 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cyclohexane oxidation using Au/MgO: an investigation of the reaction mechanism
Marco
Conte
*a,
Xi
Liu
a,
Damien M.
Murphy
a,
Keith
Whiston
b and
Graham J.
Hutchings
*a
aCardiff Catalysis Institute, School of Chemistry, Cardiff University, Cardiff, CF10 3AT, UK. E-mail: ConteM1@cardiff.ac.uk; Hutch@cardiff.ac.uk
bINVISTA Textiles (UK) Limited, P.O. Box 2002, Wilton, Redcar, TS10 4XX, UK
First published on 29th October 2012
Abstract
The liquid phase oxidation of cyclohexane was undertaken using Au/MgO and the reaction mechanism was investigated by means of continuous wave (CW) EPR spectroscopy employing the spin trapping technique. Activity tests aimed to determine the conversion and selectivity of Au/MgO catalyst showed that Au was capable of selectivity control to cyclohexanol formation up to 70%, but this was accompanied by a limited enhancement in conversion when compared with the reaction in the absence of catalyst. In contrast, when radical initiators were used, in combination with Au/MgO, an activity comparable to that observed in industrial processes at ca. 5% conversion was found, with retained high selectivity. By studying the free radical autoxidation of cyclohexane and the cyclohexyl hydroperoxide decomposition in the presence of spin traps, we show that Au nanoparticles are capable of an enhanced generation of cyclohexyl alkoxy radicals, and the role of Au is identified as a promoter of the catalytic autoxidation processes, therefore demonstrating that the reaction proceeds via a radical chain mechanism.
1. Introduction
The partial oxidation of cyclohexane to cyclohexanone and cyclohexanol is a major process in industrial chemistry since these two products are chemical precursors for the manufacture of nylon-6 and nylon 6,6 fibres via oxidation to adipic acid.1 Typically this reaction is carried out in the liquid phase under aerobic conditions using air as oxidant at 125–160 °C and 3–15 bar, normally using cobalt-based homogeneous catalysts, such as cobalt(II)-naphthenate or cobalt acetylacetonate.2,3 The reaction is known to proceed by a free radical autoxidation mechanism.4,5 Heterogeneous catalysts such as MoO3, Cr2O3 and WO3 have also been reported,6 and gas phase catalytic oxidation can be used.7 However, the major drawback of these processes lies in the poor selectivity control to cyclohexanol and cyclohexanone. In fact, in order to avoid the formation of a high organic acid content, and to preserve a high selectivity to the alcohol and the ketone (>70%), conversion values are industrially limited to 4–12%.8,9 This prompted several research groups to explore the possibility of developing new catalysts, for example using gold-based catalysts for the liquid phase oxidation of cyclohexane10–14 because of the efficiency of these materials in a vast array of selective oxidation reactions.15–17However, it is currently debated if gold-based materials are real catalytic systems or rather act as promoters of the autoxidation pathways for the cyclohexane oxidation reaction.18 SiO2 supported gold catalysts, modified by doping with TiO2, were reported to be capable of high conversion,19,20 relative to the industrial catalyst, of ca. 10% and selectivity to the alcohol and ketone (K/A oil) > 70%, which was not observed in the absence of supported gold nanoparticles; the conclusion reached was that this was a real catalyst for cyclohexane oxidation. On the other hand, investigation of the cyclohexane oxidation over Au/Al2O3, Au/TiO2 and Au/SBA-1521 showed that the reaction proceeds via a pure radical pathway with products typical of autoxidation and the reaction could be fully inhibited by means of radical scavengers. Finally, it has also been shown that gold nanoclusters supported on hydroxyapatite were capable of displaying high activity towards cyclohexane22 and that no reaction was taking place in the absence of gold, although the reaction required the presence of radical initiators, typically tert-butylhydroperoxide (TBHP).
This lack of unambiguous evidence on the true catalytic role of gold in the cyclohexane oxidation, prompted us to carry out a mechanistic study using Au/MgO because of its excellent selective oxidation properties.23–25 To test if the cyclohexane oxidation reaction proceeds via a radical mechanism, we employed X-band EPR spectroscopy combined with the spin trapping technique26–28 as well as radical scavengers. The principle of the spin-trapping methodology relies on the fast selective addition, i.e. trapping, of short-lived radicals to a diamagnetic spin trap, usually a nitrone or a nitroso compound, such as 5,5-dimethyl-1-pyrroline-N-oxide (DMPO). The product of this addition, known as the spin adduct, is a persistent free nitroxide radical with a sufficiently long lifetime to enable detection by conventional EPR spectroscopy (Scheme 1).29 Because of the hyperfine coupling between the unpaired electron in the spin adduct and the 1H in the beta position for the chosen spin trap, it is often possible to assign the structure of the original short-lived radicals due to changes in the 14N and 1H hyperfine coupling constants of the spin trap molecule.30 In this paper we present the results of this mechanistic study.
 | ||
| Scheme 1 Spin trapping mechanism for DMPO with a free radical (R˙). | ||
2. Materials and methods
2.1 Chemicals
DMPO, toluene, dichloromethane, chloroform and other chemicals were purchased from Aldrich and used without further purification unless otherwise specified.2.2 EPR experiments
X-band continuous wave (CW) EPR spectra were recorded at room temperature in deoxygenated cyclohexane, using a Bruker EMX spectrometer. The typical instrument parameters were: centre field 3487 G, sweep width 100 G, sweep time 55 s, time constant 10 ms, power 5 mW, modulation frequency 100 kHz, and modulation width 1 G. Quantitative spectral analysis was carried out using WinSim software.31The spin trapping experiments were performed using the following procedure: 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) (0.1 mL of 0.1 M solution in cyclohexane) was added to the substrate (0.1 mL of 2.5 molar% solution of cyclohexyl-hydroperoxide – hereafter abbreviated CHHP – in cyclohexane), in an EPR sample tube. The mixture was deoxygenated by bubbling N2 for 1 min prior to recording the EPR spectra in order to enhance the signal intensity.30 For the samples containing the Au/MgO catalyst, deoxygenation was carried out at room temperature, 5 min after the mixing of the catalyst with the reaction mixture.
2.3 Catalyst activity studies and product analysis
Catalytic oxidation of cyclohexane (Alfa Aesar, 8.5 g, HPLC grade) was carried out in a glass bench reactor using 6 mg of catalyst in 10 mL of cyclohexane. The reaction mixture was magnetically stirred at 140 °C under 3 bar O2 for 17 hours. Samples of the reaction mixture were periodically analyzed by gas chromatography (Varian 3200) with a CP-Wax 42 column. Adipic acid was converted to its corresponding ester for quantification purposes and chlorobenzene added as internal standard.2.4 Catalyst preparation
Au/MgO catalysts were prepared via impregnation of an aqueous solution (1 mL) of HAuCl4·3H2O (JM, assay 49%,) over MgO (BDH, 1 g), in order to obtain a gold loading of 1 wt%. The suspension was continuously stirred for 30 min. The sample was dried at 120 °C overnight and consecutively calcined at 300 °C for two hours. The same procedure was applied for the preparation at 0.1 and 0.01 wt% Au loading, adjusting accordingly the Au amount in the starting aqueous solution.2.5 Catalyst characterization by XRPD
X-ray powder diffraction patterns (XRPD) were acquired using a X'Pert PANalytical diffractometer operating at 40 kV and 40 mA selecting the Cu-Kα radiation. Analysis of the patterns was carried out using X'Pert HighScore Plus software. In order to enhance the signal for the assignment and determination of line broadening in the Au peaks, XRPD patterns were base line corrected, and the signal-to-noise ratio was increased using a Gaussian filter. The peak identification was carried out using a second derivative algorithm to further enhance the signal. Crystallite sizes for the metal and metal oxide clusters was determined using the Scherrer equation32 assuming spherical particle shapes and a K factor of 0.89. The line broadening was determined using a Voigt profile function,33 convoluting the Gaussian and Lorentzian profile part of the reflection peak and the instrumental broadening for the Bragg–Brentano geometry used was estimated to be 0.06° 2θ.2.6 Catalyst characterization by DR-UV
UV-Vis diffuse reflectance spectra were collected using a Harrick Praying Mantis cell mounted on a Varian Cary 4000 spectrophotometer. The spectra were collected from 900 to 200 nm at a scan speed of 60 nm min−1. Background correction was carried out using teflon powder (Spectralon). The sample was mounted on a 3 mm diameter diffuse reflectance sampling cup.3. Results and discussion
3.1 Catalytic tests
Au/MgO was investigated for the cyclohexane oxidation using different gold loadings in the presence and absence of radical initiators (Table 1). It is possible to observe that in the absence of radical initiators (entries 1–3), Au/MgO displays little activity; ca. 2% or less, when compared to autoxidation, which was in the range of ca. 1.1% (entry 4). However, it is evident the effect of gold on the selectivity of the reaction with enhanced cyclohexanol formation up to 50%; an effect that is lost when the amount of gold present in the catalyst is reduced. This is also reflected in variations in the amount of cyclohexyl hydroperoxide (CHHP). In fact, when the gold loading is reduced from 1 to 0.01 wt%, the selectivity to CHHP increases from 7% to ca. 27%, while in the absence of catalyst the selectivity to CHHP was about 57%. These data clearly indicate the influence of gold in the oxidation process, even when the metal is present in low concentrations, and including CHHP decomposition34 and the possible quenching of some radicals intermediates by gold.35,36 This relates to the catalytic decomposition of CHHP by a gold surface, which would drive the selectivity towards cyclohexanol. In fact, if only autoxidation was operative, an increase in conversion would always be accompanied by a loss of selectivity to the alcohol.37 This is due to the fact that when autoxidation is operating, it always leads to the ketone (see Section 3.3).| Entry | Catalyst | Au loading (wt%) | Initiator | Conversiona (%) | Selectivityb (%) | K/A ratio | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| K | A | CHHP | AA | Total | ||||||
| a We detected a closed carbon mass balance within an experimental error of 5%. b The missing components are carboxylic acids as ring opening products. | ||||||||||
| 1 | Au/MgO-Imp | 1 | — | 1.9 | 30 | 51 | 7 | 0 | 88 | 0.58 |
| 2 | Au/MgO-Imp | 0.1 | — | 1.7 | 35 | 44 | 15 | 0 | 94 | 0.8 |
| 3 | Au/MgO-Imp | 0.01 | — | 1.3 | 28 | 36 | 27 | 0 | 91 | 0.78 |
| 4 | Autoxidation | — | — | 1.1 | 19 | 22 | 57 | 0 | 98 | 0.86 |
| 5 | MgO | — | — | 1.4 | 36 | 25 | 4 | 0 | 65 | 1.44 |
| 6 | Au/MgO-Imp | 1 | AIBN | 4.5 | 34 | 51 | 6 | 0 | 91 | 0.67 |
| 7 | Autoxidation | — | AIBN | 6.7 | 32 | 57 | 2 | 0 | 91 | 0.56 |
| 8 | Au/MgO-Imp | 1 | TBHP | 5.0 | 29 | 50 | 0 | 19 | 98 | 0.58 |
| 9 | Autoxidation | — | TBHP | 6.6 | 33 | 52 | 0 | 15 | 100 | 0.63 |
However, because the conversion values are just above those obtained by autoxidation, these data suggest that the reaction still proceeds via a radical chain mechanism. A control experiment using MgO in the absence of gold (entry 5) showed that the conversion is higher than the one observed for autoxidation, but lower than that observed for the gold containing catalyst, where the selectivity is shifted to the ketone.
In view of this, the effect of initiators such as azo-bis-isobutyronitrile (AIBN) and tert-butylhydroperoxy radical (TBHP) were evaluated (Table 1). When AIBN was used (entries 6 and 7), the activity of Au/MgO catalyst increased to ca. 4% and with selectivity values quite similar to those obtained for the catalyst in absence of initiators (entry 1). However, despite this apparent increase in conversion, this is less than the value obtained in the presence of AIBN only (ca. 7%). This is consistent with the role of Au as an inhibitor for the reaction. In contrast, if TBHP was used (entries 8 and 9), the activity of Au/MgO increased, (up to ca. 5%) but, also in this case, it was lower than that for the reaction carried out in presence of TBHP only (ca. 6.6%). Moreover, the selectivity control to cyclohexanol and cyclohexanone was limited, and a significant amount of adipic acid was detected, an effect that should be considered a direct consequence of the large amount of peroxides in solution.38
3.2 Nature of the catalyst
The catalyst was obtained by impregnating an aqueous solution of HAuCl4 into MgO as support. In view of this preparation procedure, and the well known properties of MgO, the final material is a mixture of MgO and Mg(OH)2. This was confirmed by XRPD (Fig. 1) where the following phases were identified: MgO (periclase),39 Mg(OH)2 (brucite),40 with traces amounts of MgCO3 (magnesite).41 However, this does not affect our results and this material is referred to hereafter as Au/MgO.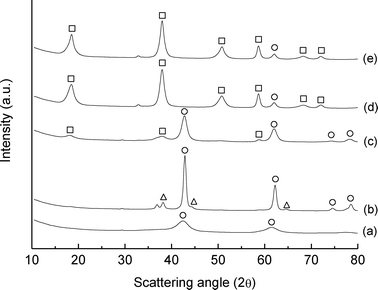 | ||
| Fig. 1 XRPD patterns of: (a) MgO starting material, (b) Au/MgO catalyst 1 wt%, (c) Au/MgO catalyst 0.1 wt%, (d) Au/MgO catalyst 0.01 wt% and (e) MgO support impregnated with water. The symbols used indicate: (○) MgO – periclase (□) Mg(OH)2 – brucite, and (△) Au. | ||
From the XRPD pattern it was also possible to estimate the particle size of gold, using the Au(111) reflection at 38.2° 2θ.42 These were estimated to be ca. 17 and 8 nm for the materials containing 1 and 0.1 wt% Au respectively. In contrast, no gold reflection for the 0.01 wt% Au sample was detected, indicating a particle size below the detection limit of the XRD method, which is ca. 4 nm. Gold nanoparticles were also well identified by means of plasmon resonance43via diffuse reflectance UV-Vis spectroscopy (Fig. 2), and the band intensity of the spectra is consistent with the particles size estimation obtained by XRPD; i.e., with gold particles less than 4 nm diameter for the 0.01 wt% Au sample.
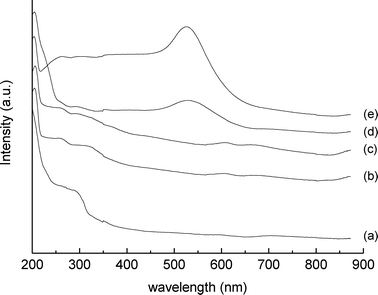 | ||
| Fig. 2 Diffuse reflectance UV-Vis spectra of: (a) MgO starting material, (b) MgO support impregnated with water, (c) Au/MgO catalyst 0.01 wt%, (d) Au/MgO catalyst 0.1 wt% and (e) Au/MgO catalyst 1 wt%. | ||
A detailed analysis of the diffuse reflectance spectra shows that MgO, even for the untreated sample, presents absorption bands in the UV range at ca. 210 and 280–300 nm. While MgO is commonly regarded as a white standard for DR-UV in the visible region,44 absorption bands at 213 nm and 282 nm are associated with the excitation of four-fold and three-fold coordinated surface O2− anions at edge and corner positions of MgO crystals respectively.45,46 This is a consequence of the use of a commercial purity grade MgO as catalyst support, rather than the use of an optically purity grade MgO.
3.3 Free radical chain mechanism
In order to rationalize the results described above on the behaviour of Au/MgO catalyst, it is important to consider the accepted free radical chain reaction mechanism in the autoxidation case, and the different decomposition pathways for AIBN and TBHP. The commonly accepted radical chain pathway in the formation of cyclohexanol and cyclohexanone is reported in Scheme 2 (eqn (1)–(7)).47,48 | ||
| Scheme 2 Radical chain pathway in the formation of cyclohexanol and cyclohexanone during the oxidation of cyclohexane. | ||
The first step of the oxidation is the initiation, which involves activation of the C–H bond via abstraction of an H atom. This can occur by a number of events, including: (i) cleavage by an unsaturated metal centre,49 (ii) H abstraction by a peroxide species (either peroxyl or alkoxy radicals present in solution),28 or (iii) H abstraction by a superoxide species (O2−) bound to metal centres or metal oxides.50 For each of these processes this results in the formation of a carbon centred parent radical (C6H11˙). It is well known that carbon-centred radicals are extremely reactive51 and they immediately react with O2 to give peroxyl radicals, in our case cyclohexyl peroxyl radical (C6H11–OO˙) according to (eqn (2)). In principle, the oxygen incorporated into the products can originate from oxygen dissolved in solution or from adsorbed oxygen species on the metal oxide surface.52 C6H11–OO˙ can react further with cyclohexane to give cyclohexyl hydroperoxide (CHHP) and another C6H11˙ radical, thus ensuring propagation of the reaction (eqn (3)). It should be stressed that, in this scheme C6H11–OO˙ is the main radical chain carrier, with CHHP reacting in a sequence that finally yields cyclohexanone (eqn (4) and (5)).
In contrast, cyclohexanol can be obtained by hydrogenation of cyclohexyl alkoxy radical C6H11–O˙ (eqn (6)), cleavage of CHHP (eqn (7)) or via recombination of two peroxyl radicals (eqn (8)).53 In principle, cyclohexanol can originate from insertion of lattice oxygen from the metal oxide to the C6H11˙ parent radical adsorbed over the catalyst surface.54 However, when MgO only was tested, no activity was detected which therefore rules out this latter possible route, and this is also not related to the purity level of MgO.
From this model, it is evident that the ketone is always obtained (eqn (5) and (8)) if autoxidation is operating. Therefore selectivity control, if any, can occur only in the decomposition step of CHHP (eqn (7)). More recently, solvent-cage models have also been proposed for the autoxidation pathways,55 to explain the alcohol and ketone formation.
3.4 Radical initiators
In this context, radical initiators were used to promote the oxidation reaction. However, it is necessary to emphasise that AIBN and TBHP, despite both being radical initiators, operate through a different decomposition pathway and this can help to explain the differences in reactivity observed in the present study. AIBN is thermally decomposed to two cyanopropyl radicals and N2 (Scheme 3).51 Carbon centred radicals are extremely reactive and they can instigate H abstraction, or they can react further with the oxygen present in the reaction media to form peroxy radicals. The new cyanopropyl peroxy radical can also act as a H abstractor, and therefore initiate the reaction acting on eqn (1).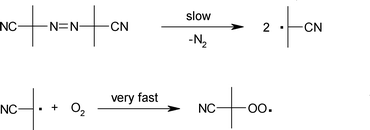 | ||
| Scheme 3 AIBN decomposition pathway. | ||
In contrast TBHP can undergo homolytic cleavage of the O–O bond and from this to peroxyl condensation via the set of equations ((9)–(11)) reported in Scheme 4.56
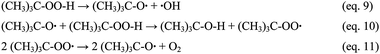 | ||
| Scheme 4 TBHP decomposition pathway. | ||
Considering the mechanisms of action for these initiators and the results obtained in the current case (i.e., an enhanced oxidation when initiators are used, but a decrease when combined with Au/MgO) it would be possible to conclude that Au/MgO can actually quench some of the radicals generated by the initiators, such as cyanopropyl radicals,57,58 but at the same time acting as a promoter for the O–O cleavage in the TBHP decomposition;34–36 this could also explain the enhanced selectivity to cyclohexanol. It is possible that quenching comes at least in part from the support, with the activation role from the metal nanoparticle counterpart. Many metal oxides, such as MgO, present neutral oxygen vacancies59,60 that could possibly aid in the localisation of peroxyl species, and so partially inhibit the reaction when initiators are used. A similar quenching effect was observed for ZnO in the aldehyde oxidation by Au/ZnO catalysts.61 In fact, regardless of the insulator properties of MgO, compared to the semi-conducting properties of ZnO which may increase the tendency to radical quenching, the presence of defects in or on the MgO crystals may facilitate radical trapping and stabilisation.62 This effect has been experimentally observed for methyl radicals in gas phase.63
3.5 EPR spin trapping experiments
In order to explain the enhanced selectivity to the alcohol when Au is used, spin trapping was carried out in presence of CHHP using 5,5-dimethyl-1-pyrroline-N-oxide (DMPO) as the spin trap. CHHP was chosen for two reasons: it is a peroxide involved in the cyclohexane oxidation, and it can be prepared free of water, which could interfere in the determinations.When the reaction was carried out at room temperature in the presence of DMPO as spin trap, CW EPR spectra were acquired in the presence (Fig. 3) and absence (Fig. 4) of Au/MgO. Simulation of the spectrum and comparison with literature values allowed the identification of all the radical intermediates which are expected in the autoxidation pathway of cyclohexane to cyclohexanone and cyclohexanol. In particular these species include: di-tert-butyl-nitroxide derivative,64 a DMPO–O–C6H11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 65 and a DMPO–OOC6H11 spin adduct,66 a DMPO–C6H11 carbon-centred adduct characteristic of the parent radical C6H11˙67 and a carbon centred adduct which is possibly a DMPO–C(OH)R2 species.68 The spin Hamiltonian parameters of these spin adducts are reported in Table 2.
65 and a DMPO–OOC6H11 spin adduct,66 a DMPO–C6H11 carbon-centred adduct characteristic of the parent radical C6H11˙67 and a carbon centred adduct which is possibly a DMPO–C(OH)R2 species.68 The spin Hamiltonian parameters of these spin adducts are reported in Table 2.
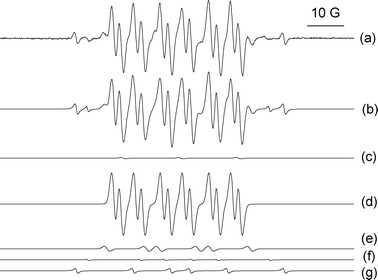 | ||
| Fig. 3 Deconvoluted EPR spectra of DMPO spin adducts obtained during the decomposition of CHHP in cyclohexane in the presence of Au/MgO: (a) experimental spectrum and (b) simulated spectrum; (c) di-tert-butyl-nitroxide derivative, (d) DMPO–O–C6H11 spin adduct, (e) a DMPO–OO–C6H11 adduct, (f) DMPO–C6H11 carbon centred adduct, and (g) carbon centred adduct, which is possibly a DMPO–C(OH)R2 species. | ||
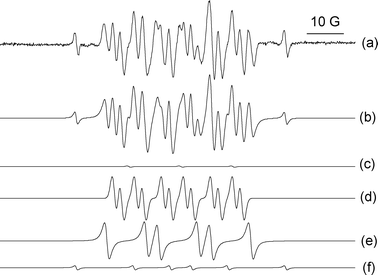 | ||
| Fig. 4 Deconvoluted EPR spectra of the DMPO spin adducts obtained during cyclohexane autoxidation at room temperature in the presence of CHHP: (a) experimental spectrum and (b) simulated spectrum; (c) di-tert-butyl-nitroxide derivative, (d) DMPO–O–C6H11 spin adduct, (e) DMPO–OO–C6H11 adduct, and (f) carbon centred adduct which is possibly a DMPO–C(OH)R2 species. | ||
| Radical | a N /G | a H(β)/G | a H(γ)/G |
|---|---|---|---|
| tert-Butyl nitroxide derivative | 14.2 (14.2) | — | — |
| C6H11–O˙ | 13.4 (13.4) | 6.00 (6.00) | 1.80 (1.90) |
| C6H11–OO˙ | 14.3 (14.2) | 10.8 (10.8) | |
| C6H11˙ | 14.3 (n.d.) | 21.2 (n.d.) | |
| R2(OH)C˙ | 15.6 (15.8) | 25.9 (25.7) |
It should be noted that the spin trapping technique only allows for semi-quantitative determination of the adducts detected. This is a consequence of the life-time of the spin adduct, the nature of the solvent, the temperature and the efficiency of the capture reaction which is different for each radical.28,30 With these limitation in mind, the simulation revealed the following semi-quantitative values: di-tert-butyl-nitroxide derivative (2%); DMPO–O–C6H11 (81%); DMPO–OO–C6H11 (8%); DMPO–C6H11 (4%); and possible DMPO–C(OH)R2 adduct (5%).
When the same experiment was carried out in the absence of Au/MgO, to assess the CHHP decomposition via the autoxidation pathway, the following species were obtained (Fig. 4): di-tert-butyl-nitroxide derivative, DMPO–O–C6H11 and DMPO–OO–C6H11 spin adducts and the possible DMPO–C(OH)R2 adduct. The hyperfine splitting constants of these spin adducts are reported in Table 2. These species were quantified as follow: di-tert-butyl-nitroxide derivative (2%); DMPO–O–C6H11 (50%); DMPO–OO–C6H11 (43%); and DMPO–C(OH)R2 (5%).
The species trapped in the presence and absence of Au/MgO are basically the same in both sets of experiments, but the parent radical is not detected in the autoxidation case. In contrast, the most remarkable difference between the two sets is an increased quantity of alkoxy (C6H11–O˙) species, from 50% to 80% when gold is used. This indicates that Au/MgO is capable of enhancing alcohol formation by cleavage of the O–O bond of CHHP.
However, as the spin trapping technique is prone to artifacts, control tests were necessary and we carefully carried out. In fact, spin adducts can be formed not just by the radical addition to a spin trap, but also by nucleophilic addition followed by oxidation of the spin adduct69,70 or by oxidation of the spin trap followed by nucleophilic addition, in our case by species such as −OH. Control tests in the presence of Au/MgO and the spin trap, but in the absence of substrate, did not reveal any trace of the DMPOX oxidation product.71 Moreover, the trace amount of di-tert-butyl-nitroxide derivative should be considered ubiquitous in these types of experiments and can be discounted.28,61
On the other hand, no DMPO–OH adduct was detected in the tests we carried out. If homolytic cleavage of a substrate is considered, it is not unprecedented, by using spin trapping, to detect just one of the two expected partners (in our case RO˙). This could be due to a failure of the spin trap molecule to capture the ˙OH species under the reaction conditions used, or by termination of ˙OH on the metal surface. In addition, it is known that ˙OH is among the most reactive known radical species,72 and therefore difficult to trap.
This still does not preclude a redox cycle mediated by the metal centre. At present, this has been accredited in the case of oxidation by means of Co(III) salts in agreement with the Haber–Weiss cycle,73 (Scheme 5, eqn (12)–(14)).
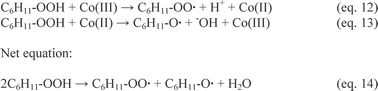 | ||
| Scheme 5 Haber–Weiss cycle for the oxidation of cyclohexane mediated by Co(III). | ||
No adduct from nucleophilic attack and oxidation was detected when Au was present. Moreover, systems like those in eqn (12)–(14) have a K/A ratio in the range of 1.5,2,21 while in our case the K/A ratio is in the range of 0.6 with clear selectivity to the alcohol, therefore supporting the conclusion that gold has to operate a homolytic cleavage of CHHP in agreement with eqn (7).
3.6 Effect of CBrCl3 as radical scavenger
In view of the data reported so far, supported gold nanoparticles appear to be capable of accelerating the reaction rate (although to a minor extent) but within the autoxidation pathway, i.e., without inducing alternative reaction mechanisms or intermediates from those that would be expected from the free radical chain mechanism. In order to test this hypothesis, a reaction in the presence of CBrCl3 was carried out. CBrCl3 can act as radical scavenger by cleavage of the C–Br bond by carbon centred radicals.74 Therefore, CBrCl3 is capable of reacting with the parent C6H11˙ radical to yield to C6H11–Br and thus quench the reaction.When CBrCl3 was used (Fig. 5) the reaction was completely inhibited at the initial stage (first 2 hours). Then as long as CBrCl3 was consumed in the reaction media, with consequent C6H11–Br formation which is detected after ca. 30 min, product formation is observed. In particular, when all CBrCl3 was consumed (after 2 hours), the oxidation reaction newly started as in the absence of inhibitor, thus demonstrating that Au/MgO promotes the decomposition of CHHP but within a free radical-chain reaction mechanism.
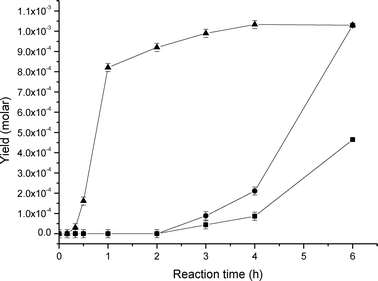 | ||
| Fig. 5 Product evolution in the liquid phase oxidation of cyclohexane over Au/MgO in presence of CBrCl3 as radical scavenger at 140 °C under 3 bar of O2: (▲) bromocyclohexane, (●) cyclohexanol and (●) cyclohexanone. | ||
4. Conclusions
The current literature on the use of Au based catalysts in cyclohexane oxidation describes the role of Au to be that of either a true catalyst or a mere promoter for the reaction. The present study shows a situation which should be considered as intermediate within these two extremes. In fact, Au is capable of accelerating the reaction, without the need for initiators, and so it is by definition a catalyst for the cyclohexane oxidation, but this acceleration occurs by increasing the concentration of species (through C6H11–OOH or C5H11–OO˙) which are chain carriers in the radical pathway of the reaction and therefore promote catalytic autoxidation processes via a radical-chain mechanism. These findings can shed light on the use of gold catalysts for alkane oxidations showing that selectivity control is possible but with a limited effect on the conversion of the reaction.Acknowledgements
The authors thank INVISTA for financial support.Notes and references
- J. G. Speight, Chemical and Process Design Handbook, McGraw-Hill, New York, 2002, p 2.185 Search PubMed.
- A. Ramanathan, M. S. Hamdy, R. Parton, T. Maschmeyer, J. C. Jansen and U. Hanefeld, Appl. Catal., A, 2009, 355, 78–82 CrossRef CAS.
- H. C. Shen and H. S. Weng, Ind. Eng. Chem. Res., 1988, 27, 2254–2260 CrossRef CAS.
- A. F. Masters, J. K. Beattie and A. L. Roa, Catal. Lett., 2001, 75, 159–162 CrossRef CAS.
- V. Govindan and A. K. Suresh, Ind. Eng. Chem. Res., 2007, 46, 6891–6898 CrossRef CAS.
- C. Hettige, K. R. R. Mahanama and D. P. Dissanayake, Chemosphere, 2001, 43, 1079–1083 CrossRef CAS.
- M. Conte and V. Chechik, Chem. Commun., 2010, 46, 3991–3993 RSC.
- U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. de Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinace and E. L. Fires, Appl. Catal., A, 2001, 211, 1–17 CrossRef CAS.
- G. P. Chiusoli and P. M. Maitlis, Metal-catalysis in Industrial Organic Processes, RSC Publising, Cambridge, 2008, p 29 Search PubMed.
- L. X. Xu, C. H. He, M. Q. Zhu and S. Fang, Catal. Lett., 2007, 114, 202–205 CrossRef CAS.
- G. M. Lu, D. Ji, G. Qian, Y. X. Qi, X. L. Wang and J. S. Suo, Appl. Catal., A, 2005, 280, 175–180 CrossRef.
- K. K. Zhu, J. C. Hu and R. Richards, Catal. Lett., 2005, 100, 195–199 CrossRef CAS.
- L. Li, C. Jin, X. C. Wang, W. J. Ji, Y. Pan, T. van der Knaap, R. van der Stoel and C. T. Au, Catal. Lett., 2009, 129, 303–311 CrossRef CAS.
- Y.-J. Xu, P. Landon, D. Enache, A. Carley, M. Roberts and G. Hutchings, Catal. Lett., 2005, 101, 175–179 CrossRef CAS.
- C. Della Pina, E. Falletta, L. Prati and M. Rossi, Chem. Soc. Rev., 2008, 37, 2077–2095 RSC.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef.
- M. D. Hughes, Y.-J. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS.
- C. Della Pina, E. Falletta and M. Rossi, Chem. Soc. Rev., 2012, 41, 350–369 RSC.
- L. X. Xu, C. H. He, M. Q. Zhu, K. J. Wu and Y. L. Lai, Catal. Lett., 2007, 118, 248–253 CrossRef CAS.
- L. X. Xu, C. H. He, M. Q. Zhu, K. J. Wu and Y. L. Lai, Catal. Commun., 2008, 9, 816–820 CrossRef CAS.
- P. C. Hereijgers and B. M. Weckhuysen, J. Catal., 2010, 270, 16–25 CrossRef.
- Y. Liu, H. Tsunoyama, T. Akita, S. Xie and T. Tsukuda, ACS Catal., 2011, 1, 2–6 CrossRef CAS.
- G. L. Brett, Q. He, C. Hammond, P. J. Miedziak, N. Dimitratos, M. Sankar, A. A. Herzing, M. Conte, J. A. Lopez-Sanchez, C. J. Kiely, D. W. Knight, S. H. Taylor and G. J. Hutchings, Angew. Chem., Int. Ed., 2011, 50, 10136–10139 CrossRef CAS.
- J. Guzman and B. C. Gates, J. Am. Chem. Soc., 2004, 126, 2672–2673 CrossRef CAS.
- M. S. Chen and D. W. Goodman, Catal. Today, 2006, 111, 22–33 CrossRef CAS.
- P. Ionita, B. C. Gilbert and V. Chechik, Angew. Chem., Int. Ed., 2005, 44, 3720–3722 CrossRef CAS.
- A. Burt, M. Emery, J. Maher and B. Mile, Magn. Reson. Chem., 2001, 39, 85–88 CrossRef CAS.
- M. Conte, K. Wilson and V. Chechik, Org. Biomol. Chem., 2009, 7, 1361–1367 CAS.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, J. Am. Chem. Soc., 2009, 131, 7189–7196 CrossRef CAS.
- P. Ionita, M. Conte, B. C. Gilbert and V. Chechik, Org. Biomol. Chem., 2007, 5, 3504–3509 CAS.
- Simulations were carried out using WinSim software: http://www.niehs.nih.gov/research/resources/software/tox-pharm/tools/index.cfm.
- B. D. Cullity and S. R. Stock, Elements of X-Ray Diffraction, Prentice-Hall Inc., Upper Saddle River, 3rd edn, 2001, p. 161 Search PubMed.
- J. I. Langford, J. Appl. Crystallogr., 1978, 11, 10–14 CrossRef CAS.
- J. R. Sanderson, M. A. Mueller and Y.-H. E. Sheu, US Patent, 5401889, 1995 Search PubMed.
- J. D. Druliner, K. Kourtakis and L. E. Manzer, International Patent, WO 98/34894, 1998 Search PubMed.
- N. Herron, S. Schwarz and J. D. Druliner, International Patent, WO 02/16296, 2002 Search PubMed.
- N. Turrà, A. B. Acuņa, B. Schimmöller, B. Mayr-Schmölzer, P. Mania and I. Hermans, Top. Catal., 2011, 54, 737–745 CrossRef.
- K. Sato, M. Aoki and R. Noyori, Science, 1998, 281, 1646–1647 CrossRef CAS.
- A. Kern, R. Doetzer and W. Eysel, ICDD Grant-in-Aid, 1993, in International Centre for Diffraction Data, Powder Diffraction File, Entry 45-946, 1996.
- US Natl. Bur. Stand., 1956, in International Centre for Diffraction Data, Powder Diffraction File, Entry 7-239, 1996.
- US Natl. Bur. Stand., 1957, in International Centre for Diffraction Data, Powder Diffraction File, Entry 8-479, 1996.
- US Natl. Bur. Stand., 1953, in International Centre for Diffraction Data, Powder Diffraction File, Entry 4-784, 1996.
- W. W. Weare, S. M. Reed, M. G. Warner and J. E. Hutchison, J. Am. Chem. Soc., 2000, 122, 12890–12891 CrossRef CAS.
- R. G. J. Strens and B. J. Wood, Mineral. Mag., 1979, 43, 347–354 CAS.
- F. Gu, C. Li, H. Cao, W. Shao, Y. Hu, J. Chen and A. Chen, J. Alloys. Compd., 2008, 453, 361–365 CrossRef CAS.
- S. Stankic, M. Müller, O. Diwald, M. Sterrer, E. Knözinger and J. Bernardi, Angew. Chem., Int. Ed., 2005, 44, 4917–4920 CrossRef CAS.
- C. A. Tolman, J. D. Druliner, M. J. Nappa and N. Herron, Activation and Functionalization of Alkanes, Wiley, New York, 1989, p. 303 Search PubMed.
- L. Vereecken, T. L. Nguyen, I. Hermans and J. Peeters, Chem. Phys. Lett., 2004, 393, 432–436 CrossRef CAS.
- S. Bhaduri and D. Mukesh, Homogeneous Catalysis: Mechanisms and Industrial Applications, John Wiley & Sons Inc., New York, 2000, p. 179 Search PubMed.
- D. Mandon, H. Jaafar and A. Thibon, New J. Chem., 2011, 35, 1986–2000 RSC.
- M. Conte, Y. Ma, C. Loyns, P. Price, D. Rippon and V. Chechik, Org. Biomol. Chem., 2009, 7, 2685–2687 CAS.
- R. Schlögl, A. Knop-Gericke, M. Hävecker, U. Wild, D. Frickel, T. Ressler, R. E. Jentoft, J. Wienold, G. Mestl, A. Blume, O. Timpe and Y. Uchida, Top. Catal., 2001, 15, 219–228 CrossRef.
- I. Hermans, P. Jacobs and J. Peeters, Chem.–Eur. J., 2006, 13, 754–761 CrossRef.
- B. Modén, B.-Z. Zhan, J. Dakka, J. G. Santiesteban and E. Iglesia, J. Catal., 2006, 239, 390–401 CrossRef.
- I. Hermans, T. L. Nguyen, P. A. Jacobs and J. Peeters, ChemPhysChem, 2005, 6, 637–645 CrossRef CAS.
- C. Walling and L. Heaton, J. Am. Chem. Soc., 1965, 87, 38–47 CrossRef CAS.
- M. Álvaro, C. Aprile, A. Corma, B. Ferrer and H. García, J. Catal., 2007, 245, 249–252 CrossRef.
- K. L. Mcgilvray, M. R. Decan, D. Wang and J. C. Scaiano, J. Am. Chem. Soc., 2006, 128, 15980–15981 CrossRef CAS.
- L. Dall’Acqua, I. Nova, L. Lietti, G. Ramis, G. Buscac and E. Giamello, Phys. Chem. Chem. Phys., 2000, 2, 4991–4998 RSC.
- G. Mestl, N. F. D. Verbruggen, E. Bosch and H. Knözinger, Langmuir, 1996, 12, 2961–2968 CrossRef CAS.
- M. Conte, H. Miyamura, S. Kobayashi and V. Chechik, Chem. Commun., 2010, 46, 145–147 RSC.
- D. Ricci, G. Pacchioni, P. V. Sushko and A. L. Shluger, J. Chem. Phys., 2002, 117, 2844–2851 CrossRef CAS.
- D. J. Driscoll, W. Martyr, J.-X. Wang and J. H. Lunsford, J. Am. Chem. Soc., 1985, 107, 58–63 CrossRef CAS.
- M. Novak and B. A. Brodeur, J. Org. Chem., 1984, 49, 1142–1144 CrossRef CAS.
- S. L. Baum, I. G. M. Anderson, R. R. Baker, D. M. Murphy and C. C. Rowlands, Anal. Chim. Acta, 2003, 481, 1–13 CrossRef CAS.
- M. J. Davies and T. F. Slater, Biochem. J., 1986, 240, 789–795 CAS.
- E. G. Janzen, C. A. Evans and J.-P. Liu, J. Magn. Reson., 1973, 9, 513–516 CAS.
- M. Conte, K. Wilson and V. Chechik, Rev. Sci. Instrum., 2010, 81, 104102 CrossRef.
- P. Ionita, B. C. Gilbert and A. C. Whitwood, J. Chem. Soc., Perkin Trans. 2, 2000, 2436–2440 RSC.
- P. Ionita, B. C. Gilbert and A. C. Whitwood, Lett. Org. Chem., 2004, 1, 70–74 CrossRef CAS.
- V. Chechik, M. Conte, T. Dransfield, M. North and M. Omedes-Pujol, Chem. Commun., 2010, 46, 3372–3374 RSC.
- W.-F. Wang, M. N. Schuchmann, H.-P. Schuchmann, W. Knolle, J. von Sonntag and C. von Sonntag, J. Am. Chem. Soc., 1999, 121, 238–245 CrossRef CAS.
- R. P. Houghton and C. R. Rice, Polyhedron, 1996, 15, 1893–1897 CrossRef CAS.
- S. V. Dvinskikh, A. V. Yurkovskaya and H.-M. Vieth, J. Phys. Chem., 1996, 100, 8125–8130 CrossRef CAS.
| This journal is © the Owner Societies 2012 |