A sustainable route from the renewable myrcene to methyl ethersvia direct hydroalkoxylation†
Arno
Behr
*,
Leif
Johnen
and
Peter
Neubert
Technische Universität Dortmund, Lehrstuhl für Technische Chemie A, Emil-Figgestr. 66, 44227 Dortmund, Germany. E-mail: behr@bci.tu-dortmund.de; Fax: +49-231-7552311
First published on 8th November 2011
Abstract
A direct route to odoriferous methyl ethers from the monoterpene myrcene is presented. Investigating the activity of different catalytically active Lewis acids, the non-toxic and low-cost AlCl3 appeared to be very suitable. Thus, the common route to ethersvia the corresponding alcohols was reduced from four steps to one.
Terpenes are a large and useful class of hydrocarbons that are rarely taken into consideration in ever-present discussion about a new product platform derived from renewable biological feedstocks. They have been known for hundreds of years as components of essential oils obtained from leaves, flowers, and fruits of many plants. Perfumes, fragrances, and resins are the most common applications of pure or crude terpenes, but they are also used as starting materials in pharmaceutical syntheses.1 Recent review articles on catalytic conversions of terpenes to high-value fine chemicals were written by Swift and Monteiro.2
The purpose of our ongoing investigations is to use particularly the monoterpene myrcene as a renewable feedstock for green processes. Recently, we reported on amine derivatives which can be derived from homogeneous-catalysed hydroamination and telomerisation.3Myrcene appears to be a suitable renewable resource because it is an unsaturated renewable hydrocarbon which allows functionalization similar to the well-established petrochemistry. In addition, myrcene can be easily obtained from pyrolysis of β-pinene, a key compound in the crude resin of pines, and is already used in many industrial processes (Fig. 1).4
 | ||
| Fig. 1 Structure of myrcene. | ||
Thus, we are still focusing on the development of catalytic and atom-economical processes for adding value to the myrcene molecule. Herein, we present our investigations concerning catalysed hydroalkoxylation of myrcene with methanol leading directly to methyl ethers. As Boelens et al. have already shown in olfactory characterisations of homologous myrcene derivatives (alcohol, ether, acetate, and acetone), the resulting methyl ethers provide similar odour properties to the top-selling flavours geraniol and nerol.5 Until now, the ethers have been prepared from the corresponding alcohols, which in turn are obtained industrially from a laborious multistep synthesis (myrcene hydrochlorination, esterification with sodium acetate, and final hydrolysis). Thus, direct addition of an alcohol to myrcene would reduce the synthesis from four steps to one.
The analogous reaction of an amine, referred to as hydroamination, has been described in several protocols offering a large number of efficient and powerful catalysts.6 However, there are only a few contributions on the addition of an alcohol to non-activated alkenes, although this method generates higher-valued building blocks from simple alkenes. Currently huge efforts are spent to modify the electronic properties of the starting compounds in order to facilitate the direct addition.
By using catalytic amounts of Brønsted acids a cation is generated that can be easily attacked by a nucleophile. The low catalyst costs and in particular the non-toxicity of the acid are advantageous; however, regioselectivity depends on the position of the most stable cation (see industrial synthesis of methyl tert-butylether). Indeed, Rummelsburg reported on phosphoric acid and Milas used sulfuric acid as a suitable catalyst for hydroalkoxylation of myrcene, but the main products were ethers derived from a tertiary cation.7 Particularly, when using 1,3-dienes in the presence of acids, a variety of diene dimers and oligomers as well as dialkyl ethers with non-significant applications were obtained.
The ability of Lewis acids to receive electron pairs can also be used in hydroalkoxylation reactions. Dunach et al. conducted an intramolecular cyclisation of γ,δ-unsaturated alcohols using 5 mol% aluminium triflate.8 Adrio and Hii achieved good results when using copper(II) triflate as a catalyst. Phenol derivatives could be added to 1,3-dienes9 and norbornene10 with good yields. Nevertheless, the role of the Lewis acidic metal could not be entirely explained, in particular due to ligand exchange at the copper, which probably releases catalytically active trifluoromethanesulfonic acid.
The use of catalysts based on transition metals may provide a regioselective addition and a higher tolerance against functional groups. Hitherto, nickel,11rhodium,12 and palladium13 have been the prevalent metals which are used in ether syntheses starting directly from alkenes. For example, Fache and Mercier described a rhodium catalysed hydroalkoxylation of isoprene.12 In the presence of 1.5 mol% RhCl3 a good yield of 66% methyl ether was obtained. It was observed that longer reaction times (>20 h) led to back formation of the starting compounds from the evidently unstable methyl ethers. Similar evidence of an equilibrium-limited reaction was given by Utsunomiya et al. for the palladium-catalysed addition of phenol to cyclohexadiene.13a Concerning myrcene, Suzuki et al. reported on an isolated myrcene–palladium complex which can be reduced with NaOH or NaBH4 to palladium(0) and the desired methyl ethers.13e,f Another approach to direct ether synthesis from myrcene was described by Watanabe et al.; however, the reaction of myrcene with phenol catalysed by a palladium complex gave only a mixture of 17% phenyl ether.13d
Our investigations of the hydroalkoxylation reaction of myrcene with methanol started with a screening of different Lewis acids. As yet unpublished investigations of the telomerisation reaction with methanol, which are analogous to our protocol using amines,3b contained a screening of different palladium precursors and phosphine ligands. As we had not observed methanol–myrcene telomers we subsequently added small amounts of the Lewis acid BF3·OEt2 to increase the nucleophilicity of methanol. Surprisingly, this led to formation of the methyl ethers presented in this contribution. It was striking that these compounds were only obtained in the presence of the Lewis acid. These experiments indicated that the additive plays a central role in the hydroalkoxylation reaction and encouraged us to have a closer look into this reaction. The initial screening was performed with relatively high amounts of Lewis acids (10 mol%) and a distinct excess of methanol (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10). As shown in Scheme 1, different products and reaction pathways were observed under the investigated reaction conditions. Compounds 1–3 are the desired products, which were separately synthesised as pure compounds via Williamson etherification of the commercially available alcohols, characterised by NMR, and finally assigned by GC (see ESI†). Further methyl ethers were verified by GC/MS coupling and the yields were combined in the following experiments (4).
:10). As shown in Scheme 1, different products and reaction pathways were observed under the investigated reaction conditions. Compounds 1–3 are the desired products, which were separately synthesised as pure compounds via Williamson etherification of the commercially available alcohols, characterised by NMR, and finally assigned by GC (see ESI†). Further methyl ethers were verified by GC/MS coupling and the yields were combined in the following experiments (4).
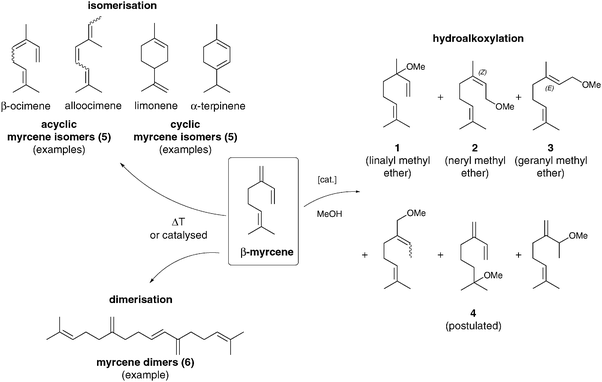 | ||
| Scheme 1 Desired products and observed side reactions in the hydroalkoxylation reaction of myrcene with methanol. | ||
Due to the high reactivity of myrcene, isomerisation to acyclic (e.g. β-ocimene, alloocimene) or cyclic (e.g.limonene, α-terpinene) monoterpenes (5) and dimerisation of myrcene to diterpenes (6) were also observed. Both reaction pathways are favoured in the case of harsh conditions, such as temperatures above 100 °C or an acidic environment. They were verified by GC/MS coupling. All primary screening experiments were carried out in a 10 ml multiplex reactor developed by our group.14 This set of six parallel reactors allows efficient high-throughput screening under inert and cost effective conditions. The reactor was charged with an argon pressure of 5 bar to keep the volatile methanol in solution. The screening proved that electron-deficient compounds BF3·OEt2 and AlCl3 were able to catalyse the desired reaction satisfactorily, as shown in Table 1. The methyl ethers 1–3 were generated with a total yield of 16% and 25%, respectively (entries 2 and 3). The selectivity towards all ethers was 51% when using AlCl3 as a catalyst. In contrast, the ‘softer’ Lewis acid InCl3 showed no activity (entry 4). Also the classic Lewis acids FeCl3 and ZnCl2 do not catalyse the addition of methanol to myrcene (entries 5 and 6).
| Entry | Lewis acid | X b (%) | Yields (%) | S c (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1–4 | |||
|
a Reaction conditions: 10.0 mol% Lewis acid, 7.5 mmol of myrcene, myrcene:methanol = 1:10, t = 4 h, T = 100 °C, 5 bar argon.
b Conversion of myrcene.
c Selectivity towards all ethers.
|
|||||||||
| 1 | — | 5 | — | — | — | — | 2 | 3 | — |
| 2 | BF3·OEt2 | 96 | 1 | 3 | 12 | 4 | 23 | 52 | 21 |
| 3 | AlCl3 | 70 | 3 | 6 | 16 | 11 | 14 | 20 | 51 |
| 4 | InCl3 | 12 | — | — | — | — | 4 | 8 | — |
| 5 | FeCl3 | 11 | — | — | — | — | 1 | 10 | — |
| 6 | ZnCl2 | 20 | — | — | — | — | 3 | 17 | — |
The following experiments were conducted only with AlCl3, because this inorganic salt is very competitive, non-toxic, and in particular much easier to handle than the also active BF3·OEt2. In addition, undesired side reactions giving isomers and dimers were less favoured. Although the solvent is not directly involved as a reagent, it can significantly influence the state of the Lewis acid/base equilibrium through solvation effects. Thus, solvents of different polarity were investigated concerning catalyst activity and yield of ethers (Table 2).
| Entry | Solvent b | X c (%) | Yields (%) | S d (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1–4 | |||
|
a Reaction conditions: 10.0 mol% AlCl3, 7.5 mmol of myrcene, myrcene:methanol = 1:10, t = 4 h, T = 100 °C, 5 ml solvent, 5 bar argon.
b
DMF = N,N-Dimethylformamide, DMPU = N,N′-dimethylpropylene urea, THF = tetrahydrofuran, DMSO = dimethyl sulfoxide, PC = propylene carbonate.
c Conversion of myrcene.
d Selectivity towards all ethers.
e No additional solvent was used.
f Conversion not quantifiable due to overlay of solvent peak with myrcene peak in the gaschromatogram.
|
|||||||||
| 1 | —e | 70 | 3 | 6 | 16 | 11 | 14 | 20 | 51 |
| 2 | DMF | 3 | — | — | — | — | 1 | 2 | — |
| 3 | DMPU | 14 | — | 2 | 2 | 3 | 6 | 1 | 50 |
| 4 | THF | 21 | — | 2 | 8 | 7 | 3 | 1 | 81 |
| 5 | DMSO | 26 | 1 | 3 | 6 | 5 | 9 | 2 | 58 |
| 6 | 1,3-Dioxane | 36 | 1 | 3 | 9 | 7 | 11 | 5 | 56 |
| 7 | Acetonitrile | 52 | 3 | 4 | 14 | 9 | 10 | 12 | 58 |
| 8 | PC | —f | 1 | 3 | 11 | 6 | 15 | 18 | — |
| 9 | Acetone | 60 | 3 | 6 | 16 | 13 | 14 | 6 | 63 |
| 10 | Isopropanol | 65 | 2 | 4 | 12 | 11 | 21 | 15 | 45 |
Synthesis of methyl ethers using solvents containing an amide group such as N,N-dimethylformamide and N,N′-dimethylpropylene urea is not feasible under the chosen conditions (entries 2 and 3). Upon varying the solvent, the best yields of the desired compounds 1–3 were achieved in acetone, with a total yield of 25%. Using acetone as solvent did not actually increase the yield compared to the solvent-free experiment, but fewer dimers were obtained, which results in higher selectivity towards methyl ethers of 63%. Besides, acetone is easy to remove from the product mixture.
Investigations of the catalyst amount c showed that the Lewis acid was able to sufficiently catalyse the direct addition even with a lower amount of 1.0 mol% (Table 3). Amounts higher than 5.0 mol% did not result in better yields (entries 4 and 5). At a concentration of 10.0 mol% the selectivity towards methyl ethers was even lower, at 63%, than when using an amount of 5.0 mol% (69%).
| Entry | c b (mol%) | R c | X d (%) | Yields (%) | S e (%) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1–4 | ||||
| a Reaction conditions: 7.5 mmol of myrcene, t = 4 h, T = 100 °C, 5 ml acetone, 5 bar argon. b Catalyst concentration. c Molar ratio of myrcene to methanol. d Conversion of myrcene. e Selectivity towards all ethers. | ||||||||||
| 1 | 0.2 | 1:10 |
5 | — | — | — | — | 2 | 3 | — |
| 2 | 0.5 | 1:10 |
8 | — | — | 1 | — | 3 | 4 | 13 |
| 3 | 1.0 | 1:10 |
36 | 2 | 5 | 9 | 9 | 8 | 3 | 69 |
| 4 | 5.0 | 1:10 |
54 | 2 | 5 | 16 | 14 | 10 | 5 | 69 |
| 5 | 10.0 | 1:10 |
60 | 3 | 6 | 16 | 13 | 14 | 6 | 63 |
| 6 | 5.0 | 1:1 |
68 | — | — | — | 1 | 25 | 42 | 1 |
| 7 | 5.0 | 1:4 |
51 | — | — | 2 | 3 | 21 | 25 | 10 |
| 8 | 5.0 | 1:8 |
51 | 1 | 3 | 12 | 9 | 14 | 12 | 49 |
| 9 | 5.0 | 1:10 |
60 | 4 | 5 | 16 | 14 | 14 | 7 | 65 |
| 10 | 5.0 | 1:15 |
64 | 3 | 4 | 14 | 11 | 17 | 9 | 50 |
Another important influencing factor is the substrate ratio R. Until now, a high excess of methanol (1:10) was used to allow sufficient shifting of the equilibrium. A variation of the molar ratio of both substrates in the presence of 5.0 mol% AlCl3 showed that the substrate ratio had a stronger influence on ether yields than on conversion. For example, an equimolar composition led to nearly the same conversion of 64–68% as was observed with a methanol excess of 1:15 (entries 6 and 10). Nevertheless, the total methyl ethers yields differed significantly between the experiments, being 1% and 32% respectively. With a molar ratio of 1:10 a yield of 39% methyl ethers and a selectivity of 65% were obtained. Increasing the ratio to 1:15 did not lead to a higher yield of methyl ethers. This confirmed that the initial choice of a molar ratio of 1:10 in the primary screening was very reasonable.
In experiments investigating the influence of temperature it was observed that at room temperature only 1% methyl ethers were obtained and the total conversion (4%) was relatively low (Table 4, entry 1). It can be seen that the yields increased at higher temperatures. The highest yields of methyl ethers were observed at 80 °C and 100 °C (about 37%, entries 4 and 5). At a temperature of 80 °C the selectivity was 88% and thus higher than at 100 °C (65%). It is obvious that temperatures above 80 °C lead to decreased selectivities as a result of thermally induced isomerisation and dimerisation. Finally, the reaction course was monitored using time dependent hydroalkoxylation experiments (Fig. 2). The reaction was carried out under the optimised conditions at a twenty-fold scale. A bigger autoclave (300 ml) was used to allow continuous sampling. The turnover number (TON) and turnover frequency (TOF) were calculated to determine catalyst activity. As a reference, the TOF was calculated when a constant conversion was observed. Accordingly, the hydroalkoxylation of myrcene with AlCl3 as catalyst provides a TON of 9.1 and a TOF of 2.5 h−1. Within 225 min, the yield of methyl ethers increased up to 42%. At the same time conversion was 46%, giving a selectivity of 91% towards methyl ethers. Afterwards no further increase in yield was observed and also the yield of side products remained at 8%, so the product distribution could not be controlled by means of different reaction times.
| Entry | T/°C | X b (%) | Yields (%) | S c (%) | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 1–4 | |||
|
a Reaction conditions: 5.0 mol% AlCl3, 7.5 mmol of myrcene, myrcene:methanol = 1:10, t = 4 h, 5 ml acetone, 5 bar argon.
b Conversion of myrcene.
c Selectivity towards all ethers.
|
|||||||||
| 1 | 26 | 4 | — | — | — | 1 | 3 | — | 25 |
| 2 | 60 | 21 | 1 | 3 | 4 | 5 | 3 | 5 | 62 |
| 3 | 70 | 25 | 2 | 3 | 4 | 6 | 3 | 7 | 60 |
| 4 | 80 | 43 | 3 | 6 | 15 | 14 | 3 | 2 | 88 |
| 5 | 100 | 60 | 4 | 5 | 16 | 14 | 14 | 7 | 65 |
| 6 | 120 | 58 | 3 | 7 | 12 | 14 | 14 | 8 | 62 |
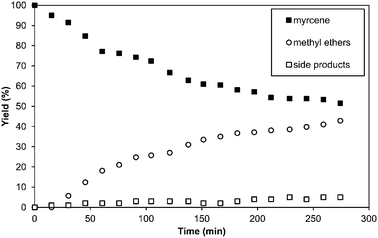 | ||
| Fig. 2 Activity of AlCl3 as catalystversus time. Reaction conditions: 5.0 mol% AlCl3, 150 mmol of myrcene, myrcene:methanol = 1:10, T = 80 °C, 100 ml acetone, 5 bar argon. | ||
Various protocols, particularly from Utsunomiya et al.,13a showed that the value of ΔG for the hydroalkoxylation reaction is very close to zero, so that even small modifications of the structure and substrate concentrations have a significant influence on the conversion. This information and our considerations about the modest yield of 42% indicate that the addition is reversible and that thermodynamic factors influence the conversion. This encouraged us to investigate the behaviour of neryl methyl ether in the presence of AlCl3 under the conditions chosen above. If the reaction was limited by equilibrium, we expected that the methyl ether would decompose as shown in Scheme 2.
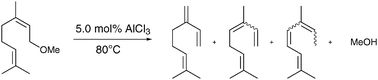 | ||
| Scheme 2
Decomposition reaction of neryl methyl ether. Reaction conditions: 5.0 mol% AlCl3, c(neryl methyl ether) = 1.5 mol l−1, neryl methyl ether:methanol 1:9, 4 h, 80 °C, acetone, 5 bar argon. | ||
In the hydroalkoxylation reaction under optimised conditions the conversion of myrcene was usually nearly completed and the molar ratio of methyl ethers to methanol was 1:9. Therefore neryl methyl ether was used with the same substrate ratio in the presence of 5.0 mol% AlCl3 at a temperature of 80 °C. After 4 h a conversion of 83% of the neryl methyl ether was observed. Major products were myrcene and myrcene isomers, as shown in Scheme 2. Furthermore, 20% of other methyl ethers were obtained, e.g.geranyl methyl ether and linalyl methyl ether. They may have resulted from a rearrangement, but it seemed more reasonable that an addition of methanol to a newly generated myrcene molecule occurred. These results additionally indicate that the reaction is equilibrium-limited. At conversions beyond this equilibrium state the products decomposed and the system seemed to be in a steady state concerning the ether yields. Obviously, decomposition of methyl ethers leads not only to myrcene but also to various isomers. Therefore, a longer reaction time has a negative effect on the selectivity of the reaction.
Nevertheless, the yield of methyl ethers is actually only moderate due to the limitations of thermodynamics. Classic methods, for example distilling off a low-boiling point co-product during the reaction or continuously extracting one product from the reaction mixture, are also not feasible for shifting the equilibrium to higher yields in this case. With the aim of performing a more sufficient synthesis of methyl ethers we propose a process flow sheet as shown in Scheme 3.
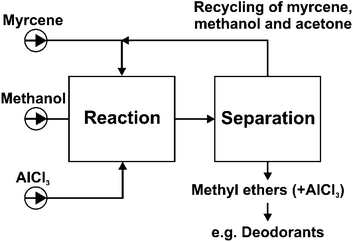 | ||
| Scheme 3 Proposal of a process flow sheet for the synthesis of methyl ethers from myrcene. | ||
After the reaction, the unreacted starting compounds (methanol bp 65 °C, myrcene and isomers bp 167 °C) and the solvent acetone (bp 56 °C) were distilled off and recycled to the initial reaction vessel. The residue containing flowery odorant methyl ethers (bp about 184–194 °C) and the catalyst aluminium chloride can be used directly as additives for deodorants in particular, because AlCl3 is added to many deodorants due to its antiperspirant effect.16 So the catalyst may advantageously remain in the product mixture and could even find an application in the end product. If a catalyst-free product is more preferred, the methyl ethers can be separated by an additional distillation of the mixture.
Conclusions
In summary, the present investigations showed that Lewis acids were capable catalysts for the direct addition of methanol to myrcene. Besides BF3·OEt2 the non-toxic and low-cost AlCl3 appeared to be very active. Systematic optimisation concerning solvent, substrate ratio, catalyst concentration, and temperature led to the best yield of 42% methyl ethers, which was reached in the presence of 5.0 mol% AlCl3 in acetone at a temperature of 80 °C within 225 min. Experimental investigation of the back reaction indicated an equilibrium-limited reaction. Using aluminium chloride as a catalyst allows a feasible and low-cost15 preparation of flowery odorant methyl ethers from myrcene. The prepared fragrances could be used in deodorants, particularly because the catalyst AlCl3 may remain in the product mixture due to its antiperspirant effect.Notes and references
- (a) A. A. Newman, Chemistry of Terpenes and Terpenoids, Academic Press Inc., London, 1972 Search PubMed; (b) J. Verghese, Terpene Chemistry, McGraw-Hill Publishing Company, New Delhi, 1982 Search PubMed; (c) E. Breitmaier, Terpenes, Wiley-VCH, Weinheim, 2006 Search PubMed.
- (a) K. A. D. Swift, Top. Catal., 2004, 27, 143 CrossRef CAS; (b) J. L. F. Monteiro and C. O. Veloso, Top. Catal., 2004, 27, 169 CrossRef CAS.
- (a) A. Behr, L. Johnen and N. Rentmeister, Adv. Synth. Catal., 2010, 352, 2062 CrossRef CAS; (b) A. Behr, L. Johnen and A. J. Vorholt, ChemCatChem, 2010, 2, 1271 CrossRef CAS.
- A. Behr and L. Johnen, ChemSusChem, 2009, 2, 1072 CrossRef CAS.
- H. Boelens, R. Ter Heide and F. Rijkens, Am. Perfum. Cosmet., 1968, 83, 27 CAS.
- (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675 CrossRef; (b) T. E. Müller, K. C. Hultzsch, M. Yus, F. Foubelo and M. Tada, Chem. Rev., 2008, 108, 3795 CrossRef; (c) J. Seayad, A. Tillack, C. G. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795 CrossRef CAS; (d) U. Dzhemilev, G. Tolstikov and R. Khusnutdinov, Russ. J. Org. Chem., 2009, 45, 957 CrossRef CAS.
- (a) US Pat., 2467330, 1949; (b) US Pat., 2388765, 1945.
- L. Coulombel, M. Rajzmann, J.-M. Pons, S. Olivero and E. Dunach, Chem.–Eur. J., 2006, 12, 6356 CrossRef CAS.
- L. A. Adrio and K. K. Hii, Chem. Commun., 2008, 2325 RSC.
- J. G. Taylor, N. Whittall and K. K. M. Hii, Chem. Commun., 2005, 5103 RSC.
- US Pat., 4843180, 1989.
- (a) E. Fache and C. Mercier, J. Mol. Catal., 1993, 78, 21 CrossRef CAS; (b) K. C. Dewhirst, J. Org. Chem., 1967, 32, 1297 CrossRef CAS; (c) H. Kawazura, T. Takagaki and Y. Ishii, Bull. Chem. Soc. Jpn., 1975, 48, 1949 CrossRef CAS.
- (a) M. Utsunomiya, M. Kawatsura and J. F. Hartwig, Angew. Chem., Int. Ed., 2003, 42, 5865 CrossRef CAS; (b) W. Gaube and H. Stegemann, J. Prakt. Chem., 1984, 326, 947 CrossRef CAS; (c) P. W. Jolly and N. Kokel, Synthesis, 1990, 771 CrossRef CAS; (d) S. Watanabe, K. Suga and K. Hijikata, Isr. J. Chem., 1971, 9, 273 CAS; (e) M. Takahashi, H. Suzuki, Y. Morooka and T. Ikawa, Chem. Lett., 1979, 53 CrossRef CAS; (f) M. Takahashi, H. Urata, H. Suzuki, Y. Morooka and T. Ikawa, J. Organomet. Chem., 1984, 266, 327 CrossRef CAS.
- A. Behr, G. Henze, L. Johnen and S. Reyer, J. Mol. Catal. A: Chem., 2008, 287, 95 CrossRef CAS.
- Prices in 2009: 0.29–0.31 € kg−1, source: www.icispricing.com.
- (a) US Pat., 5143718, 1992; (b) E. Hölzle and A. M. Kligman, J. Soc. Cosmet. Chem., 1979, 30, 279 Search PubMed; (c) W. B. Shelley and H. J. Hurley, Acta Derm.-Venereol., 1975, 55, 241 CAS; (d) Ger Pat., 3722674, 1987.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, general procedure, synthesis of pure compounds, 1H and 13C NMR. See DOI: 10.1039/c1cy00359c |
| This journal is © The Royal Society of Chemistry 2012 |