the silica framework, as in bridged polysilsesquioxanes which can be prepared via the co-condensation of 1,4-bis(triethoxysilyl)benzene (BTEB), or 1,2-bis(trimethoxysilyl)-ethane (BTME), with TEOS and MPTS in the sol–gel process.132–135 This can increase activity towards small molecular esterification136 and etherification.137 The incorporation of organic spectator groups (e.g. phenyl, methyl or propyl) during the sol–gel syntheses of SBA-15138 and MCM-41139 sulphonic acid silicas can be achieved via co-grafting or simple addition of the respective alkyl or aryl trimethoxysilane during co-condensation preparations. Such approaches to facilitating adsorption of hydrophobic can significantly enhance esterification rates. Indeed, methyl and phenyl modification of MCM-41 sulphonic acids140 improves glycerol esterification with lauric acid. The hydrophobicity of SBA-15 propylsulphonic acids, co-functionalisation with methyl, ethyl or phenyl groups has also been investigated in palmitic acid esterification.68 Although some differences in initial rates of reaction were reported, changes in the textural properties of SBA-15 across the series made it difficult to draw firm conclusions about the effect of apolar spectator groups.
To address these issues, an experimental and computational study of a series of sulphonic acid functionalised MCM-41 materials was reported in which special attention was been paid to the effect of both acid site density and surface hydrophobicity on the overall acidity and catalytic performance. 141 MCM-41 was selected as the support due to the availability of accurate models for the pore structure generated using kinetic Monte Carlo simulations.142 This MCM-41 pore model can also be readily modified with surface groups thus allowing dynamic simulation of sulphonic acid and octyl groups attached within the MCM pores. Two series of catalysts were investigated in which the propylsulphonic acid coverage was varied over the range θ(RSO3H) = 0–100% ML (MCM-SO3H), and an octyl co-grafted series with varying octyl coverage (MCM-Oc-SO3H) as illustrated in Scheme 7. This family of materials allows the effect of lateral interactions between acid head groups and role of inert hydrophobic octyl chain ‘spectator groups’ on acid strength and catalyst activity to be separately probed. To ensure there were no complications in the kinetic analysis from diffusion limitation, esterification of a short chain alcohol, butanol (Fig. 10), with acetic acid was selected as a model reaction.
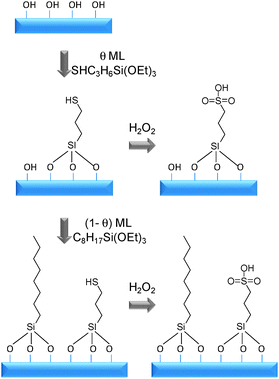 | ||
| Scheme 7 Preparation of sulfonic acid and octyl co-functionalised sulfonic acid MCM-41 materials (Reproduced from ref. 141 with permission of the Royal Society of Chemistry). | ||
 | ||
| Fig. 10 Activity of sulfonic acid silicas in butanol esterification with acetic acid, showing the impact of acid site density and increased hydrophobicity on catalyst turnover frequency. (Adapted from ref. 141 with permission of the Royal Society of Chemistry). | ||
For the MCM-SO3H series, NH3 calorimetry revealed acid strength increases as a function of sulphonic acid loading, with −ΔHads(NH3) increasing from 87 to 118 kJ mol−1. In contrast, MCM-Oc-SO3H exhibits a dramatic enhancement of acid strength for submonolayer SO3H coverages, with −ΔHads(NH3) found to increase to 103 kJ mol−1. In line with these acid strength measurements the per site activity of the MCM-SO3H series in the esterification of butanol with acetic acid was found to increase with SO3H content. Incorporation of octyl groups further promotes esterification activity of all the samples within the MCM-Oc-SO3H series, such that the turn over frequency of the lowest loading SO3H sample more than doubles. Molecular dynamic simulations indicate that the interaction of isolated sulphonic acid groups with the pore walls is the primary cause of decreased acid strength and activity of submonolayer samples within the MCM-SO3H series. Octyl groups increased surface hydrophobicity and lateral interactions between adjacent sulphonic acid head groups, resulting in a striking enhancement of acid strength and esterification activity.
3.4 Effect of pore architecture
Although numerous solid acids and bases have been explored for biodiesel synthesis,21,45,92,143 most materials described so far exhibit micro and/or mesoporosity which are not optimal for accommodating bulky and viscous C16–C18 TAGs. Such effects are evident from the application of hierarchical H-β-zeolite144 which shows increased catalytic activity compared to conventional β-zeolite due to presence of a secondary micro-mesoporosity that reduces the diffusion limitations. Likewise, kinetic studies of basic resins also reveal significant major mass transfer limitation during coconut oil transesterification.145 To alleviate diffusional problems, templated mesporous materials have attracted much interest as catalysts supports, with SBA-15 derived catalysts proving particularly popular candidates for biodiesel synthesis.69,70,146–148 However, these templated supports possess long, isolated 5 nm diameter parallel channels which are not conducive to efficient in-pore diffusion and result in poor catalytic turnover. More effort is thus required to tailor catalyst porosity to optimise mass-transport of theses bulky and viscous C16–C18 TAGs or FFAs typical of plant oils. Simulations demonstrate that in the Knudsen diffusion regime,149 where reactants/products are able to diffuse into/out of mesopores but experience moderate diffusion limitation, construction of hierarchical pore structures would significantly improve catalyst activity. Methods to prepare materials with interpenetrating bimodal meso-macropore networks150–154 using microemulsion152 or co-surfactant155 templating routes have been reported, and are particularly attractive for developing materials for use in liquid flow reactors where rapid pore diffusion is required. Using a combination of liquid crystalline and physical templating methods, highly organized macro-mesoporous ‘SBA-15 like’ silica materials have been developed (Scheme 8), in which both macro and mesopore diameters can be tuned over the range 200–500 and 5–20 nm respectively.156 | ||
| Scheme 8 Liquid crystal templating route to form mesoporous silica and combined physical templating method using polystyrene microspheres to introduce a macropore network. | ||
Fig. 11 shows SEM and HRTEM of the macropore and mesopore network of a resulting propylsulfonic acid functionalised, macro-mesoporous solid acid catalyst. The incorporation of such macropores is found to confer a striking enhancement in both the rate of transesterification of tricaprylin and esterification of palmitic acid, with methanol, (Fig. 12) which is attributed to the greater accessibility of SO3H sites within the mesopores due to macropores acting as rapid transport conduits to the active sites and enhancing mass-transport. Recent exciting reports have also demonstrated a dramatic impact of macropore incorporation on the activity of hydrotalcite materials, with macropores conferring a 10-fold rate enhancement over conventional hydrotalcites in triolein transesterification.157
 | ||
| Fig. 11 (a) SEM and (b, c) low and high magnification TEM images for hierarchical macro-mesoporous SBA-15. (Adapted from ref. 156 with permission of the Royal Society of Chemistry). | ||
 | ||
| Fig. 12 Catalytic activity of hierarchical macro-mesoporous SBA-15 sulfonic acid catalysts in the transesterification of tricaprylin and esterification of palmitic acid with methanol at 60 °C. (Adapted from ref. 156 with permission of the Royal Society of Chemistry). | ||
In an alternative approach, the effect of mesopore channel length on acid site accessibility was explored via sulfonic acid-functionalised platelet SBA-15 materials158 with ordered short mesochannels (7 nm diameter × 150–350 nm long). These materials were efficient catalysts for FFA esterification with methanol, exhibiting higher catalytic activities than SBA catalysts with conventional rod-like or fibre-like morphologies, which was attributed to better in-pore molecular diffusion. These short pore sulphonic acid SBA-15 materials also exhibit a higher per site activity than Amberlyst 15 resin, in line with macroporous-mesoporous solid acids, highlighting the poor accessibility of acid sites within resin based catalysts.
While there are clear benefits of breaking up the porous network of SBA materials either by incorporating 300–500 nm macropores or shortening the mesopore length, mesopores diameters typically remain close to the molecular dimensions of bulky TAGs (∼5 nm), hence, there is further scope for improving catalyst performance by also enlarging the mesopores. Porogens including trimethylbenzene,159,160 triethylbenzene or triisopropylbenzene161 have been employed to swell the Pluronic P123 micelles used to produce SBA-15, enabling formation of mesopores with diameters 5 to 30 nm. Surprisingly, the potential catalytic applications of the resulting pore-expanded materials have rarely been exploited, with MCM-41,162 sulfonic acid163 and polystyrene sulfonic acid composites164 within an ultra-large pore SBA-15 among the few reported examples. While sulfonic acid-functionalized mesoporous materials have been previously tested in biodiesel synthesis,69,165,166 the pore sizes of such catalysts fall ≤6.5 nm and the relationship between pore diameter and mass transport properties remains unexplored. Large pore, mesostructured sulfonic acid silicas have been studied for the esterification of palmitic acid, and separate transesterification of tricaprylin and triolein, with methanol (Fig. 13).163
 | ||
| Fig. 13 Reactivity of Amberlyst and sulfonic acid derivatised SBA-15-6; SBA-15-8 and SBA-15-14 catalysts in palmitic acid esterification and transesterification of tricaprylin and triolein. (Reproduced from ref. 163 with permission of the Royal Society of Chemistry). | ||
Steric factors and associated diffusion limitations render tricaprylin and triolein difficult to transesterify via conventional solid acid catalysts,167 making them interesting targets for mesostructured solid acids in their own right. The C18 triolein is also the major component of olive and Jatropha oil, and thus an excellent model TAG with which to assess the potential benefits of mesopore expansion in biodiesel synthesis. Fig. 13 shows the corresponding Turnover Frequencies (TOFs, initial rates normalised per acid site) which indicate that SBA-SO3H materials significantly outperform Amberlyst. Pore-expansion enhances TOF from 23 h−1 for conventional sulfonic acid SBA-15 to 120 h−1 for the analogous SBA-15-14 material. Similar enhancement was seen for tricaprylin and triolein transesterification with increasing pore diameter. The improved performance in both reactions was attributed to the greater accessibility of sulfonic acid sites within larger mesopores. These pore-expanded SBA-15 sulfonic acids also outperformed hierarchical sulfonic acid silicas (RSO3H-MM-SBA-4),156 suggesting that mesopore diffusion may have been only partially alleviated by macropore conduits. This work highlights the significant benefits achievable through careful nanoengineering of materials for FAME synthesis. Molecular dynamic and Monte Carlo simulations are required to further optimise these pore-expanded silicas and assess the potential benefits of macropore integration to generate hierarchical pore-expanded networks.
Work tailoring the pore architectures of inorganic solid acid catalyst highlights that despite their widespread usage commercial resins are inherently inefficient due to their poor acid site accessibility. The hydrophobic nature of resin based catalysts can however be advantageous, hence there has been interest in developing hybrid catalysts based around expanded SBA-15-type materials functionalized with poly(styrenesulfonic acid) moieties via surface-initiated atom transfer radical polymerization.164 The resulting hybrid organic–inorganic materials possess BJH pore sizes in the range 7–28 nm, and show comparable performance in oleic acid esterification with n-butanol as commercial SAC-13 and Amberlyst-15 catalysts. The combination of large mean pore diameters, strong acidity and increased hydrophobicity compared to conventional sulfonic acid-SBA-15 makes such hybrids interesting alternatives for acid-catalysed biodiesel synthesis.
Outlook & conclusions
This perspective highlights the significant progress made in recent years towards developing heterogeneous catalysts for biodiesel synthesis from plant oils, themselves complex feedstocks of triglycerides, free fatty acids and trace components such as phospholipids and sterols. The application of solid acid and base catalysts in esterification and transesterification is widespread, however further progress requires more detailed consideration of structure activity relationships. The development of a commercial heterogeneously catalysed process clearly requires a better understanding of how each reactant interacts with the active catalyst phase, particularly high molecular weight sterols that burn inefficiently in vehicle engines and should be removed or perhaps converted into lighter (fuel compatible) components. The design of pore networks with interconnecting macro and mesoporous channels has clear beneficial effects on reaction rates by improving in-pore diffusional properties and should be pursued.It should also be noted that despite concerns over long-term biodiesel use and associated engine wear in high-performance diesel vehicles,28 longer chain (>C18) FAMEs should be easier to implement in heavy-duty diesel engines. Crucially, widespread uptake and development of next-generation biodiesel fuels requires progressive government policies and incentive schemes to place biodiesel on a comparative footing with cheaper fossil-based fuels.168 Genetic modification, though controversial, may also assist by enabling plant biologists to engineer new higher yielding seed crops,169 that are rich in the optimum triglyceride chain length and degree of saturation and matched to the catalyst of choice, and desired fuel properties. The decision whether to use a single solid acid catalyst operating at high temperature, or a dual system coupling a solid acid FFA pre-treatment followed by a lower temperature solid base-catalysed transesterification, remains to be explored, and is inextricably coupled with the particular oil composition. The choice of feedstock may dictate the catalyst formulation required, and the ‘one type fits all’ approach is not appropriate. The increasing use of waste or low grade oil sources74 presents a challenge due to the presence of impurities which either requires improved purification technology170 or design of catalysts that are robust to these components. Oil sources may also be regional, and with the drive to use more 'non-food' oil sources, closer attention needs to be paid to the oil composition to select the most appropriate catalysts.
Solid materials capable of simultaneous esterification and transesterification under mild conditions present a future challenge for catalyst scientists, although superacids may be one solution.171 Alternatively, solid acids may be employed to first hydrolyse TAGs, and then esterify the resulting FFAs to FAME.172 Materials development, such as in the production of non-porous catalysts173 or application of surface-initiated controlled polymerization to functionalise oxide surfaces with polymeric organic species to create hybrid systems with high active site loadings, may prove valuable in the quest for catalysts with improved efficiency.
Plant-oil viscosity and poor miscibility with light alcohols continues to hamper the use of new heterogeneous catalysts for continuous biodiesel production from both materials and engineering perspectives. Process optimisation needs collaboration between catalyst chemists, chemical engineers and experts in molecular simulation to take advantage of innovative reactor designs;174 the future of biodiesel requires a concerted effort from chemists and engineers to develop catalysts and reactors in tandem. Alternative reactor technology and intensive processing such as reactive distillation175–177 and oscillatory flow reactors178–180 will enable more regional production of biodiesel. It is essential that technical advances in both materials chemistry and reactor engineering are pursued if biodiesel is to remain a key player in the renewable energy sector during the 21st century.
Acknowledgements
We thank the EPSRC for funding (EP/F063423/2). KW acknowledges The Royal Society for the award of an Industry Fellowship, and AFL thanks the EPSRC for the award of a Leadership Fellowship (EP/G007594/2).Notes and references
- P. Anastas and J. Warner, Green Chemistry: Theory and Practice, Oxford University Press, New York, 1998 Search PubMed.
- B. Walter, J. F. Gruson and G. Monnier, Oil & Gas Science and Technology-Revue De L Institut Francais Du Petrole, 2008, 63, 387–393 Search PubMed.
- I. B. T. Barker, L. Bernstein, J. E. Bogner, P. R. Bosch, R. Dave, O. R. Davidson, B. S. Fisher, S. Gupta, K. Halsnæs, G. J. Heij, S. Kahn Ribeiro, S. Kobayashi, M. D. Levine, D. L. Martino, O. Masera, B. Metz, L. A. Meyer, G.-J. Nabuurs, A. Najam, N. Nakicenovic, H.-H. Rogner, J. Roy, J. Sathaye, R. Schock, P. Shukla, R. E. H. Sims, P. Smith, D. A. Tirpak, D. Urge-Vorsatz and D. Zhou, Technical Summary. In: Climate Change 2007: Mitigation. Contribution of Working Group III to the Fourth Assessment Report of the Intergovernmental Panel on Climate Change, 2007 Search PubMed.
- UNFCCC, Fifth session, Copenhagen Accord, December, 2009.
- United Nations Environment Programme and New Energy Finance, Global Trends in Sustainable Energy Investment 2010, Analysis of Trends and Issues in the Financing of Renewable Energy and Energy Efficiency, 2010, ISBN: 978-92-807-3085-2.
- BP Energy Outlook 2030, London, January 2011.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS.
- D. W. McLaughlin, Conserv. Biol., 2011, 25, 1120 CrossRef.
- F. Danielsen, H. Beukema, N. D. Burgess, F. Parish, C. A. Bruhl, P. F. Donald, D. Murdiyarso, B. Phalan, L. Reijnders, M. Struebig and E. B. Fitzherbert, Conserv. Biol., 2009, 23, 348–358 CrossRef.
- T. M. Mata, A. A. Martins and N. S. Caetano, Renewable Sustainable Energy Rev., 2010, 14, 217–232 CrossRef CAS.
- R. Luque, Energy Environ. Sci., 2010, 3, 254–257 CAS.
- Y. Chisti, Biotechnol. Adv., 2007, 25, 294–306 CrossRef CAS.
- J. J. Cheng and G. R. Timilsina, Renewable Energy, 2011, 36, 3541–3549 CrossRef CAS.
- Enabling the next generation of biodiesel, http://nextcatinc.com/The%20NextCAT%20Solution.htm Accessed 01/01/2012.
- President Obama Signs Bill Extending Biodiesel Tax Incentive Into Law, http://www.biodiesel.org/news/taxcredit/default.shtm Accessed 01/01/2012.
- J. Hill, E. Nelson, D. Tilman, S. Polasky and D. Tiffany, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 11206–11210 CrossRef CAS.
- X. Y. Yan, O. R. Inderwildi and D. A. King, Energy Environ. Sci., 2010, 3, 190–197 CAS.
- R. Luque, L. Herrero-Davila, J. M. Campelo, J. H. Clark, J. M. Hidalgo, D. Luna, J. M. Marinas and A. A. Romero, Energy Environ. Sci., 2008, 1, 542–564 CAS.
- R. Luque, J. C. Lovett, B. Datta, J. Clancy, J. M. Campelo and A. A. Romero, Energy Environ. Sci., 2010, 3, 1706–1721 CAS.
- A. Corma, S. Iborra and A. Velty, Chem. Rev., 2007, 107, 2411–2502 CrossRef CAS.
- J.-P. Dacquin, A. F. Lee and K. Wilson, in Thermochemical Conversion of Biomass to Liquid Fuels and Chemicals, ed. M. Crocker, RSC Publishing, 2010 Search PubMed.
- Z. Helwani, M. R. Othman, N. Aziz, J. Kim and W. J. N. Fernando, Appl. Catal., A, 2009, 363, 1–10 CrossRef CAS.
- Y. C. Sharma, B. Singh and J. Korstad, Fuel, 2011, 90, 1309–1324 CrossRef CAS.
- A. Sivasamy, K. Y. Cheah, P. Fornasiero, F. Kemausuor, S. Zinoviev and S. Miertus, ChemSusChem, 2009, 2, 278–300 CrossRef CAS.
- B. Smith, H. C. Greenwell and A. Whiting, Energy Environ. Sci., 2009, 2, 262–271 CAS.
- A. F. Clarens, E. P. Resurreccion, M. A. White and L. M. Colosi, Environ. Sci. Technol., 2010, 44, 1813–1819 CrossRef CAS.
- K. Narasimharao, A. Lee and K. Wilson, J. Biobased Mater. & Bioen., 2007, 1, 19–30 Search PubMed.
- G. Knothe, Energy Environ. Sci., 2009, 2, 759–766 CAS.
- A. C. Lanser, G. R. List, R. K. Holloway and T. L. Mounts, J. Am. Oil Chem. Soc., 1991, 68, 448–449 CrossRef CAS.
- Y. C. Sharma, B. Singh and S. N. Upadhyay, Fuel, 2008, 87, 2355–2373 CrossRef CAS.
- H. J. Berchmans and S. Hirata, Bioresour. Technol., 2008, 99, 1716–1721 CrossRef CAS.
- M. G. Kulkarni and A. K. Dalai, Ind. Eng. Chem. Res., 2006, 45, 2901–2913 CrossRef CAS.
- M. Bensmira, B. Jiang, C. Nsabimana and T. Jian, Food Res. Int., 2007, 40, 341–346 CrossRef CAS.
- B. Saad, C. W. Ling, M. S. Jab, B. P. Lim, A. S. M. Ali, W. T. Wai and M. I. Saleh, Food Chem., 2007, 102, 1407–1414 CrossRef CAS.
- M.-J. Paik, H. Kim, J. Lee, J. Brand and Y.-R. Kim, J. Chromatogr., A, 2009, 1216, 5917–5923 CrossRef CAS.
- B. Freedman, E. H. Pryde and T. L. Mounts, J. Am. Oil Chem. Soc., 1984, 61, 1638–1643 CrossRef CAS.
- M. Canakci and J. Van Gerpen, Trans. of the ASAE, 1999, 42, 1203–1210 CAS.
- F. R. Ma and M. A. Hanna, Bioresour. Technol., 1999, 70, 1–15 CrossRef CAS.
- M. P. Dorado, E. Ballesteros, J. A. de Almeida, C. Schellert, H. P. Lohrlein and R. Krause, Trans. ASAE, 2002, 45, 525–529 CAS.
- R. Turck, US6538146, to At Agrar-Technik GmbH, 2002.
- E. Santacesaria, R. Tesser, M. Di Serio, M. Guida, D. Gaetano, A. G. Agreda and F. Cammarota, Ind. Eng. Chem. Res., 2007, 46, 8355–8362 CrossRef CAS.
- F. R. Ma, L. D. Clements and M. A. Hanna, Bioresour. Technol., 1999, 69, 289–293 CrossRef CAS.
- A. Demirbas, Energy Convers. Manage., 2003, 44, 2093–2109 CrossRef CAS.
- A. Demirbas, Energy Policy, 2007, 35, 4661–4670 CrossRef.
- E. Lotero, Y. J. Liu, D. E. Lopez, K. Suwannakarn, D. A. Bruce and J. G. Goodwin, Ind. Eng. Chem. Res., 2005, 44, 5353–5363 CrossRef CAS.
- B. Freedman, E. H. Pryde and T. L. Mounts, J. Am. Oil Chem. Soc., 1984, 61, 1638–1643 CrossRef CAS.
- M. M. K. Tanabe, Y. Ono and H. Hattori, Stud. Surf. Sci. Catal., 1989, 51, 1 CrossRef.
- G. R. Peterson and W. P. Scarrah, J. Am. Oil Chem. Soc., 1984, 61, 1593–1597 CrossRef CAS.
- G. Vicente, A. Coteron, M. Martinez and J. Aracil, Ind. Crops Prod., 1998, 8, 29–35 CrossRef CAS.
- G. J. Lopez DE and Bruce DA, Appl. Catal., A, 2005, 295, 97–105 CrossRef.
- T. A. Peters, N. E. Benes, A. Holmen and J. T. F. Keurentjes, Appl. Catal., A, 2006, 297, 182–188 CrossRef CAS.
- K. Suwannakarn, E. Lotero, J. G. Goodwin and C. Q. Lu, J. Catal., 2008, 255, 279–286 CrossRef CAS.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Adv. Synth. Catal., 2006, 348, 75–81 CrossRef CAS.
- D. Rattanaphra, A. P. Harvey, A. Thanapimmetha and P. Srinophakun, Renewable Energy, 2011, 36, 2679–2686 CrossRef CAS.
- N. Mizuno, Chem. Rev., 1998, 98, 199–217 CrossRef CAS.
- T. Okuhara and M. Misono, Adv. Catal., 1996, 41, 113–252 CrossRef CAS.
- M. Misono, Catal. Rev. Sci. Eng., 1987, 29, 269–321 CAS.
- A. D. Newman, D. R. Brown, P. Siril, A. F. Lee and K. Wilson, Phys. Chem. Chem. Phys., 2006, 8, 2893–2902 RSC.
- A. D. Newman, A. F. Lee, K. Wilson and N. A. Young, Catal. Lett., 2005, 102, 45–50 CrossRef CAS.
- W. Kaleta and K. Nowinska, Chem. Commun., 2001, 535–536 RSC.
- M. Kamada, H. Nishijima and Y. Kera, Bull. Chem. Soc. Jpn., 1993, 66, 3565–3570 CrossRef CAS.
- K. N. Rao, A. Sridhar, A. F. Lee, S. J. Tavener, N. A. Young and K. Wilson, Green Chem., 2006, 8, 790–797 RSC.
- T. Okuhara, T. Nishimura, H. Watanabe and M. Misono, J. Mol. Catal., 1992, 74, 247–256 CrossRef CAS.
- T. Okuhara, T. Arai, T. Ichiki, K. Y. Lee and M. Misono, J. Mol. Catal., 1989, 55, 293–301 CrossRef CAS.
- K. Narasimharao, D. R. Brown, A. F. Lee, A. D. Newman, P. F. Siril, S. J. Tavener and K. Wilson, J. Catal., 2007, 248, 226–234 CrossRef CAS.
- L. Pesaresi, D. R. Brown, A. F. Lee, J. M. Montero, H. Williams and K. Wilson, Appl. Catal., A, 2009, 360, 50–58 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin, Jr., J. Catal., 2006, 243, 221–228 CrossRef CAS.
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2005, 229, 365–373 CrossRef CAS.
- I. K. Mbaraka, D. R. Radu, V. S. Y. Lin and B. H. Shanks, J. Catal., 2003, 219, 329–336 CrossRef CAS.
- J. A. Melero, L. F. Bautista, G. Morales, J. Iglesias and D. Briones, Energy Fuels, 2009, 23, 539–547 CrossRef CAS.
- X.-R. Chen, Y.-H. Ju and C.-Y. Mou, J. Phys. Chem. C, 2007, 111, 18731–18737 CAS.
- V. Dufaud and M. E. Davis, J. Am. Chem. Soc., 2003, 125, 9403–9413 CrossRef CAS.
- I. K. Mbaraka and B. H. Shanks, J. Catal., 2006, 244, 78–85 CrossRef CAS.
- G. Morales, L. Fernando Bautista, J. A. Melero, J. Iglesias and R. Sanchez-Vazquez, Bioresour. Technol., 2011, 102, 9571–9578 CrossRef CAS.
- J. Iglesias, J. A. Melero, L. Fernando Bautista, G. Morales, R. Sanchez-Vazquez, M. Teresa Andreola and A. Lizarraga-Fernandez, Catal. Today, 2011, 167, 46–55 CrossRef CAS.
- M. Kouzu, A. Nakagaito and J. S. Hidaka, Appl. Catal., A, 2011, 405, 36–44 CrossRef CAS.
- H. Hattori, Chem. Rev., 1995, 95, 537–558 CrossRef CAS.
- N. Greenwood and A. Earnshaw, Chemistry of the Elements, Pergamon Press, Oxford, 1989 Search PubMed.
- M. C. G. Albuquerque, D. C. S. Azevedo, C. L. Cavalcante Jr, J. Santamaria-Gonzalez, J. M. Marida-Robles, R. Moreno-Tost, E. Rodreguez-Castellan, A. Jiminez-Lopez and P. Maireles-Torres, J. Mol. Catal. A: Chem., 2009, 300, 19–24 CrossRef CAS.
- S. Gryglewicz, Bioresour. Technol., 1999, 70, 249–253 CrossRef CAS.
- M. L. Granados, D. M. Alonso, A. C. Alba-Rubio, R. Mariscal, M. Ojeda and P. Brettes, Energy Fuels, 2009, 23, 2259–2263 CrossRef.
- M. Verziu, S. M. Coman, R. Richards and V. I. Parvulescu, Catal. Today, 2011, 167, 64–70 CrossRef CAS.
- P.-L. Boey, G. P. Maniam and S. Abd Hamid, Chem. Eng. J., 2011, 168, 15–22 CrossRef CAS.
- A. Demirbas, Energy Convers. Manage., 2007, 48, 937–941 CrossRef CAS.
- J. M. Montero, K. Wilson and A. F. Lee, Top. Catal., 2010, 53, 737–745 CrossRef CAS.
- R. S. Watkins, A. F. Lee and K. Wilson, Green Chem., 2004, 6, 335–340 RSC.
- C. S. MacLeod, A. P. Harvey, A. F. Lee and K. Wilson, Chem. Eng. J., 2008, 135, 63–70 CrossRef CAS.
- D. M. Alonso, R. Mariscal, M. L. Granados and P. Maireles-Torres, Catal. Today, 2009, 143, 167–171 CrossRef CAS.
- A. Corma and S. Iborra, Adv. Catal., 2006, 49, 239–302 CrossRef CAS.
- M. Verziu, B. Cojocaru, J. Hu, R. Richards, C. Ciuculescu, P. Filip and V. I. Parvulescu, Green Chem., 2008, 10, 373–381 RSC.
- K. Zhu, J. Hu, C. Kuebel and R. Richards, Angew. Chem., Int. Ed., 2006, 45, 7277–7281 CrossRef CAS.
- M. Di Serio, M. Ledda, M. Cozzolino, G. Minutillo, R. Tesser and E. Santacesaria, Ind. Eng. Chem. Res., 2006, 45, 3009–3014 CrossRef CAS.
- N. Barakos, S. Pasias and N. Papayannakos, Bioresour. Technol., 2008, 99, 5037–5042 CrossRef CAS.
- D. G. Cantrell, L. J. Gillie, A. F. Lee and K. Wilson, Appl. Catal., A, 2005, 287, 183–190 CrossRef CAS.
- D. P. Debecker, E. M. Gaigneaux and G. Busca, Chem.–Eur. J., 2009, 15, 3920–3935 CrossRef CAS.
- F. Cavani, F. Trifiro and A. Vaccari, Catal. Today, 1991, 11, 173–301 CrossRef CAS.
- J. M. Fraile, N. Garcia, J. A. Mayoral, E. Pires and L. Roldan, Appl. Catal., A, 2009, 364, 87–94 CrossRef CAS.
- H. E. Cross and D. R. Brown, Catal. Commun., 2010, 12, 243–245 CrossRef CAS.
- Y. Xi and R. J. Davis, J. Catal., 2008, 254, 190–197 CrossRef CAS.
- M. A. Olutoye and B. H. Hameed, Bioresour. Technol., 2011, 102, 6392–6398 CrossRef CAS.
- R. Rahul, J. K. Satyarthi and D. Srinivas, Ind. Eng. Chem. Res., 2011, 50, 1017–1025 CrossRef.
- H. Sun, Y. Ding, J. Duan, Q. Zhang, Z. Wang, H. Lou and X. Zheng, Bioresour. Technol., 2010, 101, 953–958 CrossRef CAS.
- S. Yan, M. Kim, S. O. Salley and K. Y. S. Ng, Appl. Catal., A, 2009, 360, 163–170 CrossRef CAS.
- L. Bournay, D. Casanave, B. Delfort, G. Hillion and J. A. Chodorge, Catal. Today, 2005, 106, 190–192 CrossRef CAS.
- G. Hillion, S. Leporq, D. L. Pennec and B. Delfort, EP1468734, to Institut Français du Pétrole, 2004 Search PubMed.
- G. Hillion, S. Leporq, D. L. Pennec and B. Delfort, US10801543, to Institut Français du Pétrole, 2004 Search PubMed.
- A. Corma, S. B. Abd Hamid, S. Iborra and A. Velty, J. Catal., 2005, 234, 340–347 CrossRef CAS.
- J. L. Shumaker, C. Crofcheck, S. A. Tackett, E. Santillan-Jimenez and M. Crocker, Catal. Lett., 2007, 115, 56–61 CrossRef CAS.
- J. L. Shumaker, C. Crofcheck, S. A. Tackett, E. Santillan-Jimenez, T. Morgan, Y. Ji, M. Crocker and T. J. Toops, Appl. Catal., B, 2008, 82, 120–130 CrossRef CAS.
- C. S. Castro, D. Cardoso, P. A. P. Nascente and J. M. Assaf, Catal. Lett., 2011, 141, 1316–1323 CrossRef CAS.
- K. Wilson, C. Hardacre, A. F. Lee, J. M. Montero and L. Shellard, Green Chem., 2008, 10, 654–659 RSC.
- O. Ilgen, Fuel Process. Technol., 2011, 92, 452–455 CrossRef CAS.
- R. Philipp and K. Fujimoto, J. Phys. Chem., 1992, 96, 9035–9038 CrossRef CAS.
- J. M. Montero, P. Gai, K. Wilson and A. F. Lee, Green Chem., 2009, 11, 265–268 RSC.
- M. L. Granados, M. D. Z. Poves, D. M. Alonso, R. Mariscal, F. C. Galisteo, R. Moreno-Tost, J. Santamaria and J. L. G. Fierro, Appl. Catal., B, 2007, 73, 317–326 CrossRef CAS.
- S. Yan, H. Lu and B. Liang, Energy Fuels, 2008, 22, 646–651 CrossRef CAS.
- J. M. Montero, D. R. Brown, P. L. Gai, A. F. Lee and K. Wilson, Chem. Eng. J., 2010, 161, 332–339 CrossRef CAS.
- J. A. D. Matthew and S. Parker, J. Electron Spectrosc. Relat. Phenom., 1997, 85, 175–178 CrossRef CAS.
- P. Ascarelli and G. Moretti, Surf. Interface Anal., 1985, 7, 8–12 CrossRef CAS.
- G. Moretti, J. Electron Spectrosc. Relat. Phenom., 1992, 58, 105–118 CrossRef CAS.
- R. Richards, R. S. Mulukutla, I. Mishakov, V. Chesnokov, A. Volodin, V. Zaikovski, N. Sun and K. J. Klabunde, Scr. Mater., 2001, 44, 1663–1666 CrossRef CAS.
- S. Utamapanya, K. J. Klabunde and J. R. Schlup, Chem. Mater., 1991, 3, 175–181 CrossRef CAS.
- D. M. Alonso, M. L. Granados, R. Mariscal and A. Douhal, J. Catal., 2009, 262, 18–26 CrossRef CAS.
- Y. Liu, E. Lotero and J. G. Goodwin, Jr., J. Catal., 2006, 242, 278–286 CrossRef CAS.
- M. J. Lee, J. Y. Chiu and H. M. Lin, Ind. Eng. Chem. Res., 2002, 41, 2882–2887 CrossRef CAS.
- R. Aafaqi, A. R. Mohamed and S. Bhatia, J. Chem. Technol. Biotechnol., 2004, 79, 1127–1134 CrossRef CAS.
- T. F. Dossin, M. F. Reyniers and G. B. Marin, Appl. Catal., B, 2006, 62, 35–45 CrossRef CAS.
- T. F. Dossin, M.-F. Reyniers, R. J. Berger and G. B. Marin, Appl. Catal., B, 2006, 67, 136–148 CrossRef CAS.
- A. Kapil, S. A. Bhat and J. Sadhukhan, Ind. Eng. Chem. Res., 2010, 49, 2326–2335 CrossRef CAS.
- D. E. Lopez, J. G. Goodwin, Jr. and D. A. Bruce, J. Catal., 2007, 245, 381–391 CrossRef CAS.
- K. Wilson, A. Renson and J. H. Clark, Catal. Lett., 1999, 61, 51–55 CrossRef CAS.
- B. Rac, P. Hegyes, P. Forgo and A. Molnar, Appl. Catal., A, 2006, 299, 193–201 CrossRef CAS.
- B. Rac, A. Molnar, P. Forgo, M. Mohai and I. Bertoti, J. Mol. Catal. A: Chem., 2006, 244, 46–57 CrossRef CAS.
- Q. H. Yang, J. Liu, J. Yang, M. P. Kapoor, S. Inagaki and C. Li, J. Catal., 2004, 228, 272 Search PubMed.
- Q. H. Yang, M. P. Kapoor, S. Inagaki, N. Shirokura, J. N. Kondo and K. Domen, J. Mol. Catal. A: Chem., 2005, 230, 85–89 CrossRef CAS.
- Q. H. Yang, M. P. Kapoor, N. Shirokura, M. Ohashi, S. Inagaki, J. N. Kondo and K. Domen, J. Mater. Chem., 2005, 15, 666–673 RSC.
- G. Morales, G. Athens, B. F. Chmelka, R. van Grieken and J. A. Melero, J. Catal., 2008, 254, 205–217 CrossRef CAS.
- D. Margolese, J. A. Melero, S. C. Christiansen, B. F. Chmelka and G. D. Stucky, Chem. Mater., 2000, 12, 2448–2459 CrossRef CAS.
- I. Diaz, C. Marquez-Alvarez, F. Mohino, J. Perez-Pariente and E. Sastre, J. Catal., 2000, 193, 283–294 CrossRef CAS.
- I. Diaz, C. Marquez-Alvarez, F. Mohino, J. Perez-Pariente and E. Sastre, J. Catal., 2000, 193, 295–302 CrossRef CAS.
- J.-P. Dacquin, H. E. Cross, D. R. Brown, T. Duren, J. J. Williams, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 1383–1391 RSC.
- C. Schumacher, J. Gonzalez, P. A. Wright and N. A. Seaton, J. Phys. Chem. B, 2006, 110, 319–333 CrossRef CAS.
- J. A. Melero, J. Iglesias and G. Morales, Green Chem., 2009, 11, 1285–1308 RSC.
- A. Carrero, G. Vicente, R. Rodriguez, M. Linares and G. L. del Peso, Catal. Today, 2011, 167, 148–153 CrossRef CAS.
- C. E. T. Co, M. C. Tan, J. A. R. Diamante, L. R. C. Yan, R. R. Tan and L. F. Razon, Catal. Today, 2011, 174, 54–58 CrossRef CAS.
- S. Garg, K. Soni, G. M. Kumaran, R. Bal, K. Gora-Marek, J. K. Gupta, L. D. Sharma and G. M. Dhar, Catal. Today, 2009, 141, 125–129 CrossRef CAS.
- E. Li and V. Rudolph, Energy Fuels, 2008, 22, 145–149 CrossRef CAS.
- D. Y. Zhao, Q. S. Huo, J. L. Feng, B. F. Chmelka and G. D. Stucky, J. Am. Chem. Soc., 1998, 120, 6024–6036 CrossRef CAS.
- S. Gheorghiu and M. O. Coppens, AIChE J., 2004, 50, 812–820 CrossRef CAS.
- W. H. Deng, M. W. Toepke and B. H. Shanks, Adv. Funct. Mater., 2003, 13, 61–65 CrossRef CAS.
- Z. Y. Yuan and B. L. Su, J. Mater. Chem., 2006, 16, 663–677 RSC.
- X. Zhang, F. Zhang and K. Y. Chan, Mater. Lett., 2004, 58, 2872–2877 CrossRef CAS.
- X. Zhang, F. Zhang and K. Y. Chan, Scr. Mater., 2004, 51, 343–347 CrossRef CAS.
- J.-P. Dacquin, J. Dhainaut, D. Duprez, S. Royer, A. F. Lee and K. Wilson, J. Am. Chem. Soc., 2009, 131, 12896–12897 CrossRef CAS.
- J. H. Sun, Z. P. Shan, T. Maschmeyer and M. O. Coppens, Langmuir, 2003, 19, 8395–8402 CrossRef CAS.
- J. Dhainaut, J.-P. Dacquin, A. F. Lee and K. Wilson, Green Chem., 2010, 12, 296–303 RSC.
- J. J. Woodford, J.-P. Dacquin, K. Wilson and A. F. Lee, Energy Environ. Sci., 2012 10.1039/C2EE02837A , advance article.
- S.-Y. Chen, T. Yokoi, C.-Y. Tang, L.-Y. Jang, T. Tatsumi, J. C. C. Chan and S. Cheng, Green Chem., 2011, 13, 2920–2930 RSC.
- D. H. Chen, Z. Li, Y. Wan, X. J. Tu, Y. F. Shi, Z. X. Chen, W. Shen, C. Z. Yu, B. Tu and D. Y. Zhao, J. Mater. Chem., 2006, 16, 1511–1519 RSC.
- N. Calin, A. Galarneau, T. Cacciaguerra, R. Denoyel and F. Fajula, C. R. Chim., 2010, 13, 199–206 CrossRef CAS.
- L. Cao, T. Man and M. Kruk, Chem. Mater., 2009, 21, 1144–1153 CrossRef CAS.
- D. D. Das, P. J. E. Harlick and A. Sayari, Catal. Commun., 2007, 8, 829–833 CrossRef CAS.
- J. P. Dacquin, A. F. Lee, C. Pirez and K. Wilson, Chem. Commun., 2012, 48, 212–214 RSC.
- A. Martin, G. Morales, F. Martinez, R. van Grieken, L. Cao and M. Kruk, J. Mater. Chem., 2010, 20, 8026–8035 RSC.
- R. Liu, X. Wang, X. Zhao and P. Feng, Carbon, 2008, 46, 1664–1669 CrossRef CAS.
- J. A. Melero, L. Fernando Bautista, G. Morales, J. Iglesias and R. Sanchez-Vazquez, Chem. Eng. J., 2010, 161, 323–331 CrossRef CAS.
- X. Mo, E. Lotero, C. Lu, Y. Liu and J. G. Goodwin, Catal. Lett., 2008, 123, 1–6 CrossRef CAS.
- S. Pinzi, I. L. Garcia, F. J. Lopez-Gimenez, M. D. Luque de Castro, G. Dorado and M. P. Dorado, Energy Fuels, 2009, 23, 2325–2341 CrossRef CAS.
- G. Knothe, Fuel Process. Technol., 2005, 86, 1059–1070 CrossRef CAS.
- T. M. Mata, N. Cardoso, M. Ornelas, S. Neves and N. S. Caetano, Energy Fuels, 2011, 25, 4756–4762 CrossRef CAS.
- R. Luque and J. H. Clark, ChemCatChem, 2011, 3, 594–597 CrossRef CAS.
- K. Suwannakarn, E. Lotero, K. Ngaosuwan and J. G. Goodwin, Ind. Eng. Chem. Res., 2009, 48, 2810–2818 CrossRef CAS.
- W. Long and C. W. Jones, ACS Catal., 2011, 1, 674–681 CrossRef CAS.
- A. C. Dimian, Z. W. Srokol, M. C. Mittelmeijer-Hazeleger and G. Rothenberg, Top. Catal., 2010, 53, 1197–1201 CrossRef CAS.
- A. A. Kiss, F. Omota, A. C. Dimian and G. Rothenberg, Top. Catal., 2006, 40, 141–150 CrossRef CAS.
- A. A. Kiss, A. C. Dimian and G. Rothenberg, Energy Fuels, 2008, 22, 598–604 CrossRef CAS.
- A. A. Kiss and G. Rothenberg, in Catalysis of Organic Reactions, Editon edn, 2009, vol. 123, pp. 291–301 Search PubMed.
- A. P. Harvey, M. R. Mackley and T. Seliger, J. Chem. Technol. Biotechnol., 2003, 78, 338–341 CrossRef CAS.
- A. N. Phan, A. P. Harvey and M. Rawcliffe, Fuel Process. Technol., 2011, 92, 1560–1567 CrossRef CAS.
- M. Zheng, R. L. Skelton and M. R. Mackley, Process Saf. Environ. Prot., 2007, 85, 365–371 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |