Alloys in catalysis: phase separation and surface segregation phenomena in response to the reactive environment
Spiros
Zafeiratos
*a,
Simone
Piccinin
b and
Detre
Teschner
*c
aLaboratoire LMSPC, UMR7515 CNRS-Université de Strasbourg, 25 rue Becquerel, 67087 Strasbourg, France. E-mail: spiros.zafeiratos@unistra.fr; Fax: +33 39024 2761; Tel: +33 36885 2755
bCNR-IOM, Istituto Officina dei Materiali, Via Bonomea 265, 34136 Trieste, Italy
cFritz-Haber Institute of the Max Planck Society, Department of Inorganic Chemistry, Faradayweg 4–6, Berlin, D-14195, Germany. E-mail: teschner@fhi-berlin.mpg.de; Fax: +49 30 8413 4676; Tel: +49 30 8413 5408
First published on 26th January 2012
Abstract
Alloys play a crucial role in several heterogeneous catalytic processes, and their applications are expected to rise rapidly. This is essentially related to the vast number of configurations and type of surface sites that multi-component materials can afford. It is well established that the chemical composition at the surface of an alloy usually differs from that in the bulk. This phenomenon, referred to as surface segregation, is largely controlled by the surface free energy. However, surface energy alone cannot safely predict the active surface state of a solid catalyst, since the contribution of other parameters such as size and support effects, as well as influence of the adsorbates, play a major role. This can lead to numerous surface configurations as for example over the length of a catalytic reactor, as the chemical potential of the gas phase changes continuously over the catalyst bed and hence different reactions may prevail at different catalyst bed segments. Thanks to the rapid improvement of the analytical surface science characterization techniques and theoretical methodologies, the potential effects induced by alloyed catalysts are better understood. For catalysis, the relevance of measurements performed on well-defined surfaces under idealized ultrahigh vacuum conditions has been questioned and studies in environments that closely resemble conditions of working alloy catalysts are needed. In this review we focus on experimental and theoretical studies related to in situ (operando) observations of surface segregation and phase separation phenomena taking place on the outermost surface layers of alloy catalysts. The combination of first principles theoretical treatment and in situ observation opens up new perspectives of designing alloy catalysts with tailored properties.
1. Introduction
Development of new materials with improved performance is one of the main objectives of research in catalysis. In heterogeneous catalysis, catalysts containing two or more metals might show significantly different catalytic properties compared to the parent metals and thus are widely utilized in several reactions including reforming,1 pollution control2 and alcohol oxidation.3 One of the most attractive aspects of using multi-metallic catalysts is the possibility of rationally designing the catalytic properties by controlling the type and composition of the elements.4,5 A detailed knowledge of the catalyst surface is a crucial first step in this approach and therefore has been the focus of numerous studies as summarized in several earlier reviews.6–13 Since the definition of different types of multimetallic materials is not very consistent in the literature, throughout this manuscript an “intermetallic compound” refers to a chemical compound of two or more metallic elements which adopts (at least partly) an ordered crystal structure that differs from those of the constituent metals. On the other hand, an “alloy” is a mixture of metals, intermetallic compounds and/or non-metals, and thus it can contain more than one phase. The term “bimetallic” refers to an alloy of two metals.Two main models have been put forward in order to explain the modification of the chemical properties of a metal in a bimetallic surface. First, the formation of the hetero-atom bonds changes the electronic environment of the metal surface, through the electronic (or ligand) effect. A second explanation is the so-called geometric (or ensemble) effect. According to this model, incorporation of atoms with different reactivity patterns on a bimetallic surface not only gives rise to strain effects, but also decreases the number of available multiple absorption sites (3-fold, 2-fold, in favour of single atom sites), modifying the chemisorption and reaction properties of adsorbates (ref. 14 and references therein). In general, differentiation between ensemble and electronic effects is rarely clear and often there is a contribution from both effects.
Although bulk alloys have been thoroughly studied and phase diagrams predicting the thermodynamic conditions for alloy formation in the bulk are well established, the composition of a bimetallic surface may be significantly different compared to the bulk material. This phenomenon is usually described as surface segregation (or just segregation). These differences are being driven by a complex interplay of both thermodynamic parameters, such as the relative surface energies, the bonding strengths of each metallic component with adsorbed atoms and molecules, the tendency of the metal components to form ordered alloy phases, and kinetic parameters such as diffusion barriers. Therefore, stable bulk alloys may exhibit phase separation on their surfaces and vice versa. For example, metals that are almost immiscible in the bulk, such as Au and Ni, showed evidence of extended bimetallic interaction on the surface (surface alloy).15 As a simple rule of thumb the element with the lowest surface energy segregates to the surface under vacuum conditions.
Alloy catalysts, however, typically operate under high pressure and high temperature conditions, and these reactive environments may substantially influence the alloy surface composition. If reactants interact strongly with one of the alloy constituents, the surface tends to be enriched in that constituent, even when it is not predominant in an inert atmosphere (adsorbate-induced surface segregation). Moreover, small excesses of one element with respect to the others in the bulk alloy compared to the nominal composition, or other bulk defects, can also play a role, especially in strongly ordered compounds.16 The combined effects of the gas-phase atmosphere and the bulk alloy properties will therefore determine the composition and oxidation state of the alloy surface. An example of the modification induced by high pressure conditions is the development of an oxide like film at the catalyst surface of several transition metals (e.g. Ru, Pd, Ag) encountered in oxidation catalysis.17–19 Reaching the active state of the catalyst may require a considerable induction period, which reflects the large modifications resulting from the interaction of the metal surface with the reaction environment.
In the following review we summarize recent experimental and theoretical work focusing on the description of bimetallic and ternary, extended and nanosized, alloy surfaces under reactive gas phase environments. Emphasis will be given to studies performed at catalytically relevant conditions (elevated temperature and pressure) using in situ applied surface sensitive techniques.
2. Methods
2.1. Experimental approaches
Traditionally, in situ studies of solids under realistic liquid or gas phase environments were performed by using experimental techniques based on photons, like for example UV-Vis, Extended X-ray Absorption Fine Structure (XAFS) and Raman spectroscopies, etc. The use of photons as a probe was implied by the fact that they do not interact strongly with matter (of course this is also related to their energy) and therefore in a large extent could go through the gas or liquid environment above the sample. These techniques are however not particularly sensitive to the surface and probe a large volume of the solid. Techniques based on low and medium energy electron probing are better suited for surface sensitive studies, but until recently most of them were confined in vacuum environments. In the last few years, several microscopic and spectroscopic techniques capable to operate in situ and deliver atomic-scale information on solid surfaces or nanoscale materials were developed and a recent review about their impact on our understanding of surface chemistry is given by Tao and Salmeron.20 Below we briefly describe aspects of selected in situ techniques cited in this article to facilitate the comprehension.2.2. Theoretical approaches
The structure and composition of bimetallic surfaces in vacuum was the focus of early first-principles calculations, based on density functional theory (DFT) using the coherent-potential approximation and on effective-medium theory24 as well as on the Green's-function linear-muffin-tin-orbital (LMTO) approach.25 In these studies the authors computed the derivatives of the surface energy versus surface concentration: the first derivative is the surface segregation energy (which determines whether an impurity segregates to the surface or not) and the second derivative is the surface mixing energy (which determines whether the two elements mix at the surface or phase-separate, forming islands). These results were used to construct a complete database of the segregation energies and the surface mixing energies for all combinations of the transition metals, which have been instrumental in rationalizing the surface composition of different bimetallic alloys in experiments as well as in designing new materials.4,5 The role of the reactive environment in the structure and composition of the alloy surfaces, however, was not investigated in these earlier studies.In this section we will briefly review two well established methodologies, both based on a DFT description of the electronic structure of the system, that have been employed to theoretically model the equilibrium structure of metal surfaces, and alloys in particular, in a reactive environment.
In the first approach, called ab initio atomistic thermodynamics, one considers a portion of the system (the surface) to be in contact with a gas phase atmosphere and with a bulk phase, and computes the surface free energy of several candidate surface structures to determine the most stable under specific conditions of temperature, partial pressures of the gas phase species and chemical potentials of the bulk phase elements.
A second approach is the lattice-gas-Hamiltonian (also called cluster expansion) method, where a polynomial expansion of the energy of the system is parametrized in terms of a set of DFT calculations. This enables us to study many more and much larger surface structures compared to the previous approach, properly accounting for disorder and configurational entropy effects, but is limited to the treatment of structures with a pre-determined lattice topology.
| γ(T, p) = 1/Asurf[G(T, p, NA, NB, NO) − NAμA − NBμB − NOμO(T, p)] | (1) |
 | (2) |
The free energy comprises several terms that need to be evaluated separately:
| G = Etotal + Fvib − TSconf + pV | (3) |
The total energy, Etotal, is normally evaluated using quantum-mechanical DFT calculations, which offer the best compromise between the accuracy required to simulate processes like the adsorption/desorption of the gas phase species and computational feasibility. Moreover, one is typically interested in computing the relative stability of different surface structures (i.e. surface energy differences) rather than absolute values of surface energies, so some degree of error cancellation can be expected. Absolute values, on the other hand, are more sensitive to the intrinsic errors due to the approximations in the exchange and correlation (XC) functional employed in DFT. It also worth mentioning that computing the value of the chemical potential of the oxygen reservoir is problematic in calculations that employ generalized gradient approximation (GGA) functionals such as PBE.31 The binding energy of the oxygen molecule is in this case overestimated by about 0.5 eV per O atom, which translates into a severe error bar in the determination of temperatures and pressures in the phase diagrams. This error, however, will be partially compensated by the analogous GGA error in the description of oxygen chemisorbed on the metal surfaces.
In these calculations the system is described using a slab geometry in small unit cells periodically repeated in space; therefore only ordered structures can be simulated in this approach. Furthermore, when dealing with surface alloys or overlayers formed by impurity atoms introduced at the surface of the hosting metal, such small unit cells cannot accommodate large deformations that might be induced by, for example, size mismatch between the host and the impurity atoms, thus leading to large stresses in the overlayers.
 | (4) |
Once the lattice-gas-Hamiltonian has been constructed, it can be used to study large ordered or disordered systems at finite temperatures with Monte Carlo simulations, either in the canonical33,34 or gran-canonical35 ensembles. In the latter case, in particular, the surface segregation properties of an alloy can be studied as a function of the environment, as we will see in Section 3.1.3.
3. Examples
3.1. Selected theoretical studies of bi-metallic alloys in an oxygen atmosphere
In the following we will review three systems that have been investigated using the theoretical methodologies described above: the Ag–Cu and the Ag3Pd alloys in an oxygen atmosphere, studied using the ab initio thermodynamics method and the Pt–Ru alloy again in an oxygen atmosphere, studied using the lattice-gas-Hamiltonian method. The first example, in particular, will show how experimental and theoretical approaches can complement each other in a synergic effort to unravel the structure of an alloy in a reactive environment.Considering the (111) surfaces of Ag and Cu, due to the fact that the surface energy of silver is considerably lower than the one of copper (experimental values: γAg(111) = 0.078 eV Å−2 and γCu(111) = 0.114 eV Å−2), the surface of the alloy is expected to expose Ag under UHV conditions. Indeed, the deposition of a Cu thin-film on Ag(111) in ultra high vacuum (UHV) leads to the formation of Cu islands encapsulated by one monolayer of Ag.38,39
On the other hand, experiments carried out on Ag–Cu alloys with 0.11% Cu content at ∼500 K show that in an oxidizing environment the Cu surface content is in the 0.1–0.75 ML range,36 much higher than expected from the bulk Cu concentration. The overall effect of the oxygen atmosphere is therefore to promote the segregation of Cu to the surface.
Using the ab initio atomistic thermodynamics approach, Piccinin et al. have studied the surface structure of the Ag–Cu alloy in the presence of oxygen.40–42 They have shown that the strength of Cu–O bond relative to the Ag–O bond drives the segregation of Cu to the surface. Their main finding is that under conditions of pressure and temperature relevant for ethylene epoxidation, a thin copper-oxide-like layer is predicted to form at the surface. Several energetically similar structures have been predicted, each of them bearing little resemblance with either bulk cuprous (Cu2O) or cupric (CuO) oxide phases (see Fig. 1). APPES experiments on copper–silver nanopowder (∼100 nm, 2.5 wt% Cu) at 520 K in O2 as well as in C2H4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :O2 (1:2) atmospheres at a total pressure of 0.5 mbar confirmed the presence of a thin copper-oxide layer at the surface of the alloy.
:O2 (1:2) atmospheres at a total pressure of 0.5 mbar confirmed the presence of a thin copper-oxide layer at the surface of the alloy.
 | ||
| Fig. 1 Surface phase diagram for the (111) facet under constrained thermodynamic equilibrium with oxygen and ethylene. Solid (dot-dashed) lines represent the stability limit with respect to the formation of Ac (EO). Each shaded area represents the region of stability of a combination of two surface structures giving a Cu coverage of 0.5 ML. The white area is the region of stability of the clean Ag(111) surface. The dashed polygon corresponds to typical values of temperature and pressures used in experiments (T = 300–600 K and pO2, pC2H4 = 10−4–1 atm). Ag1.2O and Ag1.5O are 1-layer thin oxide-like structures; CuO and CuO(b) are 1-layer thin and bulk CuO structures, respectively; p2 and p4-OCu3 are 1-layer thin Cu2O-like structures with (2 × 2) and (4 × 4) periodicity, respectively. In the latter, an OCu3 unit is removed. Reproduced with permission from ref. 42. | ||
An ab initio atomistic thermodynamics study of the Ag–Cu in the presence of both reactants in ethylene epoxidation has been carried out by Nguyen et al.,27 showing that the thin surface oxide layer is thermodynamically stable also in the presence of ethylene, at the partial pressures and temperatures typically used in practical applications. The mechanism of epoxidation catalyzed by these and other surface structures has also been investigated theoretically using DFT calculations.43–48
Not surprisingly, under UHV conditions the more noble metal, Ag, is found to segregate to the surface. As the chemical potential of oxygen is increased, however, the adsorption of oxygen drives the segregation of Pd to the surface, increasing the Pd concentration in the first two layers. As in the case of the Ag–Cu alloy, there is a competition between the tendency of segregating the more noble metal to the surface and the tendency to lower the surface free energy by segregating the element that makes a stronger bond with the adsorbed oxygen.
The authors examined also the role played by the chemical potentials of the elements in the bulk alloy. As can be seen from Fig. 2, while under Pd-rich conditions (left panel) the onset of oxygen adsorption corresponds to the segregation of Pd to the surface, under Ag-rich conditions (right panel) oxygen adsorbs first on a Ag-pure surface and Pd segregation is induced only when a higher oxygen coverage is reached. The stabilization of Pd–O structures at the surface of the alloy takes place at a higher oxygen chemical potential compared to the Pd-rich case, reflecting the higher energetic cost to remove Pd atoms from an Ag-rich bulk phase. Moreover, the sequence of surface structures obtained by sweeping the values of oxygen chemical potential is different in the two cases, stressing how both the effects of the reactive environment and the bulk alloy structure need to be accounted for.
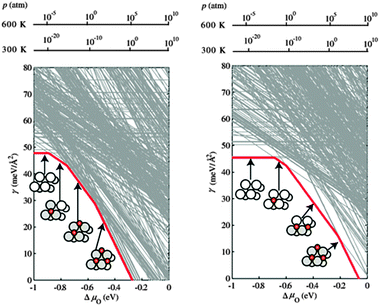 | ||
| Fig. 2 Surface free energy of Ag3Pd(111) in equilibrium with a Pd-rich Ag3Pd bulk reservoir, as a function of oxygen chemical potential. Each line corresponds to one of the tested surface configurations, and only the few configurations that result as most stable for a range of oxygen chemical potential are drawn as dark (red) lines. Additionally shown as insets are top views of the most stable surface configurations, with adsorbed O atoms shown as red small circles, Ag atoms as white circles, and Pd atoms as grey circles. The dependence on the oxygen chemical potential is translated into pressure scales for T = 300 K and T = 600 K (upper x axes). The left panel corresponds to Pd-rich Ag3Pd bulk reservoir conditions, the right panel to Ag-rich conditions. Reproduced with permission from ref. 49. | ||
One interesting point discussed by Kitchin et al. in their work is the fact that the onset of bulk oxidation of Pd in the Ag3Pd alloy, resulting in PdO, is predicted to take place at values of oxygen chemical potential that are only slightly higher than those corresponding to oxygen adsorption of the surface of the alloy. This is quite different from what happens on pure Pd surfaces, where the adsorption of oxygen takes place at values of oxygen chemical potential well below the onset of oxidation. This is due to the higher cost to segregate Pd in Ag3Pd, where Pd is scarce, compared to pure Pd. This therefore suggests that the segregation of Pd to the surface is likely to be accompanied by the formation of bulk-like oxide films at the surface.
One restriction of this approach is the fact that these types of studies are limited to a few periodic structures in a small unit cell, so that the configurational entropy contribution or intermediated coverages cannot be considered. These limitations can be circumvented by the use of the lattice-gas-Hamiltonian (or cluster expansion) method, as we will see in the next section.
At high values of Ru chemical potentials, on the other hand, the oxygen surface coverage and ruthenium surface composition are strongly coupled, both increasing abruptly above a critical value of oxygen chemical potential (see Fig. 3). One interesting finding is that while at low oxygen chemical potentials Ru segregates to the surface in the form of isolated atoms, at higher values the Ru atoms cluster together, resulting in islands of Pt surrounded by oxygen-covered Ru regions. This can be relevant for catalysis, since at the edges between Pt and Ru–O islands both Pt sites for the decomposition of methanol and oxygen for the oxidation reactions are available.
 | ||
| Fig. 3 Monte Carlo simulations and surface structure evolutions as a function of oxygen chemical potential μO, T and bulk Ru composition CRu. The conditions of (a) are T = 600 K, μRu = 1750 meV and (b) T = 1050 K, μRu = 665 meV. Reproduced with permission from ref. 35. | ||
One important consideration stressed by Han et al. in their work is the assumption of thermodynamic equilibrium as well as sufficient mobility of Ru through the bulk necessary to allow the segregation to the surface. Industrial catalysts, on the other hand, might operate far from equilibrium, since they are prepared through a series of low-medium temperature steps to retain their small particle size and avoid sintering. Moreover, diffusion of oxygen adsorbates below the surface and the formation of surface oxides have been neglected, which could be important, especially at high values of oxygen chemical potentials.
3.2. Single feed experiments
The simplest approach to understand the effect of the catalytic reaction feed (usually a mixture of two or more gases) on the surface structure and composition is to describe the interaction of an alloy with a single gas atmosphere that thought to dominate gas–solid interaction. Typically, gases can be classified as oxidant (NO, O2), reducing agent (CO, H2) or inert (He, N2), with UHV resembling reducing conditions. Below we will describe the relation between the composition, structure and chemical state at the surface of alloys upon their interaction with simple molecules like O2, H2, NO and CO. Thereafter, in Section 3.3, we go a step further and will consider the effect of mixtures of gases, thus alloys under catalytic turnover. The DFT calculations cited in following sections employ the generalized gradient approximation (GGA) for the exchange and correlation functional, using either the PBE,31 revPBE50 or RPBE51 parametrization. For more details please refer to the original papers.15 and at the same time demonstrates the significance of the so-called ‘pressure gap’ effect on surface studies. Combining DFT and atomic resolution STM under vacuum, Besenbacher et al. showed that in contrast to the predictions of the bulk phase diagram, Au and Ni can form a quite extended surface alloy, which is equally active, but less prone to deactivation compared to pure Ni.15 However, some years later, the same group using high-pressure STM showed that the AuNi surface alloy is stable only under UHV conditions or Langmuir CO exposures, while higher CO pressure induces dealloying (phase separation) and formation of monoatomic Au islands over nickel, even at room temperature.52 The rate of dealloying process is directly related to the CO pressure above the sample (Fig. 4). At CO pressure of 1000 mbar the effect was rapid, while at 7–53 mbar it was much slower and the authors were able to follow the dynamics of the de-alloying process by acquiring STM movies (sequential fast acquisition of STM images). DFT calculations revealed that formation of volatile Ni-carbonyl species at kink-sites was mainly responsible for the observed de-alloying. However the effect was reversible and annealing in UHV at 800 K restored back the initial AuNi surface alloy structure.
 | ||
| Fig. 4 (a) STM image (800 × 800 Å2) of Au/Ni(111) after annealing (alloy formation). Inset: atomic-resolution image (50 × 50 Å2) of the Au/Ni(111) surface alloy. The Au atoms are imaged as depressions. (b) STM image (1000 × 1000 Å2) of Au/Ni(111) after exposure to 1000 mbar of CO, in which case the surface is observed to be covered with islands. The line scan (indicated by the white arrow) shows islands of two different heights. Inset: atomic resolution of an area between the Au islands (60 × 60 Å2) reveals a clean Ni(111) surface. Reproduced with permission from ref. 52. | ||
Recently, Andersson et al.53,54 demonstrated in two successive publications that the expected (based on thermodynamic concepts) gas-induced surface segregation of the more reactive alloy component might be not always the case. The authors investigated the stability of CuPt near surface alloy (NSA) upon exposure to CO and O2 environments. The CuPt NSA was prepared by physical vapour deposition of one atomic layer of Cu on Pt(111) and annealing in UHV at 800 K. Combining PES, low energy electron diffraction (LEED) and ion scattering spectroscopy (ISS) techniques the authors showed that in vacuum Cu was preferentially located to the second layer leaving a monoatomic Pt ‘skin’ at the outermost surface (NSA). Upon Langmuir CO exposures at low temperatures the NSA alloy remained practically intact. In contrast, after treatments in 2 mbar CO at temperatures between 473 and 723 K, the initial surface configuration was modified in a rather unexpected way. Copper was segregated to the surface and formed a stable self-organized CuPt surface alloy, on which the CO bond strength was higher compared to the pure Pt(111) surface. Since CO bonding to pure Pt is stronger compared to pure Cu surfaces, segregation of Cu upon CO exposure contradicts at a first glance to the thermodynamic expectation for surface segregation of the more reactive surface component (in this case Pt). However, by using DFT calculations, the authors could rationalize their experimental findings showing that the Pt–CO bond energy considerably increases in the presence of Cu in the first surface layer. By exposing CO–PtCu to 200 mbar of O2, CO is removed and an ultra-thin CuOx film is built up on the surface. This process is reversible and the surface alloy is reformed upon re-exposure to CO.54
In contrast to CuPt, adsorbate-induced surface segregation of the PtFe NSA showed a rather conventional behaviour when tested under 1 × 10−6 mbar of O2 and H2.55 The PtFe NSA prepared in UHV over the Pt(111) crystal consists of a Fe subsurface layer under the Pt skin. Exposure to O2 at 850 K induced surface segregation and oxidation of Fe to FeO. As in the case of CuPt, this process is reversible and in H2 FeO is reduced back to Fe and dilute into the subsurface forming a NSA similar to UHV conditions. By DFT calculations it was confirmed that subsurface Fe was thermodynamically more stable in UHV and H2 environments, while in O2 the significantly stronger Fe–O bond, compared to Pt–O, was responsible for Fe segregation over Pt. Similarities of the PtFe surface termination between reducing gas atmospheres and acidic electrochemical environments can be derived. By using in situ electrochemical STM, Wan et al.56 observed the formation of a Pt skin layer with pronominally (111)-oriented facets on the surface of Pt–Fe alloys under potential cycling in 0.1 M HClO4 solution. In contrast to H2 gas exposure, where Pt surface segregation was due to Fe subsurface diffusion,55 in acidic electrochemical environments the formation of Pt-skin layer is caused by the dissolution of Fe into the acidic solution.56
Apart from single crystalline surface and near-surface alloys, polycrystalline bulk Pt–M (M = Sn,57 Ni58 and Co58,59) alloys have been studied upon exposure to O2 and H2 environments. In all the above cases surface segregation of Pt was observed in H2, while in O2 segregation and oxidation of the other alloy constituent were found. By using real-time APPES, Papaefthimiou et al.59 have shown that extended segregation on the PtCo bulk alloy takes place within seconds in response to the gas phase environment (Fig. 5). A thick (>5 nm) Co3O4 oxide layer is formed over Pt in 0.2 mbar O2 at 520 K. This effect was reversible, and in H2, reduction of Co3O4 followed by re-segregation of Pt and bimetallic PtCo surface was observed. Surprisingly for a bulk alloy, Pt did not have a notable effect on the reduction process of cobalt oxides. This was rationalized based on the observation that Pt was well diffused in the interior under oxidizing conditions and during reduction it reappeared on the surface only shortly after cobalt has reverted to the metallic state.
 | ||
| Fig. 5 Real-time experiment showing the evolution of Co 2p3/2 (top) and Pt 4f (bottom) spectra from a PtCo polycrystalline foil in response to the changes of the gas atmosphere in the experimental cell. The on-line mass spectrometry signal for O2 and H2 and the periods of photoemission spectra acquisition are shown in the central diagram. The total gas pressure and the temperature were kept constant at 0.2 mbar and 520 K, respectively. Reproduced with permission from ref. 59. | ||
Compared to Au and Pt-based alloys, segregation in alloys containing two or more transition metals is in general more difficult to categorize since their oxygen affinity is usually high and similar, while formation of mixed oxide phases might complicate their behaviour. Oxidation of Cu3Fe,60 Cu2.75Ni0.25Fe,60 Ni65Nb3561 and NiCo62 polycrystalline alloys has been studied by surface sensitive methods. As shown by in situ APPES and XANES studies, the surface of Cu3Fe alloys is not significantly influenced by exposure to 1 mbar O2 or H2 at 527 K. In contrast, formation of a ternary alloy by Ni addition significantly enhances Fe and Cu reduction in H2 and promotes segregation of metallic Cu. The presence of Ni also found to hinder re-oxidation of the alloy constituents upon O2 annealing.60 The surface composition of Ni65Nb35 polycrystalline alloy upon exposure to 1 bar of O2 is temperature dependent. In particular, up to 373 K, Nb segregates and forms Nb2O5, while above 523 K segregation of nickel as a mixture of metal and oxide is observed.61 The temperature dependence of the oxidation mechanism was attributed to the different kinetics in the diffusion of constituent atoms. It is interesting to note that the bulk structure of the alloy was not influenced in the course of the experiment. In a recent study of NiCo alloy oxidation, a clear effect of the pressure gap was observed.62 At low oxygen pressure (10−6 mbar), segregation and preferential oxidation of cobalt to CoO take place, while oxidation of nickel is largely suppressed. At ambient O2 pressure, alloy oxidation was temperature dependent (as in the previous case of NiNb). Up to 520 K, cobalt segregated to the surface and transformed to CoO, similar to the observations at low pressure O2 exposure. However, at higher temperatures, nickel re-segregates to the surface, at the expense of cobalt, while the remaining CoO is further oxidized to Co3O4. In this temperature range, formation of mixed Ni–Co–O spinel-like oxide was proposed based on experimental evidence.
Pd and Rh are typical elements of automobile exhaust catalysts and therefore studies of their redox behaviour might have major technological and economic impact. Bulk PdRh alloy has been studied by Grass et al. in various oxidative (O2, NO) and reducing (CO, H2) atmospheres using APPES.63 As in the cases of NiNb and NiCo, a notable effect of the annealing temperature to the surface state was observed. At temperatures below 473 K the surface oxidation state could be controlled by the redox potential of the gas phase but without significant surface segregation effects. In contrast, at high temperatures (623 K) modification of the oxidation state is followed by prominent surface reorganization. Segregation of Rh over Pd was favoured at oxidative environments, and vice versa in reducing environments. In this study a comparison between the behaviours of extended and nanosized alloys with the same nominal composition was attempted. Segregation and oxidation/reduction were found to proceed faster at lower temperatures for nanosized alloys compared to the extended bulk materials.
Since the maximum pressure at which the APPES technique can operate is limited to a few mbar, in a more recent work in situ XANES was used to monitor the oxidation state of 4 nm PtCo NPs deposited on a Au foil, at pressures up to 1 bar of O2 and H2.23 It was found that Pt catalyses the reduction of cobalt oxides in H2, while under O2 the effect of Pt on the cobalt oxidation state is pressure dependent. A stable tetrahedral Co2+ oxide was found coexisting with the octahedral Co2+ phase. In another study, the redox behaviour of 3 nm PtCo NPs prepared by physical methods (Low Energy Cluster Beam Deposition technique) and deposited on amorphous carbon was studied by a combination of APPES and XANES techniques.59 For comparison, a polycrystalline PtCo foil was measured under the same conditions in order to reveal size dependent effects. It was shown that Pt segregation took place in 0.2 mbar H2, while segregation and oxidation of cobalt were evident in 0.2 mbar O2. The presence of tetrahedral Co2+ species (being metastable as bulk phase) was confirmed by theoretical simulations of XANES peaks and it was also affirmed that its stability was related to the size, since it was not observed in the bulk samples under identical conditions. Comparison with reference monometallic cobalt samples indicated that the promotion effect of Pt to the reduction of the surface cobalt oxides is mainly pronounced in nanosized samples, while for the bulk polycrystalline foil, Pt does not markedly influence the reduction of Co oxides. However, in contrast to the findings on the PdRh63 system, nanosized PtCo was more difficult to reduce compared to the extended PtCo bulk alloy. The essential advantage of physical preparation methods used in ref. 59, compared to chemical synthesis typically employed in nano-alloys redox studies, is the capability to synthesize bare (uncoated) nano-objects well suited as model systems for fundamental studies. In contrast, using chemical synthesis methods the surface of the prepared NPs is affected by the interaction with the solution molecules, which in turn might also influence the redox behaviour.
In a much earlier investigation nanosized PtCo supported on SiO2 and prepared by spin coating method was studied by in situ XANES65 under 1–2 mbar H2. AFM images indicated relatively flat, around 2 nm thick, PtCo layers. It was suggested that the as-prepared fresh film consisted mainly of octahedral Co2+ species (however a comparison with recent results59 indicates that the Co L-edge resembles more tetrahedral Co2+), while formation of PtCo alloyed bimetallic particles was observed after reduction in H2. It is noteworthy that compared to the relatively inert Au and carbon supports employed in ref. 23 and 59, respectively, SiO2 might have a greater influence on the reduction of PtCo NPs. Apart from gas atmospheres, surface segregation of carbon supported PtCo NPs (sizes around 5 nm) was also investigated under electrochemical environments.66 An interesting comparison between samples treated in gas-phase and alkaline electrochemical environments was attempted by an in situ cycling voltammetry (CV) method. Both, ambient pressure CO exposure at 473 K, and potential cycling in the CO-saturated alkaline electrolyte, lead to the same Pt core–PtxCoy shell structure. It should be noted that cyclic voltammetry is extremely sensitive to the outermost surface layer, however the interpretation of the current–voltage plots is not straightforward and other analytical techniques should be employed complementary to CV in order to elucidate the surface structure.
Apart from PtCo, surface segregation in reducing and oxidizing environments of other Pt based supported bimetallics (Cu,67,68 Sn,69 Rh,70,71 Ni67 and Au72,73) has been studied. The surface structure of Al2O3 supported 2.5 nm PtCu nanoparticles was found sensitive to the type of reducing gas. By combing X-ray absorption (XANES), pair distribution function analysis (PDF) and Fourier Transformed infrared spectroscopy (FT-IR) in ambient H2 pressure (548 K), no core–shell structure formation was observed, but the NPs surface was enriched in Cu, while in the interior, alloy PtCu remained.68 An inverse observation was found in ambient pressure CO exposure (up to 623 K) where Pt segregation with subsequent migration of Cu in the core took place.68 This study could not confirm Cu segregation in CO atmospheres observed on single crystalline PtCu NSA.53 This indicates that support and size effects are of major importance to the surface segregation phenomena. Nanosized PtCu and PtNi were supported on Highly Ordered Pyrolytic Graphite (HOPG) and they were characterized by XPS after 1 bar of H2 or O2 treatment at 573 K in a reaction cell attached to the UHV set-up.67 Similarly to PtCo, Cu and Ni were oxidized and segregated to the surface in oxidative atmospheres, while in H2 the surface was enriched in Pt. The redox kinetics of Pt3Sn alloy NPs (mean size 15 nm) supported on Al2O3 was studied by in situ time-resolved energy dispersive XAFS (DXAFS) and quick XAFS (QXAFS).69 Oxidation at 673 K under atmospheric O2 pressure was found to proceed in 3 steps: (a) a fast step of Pt3Sn transformation to SnO2 shell–Pt core structure, (b) oxidation of the surface of Pt–core to PtO and (c) finally these transformations were followed by shape restructuring of the nanoparticles (Fig. 6). Reduction in H2 at the same temperature proceeds in one step and regenerates the initial Pt3Sn alloy.
 | ||
| Fig. 6 Proposed mechanism for the oxidation and reduction of Pt–Sn nanoparticles on γ-Al2O3. The kinetic parameters were determined by the time-resolved XAFS. Reproduced with permission from ref. 69. | ||
The effects of size and morphology of MgO supported PtRh to the oxidation process were studied by scanning photoelectron microscopy (SPEM) and low energy electron microscopy (LEEM).70 Independently from the particle size, a thick Rh-oxide layer with various Rh stoichiometric oxide phases was formed, whereas oxidation of Pt was limited to the top few atomic layers. It was also found that the degree of oxidation within a single microparticle is not homogeneous and correlates to the initial microstructure of these particles. The Pt and Rh oxidation states were systematically higher as the size of the PtRh aggregates decreased. Oxidation of PtRh alloys is not only influenced by the size but also by their composition. In particular, Pt0.9Rh0.1 and Pt0.95Rh0.05 supported on Al2O3 catalysts with mean particle size around 2 nm were annealed in 0.7% NO in N2 up to 473 K and their surface state was indirectly characterized by FT-IR using NO as a probe molecule. Both surfaces contained a mixture of oxidized Pt and Rh species, however the amount of surface Rh oxide was directly related to its nominal content.71
Tenney and coworkers investigated Pt–Au73 and Ni–Au72 nanoparticles supported on TiO2(110). They found that under Langmuir CO exposure, segregation of Ni and Pt on the surface occurred, whereas under UHV, Au is preferentially segregated over Pt and Ni. For PdAu bimetallic particles supported on TiO2 no evidence was found for any surface segregation upon annealing at 548 K in ambient pressure H2,68 however when the gas atmosphere was switched to CO, adsorbate induced surface segregation of Pd was observed. CO adsorption on PdAu bimetallic surfaces was recently addressed by Garcia-Mota and Lopez by DFT (RPBE functional) and they calculated also IR frequencies of adsorbed CO for a high number of possible configurations.74
IrNi core–shell nanoparticles (2.2 to 3.4 nm) supported on high area Vulcan carbon were found to form an Ir shell–IrNi core structure upon annealing at 873 K at ambient pressure H2.75 This catalyst was tested in an acidic electrochemical environment and found to be stable under elevated potentials (up to 1.11 V with respect to the reversible hydrogen electrode), since nickel atoms in the core were protected against oxidation and dissolution into the acidic media.
Formation of surface alloys under reactive conditions has been also observed for non-typical bimetallic systems, like oxide supported metal catalysts. Strong indications of a surface CuZn alloy was given by the group of Topsoe et al.76,77 during annealing of Cu/ZnO methanol synthesis catalysts. Exposure to strong reducing gas phases (CO and H2) at medium temperatures (500 K) induces reduction of ZnO and migration of Zn to the Cu surface where a surface CuZn alloy is formed. This procedure was found to be reversible and Cu/ZnO was re-established upon exposure in oxidative atmospheres.
3.3. Catalytic systems
In the following section, we are going to review selected systems of alloy catalysis in which segregation or phase separation has been reported.As we have discussed above, Rh0.5Pd0.5 bimetallic Pd-core Rh-shell nanoparticles undergo significant changes in the surface composition when exposed to CO, O2 and other gases.64 However, reducing gases, such as CO, interacted with Pd more strongly inducing its segregation to the surface, whereas oxygen (or oxidizing gases such as NO) reversed this trend. RhxPd1−x bimetallic catalysts exhibit the synergetic effect of increased reactivity compared to pure Pd or Rh in CO oxidation by O2 or NO.78,79 Stronger activity enhancement is observed when oxygen is in excess of CO. Segregation phenomena, again studied using APPES, have massively slowed down in CO oxidation due to the opposite effect of CO and O2. Nevertheless, it was shown that higher temperature and lower oxygen excess result in higher Pd surface concentration. Both Pd and Rh are mainly metallic, with a minor oxidized Rh component, but at a higher pO2/pCO ratio more RhOx is formed. Due to the preferential adsorption of CO on (isolated) Pd sites and the activation of O2 over Rh/RhOx, the synergetic effect of bimetallic PdRh is attributed to a bifunctional mechanism with emphasis on the Pd–RhOx interface where oxygen spillover from Rh to Pd sites assist the reaction.
AuPd alloy materials catalyze the synthesis of vinyl acetate, H2O2, selective oxidation of alcohols but also can diminish CO emission. In a series of publications, Goodman and co-workers have studied by PM-IRAS the reasons behind the enhanced reactivity of AuPd in CO oxidation.80–83 They find that well-annealed samples with low surface Pd content are essentially inactive at low pressures, whereas higher pressures facilitate the reaction. These latter conditions are linked to higher CO and O coverages enabling the segregation of Pd to the surface.84–86 Segregation of Pd was found to be O coverage dependent with more than 1/3 of the monolayer being necessary to induce segregation,86 whereas CO pressure in the range of 10−3 Torr was enough to induce Pd segregation and 10−1 Torr to form contiguous Pd sites.80,82,84 Once segregated to the surface, due to the weak interaction, Pd sites do not have the tendency to cluster,85 and thus there is need of higher pressure to make Pd pairs. By following the carbonyl stretching vibration in situ, Goodman deduced a link between the presence of contiguous Pd sites and the reactivity. Apparently, neither Au nor isolated Pd sites were capable of dissociating O2, and the reaction only occurred once adsorbate induced surface segregation reached a level of forming contiguous Pd sites. They also noted that CO can adsorb on both Au and Pd sites weaker than on monometallic Pd. These observations in conjunction with the higher reactivity and lower apparent activation energy in comparison to the monometallic Pd counterpart are attributed to a mechanism where CO inhibition does not occur and CO and O2 activation is separated on the alloy surface. It is interesting to note that AuPd was less prone to surface Pd oxidation and thus to deactivation. Somewhat differently from the results above, Alayoglu et al.87 reported that AuxPd1−x nanoparticles (8–11 nm; x = 0.25, 0.5, 0.75) synthesised at low temperature (500 K) with poly(vinyl-pyrrolidone) surfactant are enriched in Pd at the surface, but also show higher turnover rates than the parent monometallic nanoparticles. Alloy particles with 75% and 50% nominal Pd content have not shown any significant changes in the surface composition upon CO oxidation at 473 K. On the other hand, Au0.75Pd0.25 nanoparticles restructure irreversibly to Au-rich surfaces in the reaction. The authors explained this unexpected finding by the higher gain of energy (lower surface energy of Au) with Au segregation than the gain of reactant adsorption on Pd sites.
AuCu bimetallic nanoparticles supported on the mesoporous aluminosilicate SBA-15 have been reported to be highly active in CO oxidation below RT. Using multiple in situ techniques (XRD, FT-IR, XANES) Liu et al. investigated this catalyst under activation and reaction.88 They observed that the as-calcined catalyst was made up of a gold core decorated with CuO patches (Fig. 7). During the activation at 773 K, a part of CuO was reduced to Cu and diffused into the core forming an Au3Cu1 intermetallic phase, whereas the other part of CuO that was interacting with the support was only reduced to Cu+ acting as “nano-glue” between Au3Cu1 particles and the support. During CO oxidation Cu segregated from the intermetallic core to the surface forming tiny CuOx patches over Au, and so, producing an intimate contact with long perimeter between gold and CuOx. CO adsorbed on metallic gold sites was deduced to react with activated oxygen provided by the neighbouring oxidized copper sites.
 | ||
| Fig. 7 Schematic illustration of the structural changes of the Au–Cu/SBA-15 under H2 reduction and CO oxidation conditions. Reproduced with permission from ref. 88. | ||
Applying in situ Mössbauer and FT-IR spectroscopy, Margitfalvi et al. studied a SnPt/SiO2 catalyst (Sn/Pt = 0.68) active in low temperature CO oxidation.89 They found that activation in hydrogen (573 K) resulted in two bimetallic phases, while oxidized species were absent. During CO oxidation at RT, the alloy phases segregated into a PtSn (1:1) phase with additional two types of oxidized Sn4+ species. Pure CO produced no measurable bridged carbonyl band in the IR spectrum; however addition of oxygen induced the appearance of subtle but continuous growth of tiny bridged CO. It was concluded that the in situ formation of Sn4+–Pt ensemble was the key feature of this catalytic system.
In a first principle study, Su et al. investigated the structural changes of Pt-3d transition metal (Cr, Mn, Fe, Co, Ni and Cu) alloys under reductive and oxidizing conditions as well as in CO oxidation.90 Under reductive conditions (vacuum or CO) the 3d transition metal was shown to prefer subsurface sites and avoid surface sites because of its higher surface energy and smaller atomic size. On the other hand, in oxidizing conditions employing 0.25 ML O coverage, the strong interaction between the surface oxygen and the 3d metal (with the exception of Cu) induces the migration of 3d metal to the surface, which may even form oxidized 3d transition metal islands. This indicates, but also additional calculations have confirmed, that exposed 3d sites are highly facile for molecular oxygen dissociation. Therefore, enhanced CO oxidation activity can be achieved on Pt-3d transition metal ensembles, where CO adsorbs on Pt sites and O2 activation occurs on dispersed and partially oxidized 3d atoms. Overall reactivity is therefore very high, as CO poisoning, relevant on sole Pt surfaces, does not affect this Pt-3d system. In this respect it is interesting to mention that eliminating atomic oxygen from AuNi can also be a difficult step.91
 | ||
| Fig. 8 (Top) Evolution of Rh (Rh0 + Rh2y+) and Pd (Pd0 + Pd2y+) atomic fractions in the Rh0.5Pd0.5 NPs at 300 °C under oxidizing conditions (100 mTorr NO or O2) and catalytic conditions (100 mTorr NO and 100 mTorr CO) denoted in the x-axis. (Bottom) Evolution of the fraction of the oxidized Rh (left y-axis) and Pd atoms (right y-axis) in the examined region under the same reaction conditions as the top part of the figure. All atomic fractions in this figure were obtained with an X-ray energy of 645 eV for Rh3d and Pd3d, which generates photoelectrons with a mean free path of ∼0.7 nm. Schematic diagrams above the top of the figure show the reversible segregation of Rh and Pd under alternating oxidizing and catalytic conditions. The y-axis data points for reactions (1), (3), and (5) have an associated error of ±0.03; for reactions (2) and (4), the error bar is ±0.02. Reproduced with permission from ref. 64. | ||
Andersson et al.53,54 after establishing that adsorbate CO may pull the least reactive alloying component Cu to the surface whereas Pt, the other alloy component, in this way forms a much stronger bond with CO, has investigated the stability of this surface structure in various gas mixtures (CO + H2O or CO + H2) using PM-IRAS. No indication of substantial changes in surface structure was found under these conditions (up to 673 K) compared to CO alone in the gas phase. On the other hand, O2 at 200 mbar pressure and RT was capable to remove the carbonyl band and build up an ultra-thin CuOx film.
Anderson et al.92 investigated zeolite supported PdCu bimetallic catalysts and by applying XANES in the hard X-ray regime and FT-IR experiments under CO hydrogenation at reaction temperature examined how the surface and bulk phase were influenced by the reaction condition. While XANES indicated that only one type of substitutionally disordered PdCu alloy phase was formed and stayed unmodified under longer periods at reaction conditions, FT-IR suggested that adsorbate induced surface segregation of Pd occurred. Despite Pd segregation, a progressive loss of Pd threefold adsorption centers was observed as a function of time on CO hydrogenation stream.
:1 near-surface alloy exhibits high CO2 selectivity, whereas a monolayer PdZn 1:1 surface alloy, despite its identical surface composition, produced only CO and H2.
Pt-based binary and ternary systems may eliminate CO poisoning of fuel cell anode electro-catalysts. Zafeiratos et al.97 studied the methanol steam reforming reaction (MSR) in conjunction with the water gas shift (WGS) reaction over a ternary PtRuCo alloy catalyst using APPES, with the aim to identify the nature of the working catalyst surface and thus providing a basis for the role of promoters. The surface of PtRuCo catalyst undergoes significant modifications in its composition and chemical state in response to the reaction mixture. Under MSR reaction conditions Pt, Ru and Co were metallic with traces of ionic Cox+ also observed. Significant amounts of adsorbed carbon and oxygen species were attached to the surface and indications of sub-surface (dissolved) oxygen were also found. Rich in water WGS mixtures favour surface oxidation reflecting the higher oxygen chemical potential compared to the MSR mixture and induced surface reorganization. In particular, cobalt segregates to the surface and is, to a large extent, oxidized during WGS. Interestingly, traces of ionic Pt species were also observed, while Ru was mainly metallic. Compared to MSR reaction less carbon and more surface oxygen species were observed. A bifunctional active site complex with possible electronic contribution was put forward to explain the function of the material. Additional experiments with a polycrystalline Pt0.5Co0.5 foil sample under MSR indicated the formation of Pt-skin, cobalt-rich subsurface and Pt-rich core structure with carbon and oxygen based adsorbates and dissolved oxygen.
Pd surface segregation reportedly occurs at the high hydrogen pressure side of a PdAg membrane.98 Using density functional calculations, Gonzalez et al. studied the surface structure of a Pd0.8Ag0.2 alloy and its modification in the presence of atomic hydrogen.99 At equilibrium and in complete absence of adsorbates, the surface is predicted to expose mostly silver atoms, as discussed above. Increasing the coverage of adsorbed H atoms from 1/9 to 4/9 ML gradually decreases the energy gain of lower Ag surface energy, suppresses the surface segregation of Ag, and migration of all silver surface atoms into the subsurface region becomes favourable at a H coverage of ∼0.25 ML. For this Pd-skin structure with predominantly Pd atoms in the surface layer and Ag below, the propensity of H to accommodate interstitial sites below the surface vanishes. In this regard, it is interesting to mention that Teschner et al.100,101 has shown that during selective alkyne hydrogenation over monometallic Pd catalysts the subsurface interstitial sites are carbon populated, carbon being absent under nonselective reaction. Later, using DFT calculations, it has been established that subsurface carbon decreases the binding energy of adsorbed H, forms an exclusion zone for subsurface H and increases the barrier for bulk dissolved hydrogen diffusing to the surface; all these contributing to a low surface H concentration essential to avoid over-hydrogenation.102 Selectivity is also often enhanced with addition of CO to the feed, which was shown by adsorbing strongly on Pd to diminish subsurface and bulk H population and enable only low surface hydrogen concentration.103
The Pd pseudomorphic monolayer on silver has been shown recently to decrease the formation of both C and H in the subsurface.104 A somewhat different role of silver was suggested by Jin et al.105 proposing the dilution of surface Pd sites by Ag being important for improved acetylene hydrogenation selectivity. Similar improved selectivity in 1,3-butadiene hydrogenation was observed over palladium catalysts alloyed with gold.106,107 Pd0.7Au0.30 single crystal surface after vacuum annealing is strongly enriched by gold.106 Di Vece et al.108 observed changes in the size distribution and composition of bimetallic PdAu nanoparticles that have been exposed to hydrogen at RT. The effect was identified to be due to Ostwald ripening of Pd after Pd has segregated to the surface. An increased reactivity with time on stream in butadiene selective hydrogenation was attributed to adsorbate induced partial Pd segregation to the surface.106 Interestingly, Hugon et al.107 observed no comparable change in the activity of a PdAu catalyst (Au/Pd = 10), despite detecting Pd segregation in separate CO adsorption FR-IR experiments.
Very recently, Lopez and Vargas-Fuentes reviewed the role of metallic promoters in Pd-based alkyne hydrogenation and presented new insights from their DFT calculations regarding stability and segregation, adsorption properties, and the ability to form hydride and carbide phases.109
In hydrogenation chemistry, up to now we have considered mainly the effect of hydrogen on the segregation properties. Applying medium-energy ion scattering (MEIS), the effect of organic component on segregation has been explored.110–112 Noakes et al.110 used a Pd0.5Cu0.5(110) single crystal, which was slightly Pd-rich under the initial preparation condition. Adsorbing ethylene at 468 K gave rise to substantial Pd segregation. This indicates that both hydrocarbon and hydrogen would induce Pd segregation in Pd catalysts when alloyed with more noble elements like Cu, Ag or Au. On the other hand, if the organic molecule contains more electronegative functional groups, the situation may be reversed, as suggested by the adsorption of trichloroethylene over the same Pd0.5Cu0.5(110) single crystal. In this case, Cu enrichment in the first surface layer was shown by MEIS. Baddeley and co-workers using MEIS investigated the adsorption of (S)-glutamic acid111 and methylacetoacetate112 on bimetallic NiAu surfaces and found in both cases Ni segregation. From the enantioselective methylacetoacetate hydrogenation point of view, Ni segregation around the adsorption site is likely not beneficial. Conversely, if a chiral modifier, such as the (S)-glutamic acid, induces Ni enrichment, this can produce more active reaction sites and hence the reactivity can improve. Platinum surfaces, alloyed with electropositive 3d metals, are reported to increase selectivity in the hydrogenation of α,β-unsaturated aldehydes towards the desired unsaturated alcohols. Hirschl et al.113 using spin-polarized DFT calculations studied the adsorption modes of acrolein and prenal on Pt0.8Fe0.2(111) and their effect on surface segregation. As should be evident from our discussion up to now, the clean alloy surface is characterized by a strong Pt surface segregation. Nevertheless, the considerably higher aldehyde adsorption energy upon formation of O–Fe bonds can modify the segregation profile and increase the Fe content of the surface layer. As concluded, this has a clear effect on the prevailing adsorption mode and energy of the aldehyde reactant, and thus the reaction selectivity is influenced in the desired direction.
Studt et al.114 performed theoretical screening on a broad space of alloys, and with the help of scaling relations discovered new selective acetylene hydrogenation catalysts based on the nickel–zinc alloy. Recently, Bridier et al.115 identified a novel family of ternary Cu–Ni–Fe catalysts for the partial hydrogenation of propyne with selectivity near 100%. The addition of Ni was the key for the achieved selectivity. In the follow-up work,60 the surface composition and electronic state of Cu2.75Ni0.25Fe and Cu3Fe were investigated using in situ APPES and XANES to gain insight into the state of the surface and the role of Ni promotion (Fig. 9). The bulk of the calcined samples consisted of CuO as the main phase and Cux(Ni1−x)Fe2O4 spinel as the secondary phase, whereas no spinel was identified on the surface, and, instead, Fe2O3 was observed. Once the materials were reduced, XRD identified a metallic alloy phase with some Fe3O4; nevertheless the surface was still somewhat oxidized containing various oxidized states. Under propyne hydrogenation, the Cu3Fe surface composed of a Fe-lean alloy phase with additional Fe3O4/Fe2O3 oxidized surface phases. The surface contained 30% iron but mainly (∼90%) in the oxidized form, the alloy was thus very rich in Cu. The surface of reduced Cu2.75Ni0.25Fe was enriched in Cu (∼91%), but under propyne hydrogenation both Fe and Ni segregated back to the surface giving rise to near nominal surface concentration. As opposed to the Fe-lean Cu3Fe alloy surface, most (∼70%) of iron in Cu2.75Ni0.25Fe was in its metallic alloy form under hydrogenation. The in situ Ni L3-edge absorption spectra indicated a strong Ni–carbon interaction. It was concluded that Ni not only enhances the surface H coverage, as expected from the low selectivity towards oligomerization, but drastically changes the surface composition. Ni enhances Cu and Fe reduction, thus giving rise to well-diluted Cu surface sites, and hinders re-oxidation that would favour Fe segregation.
 | ||
| Fig. 9 Schematic view of the surface of Cu2.75Ni0.25Fe under propyne hydrogenation based on in situ spectroscopic (XPS/XAS) analyses. Note the depicted surface orientation has no significance here. Reproduced with permission from ref. 60. | ||
4. Conclusions and outlook
Surface segregation under reactive environment is usually underestimated in many of the catalytic studies. Typically, when in situ techniques are used, they are often not sensitive enough to the surface, or the surface sensitive methods are only applied ex situ (usually postmortem). However, our review illustrates that these effects readily occur, and, as catalysis is a surface event, they are obviously critical. Due to the complexity of the systems (composition of alloys, secondary effects like particle size/support, and external control parameters), segregation and the surface structure are often difficult to predict by general phenomenological models. The pressure and material gap can be very important as shown in many examples, and studies under elevated pressure are essential to establish the surface composition under operating conditions and correlate the surface state to the catalytic performance.One of the most promising surface sensitive in situ tools, APPES, is still confined to few mbar pressures and hence not capable to get utilization at real atmospheric pressure, or above. Clearly, further developments (X-ray source with higher brilliance, more effective electron collection and detection) are required in this respect. New developments and new techniques are essential to go beyond our current understanding, especially related to spatial and temporal resolution during segregation events. Extending the intrinsically bulk sensitive EXAFS towards the surface by implementing modulation excitation spectroscopy and phase sensitive detection116 may open up new possibilities in studying the structural dynamics of alloy surfaces.
State-of-the-art theoretical treatments, as we have illustrated here, are at least in principle capable to describe the complex interplay of parameters influencing segregation and to predict surface structures under realistic conditions. Once the working state of an alloy surface is established, a standard DFT treatment can be utilized to calculate reaction energy profiles and to perform a microkinetic analysis, thus providing a full picture of a catalytic cycle promoted by a particular alloy. In those systems where the catalytic performance (activity and selectivity) of a material can be rationalized in terms of a universal descriptor (e.g. binding energy of one of the intermediates), theoretical methods can be used to quickly screen a large number of alloys and to computationally design new catalysts with improved performance.117 Clearly, this type of computational screening needs to include the evaluation of stability against segregation and phase separation phenomena.
One needs to keep in mind, though, that catalysis is a non-equilibrium process and the structure of the catalyst surface obtained under the assumption of thermodynamic equilibrium may differ from the one active under reaction conditions. Accounting for these effects requires a full dynamical model of the process, and hence describing the interplay between all the elementary processes (adsorption, desorption, associations, dissociations, etc.) involved in the catalytic cycle. Recent advances have brought us closer to this goal through the use of the kinetic Monte Carlo method to simulate the steady state of the catalyst during the reaction, coupled to a first principle description of each elementary step to derive the corresponding rate.118 This approach has been applied to the oxidation of CO on RuO2(111) and Pd(100),119,120 showing that indeed the surface of the catalyst continuously evolves during the reaction cycle, and at the steady state differences from the thermodynamically stable configuration are appreciable.
Alloys have the great advantage to offer access to multifunctional active sites needed for complex reactions and for reactions with selectivity challenges. Moreover, alloys often find application in electrochemical systems, in which, however, surface segregation might be additionally fuelled by selective dissolution of constituents into the liquid phase. Due to the increased difficulty in studying solid–liquid interfaces in situ, these systems are somewhat less understood. Nevertheless, the important challenges we are facing in e.g. energy conversion, sustainable production, etc. are essentially connected to engineering solid–gas and solid–liquid interfaces to the desired needs. Due to the unlimited possibilities offered by multi-component systems, the rational design of alloys when theory and in situ experimentation works hand-in-hand can offer great opportunities.
Acknowledgements
S.Z. gratefully acknowledges financial support from the FP7 Fuel Cells and Hydrogen Joint Undertaking program, under ROBANODE, IRAFC and DEMMEA projects.Notes and references
- M. Ni, D. Y. C. Leung and M. K. H. Leung, Int. J. Hydrogen Energy, 2007, 32, 3238–3247 CrossRef CAS.
- F. Garin, Catal. Today, 2004, 89, 255–268 CrossRef CAS.
- D. I. Enache, J. K. Edwards, P. Landon, B. Solsona-Espriu, A. F. Carley, A. A. Herzing, M. Watanabe, C. J. Kiely, D. W. Knight and G. J. Hutchings, Science, 2006, 311, 362–365 CrossRef CAS.
- J. Greeley, T. F. Jaramillo, J. Bonde, I. Chorkendorff and J. K. Norskov, Nat. Mater., 2006, 5, 909–913 CrossRef CAS.
- J. Greeley and M. Mavrikakis, Nat. Mater., 2004, 3, 810–815 CrossRef CAS.
- J. H. Sinfelt, Bimetallic Catalysts: Discoveries, Concepts and Application, Wiley, New York, 1983 Search PubMed.
- J. A. Rodriguez, Surf. Sci. Rep., 1996, 24, 225–287 CrossRef.
- N. Toshima and T. Yonezawa, New J. Chem., 1998, 22, 1179–1201 RSC.
- J. G. Chen, C. A. Menning and M. B. Zellner, Surf. Sci. Rep., 2008, 63, 201–254 CrossRef CAS.
- J. R. Kitchin, J. K. Norskov, M. A. Barteau and J. G. Chen, Phys. Rev. Lett., 2004, 93, 4 CrossRef.
- O. S. Alexeev and B. C. Gates, Ind. Eng. Chem. Res., 2003, 42, 1571–1587 CrossRef CAS.
- R. Ferrando, J. Jellinek and R. L. Johnston, Chem. Rev., 2008, 108, 845–910 CrossRef CAS.
- P. Liu and J. K. Norskov, Phys. Chem. Chem. Phys., 2001, 3, 3814–3818 RSC.
- V. Ponec, Appl. Catal., A, 2001, 222, 31–45 CrossRef CAS.
- F. Besenbacher, I. Chorkendorff, B. S. Clausen, B. Hammer, A. M. Molenbroek, J. K. Norskov and I. Stensgaard, Science, 1998, 279, 1913–1915 CrossRef CAS.
- V. Blum, L. Hammer, C. Schmidt, W. Meier, O. Wieckhorst, S. Müller and K. Heinz, Phys. Rev. Lett., 2002, 89, 266102 CrossRef CAS.
- J. Nørskov, M. Scheffler and H. Toulhoat, MRS Bull., 2006, 31, 669–674 CrossRef.
- C. Stampfl, A. Soon, S. Piccinin, H. Shi and H. Zhang, J. Phys.: Condens. Matter, 2008, 20, 184021 CrossRef.
- C. Stampfl, M. Veronica Ganduglia-Pirovano, K. Reuter and M. Scheffler, Surf. Sci., 2002, 500, 368–394 CrossRef CAS.
- F. Tao and M. Salmeron, Science, 331, 171–174 CrossRef CAS.
- M. Salmeron and R. Schlogl, Surf. Sci. Rep., 2008, 63, 169–199 CrossRef CAS.
- A. Knop-Gericke, E. Kleimenov, M. Haevecker, R. Blume, D. Teschner, S. Zafeiratos, R. Schloegl, V. I. Bukhtiyarov, V. V. Kaichev, I. P. Prosvirin, A. I. Nizovskii, H. Bluhm, A. Barinov, P. Dudin and M. Kiskinova, Advances in Catalysis, 2009, vol. 52, pp. 213–272 Search PubMed.
- F. Zheng, S. Alayoglu, J. H. Guo, V. Pushkarev, Y. M. Li, P. A. Glans, J. L. Chen and G. Somorjai, Nano Lett., 2011, 11, 847–853 CrossRef CAS.
- A. Christensen, A. V. Ruban, P. Stoltze, K. W. Jacobsen, H. L. Skriver, J. K. Norskov and F. Besenbacher, Phys. Rev. B: Condens. Matter, 1997, 56, 5822–5834 CrossRef CAS.
- A. V. Ruban, H. L. Skriver and J. K. Norskov, Phys. Rev. B: Condens. Matter, 1999, 59, 15990–16000 CrossRef.
- Q. Sun, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter, 2003, 67, 205424 CrossRef.
- N. L. Nguyen, S. Piccinin and S. de Gironcoli, J. Phys. Chem. C, 2011, 115, 10073–10079 CAS.
- D. R. Stull and H. Prophet, JANAF Thermochemical Tables, U.S. National Bureau of Standards, Washington, DC, 2nd edn, 1971 Search PubMed.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter, 2001, 65, 035406 CrossRef.
- K.-P. Bohnen, R. Heid and O. de la Pena Seaman, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 081405 CrossRef.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS.
- A. Zunger, in Statics and Dynamics of Alloy Phase Transformations, ed. P. E. A. Turchi and A. Gonis, Springer, New York, 1994, pp. 361–419 Search PubMed.
- S. Piccinin and C. Stampfl, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 8 CrossRef.
- Y. Zhang, V. Blum and K. Reuter, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 235406 CrossRef.
- B. C. Han, A. Van der Ven, G. Ceder and B. J. Hwang, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 205409 CrossRef.
- J. T. Jankowiak and M. A. Barteau, J. Catal., 2005, 236, 366–378 CrossRef CAS.
- S. Linic, J. Jankowiak and M. A. Barteau, J. Catal., 2004, 224, 489–493 CrossRef CAS.
- F. Bocquet, C. Maurel, J.-M. Roussel, M. Abel, M. Koudia and L. Porte, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 075405 CrossRef.
- C. Maurel, M. Abel, M. Koudia, F. Bocquet and L. Porte, Surf. Sci., 2005, 596, 45–52 CrossRef CAS.
- S. Piccinin, C. Stampfl and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075426 CrossRef.
- S. Piccinin, C. Stampfl and M. Scheffler, Surf. Sci., 2009, 603, 1467–1475 CrossRef CAS.
- S. Piccinin, S. Zafeiratos, C. Stampfl, T. W. Hansen, M. Haevecker, D. Teschner, V. I. Bukhtiyarov, F. Girgsdies, A. Knop-Gericke, R. Schloegl and M. Scheffler, Phys. Rev. Lett., 2010, 104, 035503 CrossRef.
- S. Piccinin, N. L. Nguyen, C. Stampfl and M. Scheffler, J. Mater. Chem., 2010, 20, 10521–10527 RSC.
- S. Linic and M. A. Barteau, J. Am. Chem. Soc., 2003, 125, 4034–4035 CrossRef CAS.
- M. Mavrikakis, D. J. Doren and M. A. Barteau, J. Phys. Chem. B, 1998, 102, 394–399 CrossRef CAS.
- M.-L. Bocquet, A. Michaelides, D. Loffreda, P. Sautet, A. Alavi and D. A. King, J. Am. Chem. Soc., 2003, 125, 5620–5621 CrossRef CAS.
- M.-L. Bocquet and D. Loffreda, J. Am. Chem. Soc., 2005, 127, 17207–17215 CrossRef CAS.
- D. Torres, N. Lopez and F. Illas, J. Catal., 2006, 243, 404–409 CrossRef CAS.
- J. R. Kitchin, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 075437 CrossRef.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890–890 CrossRef CAS.
- B. Hammer, L. B. Hansen and J. K. Nørskov, Phys. Rev. B: Condens. Matter, 1999, 59, 7413–7421 CrossRef.
- E. K. Vestergaard, R. T. Vang, J. Knudsen, T. M. Pedersen, T. An, E. Laegsgaard, I. Stensgaard, B. Hammer and F. Besenbacher, Phys. Rev. Lett., 2005, 95, 126101 CrossRef.
- K. J. Andersson, F. Calle-Vallejo, J. Rossmeisl and L. Chorkendorff, J. Am. Chem. Soc., 2009, 131, 2404–2407 CrossRef CAS.
- K. J. Andersson and I. Chorkendorff, Surf. Sci., 2010, 604, 1733–1736 CrossRef CAS.
- T. Ma, Q. Fu, H. Y. Su, H. Y. Liu, Y. Cui, Z. Wang, R. T. Mu, W. X. Li and X. H. Bao, ChemPhysChem, 2009, 10, 1013–1016 CrossRef CAS.
- L. J. Wan, T. Moriyama, M. Ito, H. Uchida and M. Watanabe, Chem. Commun., 2002, 58–59 RSC.
- Z. Paal, A. Wootsch, D. Teschner, K. Lazar, I. E. Sajo, N. Gyorffy, G. Weinberg, A. Knop-Gericke and R. Schloegl, Appl. Catal., A, 2011, 391, 377–385 CrossRef CAS.
- C. A. Menning and J. G. Chen, J. Power Sources, 2010, 195, 3140–3144 CrossRef CAS.
- V. Papaefthimiou, T. Dintzer, V. Dupuis, A. Tamion, F. Tournus, D. Teschner, M. Haevecker, A. Knop-Gericke, R. Schloegl and S. Zafeiratos, J. Phys. Chem. Lett., 2011, 2, 900–904 CrossRef CAS.
- B. Bridier, J. Perez-Ramirez, A. Knop-Gericke, R. Schloegl and D. Teschner, Chem. Sci., 2011, 2, 1379–1383 RSC.
- Z. Song, D. L. Tan, F. He and X. H. Bao, Appl. Surf. Sci., 1999, 137, 142–149 CrossRef CAS.
- Y. T. Law, T. Dintzer and S. Zafeiratos, Appl. Surf. Sci., 2011, 258, 1480–1487 CrossRef CAS.
- M. E. Grass, M. Park, F. Aksoy, Y. W. Zhang, M. Kunz, Z. Liu and B. S. Mon, Langmuir, 2010, 26, 16362–16367 CrossRef CAS.
- F. Tao, M. E. Grass, Y. W. Zhang, D. R. Butcher, J. R. Renzas, Z. Liu, J. Y. Chung, B. S. Mun, M. Salmeron and G. A. Somorjai, Science, 2008, 322, 932–934 CrossRef CAS.
- A. Borgna, B. G. Anderson, A. M. Saib, H. Bluhm, M. Havecker, A. Knop-Gericke, A. E. T. Kuiper, Y. Tamminga and J. W. Niemantsverdriet, J. Phys. Chem. B, 2004, 108, 17905–17914 CrossRef CAS.
- K. J. J. Mayrhofer, V. Juhart, K. Hartl, M. Hanzlik and M. Arenz, Angew. Chem., Int. Ed., 2009, 48, 3529–3531 CrossRef CAS.
- R. T. Mu, Q. Fu, H. Y. Liu, D. L. Tan, R. S. Zhai and X. H. Bao, Appl. Surf. Sci., 2009, 255, 7296–7301 CrossRef CAS.
- S. M. Oxford, P. L. Lee, P. J. Chupas, K. W. Chapman, M. C. Kung and H. H. Kung, J. Phys. Chem. C, 2010, 114, 17085–17091 CAS.
- Y. Uemura, Y. Inada, K. K. Bando, T. Sasaki, N. Kamiuchi, K. Eguchi, A. Yagishita, M. Nomura, M. Tada and Y. Iwasawa, J. Phys. Chem. C, 2011, 115, 5823–5833 CAS.
- M. Dalmiglio, M. Amati, L. Gregoratti, T. O. Mentes, M. A. Nino, L. Felisari and M. Kiskinova, J. Phys. Chem. C, 2010, 114, 16885–16891 CAS.
- P. S. Dimick, J. L. Kross, E. G. Roberts, R. G. Herman, H. G. Stenger and C. E. Lyman, Appl. Catal., B, 2009, 89, 1–11 CrossRef CAS.
- S. A. Tenney, W. He, C. C. Roberts, J. S. Ratliff, S. I. Shah, G. S. Shafai, V. Turkowski, T. S. Rahman and D. A. Chen, J. Phys. Chem. C, 2011, 115, 11112–11123 CAS.
- S. A. Tenney, J. S. Ratliff, C. C. Roberts, W. He, S. C. Ammal, A. Heyden and D. A. Chen, J. Phys. Chem. C, 2010, 114, 21652–21663 CAS.
- M. Garcia-Mota and N. Lopez, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 9 Search PubMed.
- K. Sasaki, K. A. Kuttiyiel, L. Barrio, D. Su, A. I. Frenkel, N. Marinkovic, D. Mahajan and R. R. Adzic, J. Phys. Chem. C, 2011, 115, 9894–9902 CAS.
- N. Y. Topsoe and H. Topsoe, J. Mol. Catal. A: Chem., 1999, 141, 95–105 CrossRef CAS.
- J. D. Grunwaldt, A. M. Molenbroek, N. Y. Topsoe, H. Topsoe and B. S. Clausen, J. Catal., 2000, 194, 452–460 CrossRef CAS.
- J. R. Renzas, W. Y. Huang, Y. W. Zhang, M. E. Grass, D. T. Hoang, S. Alayoglu, D. R. Butcher, F. Tao, Z. Liu and G. A. Somorjai, Phys. Chem. Chem. Phys., 2011, 13, 2556–2562 RSC.
- J. R. Renzas, W. Y. Huang, Y. W. Zhang, M. E. Grass and G. A. Somorjai, Catal. Lett., 2011, 141, 235–241 CrossRef CAS.
- F. Gao, Y. L. Wang and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 14993–15000 CAS.
- F. Gao, Y. L. Wang and D. W. Goodman, J. Catal., 2009, 268, 115–121 CrossRef CAS.
- F. Gao, Y. L. Wang and D. W. Goodman, J. Am. Chem. Soc., 2009, 131, 5734–5735 CrossRef CAS.
- F. Gao, Y. L. Wang and D. W. Goodman, J. Phys. Chem. C, 2010, 114, 4036–4043 CAS.
- M. Garcia-Mota and N. Lopez, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 075411 CrossRef.
- V. Soto-Verdugo and H. Metiu, Surf. Sci., 2007, 601, 5332–5339 CrossRef CAS.
- H. Guesmi, C. Louis and L. Delannoy, Chem. Phys. Lett., 2011, 503, 97–100 CrossRef CAS.
- S. Alayoglu, F. Tao, V. Altoe, C. Specht, Z. W. Zhu, F. Aksoy, D. R. Butcher, R. J. Renzas, Z. Liu and G. A. Somorjai, Catal. Lett., 2011, 141, 633–640 CrossRef CAS.
- X. Y. Liu, A. Q. Wang, L. Li, T. Zhang, C. Y. Mou and J. F. Lee, J. Catal., 2011, 278, 288–296 CrossRef CAS.
- J. L. Margitfalvi, I. Borbath, K. Lazar, E. Tfirst, A. Szegedi, M. Hegedus and S. Gobolos, J. Catal., 2001, 203, 94–103 CrossRef CAS.
- H. Y. Su, X. K. Gu, X. F. Ma, Y. H. Zhao, X. H. Bao and W. X. Li, Catal. Today, 2011, 165, 89–95 CrossRef CAS.
- M. Garcia-Mota and N. Lopez, Phys. Chem. Chem. Phys., 2011, 13, 5790–5797 RSC.
- J. A. Anderson, M. Fernandez-Garcia and G. L. Haller, J. Catal., 1996, 164, 477–483 CrossRef CAS.
- N. Iwasa, S. Masuda, N. Ogawa and N. Takezawa, Appl. Catal., A, 1995, 125, 145–157 CrossRef CAS.
- K. Fottinger, J. A. van Bokhoven, M. Nachtegaal and G. Rupprechter, J. Phys. Chem. Lett., 2011, 2, 428–433 CrossRef CAS.
- C. Rameshan, W. Stadlmayr, C. Weilach, S. Penner, H. Lorenz, M. Hävecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlögl, N. Memmel, D. Zemlyanov, G. Rupprechter and B. Klötzer, Angew. Chem., Int. Ed., 2010, 49, 3224–3227 CrossRef CAS.
- C. Rameshan, C. Weilach, W. Stadlmayr, S. Penner, H. Lorenz, M. Havecker, R. Blume, T. Rocha, D. Teschner, A. Knop-Gericke, R. Schlogl, D. Zemlyanov, N. Memmel, G. Rupprechter and B. Klotzer, J. Catal., 2010, 276, 101–113 CrossRef CAS.
- S. Zafeiratos, F. Paloukis, G. Papakonstantinou, D. Teschner, M. Havecker, E. Vass, P. Schnorch, A. Knop-Gericke, R. Schlogl, B. Moreno, E. Chinarro, J. R. Jurado and S. G. Neophytides, Catal. Today, 2010, 157, 250–256 CrossRef CAS.
- J. Shu, B. E. W. Bongondo, B. P. A. Grandjean, A. Adnot and S. Kaliaguine, Surf. Sci., 1993, 291, 129–138 CrossRef CAS.
- S. Gonzalez, K. M. Neyman, S. Shaikhutdinov, H. J. Freund and F. Illas, J. Phys. Chem. C, 2007, 111, 6852–6856 CAS.
- D. Teschner, J. Borsodi, A. Wootsch, Z. Revay, M. Haevecker, A. Knop-Gericke, S. D. Jackson and R. Schloegl, Science, 2008, 320, 86–89 CrossRef CAS.
- D. Teschner, Z. Revay, J. Borsodi, M. Haevecker, A. Knop-Gericke, R. Schloegl, D. Milroy, S. D. Jackson, D. Torres and P. Sautet, Angew. Chem., Int. Ed., 2008, 47, 9274–9278 CrossRef CAS.
- D. Teschner, J. Borsodi, Z. Kis, L. Szentmiklosi, Z. Revay, A. Knop-Gericke, R. Schloegl, D. Torres and P. Sautet, J. Phys. Chem. C, 2010, 114, 2293–2299 CAS.
- M. Garcia-Mota, B. Bridier, J. Perez-Ramirez and N. Lopez, J. Catal., 2010, 273, 92–102 CrossRef CAS.
- P. Tiruppathi, J. J. Low, A. S. Y. Chan, S. R. Bare and R. J. Meyer, Catal. Today, 2011, 165, 106–111 CrossRef CAS.
- Y. M. Jin, A. K. Datye, E. Rightor, R. Gulotty, W. Waterman, M. Smith, M. Holbrook, J. Maj and J. Blackson, J. Catal., 2001, 203, 292–306 CrossRef CAS.
- L. Piccolo, A. Piednoir and J. C. Bertolini, Surf. Sci., 2005, 592, 169–181 CrossRef CAS.
- A. Hugon, L. Delannoy, J. M. Krafft and C. Louis, J. Phys. Chem. C, 2010, 114, 10823–10835 CAS.
- M. Di Vece, S. Bals, J. Verbeeck, P. Lievens and G. Van Tendeloo, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 80, 125420 CrossRef.
- N. Lopez and C. Vargas-Fuentes, Chem. Commun., 2012, 48, 1379–1391 RSC.
- T. C. Q. Noakes, P. Bailey, S. Laroze, L. H. Bloxham, R. Raval and C. J. Baddeley, Surf. Interface Anal., 2000, 30, 81–84 CrossRef CAS.
- A. G. Trant, T. E. Jones, T. C. Q. Noakes, P. Bailey and C. J. Baddeley, Surf. Sci., 2010, 604, 300–307 CrossRef CAS.
- T. E. Jones, T. C. Q. Noakes, P. Bailey and C. J. Baddeley, Surf. Sci., 2004, 569, 63–75 CrossRef CAS.
- R. Hirschl, F. Delbecq, P. Sautet and J. Hafner, J. Catal., 2003, 217, 354–366 CAS.
- F. Studt, F. Abild-Pedersen, T. Bligaard, R. Z. Sorensen, C. H. Christensen and J. K. Norskov, Science, 2008, 320, 1320–1322 CrossRef CAS.
- B. Bridier and J. Perez-Ramirez, J. Am. Chem. Soc., 2010, 132, 4321–4327 CrossRef CAS.
- D. Ferri, M. S. Kumar, R. Wirz, A. Eyssler, O. Korsak, P. Hug, A. Weidenkaff and M. A. Newton, Phys. Chem. Chem. Phys., 2010, 12, 5634–5646 RSC.
- J. K. Norskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef CAS.
- K. Reuter, in Modelling Heterogeneous Catalytic Reactions: From the Molecular Process to the Technical System, ed. O. Deutschmann, Wiley-VCH, Weinheim, 2011 Search PubMed.
- K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73, 045433 CrossRef.
- J. Rogal, K. Reuter and M. Scheffler, Phys. Rev. B: Condens. Matter Mater. Phys., 2008, 77, 155410 CrossRef.
| This journal is © The Royal Society of Chemistry 2012 |