Kinetics of carbon monoxide oxidation with Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co) catalysts†
Received
13th October 2011
, Accepted 18th November 2011
First published on 18th November 2011
Abstract
Base metal substituted Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co) catalysts were synthesized by the solution combustion method and characterized by XRD, XPS, TEM and BET surface area analysis. The catalytic activities of these materials were investigated by performing CO oxidation. The rates and the apparent activation energies of the reaction for CO oxidation were determined for each catalyst. All the substituted catalysts showed high rates and lower activation energies for the oxidation of CO as compared to unsubstituted SnO2. The rate was found to be much higher over copper substituted SnO2 as compared to other studied catalysts. 100% CO conversion was obtained below 225 °C over this catalyst. A bifunctional reaction mechanism was developed that accounts for CO adsorption on base metal and support ions and O2 dissociation on the oxide ion vacancy. The kinetic parameters were determined by fitting the model to the experimental data. The high rates of the CO oxidation reactions at low temperatures were rationalized by the high dissociative chemisorption of adsorbed O2 over these catalysts.
1. Introduction
Carbon monoxide, emitted from many industrial processes and transportation, is considered as an important air pollutant. Catalytic oxidation is an efficient way to convert CO to CO2 and has been studied using various simple and mixed oxide catalysts.1–6 Precious metals such as Pt, Pd, Au supported on various metal oxides or combination of metal oxides are well known oxidation catalysts.7–9 The supports used are mainly Al2O3, CeO2, ZrO2, TiO2 and mixed oxides with high dispersion of Pt, Pd and Au. However, the high cost of precious metals and their sensitivity to sulfur poisoning have motivated the research into base metal substituted catalysts. It has been well documented that high activity and resistance to poisoning can be obtained by doping Cu, Co, Ni oxides.10,11 The reaction rate and mechanism are highly influenced by the identity of supports.12,13 For instance, non-reducible Al2O3 shows lower activity than reducible oxides such as CeO2. Reducible supports such as CeO2, TiO2, Fe2O3 are often used for CO oxidation studies and have shown higher activity at low temperatures as compared to their non-reducible oxide counterparts.14 The difference in the reducibility of supports was found to be responsible for the stabilization of various intermediate species and different reaction pathways.15,16
SnO2 has been extensively used as an oxidation catalyst as it can reversibly undergo the Sn4+ ↔ Sn2+ reaction at relatively lower temperatures (∼200 °C). It is one of the most widely used semiconductor oxides for the manufacturing of sensors.17 A significant lowering in the activation energy for CO oxidation reaction over SnO2 as compared to CeO2 is indicative of the ease of availability of lattice oxygen in SnO2. It has been reported18 that the apparent activation energy for tin oxide and cerium oxide is 4.7 kcal mol−1 and 13.2 kcal mol−1, respectively. This shows that tin oxide is an interesting material having higher activity as compared to ceria and can catalyze the oxidation reaction at relatively low temperatures. Hence, it is of interest to study the catalytic activity of SnO2 towards CO oxidation. Attempts have been made to improve the low temperature reducibility and enhance the oxygen storage capacity of SnO2 by substituting a part of Sn4+ by other suitable cations. Pt, Pd, Au impregnated SnO2 were reported as active catalysts for CO or CH4 oxidation.19,20 Grass and Lintz15 suggested a probable pathway of CO oxidation over the Pt/SnO2 catalyst, which assumed adsorption of oxygen on Pt followed by its migration to the reaction sites situated at the periphery between oxide and Pt metal. Based on the comparative evaluation of the catalytic activity of Fe2O3, Mn2O3 and SnO2 and their mixed oxides, Kulshreshtha et al. showed that mixed oxides (Fe2O3 + SnO2 and Mn2O3 + SnO2) show strong synergistic effects for CO oxidation.21 Depending on the support, different reaction mechanisms have been proposed for Pt, Rh and Pd.12,22 The reaction mechanism over a noble metal is well understood and the oxidation of CO over Pt is assumed to proceed via a reaction between adsorbed oxygen and adsorbed CO species in a Langmuir–Hinshelwood type of model. However, it is still unclear if CO reacts in a similar way over base metal catalysts in the adsorbed state or in the gas phase in an Eley–Rideal type of model.
Although catalysts for the oxidation of carbon monoxide have been widely investigated over supported base metals, there are very limited data on the kinetics of catalyticCO oxidation over these catalysts. The mechanistic understanding of correlation between catalyst properties and metal–support interaction is still missing. The oxidation of carbon monoxide over SnO2 catalysts was postulated to follow the mechanism that involves the adsorption of carbon monoxide, desorption of carbon dioxide and oxygen regeneration of the catalyst. The kinetics was represented by the Langmuir type rate equation. Sedmak et al. studied the kinetics of CO oxidation in excess hydrogen over a nanostructured Cu0.1Ce0.9O2−δcatalyst under preferential oxidation conditions and showed that the kinetics of the reaction was found to follow the redox mechanism represented by the Mars and van Krevelen type of rate equation.23 Dekker et al. proposed a model that involves the oxidation of reduced sites by O2 and/or N2O, followed by a reaction with CO, yielding a surface intermediate that releases CO.24 In a recent study, Kocemba and Rynkowski25 explained how oxygen adsorption affects the catalytic activity of tin dioxide towards CO oxidation.
In order to lower the cost and improve activity of catalysts, novel non-stoichiometric Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co) catalysts were synthesized by the solution combustion method using a tin oxalate precursor. The activities of these catalysts at various temperatures were determined and compared. A detailed kinetic model was proposed for describing the kinetics of the CO oxidation. Spectroscopic insights were used in determining the possible surface processes and rate mechanisms were proposed based on elementary pathways. The kinetic parameters were obtained from fitting the experimental data.
2. Experimental section
2.1. Synthesis and characterization
Transition metal ions (M = Cu, Fe, Mn, and Co) substituted SnO2 catalysts were prepared by a novel, single step solution combustion method using a tin oxalate precursor. The combustion mixture for the preparation of 5 atom% Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, and Co) catalysts contained stoichiometric amounts of tin oxalate (SnC2O4) and respective metal nitrates like Mn(NO3)2·4H2O, Fe(NO3)3·9H2O, Co(NO3)2·6H2O and Cu(NO3)2·3H2O and glycine (CH2NH2COOH) as fuel (all from S.D. Fine Chem, India). SnC2O4 was prepared from SnCl2 and oxalic acid. In a typical procedure, 50 ml of 0.5 M solution of SnCl2·2H2O was slowly added to 50 ml of 0.5 M solution of oxalic acid at 70 °C under stirring, giving SnC2O4 crystals. The precipitate was filtered off, washed with distilled water several times until chloride ions were completely removed, and finally dried at 75 °C for 15 h. The formation of pure crystalline SnC2O4 was confirmed by X-ray diffraction (XRD). SnC2O4 was dissolved in nitric acid and Sn2+ was oxidized to Sn4+ (as confirmed by the HgCl2 test).26
In the solution combustion method, SnC2O4, Cu(NO3)2·3H2O and CH2NH2COOH were taken in the following molar ratio 0.95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0.05:2.166 to make 5 atom% Sn0.95Cu0.05O2−δ. The solution was introduced into a muffle furnace maintained at 400 °C. The solution boiled with frothing and foaming with concomitant dehydration. At the point of its complete dehydration, the redox mixture ignited yielding a voluminous finely dispersed solid product.
:0.05:2.166 to make 5 atom% Sn0.95Cu0.05O2−δ. The solution was introduced into a muffle furnace maintained at 400 °C. The solution boiled with frothing and foaming with concomitant dehydration. At the point of its complete dehydration, the redox mixture ignited yielding a voluminous finely dispersed solid product.
All the compounds were characterized by X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS), transmission electron microscopy (TEM) and BET surface analysis. The XRD patterns of the compounds were obtained from a Philips X'pert diffractometer with Cu-Kα radiation in a 6 h range of 10–80 with an interval of 0.02. TEM analysis was performed on a JEOL JEM-200CX, operating at 200 kV. The samples were dispersed in ethanol and treated with ultrasound for 5 min, and then deposited on a copper grid coated with a preformed holey carbon film. BET surface area was determined by a nitrogen adsorption–desorption method at liquid nitrogen temperature using a Quantachrome NOVA 1000 surface area analyzer. X-Ray photoelectron spectra of transition metal substituted oxides were recorded on a Thermo Fisher Scientific Multilab 2000 (England) instrument with Al K radiation (1486.6 eV). The binding energies reported are with reference to graphite at 284.5 eV. Oxide samples were ground with 30 wt% graphite powder, made into thin pellets at room temperature, and XPS spectra were recorded. There was no charging of oxide samples. The composition of the copper substituted catalyst was confirmed by inductively coupled plasma-optical emission spectroscopy (ICP-OES, Thermo-iCAP 6000 series). The compounds were dissolved in acidic solution and heated in a water bath at 80 °C for 24 h. The clear solutions were then diluted with deionized water and used for the analysis.
2.2.
CO
oxidation experiments
CO
oxidation experiments were carried out in an 8.5 mm ID and 35 cm long quartz tube reactor. The powdered catalyst of different weights was diluted with glass beads (180 μm) to make a bed of length 1.1 cm. The catalysts were packed between two glass wool plugs in the center of the reactor. The temperature of the catalytic section was maintained using a PID controller with the thermocouple placed at the center of the catalyst bed. Before the experiments, the as-prepared catalyst was heated in O2 flow at 300 °C for 1 h followed by degassing in N2 flow to the experimental temperature to remove the residual moisture. A mixture of CO, O2, and N2 (all from Chemix Speciality Gases, Bangalore, India) was sent through flow controllers to the reactor. Reactions were carried out with a feed mixture consisting of 2.4 vol% CO, 2.4 vol% O2 and balance N2. All of the reactions were carried out under isothermal conditions with a total flow of 80 cm3 min−1 (GHSV of 28210 h−1 based on the catalyst bed volume of 0.17 cm3 for 200 mg of the catalyst). The spent catalyst was characterized after the reaction using XPS. The products of the reaction were analyzed using an online gas chromatograph (Mayura Analytical, India). CO and CO2 were separated using a molecular sieve column and were detected using a flame ionization detector. Because the signal of CO2 in FID is poor, CO2 is converted to CH4 using a methanizer in the presence of Rh catalyst at 350 °C. This peak is measured and calibrated with standard gases. The nonlinear regression technique was used to determine the optimized kinetic parameters.
3. Results
3.1. Structural studies
X-Ray diffraction spectra of all base metal substituted catalysts along with unsubstituted SnO2 are recorded at room temperature and shown in Fig. 1a. SnO2 based catalysts were found to crystallize in TiO2 like rutile structure. No additional phases such as the SnO2 orthorhombic phase, corresponding transition metal oxide or other SnO based phases were observed. This showed the formation of single phase solid solutions that can be represented by the formula Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co) where M represents the transition metal. The peaks corresponding to metals were absent showing substitution of metals in the lattice. The patterns show broad X-ray line width. The substitution of metals in the lattice results in a change in lattice parameters due to the difference in ionic radii of Sn4+ and M2+/M3+. Therefore, profile refinement analysis of the spectra was carried out to determine the structural parameters.
 |
| | Fig. 1 (a) XRD of SnO2 and Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co). (b) Profile refinement of Sn0.95Cu0.05O2−δ. | |
The XRD data were refined using the JANA 2000 suite program. The profile refinement of Sn0.95Cu0.05O2−δ is shown in Fig. 1b. The symbols in the Fig. 1b show the experimental XRD data and the solid lines show the predicted XRD pattern. The difference between the actual and theoretical patterns is shown by the light line at the bottom. Fitting of the data to the tetragonal structure gave satisfactory values of the reliability data. The rest of profile refinements of the catalyst are given in Fig. S1a–c (see ESI†). The presence of wide peaks is an indication of nanometre sized crystallites. The crystallite sizes of the compounds were calculated using the Scherrer formula from the most intense peak. The crystallites were found to be in the range of 5 to 10 nm. The crystallite size of Sn0.95Cu0.05O2−δ was the smallest while the crystallite size of Sn0.95Co0.05O2−δ was the largest among studied compounds. Table 1 shows the cell parameters, crystallite sizes and the refinement reliability data for all the compounds. It was found that a decrease in lattice parameters occurred on substitution for all compounds.
Table 1 Profile refined parameters of Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co)
|
Catalysts
|
a/b (Å) |
c (Å) |
Cell volume (Å3) |
wRp |
R
p
|
χ
2
|
Crystallite size (nm) |
Surface area (m2 g−1) |
| Sn0.95Cu0.05O2−δ |
4.7356 |
3.1854 |
71.43 |
7.2 |
5.7 |
1.02 |
5.5 |
33 |
| Sn0.95Fe0.05O2−δ |
4.7187 |
3.1735 |
70.66 |
9.1 |
6.8 |
1.05 |
8.6 |
31 |
| Sn0.95Mn0.05O2−δ |
4.7330 |
3.1862 |
70.94 |
10.4 |
7.9 |
0.84 |
7.8 |
32 |
| Sn0.95Co0.05O2−δ |
4.7263 |
3.1776 |
70.98 |
7.5 |
6.8 |
0.95 |
9.1 |
31 |
The TEM images and the ring type diffraction patterns of Sn0.95Cu0.05O2−δ and unsubstituted SnO2 are shown in Fig. 2 and Fig. S2 (see ESI†), respectively. In the TEM image of Sn0.95Cu0.05O2−δ, the absence of Cu metal/oxide particle fringes confirmed the substitution of Cu ions in the SnO2 matrix. Pure SnO2 synthesized by the solution combustion method crystallizes in the rutile phase with particle sizes of 13 to 27 nm. On substitution of copper into the SnO2 matrix, the particle size decreased and the particle sizes for Sn0.95Cu0.05O2−δ measured from the image are in the range of 9 to 18 nm. The BET surface areas for all substituted SnO2 catalysts are in the range of 31 to 33 m2 g−1, as shown in Table 1. ICP measurements gave the composition of Sn0.95Cu0.05O2−δ and the amount of Cu in the catalyst was found to be 4.8 at%.
 |
| | Fig. 2 (a) TEM image and (b) the electron diffraction pattern of Sn0.95Cu0.05O2−δ. | |
The oxidation states of the base metals in the compounds were determined using XPS. The spectra were recorded both before and after the reaction. After the experiment, the flow of the reactant gases was stopped and the catalysts were allowed to stabilize at room temperature in a stream of N2. The catalyst was quickly transferred to the vacuum chamber of the spectrometer to avoid the formation of possible surface oxide species. Different spent samples of the same catalyst obtained after different reaction runs were analyzed in the same manner and reproducible results were obtained.
Fig. 3 shows the Cu 2p spectra and Sn 3d spectra in Sn0.95Cu0.05O2−δ before and after the reaction. The binding energies were calibrated with respect to the binding energy of graphite observed at 284.5 eV for all catalysts. Core level Cu (3d3/2, 1/2) spectra of the as-prepared catalyst were resolved into sets of spin orbit doublets. Accordingly, the Cu (2p3/2, 1/2) peaks at 934.0 and 953.8 eV with satellites at 9 eV below the main peak in Sn0.95Cu0.05O2−δ can only be attributed to Cu in the 2+ oxidation state. Core level Sn (3d) spectra of the as-prepared catalyst have binding energy peaks at 486.3 eV, which correspond to the Sn4+ state and a small percentage of Sn is in the 2+ oxidation state. Sn (3d) peaks at lower binding energy of 485.3 eV are due to the 2+ oxidation state of Sn.27 Taking into consideration the full width at half maxima, it was found that Sn is present partially in the 2+ oxidation state in the as-prepared sample. This is possible because SnO2 can easily be reduced to SnO.22
 |
| | Fig. 3 Core level XPS of (a) Cu 2p and (b) Sn 3d in Sn0.95Cu0.05O2−δ. | |
The presence of Sn2+ can be explained by the substitution of Sn4+ ions by base metal ions whose oxidation state is different from the parent metal oxide. During this process, oxygen vacancies may arise to maintain the charge neutrality when some Sn4+ reduces to Sn2+. A small amount of cerium was observed in the 3+ state when 2% atom Pt was substituted in the CeO2 matrix.28 The XPS spectra of the used sample after reaction were very wide. The presence of wide peaks spanning binding energy more than 10 eV clearly showed the presence of multiple oxidation states. The spectra having non-zero intensities from 930 to 940 eV showed the presence of Cu2+ and Cu1+ states. For each oxidation state, two peaks corresponding to Cu 2p3/2, 1/2 having an intensity ratio of 3:2 should be present. These peaks were approximated as the standard Gaussian peaks. The peak positions corresponding to the different oxidation states of Cu were found from the literature.29 An increase in the Sn2+ oxidation state relative to the as-prepared catalyst was observed in all compounds. Further reduction to the metallic Sn0 state was not observed under the mentioned reaction conditions.
Fig. 4 shows the XPS spectra of the Sn 3d and Mn 2p electrons for the Sn0.95Mn0.05O2−δ after and before reaction. The Mn 2p core level spectra, as seen in Fig. 4a, are split into 2p3/2 and 2p1/2 components due to the spin orbit coupling. From the smoothed spectra, the core level binding energies of 2p3/2 and 2p1/2 for Sn0.95Mn0.05O2−δ were estimated to be about 641.7 and 653.4 eV, respectively, and the difference in binding energies between Mn 2p3/2 and Mn 2p1/2 is 11.7 eV.30 Based on above analysis, we confirmed that Mn ions have a chemical valence of 3+ in as-synthesized samples. The binding energy values of Sn 3d show that all Sn is in the 4+ state. During the oxidation reaction, several oxidation states can be formed and coexist or progressively change one into the other. Mn 2p3/2spectra are quite broad and the binding energy shifts are not large enough to clearly distinguish between the different oxides, mainly when two or more species are simultaneously present. From the analysis of the broad Mn 2p main peaks, it is not possible to have detailed information on the intermediate oxidation steps. The corresponding Mn 2p3/2 satellites cannot be used, since for Mn2O3, they obviously overlap with the Mn 2p1/2 peak resulting in a high intensity of the Mn 2p1/2 peak. However, only the binding energy shifts are sufficient to identify chemical states. The peak position corresponding to binding energies of Mn 2p3/2 was assigned to 3+ and 2+ oxidation states.30
 |
| | Fig. 4 Core level XPS of (a) Mn 2p and (b) Sn 3d in Sn0.95Mn0.05O2−δ. | |
The Co 2p spectrum, displayed in Fig. S3 (see ESI†), shows a main binding energy peak of Co 2p3/2 corresponding to 780.5 eV and a satellite peak at 786.4 eV in the as-synthesized catalyst.31 The binding energy corresponding to the 778.10 eV peak represents metallic Co. Co2+ and Co3+ have very similar Co 2p binding energy, which makes identification of the oxidation state on account of the binding energy alone unreliable. There are, however, clear features in the 2p core level photoemission spectra that allow for an unambiguous differentiation between Co2+ and Co3+. Firstly, the presence of a strong shake-up satellite peak of Co2+-2p towards higher binding energy in contrast to Co3+-2p that exhibits only a weak satellite eliminates possibility of cobalt being in the 2+ oxidation state. The XPS spectrum of Co 2p region suggests that Co is in the 2+ oxidation state.31 The spectra of Sn manifest clearly that most of Sn is in the 4+ state along with a small amount of the 2+ state. After reaction, the presence of the broad peak indicates multiple oxidation states.
The Fe 3p1/2XPS spectral region of as-synthesized Sn0.95Fe0.05O2−δ is shown in Fig. S4 (see ESI†). The binding energy corresponding to the different oxidation states of Fe was found from the literature.32 The XPS peak of hematite phases occurs at 710.8 eV; however, the core level peak of Fe in Sn0.95Fe0.05O2−δ showed a slight shift to higher binding energies compared to the Fe oxides, indicating the difference in the atomic environment surrounding the incorporated Fe ions. A detailed analysis of the spectrum demonstrated that the peak is broadened, which can be attributed to the presence of multiple oxidation states of Fe. The binding energies of a peak corresponding to Fe 2p3/2 are located at 711.30 eV and 710.30 eV, respectively; suggesting that Fe exists mainly in the 3+ state along with a small amount of the 2+ state. The positions of the Sn 3d5/2 and Sn 3d3/2 peak maxima correspond to SnO2. The presence of a small amount of Sn2+ was also observed. After reaction, the binding energies of 710.9 eV for Fe 2p3/2 can be attributed to iron being in the tervalent states (Fe3+, Fe2+ and Fe0).
3.2.
Catalytic reactions
CO
oxidation by O2 over base metal substituted SnO2 was carried out with the inlet gas flow consisting of 2.4% vol CO, 2.4% vol O2 with total gas flow of 80 cm3 min−1 made up with N2 over 200 mg catalysts. Fig. 5 shows the comparison of the catalysts for CO conversion. 100% CO conversion was obtained at below 225 °C over the Sn0.95Cu0.05O2−δ and below 350 °C over Sn0.95Co0.05O2−δ. The T50 (corresponding to 50% CO conversion temperature) over catalysts Sn0.95Cu0.05O2−δ, Sn0.95Fe0.05O2−δ, Sn0.95Mn0.05O2−δ and Sn0.95Co0.05O2−δ are 177, 279, 282 and 296 °C, respectively. Experiments were also carried out with a mixture of gases and different amounts of catalysts loading. The reactant mixture had a gas composition of 2.4% CO, 2.4% O2 and balance N2 with the total gas flow of 80 cm3 min−1. Fig. 6 shows the variation of W/FCO (W is the weight of the catalyst and FCO is the flow rate of CO) with the fractional conversion of CO for all catalysts. In the cases of Sn0.95Cu0.05O2−δ and Sn0.95Mn0.05O2−δ, it can be observed that at higher temperatures, the conversions did not vary linearly with W/FCO. This shows that the reaction is mass transfer limited at high W/FCO and shows two different reaction regimes for these catalysts.33 In all cases, the rates of CO oxidation were calculated at various temperatures from the slope of the linear portion. Thus the slope was calculated from the plot (Fig. 6) of conversion with W/FCO, where the conversions were lesser than 15%. The rate of CO oxidation reaction over the copper substituted catalyst is found to be very high at low temperatures among the reported catalysts34–39 (Table 2). The apparent activation energies of CO oxidation over different catalysts were calculated by the Arrhenius plot (Fig. 7) for all substituted catalysts. The activation energies for Cu, Fe, Mn, Co doped catalysts are 50, 53, 59 and 69 kJ mol−1, respectively.
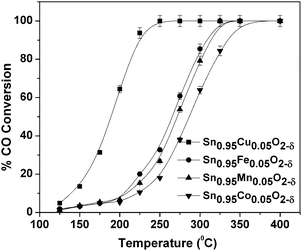 |
| | Fig. 5 Effect of temperature on CO conversion over Sn0.95M0.05O2−δ. | |
 |
| | Fig. 6 Variation of fractional conversion of CO with the weight of catalyst for (a) Sn0.95Cu0.05O2−δ, (b) Sn0.95Fe0.05O2−δ, (c) Sn0.95Mn0.05O2−δ and (d) Sn0.95Co0.05O2−δ. | |
Table 2 Comparison of rates and activation energies for the CO oxidation reaction over various catalysts
|
Catalysts
|
Rate (μmol g−1 s−1) (°C) |
E
a (kJ mol−1) |
Ref. |
| 5 wt% Ru/SiO2 |
1.00 (110) |
94 |
34
|
| 5 wt% Rh/SiO2 |
0.0251 (110) |
103 |
34
|
| 5 wt% Pd/SiO2 |
0.316 (143) |
103 |
34
|
| 5 wt% Pt/SiO2 |
0.32 (115) |
56 |
34
|
|
25Au75Pd/SiO2 |
0.128 (140) |
— |
35
|
| 0.014 wt% Rh/Al2O3 |
0.6 (196) |
115.5 |
35
|
|
Pd/CeO2/Al2O3 |
38.0 (250) |
84 |
36
|
| Ce0.98Pd0.02O1.98 |
3.9 (120) |
121 |
37
|
| 12 wt% Cu/δ-Al2O3 |
0.09 (150) |
90 |
38
|
| 10% wt CuO/SiO2 |
0.04 (250) |
59 |
39
|
| Sn0.95Cu0.05O2−δ |
15 (225) |
50 |
Present study |
 |
| | Fig. 7 Arrhenius plot of CO oxidation to determine the activation energy. | |
3.3. Kinetic model
Ionic substitution into a reducible support enhances the oxygen and hydrogen storage capacities40 due to the formation of solid defects. This is given by the formula Sn1−X4+M2+XO2−2−δ□δ where δ < x at room temperature and □ is an oxygen vacancy. These metals (M) have variable and lower oxidation states than that of Sn in SnO2. These metals induce redox couples in the substituted base metals as well as in the SnO2 matrix and render strong metal–support interactions. An ionic substitution in the form of M2+/M3+ ions for the Sn4+ site in the SnO2 lattice also creates an oxide ion vacancy. These oxide vacancies in SnO2 are crucial for oxygen adsorption and dissociation.41 The oxide vacancies are formed around the M2+/M+3 ions offering sites for O2 adsorption.42,43 However, there is experimental evidence that oxygen adsorbs in a molecular form like the superoxide O2− species, preferentially on oxygen vacancies.49 These sites are abundant in the proximity of the M2+/M3+ particles due to the Schottky junction between the M2+/M3+ and the n-type semiconducting oxides.44 Therefore, the sites along the perimeter of metal ions–support interface would be the best sites for the reaction between CO adsorbed on oxide and the oxygen adsorbed on the vacancies. During the reduction of this solid solution, the reduced ionic substituted metal cations (M) are reoxidized by reduction of Sn4+ in the vicinity of Sn2+. The redox equilibrium can be represented as Sn4+ + M1+ ⇔ Sn2+ + M2+. The reducibility of Sn4+ to Sn2+ enhances the flexibility of metal ions to adapt to a different oxidation state by maintaining the electronic neutrality of lattice.
Several explanations of observed synergism between supported noble metals and support are given in the literature. Bifunctional mechanisms with or without spillover of adsorbed CO and O2 from noble metals to tin oxide have been proposed.45,46 The (i) role of metal promoter and support, (ii) active site for adsorption of CO and O2, and (iii) interaction of support and promoter (active phase) are not clearly understood. In the case of base metal supported catalysts, it has been observed that the rate of CO oxidation has weak dependence on the partial pressure of oxygen (PO2) and has positive order dependence from 0 to 1 in the partial pressure of CO (PCO).24,47–49 Both Eley–Rideal and Langmuir–Hinshelwood models have been proposed for CO oxidation over Cu based catalysts.
Fig. 8 shows the variation in the rates of CO oxidation over Sn0.95Cu0.05O2−δ under CO and O2 partial pressures at different temperatures. The reaction rates increase with CO and O2 partial pressures, but saturate at high partial pressures. Fig. 8 shows that the reaction rate rapidly increases with PO2 than with PCO at low partial pressures. The reaction order of PCO seems to decrease from one to zero as PCO increases. Since there are no maxima in the reaction rate, when either CO or O2 partial pressures were varied, this indicates that these two reactants do not adsorb competitively on the surface of catalyst. The reaction rate has the shape of a Langmuir isotherm when CO and O2 partial pressures are varied. CO oxidation mainly occurs through the lattice oxygen incorporation and the O2 depleted oxide surface get rejuvenated due to the presence of O2 in the reactant mixture. These results are consistent with our XPS results in which substituted base metals and Sn4+ ions undergo reduction, releasing lattice oxygen to maintain electrical neutrality. Based upon the above arguments, we proposed several plausible mechanism steps for CO reaction over base metals substituted SnO2. Similar kinds of bifunctional mechanisms with or without spillover have been proposed for supported noble metals over reducible supports.12,22,48,51,52 Various possible steps involved in the mechanism are
| | | COS + “O” → CO2 + S + “V” | (3) |
S, “V”, “O” refer to the vacant sites on support,
oxide ion vacancy on support and oxygen species on the support, respectively. In this mechanism, it is assumed that CO is adsorbed on metal ions (Cu, Fe, Mn, Co) and Sn ions and oxygen is dissociatively chemisorbed on the
oxide vacancies. The chemical nature of adsorbed CO species on metal ions and Sn ions has been treated to be the same.
Fig. 8 suggests that CO
adsorption can be described by the Langmuir isotherm. In the case of supported
tin oxide on
alumina, the best correlation could be obtained
50 using the Langmuir type rate equation that involved irreversible CO
oxidation and CO
2 desorption steps and fast oxygen regeneration of the
catalyst. Kulshreshtha
et al. showed better mobility of oxygen in the case of mixed
oxide (Mn
2O
3 + SnO
2) as compared to SnO
2.
21 It was shown that the rate of CO
oxidation on the SnO
2 catalyst under steady state conditions is proportional to the fraction of (OSnOCO) sites.
3where

,
θCO,
θV1 represent the adsorption equilibrium for CO on the
catalyst, the fraction of sites occupied by CO molecules on the metal surface, fraction unoccupied by CO molecules on the metal surface and SnO
2, respectively. The site balance on the metal support is given by
Note that oxygen
adsorption is not included in this balance because CO and O
2 do not adsorb competitively. The CO coverage is
| |  | (6) |
The rate of
adsorption of O
2 on the support can be written as
| |  | (7) |
θV2 is the fraction of unoccupied oxygen vacancies. The steady state balance over
θO species is,
| |  | (8) |
Oxygen atom and
oxide ion vacancy balance on the support can be written as
where
θO is a fraction of O
2 on
oxide vacancy. The rate of CO
2 formation is
After substitution of
θCO and
θO, the final form of rate equation is given by
| |  | (11) |
This mechanism is a bifunctional mechanism in which the adsorbed CO on metal sites and oxygen adsorbed on lattice vacancies react to form CO
2. No step was assumed to be the rate determining step while deriving this expression. Note that for this mechanism, it is implicitly assumed that the reaction takes place in adjacent metal sites and tin active sites (interfacial sites). This model is tested for all
catalysts and the values of rate coefficient were determined by the nonlinear regression. The optimized rate parameters are given in
Table 3.
Fig. 9a shows the variation of the rate with temperature and this satisfactorily correlates the experimental data.
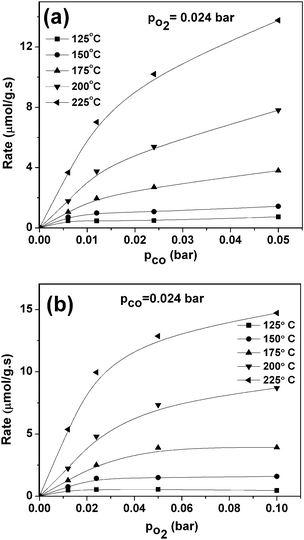 |
| | Fig. 8 Variation of the rate of CO2 formation with the (a) partial pressure of CO at constant PO2 and (b) partial pressure of O2 at constant PCO. | |
Table 3 Rate parameters used in the model for the CO oxidation reaction
| Parameter |
Sn0.95Cu0.05O2−δ |
Sn0.95Fe0.05O2−δ |
Sn0.95Mn0.05O2−δ |
Sn0.95Co0.05O2−δ |
|
K
1
|
96√Texp(4400/T) |
86√Texp(4300/T) |
83√Texp(4330/T) |
99√Texp(4400/T) |
|
K
2
|
3.6 × 106√T |
2 × 106√T |
2 × 106√T |
3.5 × 104√T |
|
K
3
|
7.3exp(−3730/T) |
9.3exp(−4410/T) |
8.9exp(−5000/T) |
11.7exp(−5620/T) |
|
K
4
|
2.9 × 105exp(−4989/T) |
1 × 105exp(−5387/T) |
2.54 × 105exp(−6100/T) |
4.85 × 105exp(−6724/T) |
|
K
5
|
1.0 × 104exp(6196/T) |
1.12 × 104exp(5997/T) |
2.9 × 104exp(6203/T) |
2.9 × 104exp(6724/T) |
|
n
|
0.18 |
0.19 |
0.21 |
0.23 |
|
K
6
|
1022exp(−5869/T) |
1114exp(−5962/T) |
3754exp(−7459/T) |
4631exp(−7795/T) |
|
K
7
|
5011exp(1003/T) |
2160exp(741/T) |
5969exp(729/T) |
6451exp(819/T) |
|
m
|
0.21 |
0.24 |
0.22 |
0.25 |
 |
| | Fig. 9 Variation of the rate of CO oxidation with temperature. The lines represent the fit based on the (a) proposed model, (b) MVK model and (c) LFS model. | |
Though the proposed model satisfactorily fits the experimental data, it was compared to existing models. Among the current models available in the literature, the Mars–van Krevelen model (MVK) and Langmuir–Hinshelwood type (LFS) model proposed by Liu and Flytzani-Stephanopoulos are often used to describe the kinetics of CO oxidation over Cu/CeO2 surfaces.23,55
The MVK model consists of two steps namely the reduction of oxidized catalyst surface along with formation of CO2 and reoxidation of the catalyst surface with O2. Thus, in this case, the catalyst undergoes reduction while the reductant is oxidized. The kinetic equation derived on the basis of above mentioned redox steps is23
| |  | (12) |
The parameters
K4 and
K5 in the above equation are the reaction rate constants for the reduction of
catalyst by CO and reoxidation of the
catalyst by O
2, respectively. The parameters
K4,
K5 and
n obtained by fitting the reaction rate with
eqn (12) are given in
Table 3. The model fit and the experimental data are shown in
Fig. 9b.
The LFS model assumes that there is no involvement of lattice oxygen in the reaction and atomic O2 is provided by the amount adsorbed onto the catalyst surface. The equation derived based on this model55 is
| |  | (13) |
The parameters
K6 and
K7 represent the surface reaction rate constant and CO
adsorption equilibrium constant, respectively. The parameters obtained by fitting
eqn (13) are given in
Table 3 and the experimental data are compared with the model fit in
Fig. 9c.
4. Discussion
The value of the parameter, K1, over Sn0.95Cu0.05O2−δ is comparable to that of CO adsorption on metallic copper.53,54 The smaller value of K1 over Sn0.95Fe0.05O2−δ and Sn0.95Mn0.05O2−δ is manifestation of lower CO adsorption on these catalysts. The Cu+ species for the Sn0.95Cu0.05O2−δcatalyst after reaction were observed by XPS. This indicates that CO adsorption sites are highly dispersed on the catalyst surface and the Cu+ surface provides strong CO adsorption sites.23,58
Several spectroscopic observations were used to characterize various oxygen species chemisorbed on SnO2.25 In the case of transition metal oxides supported on the reducible metal oxide, surface lattice oxygen is involved in the oxidation reaction.55 Our XPS studies also show involvement of lattice oxygen in the reaction. If CO reacts with adsorbed oxygen rather than oxygen, the formation of oxygen vacancies by CO will not be the rate determining step.56 This is indeed observed in our proposed mechanism. If all steps in the mechanism are compared, O2 dissociation on oxide vacancy is the slowest step and confirms the involvement of lattice oxygen during reaction. The oxygen dissociation over Sn0.95Cu0.05O2−δ is much faster than any other studied catalyst. This shows that reducibility of Sn4+ to Sn2+ enhances the flexibility of copper ions to adapt to different oxidation states by maintaining the neutrality of lattice much more easily than any other catalyst. Though the adsorption of CO on Sn0.95Co0.05O2−δ is comparable to Sn0.95Cu0.05O2−δ, the reducibility of Sn0.95Co0.05O2−δcatalyst is poor (observed from the value of K2) resulting in the lower catalytic activity of Sn0.95Co0.05O2−δ.
Fig. 9a–c represent the model fit for the proposed model, MVK model and LFS model for CO oxidation over Sn0.95M0.05O2−δ (M = Cu, Fe, Mn, Co). It can be observed that all of the models satisfactorily fit the experimental data, with the regression correlation coefficient of above 0.98. Therefore, it is not possible to discriminate among the three models on the basis of the correlation with experimental data. The derivation of the MVK rate equation for redox reactions experiences a number of inconsistencies and this rate equation must be viewed only as a mathematical data-fitting function.57 Further, this equation is applicable only for a reaction involving molecular oxygen adsorbed on a single site and not for lattice O ions/atoms that react with the reductant. The main assumption of the LFS model is that no lattice oxygen is involved in the CO oxidation reaction over the Cu/CeO2 catalyst. This is in contrast with the findings of the spectroscopic studies,58,59 where involvement of lattice oxygen was observed. Our XPS results also support the utilization of lattice oxygen during reaction. Based on the above discussion, though all the three models correlate the experimental data reasonably well, the proposed model gives insights into the rates of the actual processes that are likely to occur during the reaction.
5. Conclusions
The ionically substituted base metals (Cu, Fe Mn, CO) in the SnO2 matrix were synthesized by a single step solution combustion method and these materials showed high catalytic activity compared to unsubstituted SnO2. The order of activity for CO oxidation followed the order: Sn0.95Cu0.05O2−δ > Sn0.95Fe0.05O2−δ > Sn0.95Mn0.05O2−δ > Sn0.95Co0.05O2−δ. Oxide ion vacancy was found to be the main factor that governs the high rate of CO oxidation over Sn0.95Cu0.05O2−δ. The significant enhancement of catalytic activity in the case of Sn0.95Cu0.05O2−δ is mainly because of the efficiency of the Cu2+/Cu1+ couple in this process. A reaction model based on a bifunctional mechanism was proposed and used to correlate experimental data. The reaction rate parameters were determined from this model.
Acknowledgements
The authors thank the Department of Science and Technology, India, for financial support.
References
- B. Mguig, M. Calatayud and C. Minot, J. Mol. Struct. (THEOCHEM), 2004, 709, 73–78 CrossRef CAS.
- J. L. Ayastuy, M. P. González-Marcos, J. R. González-Velasco and M. A. Gutiérrez-Ortiz, Appl. Catal., B, 2007, 70, 532–541 CrossRef CAS.
- M. J. Fuller and M. E. Warwick, J. Catal., 1973, 29, 441–450 CrossRef CAS.
- H. Zhu, Z. Qin, W. Shan, W. Shen and J. Wang, Catal. Today, 2007, 126, 382–386 CrossRef CAS.
- P. M. Sreekanth and P. G. Smirniotis, Catal. Lett., 2008, 122, 37–42 CrossRef CAS.
- P. Ciambelli, S. Cimino, G. Lasorella, L. Lisi, S. De Rossi, M. Faticanti, G. Minelli and P. Porta, Appl. Catal., B, 2000, 37, 231–241 CrossRef.
- X. S. Huang, H. Sun, L. C. Wang, Y. M. Liu, K. N. Fan and Y. Cao, Appl. Catal., B, 2009, 90, 224–232 CrossRef CAS.
- M. Luo, Z. Y. Hou, X. X. Yuan and X. M. Zheng, Catal. Lett., 1998, 50, 205–209 CrossRef CAS.
- J. L. Cao, Q. F. Deng and Z. Y. Yuan, J. Mater. Sci., 2009, 44, 6663–6669 CrossRef CAS.
- T. J. Huang, T. C. Yu and S. H. Chang, Appl. Catal., 1989, 52, 157–163 CrossRef CAS.
- W. Liu, A. F. Sarofim and M. F. Stephanopoulos, Appl. Catal., B, 1994, 4, 167–186 CrossRef CAS.
- S. H. Oh, G. B. Fisher, J. E. Carpenter and D. W. Goodman, J. Catal., 1986, 100, 360–376 CrossRef CAS.
- M. M. V. M. Souza, N. F. P. Ribeiro and M. Schmal, Int. J. Hydrogen Energy, 2007, 32, 425–429 CrossRef CAS.
- P. Bera, K. C. Patil, V. Jayaram, G. N. Subbanna and M. S. Hegde, J. Catal., 2000, 196, 293–301 CrossRef CAS.
- K. Grass and H. G. Lintz, J. Catal., 1997, 172, 446–452 CrossRef CAS.
- U. Oran and D. Uner, Appl. Catal., B, 2004, 54, 183–191 CrossRef CAS.
- W. Fliegel, G. Behr, J. Werner and G. Krabbes, Sens. Actuators, B, 1994, 18–19, 474 CrossRef.
- R. Sasikala, N. M. Gupta and S. K. Kulshreshtha, Catal. Lett., 2001, 71, 69–73 CrossRef CAS.
- R. Schryer, B. T. Upchurch, B. D. Sidney, K. G. Brown, G. B. Hoflund and R. K. Herz, J. Catal., 1991, 130, 314–317 CrossRef.
- T. Matsui, T. Okanishi, K. Fujiwara, K. Tsutsui, R. Kikuchi, T. Takeguchi and K. Eguchi, Sci. Technol. Adv. Mater, 2006, 7, 524–530 CrossRef CAS.
- S. K. Kulshreshtha, M. M. Gadgil and R. Sasikala, Catal. Lett., 1996, 37, 181–185 CrossRef CAS.
- S. Roy, A. Marimuthu, M. S. Hegde and G. Madras, Appl. Catal., B, 2007, 73, 300–310 CrossRef CAS.
- G. Sedmak, S. Hočevar and J. Levec, J. Catal., 2003, 213, 135–150 CrossRef CAS.
- N. J. J. Dekker, J. A. A. Hoorn, S. Stegenga, F. Kapteijn and J. A. Moulijn, AIChE J., 1992, 38, 385–396 CrossRef CAS.
- I. Kocemba and J. M. Rynkowski, Catal. Today, 2011, 169, 192–199 CrossRef CAS.
- T. Baidya, A. Gupta, P. A. Deshpande, G. Madras and M. S. Hegde, J. Phys. Chem. C, 2009, 113, 4059–4068 CAS.
- R. O. Ansell, T. Dickinson, A. F. Povey and P. M. A. Sherwood, J. Electron Spectrosc. Relat. Phenom., 1977, 11, 301–313 CrossRef CAS.
- A. Gupta and M. S. Hegde, Appl. Catal., B, 2010, 99, 279–288 CrossRef CAS.
- J. G. Jolley, G. G. Geesey, M. R. Hankins, R. B. Wright and P. L. Wichlacz, Appl. Surf. Sci., 1989, 37, 469–480 CrossRef CAS.
- V. D. Castro and G. Polzonetti, J. Electron Spectrosc. Relat. Phenom., 1989, 48, 117–123 CrossRef.
- T. J. Chuang, C. R. Brundle and D. W. Rice, Surf. Sci., 1976, 59, 413–429 CrossRef CAS.
- N. S. McIntyre and D. G. Zetaruk, Anal. Chem., 1977, 49, 1521–1529 CrossRef CAS.
- L. A. Petrov, Y. Alhamed, A. Al-Zahrani and M. Daous, J. Catal., 2011, 32, 1085–1112 CrossRef CAS.
- N. W. Cant, P. C. Hicks and B. S. Lennon, J. Catal., 1978, 54, 372–383 CrossRef CAS.
- A. M. Venezia, L. F. Liotta, G. Pantaleo, V. L. Parola, G. Deganello, A. Beck, Z. Koppány, K. Frey, D. Horváth and L. Guczi, Appl. Catal., A, 2003, 251, 359–368 CrossRef CAS.
- Y. Y. Yung-Fang, J. Catal., 1984, 87, 152–162 CrossRef.
- S. Roy, A. Marimuthu, M. S. Hegde and G. Madras, Appl. Catal., B, 2007, 71, 23–31 CrossRef CAS.
- K. I. Choi and M. A. Vannice, J. Catal., 1991, 127, 465–488 CrossRef CAS.
- R. A. Prokopowicz, P. L. Silveston, R. R. Hudgins and D. E. Irish, React. Kinet. Catal. Lett., 1988, 37, 63–70 CrossRef CAS.
- G. Wrobel, C. Lamonier, A. Bennani, A. D'Huysser and A. Aboukaïs, J. Chem. Soc., Faraday Trans., 1996, 92(11), 2001–136 RSC.
- C. T. Campbell, Science, 2003, 299, 657 CrossRef.
- P. Bera, K. R. Priolkar, P. R. Sarode, M. S. Hegde, S. Emura, R. Kumashiro and N. P. Lalla, Chem. Mater., 2002, 14, 3591–3601 CrossRef CAS.
- L. Guczi, D. Horváth, Z. Pászti, L. Tóth, Z. E. Horváth, A. Karacs and G. Petõ, J. Phys. Chem. B, 2000, 104, 3183–3193 CrossRef CAS.
- H. Liu, A. I. Kozlov, A. P. Kozlova, T. Shido, K. Asakura and Y. Iwasawa, J. Catal., 1999, 185, 252–264 CrossRef CAS.
- G. C. Bond, L. R. Molloy and M. J. Fuller, J. Chem. Soc., Chem. Commun., 1975, 796–797 RSC.
- G. S. Zafiris and R. J. Gorte, J. Catal., 1993, 143, 86–91 CrossRef CAS.
-
A. Bielanski and J. Haber, Oxygen in catalysis, Dekker, New York, 1991 Search PubMed.
-
G. I. Golodets, Heterogeneous catalytic reactions involving molecular oxygen, Studies in Surface Science and
Catalysis, vol. 15, Elsevier, Amsterdam, 1983 Search PubMed.
- K. I. Choi and M. A. Vannice, J. Catal., 1991, 131, 22–35 CrossRef CAS.
- P. T. Wierzchowski and L. W. Zagorsk, Appl. Catal., B, 2003, 44, 53–65 CrossRef CAS.
- P. Mannila, T. Salmi, H. Haario, M. Luoma, M. Härkönen and J. Sohlo, J. Catal., 1986, 100, 360–376 CrossRef.
- B. K. Cho, J. Catal., 1992, 138, 255–266 CrossRef CAS.
- D. B. Clarke, I. Suzuki and A. T. Bell, J. Catal., 1993, 142, 27–36 CrossRef CAS.
- M. J. Sandoval and A. T. Bell, J. Catal., 1993, 144, 227–237 CrossRef CAS.
- W. Liu and M. F. Stephanopoulos, J. Catal., 1995, 153, 317–332 CrossRef CAS.
- A. Tschope, W. Liu, M. F. Stephanopoulos and J. Y. Ying, J. Catal., 1995, 157, 42–50 CrossRef CAS.
- M. A. Vannice, Catal. Today, 2007, 123, 18–22 CrossRef CAS.
- P. G. Harrison, I. K. Ball, W. Azelee, W. Daniell and D. Goldfarb, Chem. Mater., 2000, 12, 3715 CrossRef CAS.
- A. Martinez-Arias, M. Fernandez-Garcia, O. Galvez, J. M. Coronado, J. A. Anderson, J. C. Conesa, J. Soria and G. Munuera, J. Catal., 2000, 195, 207 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 shows the profile refinement of (a) Sn0.95Fe0.05O2−δ, (b) Sn0.95Mn0.05O2−δ and (c) Sn0.95Co0.05O2−δ. Fig. S2 shows the TEM image and the electron diffraction pattern of SnO2. Fig. S3 shows the core level XPS of Co 2p and Sn 3d in Sn0.95Co0.05O2−δ. Fig. S4 shows the core level XPS of Fe 2p and Sn 3d in Sn0.95Fe0.05O2−δ. See DOI: 10.1039/c1cy00421b |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.